

Abstract
The StARkin superfamily comprises proteins with steroidogenic acute regulatory protein–related lipid transfer (StART) domains that are implicated in intracellular, non-vesicular lipid transport. A new family of membrane-anchored StARkins was recently identified, including six members, Lam1–Lam6, in the yeast Saccharomyces cerevisiae. Lam1–Lam4 are anchored to the endoplasmic reticulum (ER) membrane at sites where the ER is tethered to the plasma membrane and proposed to be involved in sterol homeostasis in yeast. To better understand the biological roles of these proteins, we carried out a structure-function analysis of the second StARkin domain of Lam4, here termed Lam4S2. NMR experiments indicated that Lam4S2 undergoes specific conformational changes upon binding sterol, and fluorescence-based assays revealed that it catalyzes sterol transport between vesicle populations in vitro, exhibiting a preference for vesicles containing anionic lipids. Using such vesicles, we found that sterols are transported at a rate of ∼50 molecules per Lam4S2 per minute. Crystal structures of Lam4S2, with and without bound sterol, revealed a largely hydrophobic but surprisingly accessible sterol-binding pocket with the 3-OH group of the sterol oriented toward its base. Single or multiple alanine or aspartic acid replacements of conserved lysine residues in a basic patch on the surface of Lam4S2 near the likely sterol entry/egress site strongly attenuated sterol transport. Our results suggest that Lam4S2 engages anionic membranes via a basic surface patch, enabling “head-first” entry of sterol into the binding pocket followed by partial closure of the entryway. Reversal of these steps enables sterol egress.
Keywords: fluorescence resonance energy transfer (FRET), cholesterol-binding protein, cholesterol, endoplasmic reticulum (ER), lipid transport, dehydroergosterol, Lam4, membrane contact site, oxysterol, StART domain
Introduction
Sterols play an important role in eukaryotic cells because of their ability to regulate the fluidity and barrier function of the plasma membrane (PM)4 over a wide range of temperatures and lipid compositions (1, 2). Cells acquire sterols via import or biosynthesis in the endoplasmic reticulum (ER) (3) and tightly regulate their intracellular distribution. Sterol concentrations differ roughly 7–8-fold between the PM where they constitute ∼35–40% of all lipids and the ER where their levels are much lower, ∼5% of all lipids (2, 4, 5). Non-vesicular mechanisms play a key role in distributing sterols from their point of import or synthesis as well as in maintaining their precise subcellular distribution. As the spontaneous rate of sterol exchange between membranes is low in vitro and essentially undetectable in cells (6–9), sterol transport proteins are needed to catalyze rapid non-vesicular sterol movement (1, 9, 10). Oxysterol-binding protein and some of its homologs as well as members of the steroidogenic acute regulatory protein–related lipid transfer (StART) protein superfamily (termed StARkins) have been implicated as sterol transport proteins (11–14). The latter are characterized by an α/β helix-grip fold with a β sheet forming the wall of a binding pocket for sterols or other lipids (15).
The yeast Saccharomyces cerevisiae has six proteins with StARkin domains (16–18) of which four (Lam1–Lam4) are anchored to the ER membrane and located at contact sites where the ER is tethered to the plasma membrane (11, 17). Lam1 (also known as Ysp1) and Lam3 (also known as Sip3) have a single StARkin domain each, whereas Lam2 (also known as Ysp2) and Lam4 each have two StARkin domains (11, 17). The Lam1–Lam4 proteins also each have a pleckstrin homology (PH)-like GRAM domain. Lam4 (see Fig. 1A) was originally identified in a tritium suicide selection experiment designed to elucidate mechanisms of sterol transport in yeast (19), but its specific identification as a sterol-binding StARkin family member required the application of structural bioinformatics because of the lack of sequence homology between the Lam StARkin domains and known StART domains (11, 17). The localization of Lam1–Lam4 to contact sites between the organelle where sterols are synthesized (ER) and the membrane where the majority of cellular sterol is located (PM) suggested that these proteins play an important role in intracellular sterol homeostasis, potentially by transporting and/or sensing sterol. Indeed, yeast cells lacking one or more of these proteins exhibit phenotypes characteristic of cells with sterol-related lipid dysfunctions, for example hypersensitivity to amphotericin (17).
Figure 1.
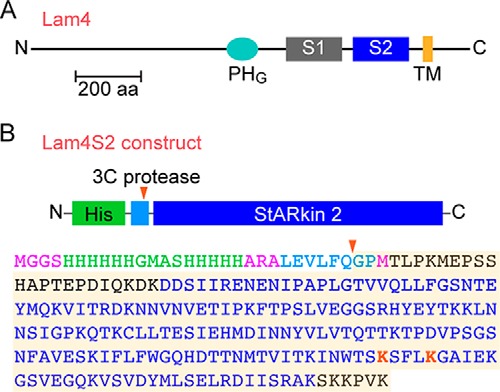
Lam4 and the Lam4S2 construct. A, schematic illustration of Lam4 (Yhr080c) indicating the PH-like GRAM domain (PHG), the two StARkin domains (S1 and S2), and transmembrane (TM) domain. The latter is strongly predicted by the OCTOPUS membrane protein topology prediction web server (http://octopus.cbr.su.se/ (Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party-hosted site.)) (48). The protein is 1345 amino acids long (∼150 kDa) and represented roughly to scale according to the indicated 200-amino acid (aa) scale bar. The transmembrane domain anchors Lam4 to the ER membrane such that its N terminus is in the cytoplasm. B, sequence of the Lam4S2 construct used in our studies. The second StARkin domain was expressed as a His-tagged protein in E. coli. The His tag (green) was attached via a linker sequence (light blue) containing a 3C protease cleavage site. The red arrowhead indicates the cleavage site. The amino acid sequence of the construct is color-coded to indicate the His tag (green), consensus sequence for 3C protease cleavage (light blue), the StARkin 2 domain (navy blue; as defined in Ref.17), and lysine residues (red) that were targeted by site-directed mutagenesis in our studies. Residues in black are part of the native Lam4 sequence, extending on either side of the StARkin 2 domain; residues in pink are part of the design of the construct. The shaded sequence corresponds to the Lam4S2 protein (predicted molecular mass, 22.7 kDa) used for crystallography with residue numbering starting with the first glycine. In this numbering scheme, the highlighted lysine residues are Lys-163 and Lys-167. The fourth residue in the shaded sequence, Thr-4, corresponds to Thr-946 in native Lam4.
To better understand the biological function of Lam proteins it is important to define structure-function characteristics of their novel StARkin domains. Here, we describe Lam4S2, a construct encompassing the second StARkin domain of Lam4. NMR experiments indicate that Lam4S2 binds sterol, undergoing specific conformational changes in the process, and fluorescence-based assays show that it catalyzes sterol transport between vesicle populations in vitro, exhibiting a preference for vesicles containing anionic lipids. Lam4S2 catalyzes sterol exchange ∼8-fold faster than the soluble mammalian sterol transporter StARD4 under similar assay conditions. We present two crystal structures of Lam4S2, with and without bound sterol. Sterol binding occurs head-down; i.e. the 3-OH of the sterol molecule is oriented to the bottom of the binding pocket. The pocket is deep and largely hydrophobic with a notable lateral opening. Entry to the pocket is posited to occur from a conserved region of the protein with a positively charged surface. Replacement of conserved lysine residues in this region severely attenuates the ability of the protein to transport sterols in vitro.
Results and discussion
Expression of Lam4S2, the StARkin 2 domain of Lam4
The Lam4S2 construct is described in Fig. 1B. Following expression in Escherichia coli, purification on Ni-NTA-agarose, and cleavage of the His tag, we obtained Lam4S2 as a single ∼25-kDa Coomassie-stained band by SDS-PAGE (WT; Fig. 2A) with a monodisperse profile on size-exclusion chromatography (not shown) and a circular dichroism (CD) spectrum indicative of a well folded structure (Fig. 2B). The protein was obtained in high yield (∼7.5 mg/liter of culture) and could be readily concentrated to ∼10 mg/ml. Similar results were obtained for a number of point mutants in which specific lysine resides in the StARkin domain were replaced with alanine or aspartic acid (Figs. 1B and 2) (see below).
Figure 2.
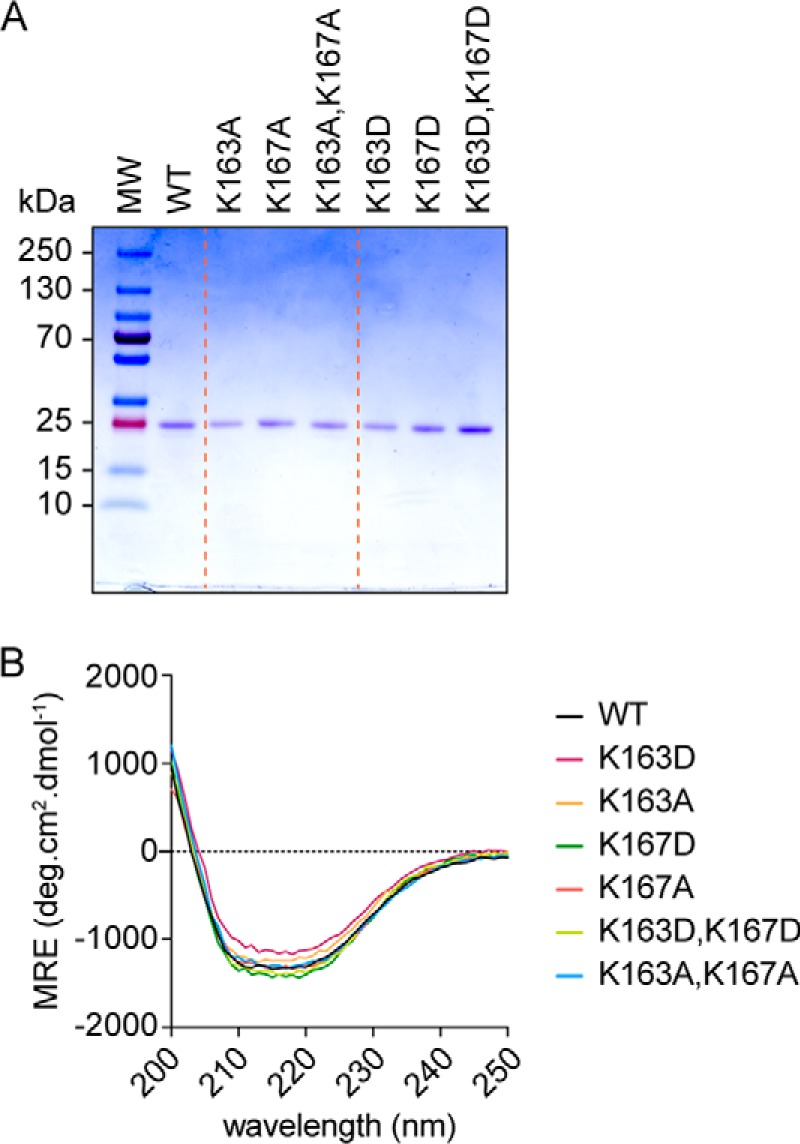
Purified Lam4S2 and point mutants. A, SDS-PAGE analysis of purified proteins. Samples (∼1 μg) were resolved on a 4–20% MiniProtean TGX gel (Bio-Rad) and visualized by Coomassie staining. Molecular mass (MW) markers (PageRulerTM Plus prestained protein ladder, 10–250 kDa) are indicated. B, circular dichroism spectra of purified proteins. Wavelength scans (200–300 nm) were performed in triplicate at ambient temperature and averaged; the value at 300 nm was subtracted before plotting. MRE, molar residue ellipticity. deg, degrees. WT corresponds to the wildtype sequence shown in Fig. 1B; the other samples correspond to single or double amino acid substitutions of lysine residues Lys-163 and Lys-167.
Conformational change in Lam4S2 upon sterol binding
The StARkin 2 domain of Lam4 was previously shown to bind sterol by two criteria (17). First, the purified domain specifically extracted cholesterol from permeabilized human leukemic HL-60 cells in which all lipids had been labeled to steady state with [14C]acetate. Second, Förster resonance energy transfer (FRET) experiments demonstrated proximity between tryptophan residue(s) and the naturally fluorescent sterol dehydroergosterol (DHE) in protein samples that had been incubated with DHE-containing liposomes. The FRET data were used to determine that the StARkin 2 domain had a submicromolar affinity for sterol, similar to other sterol-binding proteins such as Osh4/Kes1 (20). To explore sterol binding further, we carried out NMR experiments in the presence or absence of sterol. We obtained high quality 2D 1H-15N correlation spectra (heteronuclear single quantum coherence (HSQC)) of the native protein without sterol but could not obtain consistent results when we attempted to load Lam4S2 with various sterols (ergosterol, DHE, or cholesterol) that were either solubilized in ethanol or reconstituted into unilamellar phospholipid vesicles before incubation with the protein. Whereas DHE loading from vesicles revealed clear evidence of binding via the FRET readout described above, changes in the corresponding HSQC spectra were not reproducible (not shown). We therefore tested sterol loading with 25-hydroxycholesterol, an oxysterol that has a higher water solubility than the other sterols we tested, enabling convenient delivery in aqueous media. Fig. 3 shows HSQC spectra of Lam4S2 in the absence and presence of oxysterol. The two spectra have a similar overall appearance with well dispersed resonances consistent with a well folded structure. Many peaks are unchanged or move only slightly upon introduction of the oxysterol, suggesting that the overall fold and topology of the protein is not altered. Nevertheless, complete and substantial shifts in the positions of a large number of resonances indicate that the oxysterol is bound quantitatively and that binding is accompanied by a conformational rearrangement of the protein (see enlargement of a portion of the spectrum in Fig. 3). Similar changes have been observed in the 2D NMR spectra of other StART domain proteins upon ligand binding (21–23). Thus, the NMR results confirm that Lam4S2 is well folded and able to bind to 25-hydroxycholesterol and that it maintains its overall structure and topology upon ligand binding.
Figure 3.
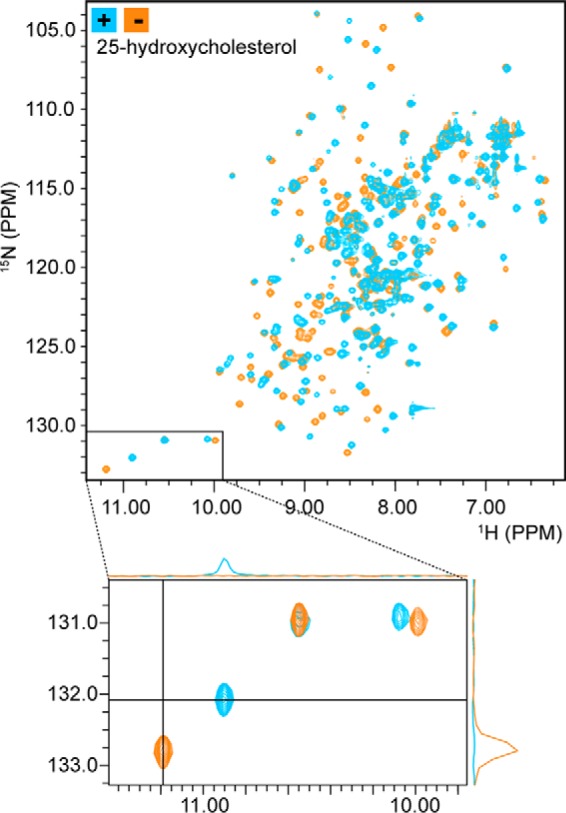
NMR spectroscopy reveals sterol binding by Lam4S2. 2D 1H-15N correlation spectra (HSQC) of Lam4S2 with and without bound sterol are shown. The spectra are well dispersed, consistent with an intact secondary and tertiary structure. Comparing spectra acquired in the absence (orange) and presence (blue) of 25-hydroxycholesterol reveals a large number of chemical shift changes, similar to those observed for other StART domains upon ligand binding and consistent with quantitative binding of 25-hydroxycholesterol. The bottom panel is an enlargement of the lower left region of the 2D HSQC spectrum, showing that resonances that shift upon sterol binding do so completely with no residual intensity at the location of the original signals. 1D projections further demonstrate the absence of free-state signals in the bound-state spectrum (upper projection) and the absence of bound-state signals in the free-state spectrum (right-hand projection).
Lam4S2 transfers DHE between vesicles
We tested the ability of Lam4S2 to transfer sterols between populations of vesicles in vitro. For this, we took advantage of a previously described assay (Fig. 4A) (10, 24) that uses FRET to measure the transfer of DHE from one population of vesicles (donor) to a second population (acceptor) that contains the fluorescent phospholipid dansyl-phosphatidylethanolamine (dansyl-PE). FRET is only observed when DHE and dansyl-PE are in the same vesicle. Both donor and acceptor vesicles were prepared from mixtures of zwitterionic (phosphatidylcholine (PC) and PE) and anionic (phosphatidylserine (PS) and phosphatidylinositol (PI)) phospholipids. Spontaneous transfer of DHE between these vesicle populations occurred slowly (Fig. 4B, trace a) but increased dramatically, ∼17-fold, upon adding Lam4S2 (Fig. 4B, trace e). Rate constants obtained by fitting the fluorescence traces are shown in Fig. 4C. Interestingly, when either the donor or acceptor vesicle population (or both) lacked anionic phospholipids, the rate of Lam4S2-mediated sterol transfer decreased ∼4–6-fold (Fig. 4, B and C). This result is reminiscent of the characteristics of sterol transfer by mammalian StARD4 protein in which a similar assay revealed that the greatest transfer rates were achieved only when both donor and acceptor vesicle populations contained anionic phospholipids (10).
Figure 4.
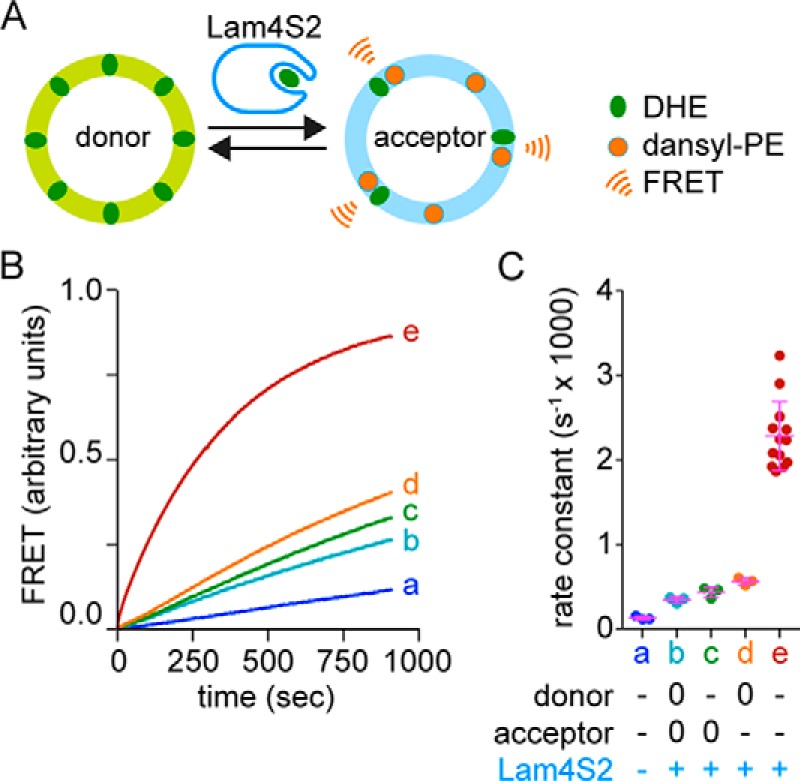
Lam4S2-mediated sterol transport: effect of anionic phospholipids. A, schematic illustration of the sterol transport assay. Donor vesicles containing DHE are mixed with acceptor vesicles containing dansyl-PE. Excitation of DHE results in FRET-mediated fluorescence emission from dansyl-PE when the two molecules are in the same vesicle. This occurs when DHE is transferred from donor to acceptor vesicles by Lam4S2 as shown. The time-dependent increase in sensitized emission from dansyl-PE is used to measure the rate at which DHE moves from donor to acceptor vesicles, either spontaneously or catalyzed by Lam4S2 or other sterol carriers such as StARD4 (see Fig. 5). B, large unilamellar donor and acceptor vesicles (100 μm phospholipid each) were combined in a stirred cuvette placed in a fluorescence spectrometer. Fluorescence was monitored using λex = 310 nm (DHE excitation) and λex = 525 nm (dansyl-PE emission) (traces were recorded at ambient temperature with a data acquisition frequency of 1 Hz) and normalized to the plateau value of FRET determined as described under “Experimental procedures”. The samples were incubated for 60 s before adding Lam4S2 (final concentration, 0.05 μm) (traces b–e) or buffer (trace a). Trace e corresponds to donor vesicles composed of DOPC:DOPE:DOPS:DHE (31:23:23:23 mol %) and acceptor vesicles composed of DOPC:DOPE:liver PI:DOPS:dansyl PE (70:7:15:5:3 mol %). Traces b–d correspond to assays in which anionic lipids (PS and PI) in the donor and/or acceptor vesicle population were replaced with PC. Thus, neutral donor vesicles were used for traces b and d, and neutral acceptor vesicles were used for traces b and c. C, rate constants deduced from monoexponential fits of traces, including those shown in B. The legend indicates whether donor or acceptor vesicles were prepared with anionic (−) or neutral (0) phospholipids. Individual measurements are shown (dots) along with the mean and S.D. (error bars).
To quantify the rate at which Lam4S2 transfers sterols, we first calibrated the FRET signal. To do this, we generated a series of acceptor vesicles that contained a fixed amount of dansyl-PE (3 mol % of total lipid) but were reconstituted with different amounts of DHE. In the range that we tested, FRET increased linearly as a function of DHE (Fig. 5A). We next carried out transport assays using different amounts of Lam4S2 (Fig. 5B) and measured the transport rate in terms of the change in FRET signal. By using the calibration plot (Fig. 5A), we could convert the FRET signal into molecules of DHE that had been transferred to acceptor vesicles. Fig. 5C shows a graph of the rate of DHE transfer (nmol·s−1) versus the amount of Lam4S2 in the assay (in nmol). The slope of this graph yields a transfer rate of ∼0.84 DHE molecules per Lam4S2 per second (Fig. 5D). For comparison, we used the same assay conditions to measure the rate of DHE transfer by mammalian StARD4 (Fig. 5, B and C). We obtained a rate of ∼0.1 DHE molecule transferred per StARD4 per second, identical to the value reported previously (25). Thus, on a per protein basis, Lam4S2 transfers DHE between anionic vesicles ∼8-fold faster than StARD4.
Figure 5.
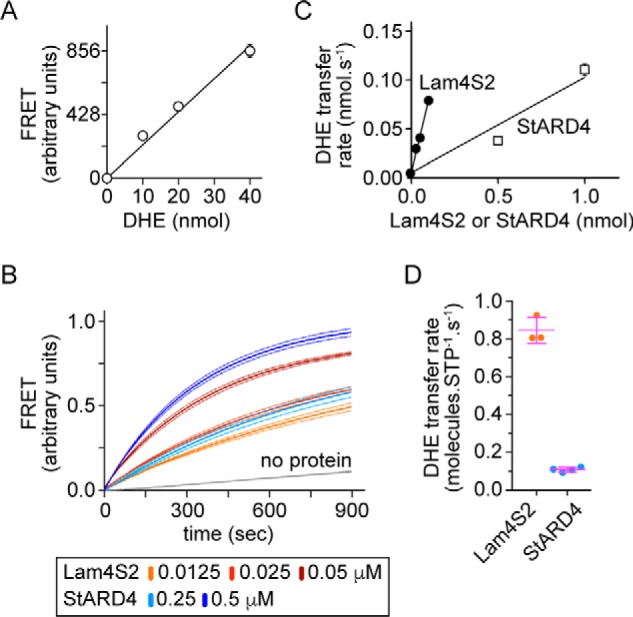
Quantification of Lam4S2-mediated DHE transport between anionic liposomes. A, calibration plot showing the extent of FRET as a function of the amount of DHE in acceptor liposomes. Acceptor liposomes containing dansyl-PE (3 mol % of total lipid) and different amounts of DHE (0–40 nmol) were placed in a cuvette (at a concentration of 100 μm phospholipid in a total volume of 2 ml), and sensitized emission of dansyl-PE was measured using λex = 310 nm and λex = 525 nm. The graph shows the recorded fluorescence emission (scaled by 1:1000) and a linear regression analysis. Data points represent the mean ± S.E. of three independent experiments; for most of the data points the error bars are smaller than the size of the symbol. B, FRET between DHE and dansyl-PE resulting from Lam4S2- or StARD4-dependent DHE transport from donor to acceptor liposomes. The traces are normalized to the plateau value of FRET determined as described under “Experimental procedures”. Dotted lines represent the mean ± S.E. of three independent experiments. C, rate of DHE transport as a function of the amount (in nmols) of Lam4s2 or StARD4. Data points represent the mean ± S.E. of three independent experiments; for most of the data points the error bars are smaller than the size of the symbol. D, comparison of transfer rate (nmol/s) per protein (nmol) between Lam4s2 and mStarD4. Individual measurements are shown (dots) along with the mean and S.D. (error bars). STP, sterol transport protein.
Crystal structures of Lam4S2 and sterol-loaded Lam4S2
We crystallized Lam4S2 in both apo and oxysterol-loaded states (Fig. 6, A and B). The structures (both at 2.2-Å resolution) display an overall fold similar to that reported for StART-domain proteins such as StARD4 and MLN64 (26, 27) except that here for the first time we could directly visualize sterol in the binding pocket of a member of this protein superfamily (only the apoprotein structures have been reported for StARD4 and MLN64 (26, 27)). The refined structure of apo-Lam4S2 (Protein Data Bank code 6BYD) has one molecule per asymmetric unit constituting residues 4–200 but lacks electron density for three residues at both the N and C termini. The overall structure is mainly composed of a β sheet with two short helices, α1 and α2, inserted between the β1 and β2 strands and a long helix, α3, at the C terminus (Fig. 6A). The deep pocket formed between the β sheet and the C-terminal helix is typical of classical StART domains (15). The loop between the β2 and β3 strands, termed the Ω1 loop, has been implicated in regulating sterol entry into the binding pocket (25, 28).
Figure 6.
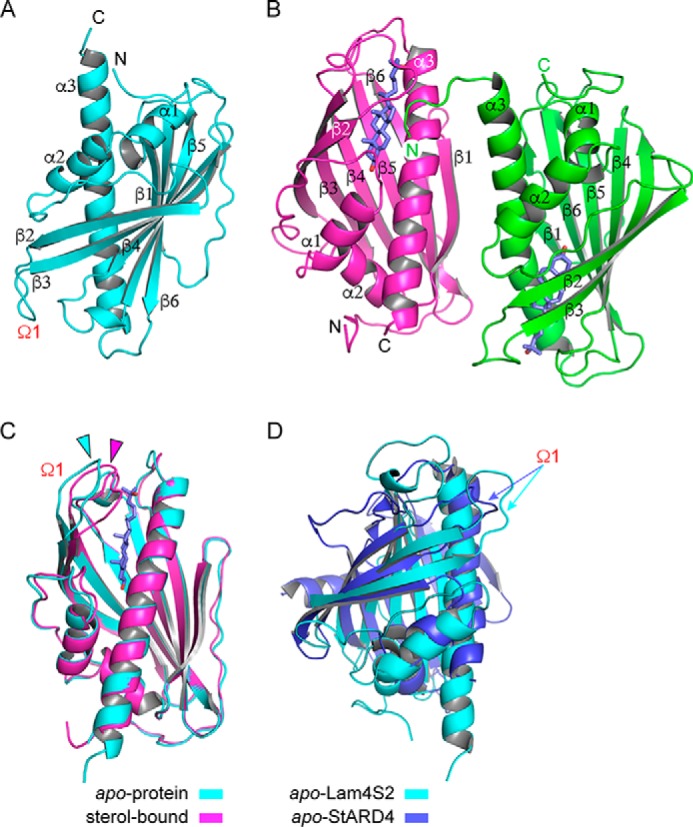
Crystal structures of Lam4S2 with and without bound sterol. A, overall structure of apo-Lam4S2 (Protein Data Bank code 6BYD), residues 4–200, shown in schematic representation. The Ω1 loop between strands β2 and β3 is indicated. B, overall structure of sterol-bound Lam4S2 (Protein Data Bank code 6BYM); monomer A (magenta) comprises residues 4–203, and monomer B (green) comprises residues 2–196. The bound 25-hydroxycholesterol molecule is shown in blue. C, overlay of apo- (cyan) and sterol-bound (magenta) Lam4S2, revealing a shift of the Ω1 loop (indicated by the correspondingly colored arrowheads). D, overlay of apo-Lam4S2 (cyan) and StARD4 (Protein Data Bank code 1JSS; blue). The Ω1 loop in each structure is indicated by a correspondingly colored arrow.
Quantitative comparison of the structures of apo-Lam4S2 and apo-StARD4 (Protein Data Bank code 1JSS) (Fig. 6D) yielded an overall root mean square deviation of 5.87 Å2 over 654 atoms, indicating a significant difference in the two structures despite a common fold. Perhaps the most striking difference between the structures is that the sterol-binding pocket in apo-StARD4 is completely occluded by the Ω1 loop and the loop between the β sheet and the C-terminal α helix. These loops are swung out of the way in apo-Lam4S2, leaving the pocket accessible. It appears likely that this opening provides a path for sterol binding and release as previously suggested based on simulations (28). Lam4S2 also lacks the long α helix and two β strands that characterize the N-terminal region of StARD4. The N-terminal helix in StARD4 runs across the external surface of the twisted β sheet that forms one wall of the binding pocket. In Lam4S2, it is replaced by an extended polypeptide stretch (residues 14–24) that runs roughly parallel to the β strands. Furthermore, Lam4S2 lacks the first β strand following the α2 helix, which is replaced with a polypeptide stretch lacking the characteristic hydrogen bonding pattern of a β sheet.
The refined oxysterol-bound Lam4S2 structure (Protein Data Bank code 6BYM) has two molecules per asymmetric unit. Monomers A and B show well resolved electron density for residues 4–203 and 2–196, respectively (Fig. 6B). The monomers are arranged as an antiparallel dimer with an interface between α3 of monomer A and β1 and α3 of monomer B. The interface is highly polar and not very well conserved, suggesting that it is unlikely to be physiologically relevant. An overlay of the apo- and sterol-bound Lam4S2 structures yields an overall root mean square deviation of 0.568 Å2 over 175 residues, showing that the structures are quite similar. Also, the sterol-binding pocket is largely preformed in the apoprotein and changes only subtly upon ligand binding. Significant differences between the structures are confined to the Ω1 loop at the entrance to the sterol-binding pocket, which shifts inward toward the pocket in the sterol-bound structure (Fig. 6C). This shift leads to the partial constriction of the entryway into the pocket but not a complete closure.
A difference electron density map (Fo − Fc) calculated for the sterol-bound structures with the ligand not included revealed a clear density for 25-hydroxycholesterol (Fig. 7A). The deep binding pocket at the core of the protein is mainly lined by hydrophobic residues (Fig. 7, B and C). The sterol fits snugly into the pocket with the 25-OH near the entryway and the intrinsic 3-OH buried at the bottom of the pocket. The latter forms a direct hydrogen bond with Gln-121 as well as water-mediated hydrogen bonds with Tyr-87 and Ser-181 (Fig. 7A). Interestingly, a number of water molecules are trapped between the sterol and the bottom of the binding pocket, suggesting that a larger polar modification at C3 could perhaps be accommodated.
Figure 7.
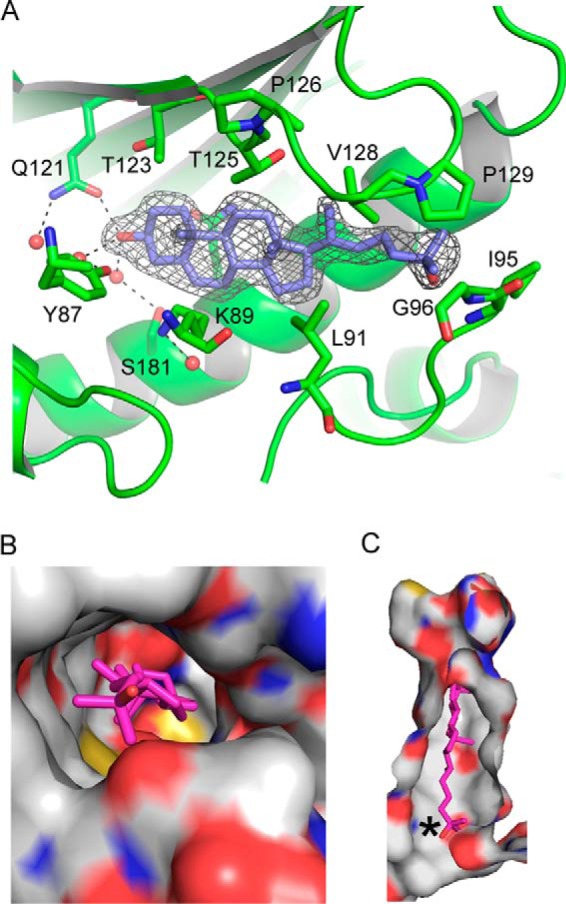
The sterol-binding cavity in Lam4S2. A, difference electron density map (Fo − Fc) calculated in the absence of the ligand for the sterol-bound structure and contoured at 3σ. Modeled 25-hydroxycholesterol is shown in stick representation with carbons colored blue. Dotted lines emphasize the hydrogen bond network surrounding the 3-OH moiety. B, top view of the sterol-binding site shown in surface representation, revealing the positioning of the 25-OH group at the entry site. The protein is colored by atom type with carbons white; bound sterol is shown in stick representation and colored magenta. C, a slice through the binding pocket. The color scheme is as in B; an asterisk marks the tip of the isooctyl tail of the sterol molecule.
Conserved lysine residues near the entry to the sterol-binding pocket are important for sterol transport
We used the ConSurf bioinformatics tool (29, 30) to estimate the evolutionary conservation of residues in the Lam4S2 domain. As expected, residues lining the sterol-binding pocket were well conserved. In addition, we noted that residues forming the protein surface near the posited entrance to the sterol-binding pocket, including the loops that occlude the pocket opening in StARD4, were highly conserved, whereas the remainder of the protein surface was much more variable (Fig. 8A). The conserved surface region contains several lysine residues, including Lys-163 and Lys-167 (Figs. 1B and 8A) that we focused on for functional studies. We replaced these lysines both individually and together with alanine or aspartic acid. The corresponding proteins were well expressed and well folded (Fig. 2). Sterol transport assays revealed that, with the exception of the K163A single mutation, which resulted in a protein with ∼2.4-fold higher transport activity than WT (Fig. 8, B and C), all other single and paired mutations resulted in attenuation of transport activity. Changes to Lys-167 resulted in the greatest effect (Fig. 8, B–E): thus, the K167D replacement, alone or in combination with K163D, resulted in proteins that were essentially inactive. Although the reason for the hyperactivity of the K163A mutant is not clear, the overall results of these mutagenesis studies are consistent with our observation that Lam4S2 transfers sterols most efficiently between anionic vesicles. The data suggest that the protein may interact with the membrane via the conserved basic residues near the sterol entry site, positioning the site optimally for sterol capture or deposition.
Figure 8.
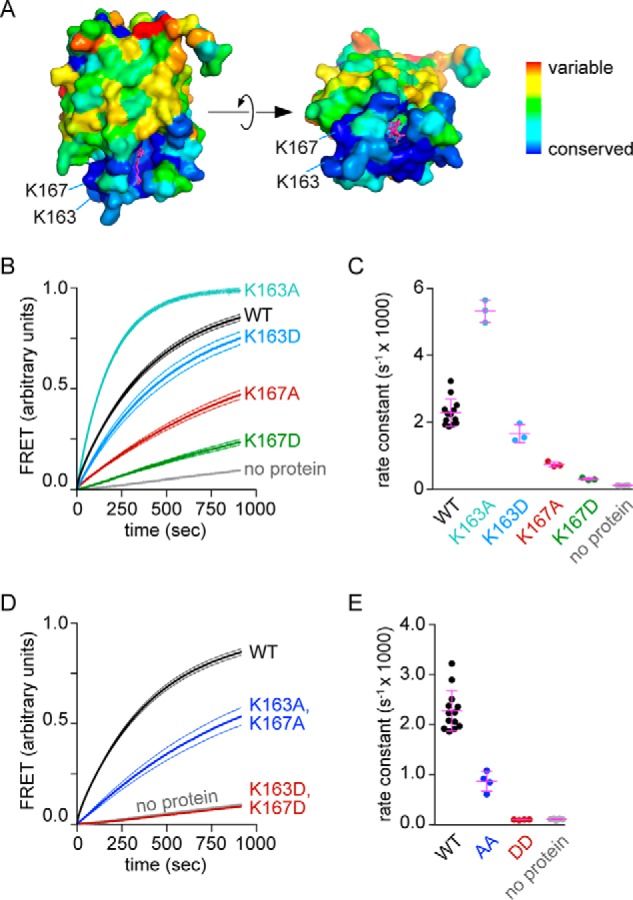
Conserved lysine residues near the entry to the sterol-binding pocket are important for Lam4S2-mediated sterol transport. A, side and top (90° rotated) views of sterol-bound Lam4S2 are shown, color-coded as indicated by the scale bar to reveal the extent of conservation determined by ConSurf analysis. The sterol ligand is shown in stick representation in magenta. The lysine residues under investigation are indicated. B, sterol transport assay with WT and single mutants of Lam4S2 using anionic donor and acceptor vesicles and 0.05 μm protein (final concentration). Assay conditions are as described in Fig. 4B, condition e. The traces are normalized to the plateau value of FRET determined as described under “Experimental procedures”. C, quantification of transport rates for single mutants. D, sterol transport assay with double mutants as for B. E, quantification of transport rates for double mutants. Dotted lines (B and D) represent S.E. of three independent experiments. Individual measurements are shown in the scatter dot plots (C and E) along with the mean and S.D. (error bars).
Conclusion
The crystal structures of apo- and sterol-bound Lam4S2 reveal a protein with the helix-grip fold characteristic of the StARkin protein family (15, 31). Similar crystal structures of the apo forms of the first and second StARkin domains of both Lam2 and Lam4 as well as the ergosterol-bound second StARkin domain of Lam2 were recently reported by Tong et al. (32). A preformed binding pocket created by strands β2–β4, loop Ω1, and helices α2 and α3 accommodates sterol in a head-down configuration, similar to predictions from in silico analyses of sterol binding by MLN64 (StARD3) and StARD4 (27, 28, 33). Sterol binding leads to a shift of the Ω1 loop in the crystal structure (Fig. 6C), resulting in partial closure of the apparent entryway to the binding pocket. Additionally, there are protein-wide subtle changes in conformation as evinced by our NMR HSQC spectra (Fig. 3) as well as NMR data reported in a recent publication (34). Notably, characteristic spectral changes observed here upon binding of 25-hydroxycholesterol are very similar to those observed upon cholesterol binding (34), confirming that both ligands induce similar structural changes. Furthermore, many of the changes were mapped to the sterol-binding pocket (34), suggesting that they reflect binding within the pocket as observed in the crystal structures. As suggested for other StARkin proteins (25), it seems likely that some of these conformational changes will play a role in regulating the exchange of sterol between the membrane and the binding pocket.
Unlike the case of StARD4, the sterol-binding pocket of Lam4S2 is accessible in both the apo and holo forms with the entryway adjacent to the Ω1 loop. This suggests that sterol uptake and release may be regulated differently in Lam StARkin domains than in the StARD4 subfamily of StART domains. This difference may reflect the fact that the Lam StARkins are localized to a specific site via their membrane anchors, constraining their target membranes, whereas the StARD4 family proteins are soluble and therefore may use additional mechanisms to regulate where and when to take up or deliver sterol. The fact that no structures of any sterol-bound StART domains have been reported to date supports the idea that sterol binding is more tightly regulated for canonical StART domains than for those of the Lam family.
Our structure-function studies of Lam4S2 demonstrate the ability of the isolated domain to bind and transfer sterols between membranes and reveal salient features of the protein that account for this activity. However, many questions remain. The S2 domain is positioned close to the membrane in native Lam4, ∼60 amino acids from the transmembrane domain (Fig. 1A). Whether the domain is oriented appropriately to exchange sterol with the ER and whether it can also function in trans to exchange sterol with the PM, which is 15–30 nm away from the ER, at contact sites requires further investigation. Also unclear are the role of the first StARkin domain and whether the two domains within a single Lam4 molecule or between individual Lam4 molecules function collaboratively. Notably, dimerization of tubular lipid-binding proteins (TULIPs), such as the SMP domain of extended synaptotagmins, has been implicated in their function (35, 36). However, the way in which these domains interact with lipids is quite different from that of the StARkins. Thus, SMP domains appear to function as grooves through which the hydrophobic portion of the lipid slides, whereas the polar headgroup remains in the aqueous space. To effect lipid transfer at a contact site, it seems likely that at least two SMP domains must cooperate (35). Although this mode of action seems unlikely for Lam4S2 given that its surface is largely unconserved and quite polar, features that argue against protein–protein interaction, it nevertheless remains a possibility that some form of StARkin domain collaboration is needed for Lam4's in vivo function.
Experimental procedures
Protein expression and purification
Lam4S2 (Fig. 1B) and corresponding point mutants (single mutants K163A, K163D, K167A, and K167D and double mutants K163A,K167A and K163D,K167D) were expressed in E. coli BL21 (DE3) cells using the isopropyl β-d-1-thiogalactopyranoside–inducible pTrcHis6A vector (Invitrogen). Briefly, cells were transformed with the vector containing the gene of interest, and a single colony was inoculated into an overnight primary culture. Cultures were grown in a shaker (220 rpm) at 37 °C. The primary culture was used to inoculate a secondary culture (typically 1 liter) that was grown to an A600 of ∼0.5–0.7 before adding 0.1 mm isopropyl β-d-1-thiogalactopyranoside (Millipore) to induce expression. After a further period of growth for 5 h, the cells were washed with phosphate-buffered saline (PBS), pelleted, and stored at −80 °C until further use.
For purification of Lam4S2, the cells generated from a 1-liter culture were thawed by resuspension in 10 ml of lysis buffer (50 mm sodium phosphate (pH 8.0), 300 mm NaCl, 10 mm imidazole, and protease inhibitor mixture (Roche Applied Science; one tablet/50 ml)). The cell suspension was placed in an ice-water bath and sonicated for 7 min in a cycle of 10 s-30 s (on-off) using a tip sonicator (Misonix 3000) set to deliver an output of 3.5. This step was followed by ultracentrifugation (Ti50.2 rotor; 33,200 rpm, 1 h, 4 °C) to pellet cell debris. The supernatant was incubated with Ni-NTA-agarose resin (Qiagen; the resin was previously equilibrated with lysis buffer) at 4 °C for 2 × 30 min with end-over-end rotation. The supernatant was removed, and the resin was washed with ∼50 ml of wash buffer (50 mm sodium phosphate (pH 8), 300 mm NaCl, 20 mm imidazole, and protease inhibitor mixture) using a disposable column and gravity-based flow. After the wash step, the resin was incubated with elution buffer (50 mm sodium phosphate (pH 8), 300 mm NaCl, and 250 mm imidazole) at 4 °C for 30 min with end-over-end rotation. The elution buffer containing Lam4S2 was recovered by gravity flow.
The purified protein was buffer-exchanged into 20 mm PIPES (pH 6.8), 3 mm KCl, and 137 mm NaCl using a prepacked PD-10 column (GE Healthcare) followed by cleavage of the affinity tag by treatment with PreScission protease (GE Healthcare) at 4 °C for 2 h (a 2:1 ratio of protein to protease was used to shorten the reaction time). The sample was then incubated with Ni-NTA-agarose to remove the tag, any uncleaved protein, and the His-tagged protease, and the resulting supernatant was subjected to gel filtration chromatography using a Superose 6 column (GE Healthcare). Gel filtration was carried out in low-salt buffer (20 mm PIPES (pH 6.8), 3 mm KCl, and 10 mm NaCl), and fractions corresponding to the protein peak were pooled, concentrated using an Amicon Ultra-4 centrifugal filter (10-kDa cutoff), and used immediately for crystallization or snap frozen and stored at −80 °C for other purposes.
Selenomethionine (SeMet)-labeled Lam4S2
Lam4S2 has six methionine residues. To generate Lam4S2 containing SeMet in place of methionine, T7 Express Crystal Competent E. coli cells (New England Biolabs) were transformed with the Lam4S2 plasmid and grown overnight in 100 ml of base medium containing l-methionine. The next day the cells were washed three times with sterile water and inoculated into 1 liter of l-SeMet medium (Molecular Dimensions) before inducing and purifying the protein as described above.
Circular dichroism
Wavelength scans (200–300 nm; bandwidth, 1 nm; time constant, 100 ms) were performed at ambient temperature on purified protein samples (20 μm) using a high-precision quartz cell (Hellma Analytics, 106-0.20-40; path length, 0.2 mm) and an AVIV 410 CD instrument. Three scans were collected for each sample, sampling per nm with an averaging time of 1 s and with no wait time between scans. Scans were averaged for each sample, and the value at 300 nm was subtracted before plotting for comparison.
NMR spectroscopy
Isotopically labeled protein was produced in E. coli grown on M9 minimal medium made using [15N]ammonium chloride and purified as described above. Purified protein was exchanged into 20 mm PIPES (pH 6.8), 3 mm KCl, and 10 mm NaCl buffer; concentrated to ∼100 μm; and supplemented with 10% D2O for NMR experiments. 1H-15N HSQC spectra were collected on a Bruker Avance 600-MHz spectrometer equipped with a cryoprobe using standard acquisition parameters. NMR data were processed with NMRPipe (37) and analyzed with NMRViewJ (38).
Crystallization of Lam4S2 with and without sterol
Initial crystal screening was set up by hanging drop using an NT8 liquid handling robot (Formulatrix) in a 1:1 ratio of 10 mg/ml SeMet apo-Lam4S2 to well solution. For the holo form, purified Lam4S2 was incubated at 4 °C for 2 h with 1 mm 25-hydroxycholesterol (Sigma-Aldrich) and centrifuged at 20,000 × g for 30 min before being taken for crystallization. The crystal screen for the native holo form was set up as for the SeMet apoprotein using 10 mg/ml protein concentration. After optimization, the best diffracting crystals of the SeMet apo-Lam4S2 grew in 0.5 m lithium chloride, 30% PEG 6000, and 0.1 m HEPES (pH 7.5). Sterol-bound Lam4S2 crystallized in 0.7 m lithium chloride, 24% PEG 6000, and 0.1 m HEPES (pH 7.5). Full-sized crystals were observed after 1 day for the apoprotein and 2 days for the holo form. These crystals were immediately flash frozen in well solution supplemented with 25% PEG 400 as a cryoprotectant and shipped to Advance Light Source at Berkeley, beamline 8.2.2, for data collection.
Data collection, structure determination, and refinement
The diffraction data for SeMet apo-Lam4S2 crystals were collected at 0.975-Å wavelength, whereas the diffraction data for the sterol-bound native Lam4S2 crystals were collected at 1.00 Å. All data were indexed, integrated, and scaled using the HKL2000 program (39). The SeMet apo-Lam4S2 structure was solved by single-wavelength anomalous dispersion using HKL2map (40) and Autobuild in PHENIX (41, 42). The structure was subjected to several rounds of manual model building using Coot (43, 44) and refinement using phenix.refine (42, 45). The native holo form structure was solved by molecular replacement with Phaser in PHENIX (42, 46) using the SeMet apo-Lam4S2 structure as the initial model. The holo form was rebuilt using Coot and refined using phenix.refine (42, 45). Data collection and refinement statistics are provided in Table 1. The coordinates of the apo- and sterol-bound proteins are available at the Protein Data Bank under codes 6BYD and 6BYM, respectively.
Table 1.
Data collection and refinement statistics for crystals of apo- and sterol-bound Lam4S2
r.m.s., root mean square; n/a, not applicable.
| Apo-Lam4a | Lam4 (sterol-bound)a | |
|---|---|---|
| Data collection | ||
| Space group | I212121 | P1 |
| Cell dimensions | ||
| a, b, c (Å) | 45.6, 67.9, 132.0 | 38.2, 47.3, 56.0 |
| α, β, γ (°) | 90, 90, 90 | 111.2, 101.6, 100.3 |
| Wavelength (Å) | 0.97494 | 1.00001 |
| Resolution (Å) | 50–2.19 (2.28–2.19) | 50.0–2.20 (2.27–2.20) |
| Rsym | 0.132 (0.664) | 0.09 (0.259) |
| I/σI | 23.6 (4.8) | 17.8 (4.8) |
| Completeness (%) | 100 (100) | 98.3 (97.1) |
| Redundancy | 14.0 (12.8) | 3.9 (3.8) |
| Refinement | ||
| Resolution (Å) | 43.1–2.19 (2.27–2.19) | 42.0–2.20 (2.28–2.20) |
| No. reflections | 10,837 | 17,131 |
| Rwork/Rfree (%) | 0.186/0.248 | 0.166/0.230 |
| No. of atoms | ||
| Protein | 1601 | 3308 |
| Ligand | n/a | 58 |
| Water | 48 | 143 |
| B-factors (Å2) | 45.2 | 30.5 |
| Protein | 45.3 | 30.6 |
| Ligands | n/a | 26.0 |
| Water | 42.0 | 30.6 |
| r.m.s. deviations | ||
| Bond lengths (Å) | 0.008 | 0.008 |
| Bond angles (°) | 0.901 | 0.999 |
| Ramachandran plot | ||
| Favored (%) | 96.9 | 98 |
| Allowed (%) | 3.1 | 2 |
| Disallowed (%) | 0.0 | 0 |
a Values in parentheses correspond to the highest resolution shell.
Liposome preparation
The composition of donor and acceptor liposomes was based on a previous study (10). Donor liposomes were composed of three dioleoyl phospholipids (DOPC, DOPE, and DOPS) and DHE (31, 23, 23, and 23 mol %), whereas acceptor liposomes were composed of DOPC, DOPE, liver PI, DOPS, and dansyl-PE (70, 7, 15, 5, and 3 mol %). For experiments where the function of anionic lipids was tested, these lipids were replaced by DOPC.
Lipids were mixed to achieve a final concentration of 2 mm in the indicated ratios and dried under a nitrogen steam at room temperature. The resulting lipid film was hydrated by resuspension in buffer (20 mm PIPES (pH 6.8), 3 mm KCl, and 10 mm NaCl) followed by agitation using a VXR basic Vibrax (IKA® Works, Inc.) for 30 min. The lipid suspension was then subjected to five freeze-thaw cycles, alternating between liquid N2 and room temperature water. This step was followed by extrusion using a hand extruder (Avanti Polar Lipids). The liposomes were passed 13 times through 200-nm-pore-size membranes, and then 13 times through 100-nm-pore-size membranes. The concentration of the resulting liposomes was measured by determining the amount of lipid phosphorus using a colorimetric assay (47). The liposomes were used immediately or after storage at 4 °C for no more than 24 h.
Sterol transport assay
The sterol transport assay was performed and analyzed as described previously (10). The assay is illustrated in Fig. 4A. All assays were carried out in 20 mm PIPES (pH 6.8), 3 mm KCl, and 10 mm NaCl. Briefly, donor and acceptor liposomes were placed in a 2-ml quartz cuvette with constant stirring. After 60 s, 200 μl of Lam4S2, StARD4 (various concentrations; StARD4 was prepared as described (10)), or buffer was added. The final concentration of each of the vesicle populations was 0.1 mm. Fluorescence was monitored using λex = 310 nm (DHE excitation) and λem = 525 nm (dansyl-PE emission).
A number of long time courses were taken (>900 s) to allow the fluorescence change to reach close to a plateau value. These data were then fit to a monoexponential function, and the mean plateau value (close to the experimentally measured amplitude) taken from a number of such traces was used to provide an amplitude constraint for the monoexponential fits of all other data as well as to normalize the FRET readout for the graphs shown in Figs. 4B, 5B, and 8, B and D.
The FRET signal was calibrated as described previously (10) to determine the number of DHE molecules that had been transferred from donor to acceptor vesicles. For calibration, a series of acceptor vesicles was generated containing a fixed amount of dansyl-PE (3 mol % of total lipid) but different amounts of DHE. The FRET signal from these vesicles (0.1 mm final concentration) was compared with their DHE content to obtain the necessary calibration plot (Fig. 5A).
Author contributions
J.-A. J., M. T., B. L., D. E., O. B., and A. K.M. conceptualization; J.-A. J., I. K., K. P., M. T., T. R., J. W., D. E., O. B., and A. K. M. data curation; J.-A. J., I. K., K. P., M. T., T. R., J. W., D. E., O. B., and A. K.M. formal analysis; J.-A. J. validation; J.-A. J., I. K., M. T., T. R., and A. K. M. investigation; J.-A. J., K. P., O. B., and A. K. M. methodology; J.-A. J., I. K., O. B., and A. K. M. writing-original draft; J.-A. J., I. K., K. P., M. T., B. L., J. W., D. E., O. B., and A. K. M. writing-review and editing; I. K., D. E., O. B., and A. K. M. visualization; B. L., D. E., O. B., and A. K. M. supervision; B. L., D. E., O. B., and A. K. M. project administration; D. E., O. B., and A. K. M. funding acquisition.
Acknowledgments
A. K. M. thanks Sam Canis and Sidse Babett Knudsen for stimulation. We thank Tim Levine, Alberto Gatta, and Louise Wong (Institute of Ophthalmology, University College London) for the parent StARkin2 domain construct on which our construct is based and for many helpful discussions. We also thank Dr. Guoan Zhang of the Weill Cornell Proteomics and Metabolomics Core Facility for analyses that enabled the design of our construct and Kiran Andra and Neha Chauhan for comments on the manuscript.
This work was supported by Graduiertenkolleg 2098, Project 11, from the Deutsche Forschungsgemeinschaft (to J.-A. J., B. L., and A. K. M.); Qatar National Research Fund Grant NPRP 7-082-1-014 (to A. K. M.); and National Institutes of Health Grants R01 GM117518 (to D. E.), R37 AG019391 (to D. E.), and R37 NS085318 (to O. B.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (codes 6BYD and 6BYM) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- PM
- plasma membrane
- ER
- endoplasmic reticulum
- dansyl-PE
- 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(5-dimethylamino-1-naphthalenesulfonyl)
- DHE
- dehydroergosterol
- DOPC
- dioleoyl phosphatidylcholine
- DOPE
- dioleoyl phosphatidylethanolamine
- DOPS
- dioleoyl phosphatidylserine
- PC
- phosphatidylcholine
- PE
- phosphatidylethanolamine
- PS
- phosphatidylserine
- PI
- phosphatidylinositol
- StART
- steroidogenic acute regulatory protein–related lipid transfer
- PH
- pleckstrin homology
- Ni-NTA
- nickel-nitrilotriacetic acid
- HSQC
- heteronuclear single quantum coherence
- SMP
- synaptotagmin-like mitochondrial lipid–binding protein.
References
- 1. Holthuis J. C., and Menon A. K. (2014) Lipid landscapes and pipelines in membrane homeostasis. Nature 510, 48–57 10.1038/nature13474 [DOI] [PubMed] [Google Scholar]
- 2. Maxfield F. R., and van Meer G. (2010) Cholesterol, the central lipid of mammalian cells. Curr. Opin. Cell Biol. 22, 422–429 10.1016/j.ceb.2010.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Goldstein J. L., and Brown M. S. (2015) A century of cholesterol and coronaries: from plaques to genes to statins. Cell 161, 161–172 10.1016/j.cell.2015.01.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Iaea D. B., and Maxfield F. R. (2015) Cholesterol trafficking and distribution. Essays Biochem. 57, 43–55 10.1042/bse0570043 [DOI] [PubMed] [Google Scholar]
- 5. Maxfield F. R., and Menon A. K. (2006) Intracellular sterol transport and distribution. Curr. Opin. Cell Biol. 18, 379–385 10.1016/j.ceb.2006.06.012 [DOI] [PubMed] [Google Scholar]
- 6. McLean L. R., and Phillips M. C. (1981) Mechanism of cholesterol and phosphatidylcholine exchange or transfer between unilamellar vesicles. Biochemistry 20, 2893–2900 10.1021/bi00513a028 [DOI] [PubMed] [Google Scholar]
- 7. Kaplan M. R., and Simoni R. D. (1985) Transport of cholesterol from the endoplasmic reticulum to the plasma membrane. J. Cell Biol. 101, 446–453 10.1083/jcb.101.2.446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Baumann N. A., Sullivan D. P., Ohvo-Rekilä H., Simonot C., Pottekat A., Klaassen Z., Beh C. T., and Menon A. K. (2005) Transport of newly synthesized sterol to the sterol-enriched plasma membrane occurs via nonvesicular equilibration. Biochemistry 44, 5816–5826 10.1021/bi048296z [DOI] [PubMed] [Google Scholar]
- 9. Dittman J. S., and Menon A. K. (2017) Speed limits for nonvesicular intracellular sterol transport. Trends Biochem. Sci. 42, 90–97 10.1016/j.tibs.2016.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mesmin B., Pipalia N. H., Lund F. W., Ramlall T. F., Sokolov A., Eliezer D., and Maxfield F. R. (2011) STARD4 abundance regulates sterol transport and sensing. Mol. Biol. Cell 22, 4004–4015 10.1091/mbc.E11-04-0372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wong L. H., and Levine T. P. (2016) Lipid transfer proteins do their thing anchored at membrane contact sites. But what is their thing? Biochem. Soc. Trans. 44, 517–527 10.1042/BST20150275 [DOI] [PubMed] [Google Scholar]
- 12. Alpy F., and Tomasetto C. (2005) Give lipids a START: the StAR-related lipid transfer (START) domain in mammals. J. Cell Sci. 118, 2791–2801 10.1242/jcs.02485 [DOI] [PubMed] [Google Scholar]
- 13. Beh C. T., McMaster C. R., Kozminski K. G., and Menon A. K. (2012) A detour for yeast oxysterol binding proteins. J. Biol. Chem. 287, 11481–11488 10.1074/jbc.R111.338400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Raychaudhuri S., and Prinz W. A. (2010) The diverse functions of oxysterol-binding proteins. Annu. Rev. Cell Dev. Biol. 26, 157–177 10.1146/annurev.cellbio.042308.113334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Iyer L. M., Koonin E. V., and Aravind L. (2001) Adaptations of the helix-grip fold for ligand binding and catalysis in the START domain superfamily. Proteins 43, 134–144 [DOI] [PubMed] [Google Scholar]
- 16. Elbaz-Alon Y., Eisenberg-Bord M., Shinder V., Stiller S. B., Shimoni E., Wiedemann N., Geiger T., and Schuldiner M. (2015) Lam6 regulates the extent of contacts between organelles. Cell Rep. 12, 7–14 10.1016/j.celrep.2015.06.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gatta A. T., Wong L. H., Sere Y. Y., Calderon-Norena D. M., Cockcroft S., Menon A. K., and Levine T. P. (2015) A new family of StART domain proteins at membrane contact sites has a role in ER-PM sterol transport. Elife 4, e07253 10.7554/eLife.07253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Murley A., Sarsam R. D., Toulmay A., Yamada J., Prinz W. A., and Nunnari J. (2015) Ltc1 is an ER-localized sterol transporter and a component of ER-mitochondria and ER-vacuole contacts. J. Cell Biol. 209, 539–548 10.1083/jcb.201502033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sullivan D. P., Georgiev A., and Menon A. K. (2009) Tritium suicide selection identifies proteins involved in the uptake and intracellular transport of sterols in Saccharomyces cerevisiae. Eukaryot. Cell 8, 161–169 10.1128/EC.00135-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Im Y. J., Raychaudhuri S., Prinz W. A., and Hurley J. H. (2005) Structural mechanism for sterol sensing and transport by OSBP-related proteins. Nature 437, 154–158 10.1038/nature03923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Barbar E., Lehoux J. G., and Lavigne P. (2009) Toward the NMR structure of StAR. Mol. Cell. Endocrinol. 300, 89–93 10.1016/j.mce.2008.12.007 [DOI] [PubMed] [Google Scholar]
- 22. Létourneau D., Bédard M., Cabana J., Lefebvre A., LeHoux J. G., and Lavigne P. (2016) STARD6 on steroids: solution structure, multiple timescale backbone dynamics and ligand binding mechanism. Sci. Rep. 6, 28486 10.1038/srep28486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Létourneau D., Lorin A., Lefebvre A., Cabana J., Lavigne P., and LeHoux J. G. (2013) Thermodynamic and solution state NMR characterization of the binding of secondary and conjugated bile acids to STARD5. Biochim. Biophys. Acta 1831, 1589–1599 10.1016/j.bbalip.2013.07.005 [DOI] [PubMed] [Google Scholar]
- 24. John K., Kubelt J., Müller P., Wüstner D., and Herrmann A. (2002) Rapid transbilayer movement of the fluorescent sterol dehydroergosterol in lipid membranes. Biophys. J. 83, 1525–1534 10.1016/S0006-3495(02)73922-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Iaea D. B., Dikiy I., Kiburu I., Eliezer D., and Maxfield F. R. (2015) STARD4 membrane interactions and sterol binding. Biochemistry 54, 4623–4636 10.1021/acs.biochem.5b00618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Romanowski M. J., Soccio R. E., Breslow J. L., and Burley S. K. (2002) Crystal structure of the Mus musculus cholesterol-regulated START protein 4 (StarD4) containing a StAR-related lipid transfer domain. Proc. Natl. Acad. Sci. U.S.A. 99, 6949–6954 10.1073/pnas.052140699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tsujishita Y., and Hurley J. H. (2000) Structure and lipid transport mechanism of a StAR-related domain. Nat. Struct. Biol. 7, 408–414 10.1038/75192 [DOI] [PubMed] [Google Scholar]
- 28. Murcia M., Faráldo-Gómez J. D., Maxfield F. R., and Roux B. (2006) Modeling the structure of the StART domains of MLN64 and StAR proteins in complex with cholesterol. J. Lipid Res. 47, 2614–2630 10.1194/jlr.M600232-JLR200 [DOI] [PubMed] [Google Scholar]
- 29. Ashkenazy H., Abadi S., Martz E., Chay O., Mayrose I., Pupko T., and Ben-Tal N. (2016) ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 44, W344–W350 10.1093/nar/gkw408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Landau M., Mayrose I., Rosenberg Y., Glaser F., Martz E., Pupko T., and Ben-Tal N. (2005) ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 33, W299–W302 10.1093/nar/gki370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Khafif M., Cottret L., Balagué C., and Raffaele S. (2014) Identification and phylogenetic analyses of VASt, an uncharacterized protein domain associated with lipid-binding domains in eukaryotes. BMC Bioinformatics 15, 222 10.1186/1471-2105-15-222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tong J., Manik M. K., and Im Y. J. (2018) Structural basis of sterol recognition and nonvesicular transport by lipid transfer proteins anchored at membrane contact sites. Proc. Natl. Acad. Sci. U.S.A. 115, E856–E865 10.1073/pnas.1719709115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Thorsell A. G., Lee W. H., Persson C., Siponen M. I., Nilsson M., Busam R. D., Kotenyova T., Schüler H., and Lehtiö L. (2011) Comparative structural analysis of lipid binding START domains. PLoS One 6, e19521 10.1371/journal.pone.0019521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gatta A. T., Sauerwein A. C., Zhuravleva A., Levine T. P., and Matthews S. (2018) Structural insights into a StART-like domain in Lam4 and its interaction with sterol ligands. Biochem. Biophys. Res. Commun. 495, 2270–2274 10.1016/j.bbrc.2017.12.109 [DOI] [PubMed] [Google Scholar]
- 35. Reinisch K. M., and De Camilli P. (2016) SMP-domain proteins at membrane contact sites: structure and function. Biochim. Biophys. Acta 1861, 924–927 10.1016/j.bbalip.2015.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Saheki Y., Bian X., Schauder C. M., Sawaki Y., Surma M. A., Klose C., Pincet F., Reinisch K. M., and De Camilli P. (2016) Control of plasma membrane lipid homeostasis by the extended synaptotagmins. Nat. Cell Biol. 18, 504–515 10.1038/ncb3339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 38. Johnson B. A., and Blevins R. A. (1994) NMR View: a computer program for the visualization and analysis of NMR data. J. Biomol. NMR 4, 603–614 10.1007/BF00404272 [DOI] [PubMed] [Google Scholar]
- 39. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 40. Pape T., and Schneider T. R. (2004) HKL2MAP: a graphical user interface for macromolecular phasing with SHELX programs. J. Appl. Crystallogr. 37, 843–844 10.1107/S0021889804018047 [DOI] [Google Scholar]
- 41. Terwilliger T. C., Grosse-Kunstleve R. W., Afonine P. V., Moriarty N. W., Zwart P. H., Hung L. W., Read R. J., and Adams P. D. (2008) Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. D Biol. Crystallogr. 64, 61–69 10.1107/S090744490705024X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Adams P. D., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., and Terwilliger T. C. (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 10.1107/S0907444902016657 [DOI] [PubMed] [Google Scholar]
- 43. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 44. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 10.1107/S0907444912001308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rouser G., Fleischer S., and Yamamoto A. (1970) Two dimensional then layer chromatographic separation of polar lipids and determination of phospholipids by phosphorus analysis of spots. Lipids 5, 494–496 10.1007/BF02531316 [DOI] [PubMed] [Google Scholar]
- 48. Viklund H., and Elofsson A. (2008) OCTOPUS: improving topology prediction by two-track ANN-based preference scores and an extended topological grammar. Bioinformatics 24, 1662–1668 10.1093/bioinformatics/btn221 [DOI] [PubMed] [Google Scholar]