

Abstract
G protein–coupled receptor (GPCR) signaling and trafficking are essential for cellular function and regulated by phosphorylation, β-arrestin, and ubiquitination. The GPCR parathyroid hormone receptor (PTHR) exhibits time-dependent reversible ubiquitination. The exact ubiquitination sites in PTHR are unknown, but they extend upstream of its intracellular tail. Here, using tandem MS, we identified Lys388 in the third loop and Lys484 in the C-terminal tail as primary ubiquitination sites in PTHR. We found that PTHR ubiquitination requires β-arrestin and does not display a preference for β-arrestin1 or -2. PTH stimulated PTHR phosphorylation at Thr387/Thr392 and within the Ser489–Ser493 region. Such phosphorylation events may recruit β-arrestin, and we observed that chemically or genetically blocking PTHR phosphorylation inhibits its ubiquitination. Specifically, Ala replacement at Thr387/Thr392 suppressed β-arrestin binding and inhibited PTHR ubiquitination, suggesting that PTHR phosphorylation and ubiquitination are interdependent. Of note, Lys-deficient PTHR mutants promoted normal cAMP formation, but exhibited differential mitogen-activated protein kinase (MAPK) signaling. Lys-deficient PTHR triggered early onset and delayed ERK1/2 signaling compared with wildtype PTHR. Moreover, ubiquitination of Lys388 and Lys484 in wildtype PTHR strongly decreased p38 signaling, whereas Lys-deficient PTHR retained signaling comparable to unstimulated wildtype PTHR. Lys-deficient, ubiquitination-refractory PTHR reduced cell proliferation and increased apoptosis. However, elimination of all 11 Lys residues in PTHR did not affect its internalization and recycling. These results pinpoint the ubiquitinated Lys residues in PTHR controlling MAPK signaling and cell proliferation and survival. Our findings suggest new opportunities for targeting PTHR ubiquitination to regulate MAPK signaling or manage PTHR-related disorders.
Keywords: G protein-coupled receptor (GPCR), receptor endocytosis, ubiquitylation (ubiquitination), mitogen-activated protein kinase (MAPK), mass spectrometry (MS), confocal microscopy, arrestin, mutagenesis in vitro, membrane trafficking, cell proliferation, parathyroid hormone receptor
Introduction
The type 1 parathyroid hormone receptor (PTHR)3 is a family B G protein–coupled receptor (GPCR). PTHR participates in calcium and phosphate homeostasis and skeletal growth and repair through actions on kidneys and bone (1). Disorders of PTHR signaling are associated with diseases of mineral-ion homeostasis and osteoporosis. Based on the cellular functions of downstream PTH signaling, modified PTH peptides or analogs are being developed and have been tested and used in treating diseases associated with abnormal PTHR signaling (2–4). Due to the variability of therapeutic effects of these hormone analogs and to the multiple signaling pathways regulated by PTHR, other modulatory events may be involved in their actions and could advance pharmaceutical development by identifying more precise or additional targets of receptor signaling.
Upon activation, PTHR signals through a combination of G protein-dependent and G protein–independent (i.e. β-arrestin–dependent) pathways (3). In the G protein-dependent pathway, PTHR responds to PTH or PTH-related peptide (PTHrP) by variously activating Gαs, Gαq, and Gα12/13, together with Gβγ, thereby triggering downstream signaling mediated by cAMP/protein kinase A, phospholipase C/inositol 1,4,5-trisphosphate/protein kinase C, or RhoA/phospholipase D (4, 5). In the G protein–independent pathway, PTHR interacts with β-arrestins and activates ERK1/2 signaling (6). After stimulation, the membrane-delimited PTHR is internalized, desensitized, and subsequently degraded or resensitized to traffic back to plasma membranes. Persistent, non-canonical endosomal cAMP signaling may also prevail (7). PTHR down-regulation is achieved by cell surface receptor desensitization as well as by receptor sequestration in endosomes and by proteolysis in lysosomes or proteasomes (8). Post-translational modifications, including protein phosphorylation and ubiquitination, play critical roles in mediating GPCR down-regulation. Early evidence established that ligand-induced PTHR phosphorylation is required for internalization (9–11) and that PTHR phosphorylated by G protein–coupled receptor kinases (GRK) (12) binds to β-arrestin, thereby physically uncoupling the receptor from its associated heterotrimeric G proteins, leading to receptor desensitization.
Ubiquitination may contribute to GPCR desensitization and down-regulation (13). Extensive evidence shows that multiple GPCRs, including the β2-adrenergic receptor (β2AR), PAR1 and PAR2 protease-activated receptors, μ- and δ-opioid receptors, CXCR chemokine receptors, V2R vasopressin receptor, D4 dopamine receptor, and PTHR (14), are ubiquitinated by the covalent addition of ubiquitin to intracellular Lys. Ubiquitination regulates internalization and trafficking of these receptors, often targeting receptor protein for degradation, either in an agonist-dependent or -independent manner (15). GPCR ubiquitination additionally contributes an important regulatory role in activating kinase cascades independent of proteasomal degradation (16).
Ubiquitin is added to protein substrates by a cascade of reactions initiated by ubiquitin activation (E1), followed by conjugation (E2) and ligation (E3) (17). Ubiquitin forms stable adducts through linear isopeptide bonds with the ϵ-amine of target Lys residues, Lys6, Lys11, Lys27, Lys29, Lys33, Lys48, or Lys63 (18), most typically Lys48 or Lys63.
Earlier studies showed that PTH(1–34) and PTH(7–34) promote PTHR ubiquitination and proteasomal degradation (19). PTHR harbors 11 intracellular Lys residues. Determining which Lys residues are targeted for ubiquitin conjugation is essential to define the molecular mechanism of PTHR ubiquitination and function. We therefore sought to identify the critical Lys residues involved in these activities. Based on the prevailing view that ubiquitination was restricted to the intracellular C terminus of GPCRs (17), we tested the effect of mutating carboxyl tail Lys residues individually or in combination or truncating most of the intracellular PTHR C terminus. These interventions, however, failed to eliminate PTH-stimulated ubiquitination. Xiao and Shenoy (20) and von Zastrow and co-workers (21) subsequently demonstrated that intracellular loops of the β2AR contain sites of ligand-induced ubiquitination. The PTHR harbors two Lys residues (Lys318 and Lys319) in the second intracellular loop, three (Lys388, Lys405, and Lys408) in the third intracellular loop, and six (Lys471, Lys472, Lys484, Lys486, Lys539, and Lys559) in the intracellular C terminus for a total of 11 cytoplasmic Lys residues (Fig. 1A). Because mutating all intracellular tail Lys or truncating virtually this entire region did not abrogate PTHR ubiquitination, we then theorized that targeted upstream Lys in the second or third loop synergize with C-terminal tail residues to account for the full program of PTHR ubiquitination. Here, we applied a combination of mass spectrometry to identify ubiquitinated di-Gly–tagged Lys residues and targeted mutagenesis to confirm their role in PTHR trafficking, signaling, and function. We now report that Lys388 in intracellular loop 3 and Lys484 in the PTHR C terminus are ubiquitinated following PTH challenge. PTHR ubiquitination did not alter receptor trafficking but significantly altered ERK and p38 signaling with attendant changes in cell proliferation and apoptosis.
Figure 1.

Functional expression of WT-PTHR and mutant PTHR. A, schematic representation of PTHR topology (43). Loops 1–3 (L1–L3) and the C terminus (CT) are marked in red. Intracellular Lys are highlighted in green and listed above. B, cAMP signaling in response to 100 nm PTH(1–34) by HEK-293 cells stably expressing luciferase-based GloSensorTM cAMP reporter and transiently expressing WT-PTHR or mutant PTHR. Error bars were omitted for clarity (**, p < 0.01, ANOVA with post hoc multiple comparison using the Bonferroni procedure (Prism). C, internalization of WT-PTHR and Lys-deficient 0K-PTHR visualized by confocal microscopy. HEK-293S GnTI− cells stably expressing TAP(HA)-WT-PTHR or TAP(HA)-0K-PTHR were incubated with anti-HA antibody conjugated to DyLight 488 to label surface receptors as detailed under “Experimental Procedures.” After washing, cells were incubated with vehicle (Control) or 100 nm PTH(1–34). After 5 min, cells were fixed, and remaining, non-internalized, surface receptors were detected with an anti-mouse secondary antibody conjugated to Alexa 594. Scale bars, 10 μm. D, quantitative analysis of WT-PTHR and 0K-PTHR from confocal images shown in C. Internalization was calculated from the ratio of post-treatment surface receptor expression (Alexa 594) to initial surface-expressed receptor (Dylight 488). Each bar represents the mean ± S.D. (error bars) (n = 11–14); *, p < 0.05. E, recycling of PTH receptors measured by cell-surface biotinylation. HEK-293S GnTI− cells stably expressing TAP(HA)-WT-PTHR or TAP(HA)-0K-PTHR were labeled with biotin as described under “Experimental procedures.” After quenching unreacted biotin and washing, cells were treated with 100 nm PTH(1–34) for 15 min at 37 °C to internalize the PTHR. The samples were then chilled, stripped, and allowed to recover at 37 °C for 0, 15, or 30 min to permit the internalized PTHR to recycle. For each time point, one dish was stripped after recycling, and one paired dish was not. These dishes represent the non-recycled and total internalized PTHR, respectively. Biotinylated receptors were immunoprecipitated with streptavidin and detected with an anti-HA antibody. Molecular weights are indicated at the left. Recycled receptor equals total internalized receptor minus non-recycled receptor. F, quantification of receptor recycling shown in E expressed as a percentage of the total receptors at the indicated time. Results are means ± S.D. (error bars) (n = 3). ANOVA indicated equivalent recycling of WT-PTHR and 0K-PTHR.
Results
Expression and signaling of wildtype and Lys-deficient PTHR
To identify the intracellular PTHR domains possessing candidate Lys residues that could be targeted for ubiquitination upon PTH stimulation, we generated four mutant receptor constructs by replacing cytosolic Lys (K) residues with Arg (R) as follows (Fig. 1A): 2K-PTHR (loop 2 Lys intact with all other intracellular Lys mutated to Arg); 3K-PTHR (loop 3 Lys protected; others replaced with Arg); CTK-PTHR (C-terminal Lys present; others converted to Arg); and 0K-PTHR (Lys-deficient construct), where all 11 Lys residues were replaced by Arg. To verify that these Lys mutant receptors were functional, the capacity of the activated receptor to promote Gs-stimulated cAMP production was examined. WT-PTHR and the various Lys mutant constructs were transiently introduced into human embryonic kidney 293 (HEK-293) cells stably expressing the GloSensor cAMP reporter (22). As shown in Fig. 1B, all mutant constructs elicited cAMP formation comparable with WT-PTHR upon PTH stimulation. Interestingly, the 0K-PTHR displayed significantly increased signaling compared with WT-PTHR or partial Lys-deficient forms of the receptor. Such a finding is in keeping with previous results, pointing to an inhibitory domain within the intracellular PTHR tail that reduces cAMP formation, which upon truncation enhances cAMP accumulation (23, 24).
Trafficking of Lys-deficient PTHR
Ubiquitination alters the trafficking of many GPCRs (25, 26). To determine the functional significance of PTHR ubiquitination, we first applied confocal microscopy to assess the effect of ligand-stimulated ubiquitination on receptor internalization. HEK-293S GnTI− cells stably expressing TAP(HA)-WT-PTHR or TAP(HA)-0K-PTHR were labeled for 1 h at room temperature with an anti-HA primary antibody directly conjugated to DyLight 488, which recognizes the HA tag in the N-terminal domain of PTHR. WT-PTHR and 0K-PTHR rapidly internalized after a 5-min treatment with 100 nm PTH(1–34), as indicated by the disappearance of plasma membrane PTHR and the formation of intracellular fluorescent puncta (Fig. 1C). Residual, non-internalized PTHRs were detected by labeling with a goat anti-mouse IgG secondary antibody conjugated to Alexa 594 that recognizes the mouse IgG heavy and light chains of the primary antibody. The fraction of internalized receptor can then be quantified by comparing the intensity of Alexa 594 staining in control and PTH-treated cells (Fig. 1D). The results show comparable reduction of WT-PTHR (33%) and 0K-PTHR (25%) after PTH stimulation (Fig. 1D). Thus, PTHR ubiquitination is not required for activity-dependent receptor internalization.
Of note, HEK-293S GnTI− cells lack N-acetylglucosaminyltransferase I (GnTI), which is required for the processing of complex N-glycans. GnTI− cells are a convenient tool for overexpressing membrane proteins for biochemical and related analyses (27). PTHR possesses four N-glycosylation sites, and the absence of GnTI restricts PTHR glycosylation to a single Man5GlcNAc2. Thus, the absence of complex N-glycans might alter PTHR localization and function. Notably, the residual glycosylation is sufficient for PTHR function, as demonstrated here by normal receptor trafficking (Fig. 1C) and signaling, as reported previously (28–30).
We complemented the imaging studies by measuring cell surface biotinylation to quantify the effect of ubiquitination on recycling of sequestered PTHR. The extent of recycled receptor is determined as the difference between internalized receptor and non-recycled receptor (Fig. 1E). The results indicated that over 50% of internalized WT-PTHR and 0K-PTHR recycled to the cell membrane within 30 min of PTH treatment (Fig. 1F). Mutant 0K-PTHR was as efficiently internalized and recycled as WT-PTHR. Thus, ubiquitination does not interfere with efficient PTHR endocytosis and is not required for PTHR recycling. Establishing that the Lys mutant receptors displayed normal signaling and trafficking permitted investigation of the consequences of PTH-induced ubiquitination independent of these biological activities.
Agonist-promoted PTHR ubiquitination and degradation
Previous work established that PTH(1–34) induced receptor ubiquitination followed by partial deubiquitination and recycling, with negligible degradation in the absence of cycloheximide to prevent de novo protein synthesis. In contrast, PTH(7–34) evoked receptor ubiquitination accompanied by degradation (19). Here, in GnTI− cells stably expressing PTHR, PTH(1–34) stimulated stable PTHR ubiquitination for at least 30 min (Fig. 2, A and C). After 45 min, ubiquitination decreased dramatically (Fig. 2A) primarily due to degradation of ubiquitinated receptors (Fig. 2B), resulting in diminished PTHR expression. After pretreatment with cycloheximide followed by 45-min PTH stimulation, WT-PTHR abundance decreased by 40% (Fig. 2, B and C), whereas the mutant 0K-PTHR was essentially refractory to metabolic degradation, with expression unchanged from control levels (Fig. 2, B and C). PTHR depletion as observed here is probably replenished by equivalent newly synthesized receptor protein because in the absence of cycloheximide, PTHR expression was stable upon PTH(1–34) stimulation (19). Thus, the time course of PTHR ubiquitination and degradation were highly correlated. Moreover, preventing receptor ubiquitination abolished degradation, as exemplified by the 0K-PTHR. These results imply that PTHR metabolism essentially proceeds in a ubiquitination-sensitive manner. Interestingly, when cells were treated with 5 μm staurosporine, a broad-spectrum protein kinase inhibitor, PTHR ubiquitination was virtually abolished (Fig. 2A, lane 1), suggesting a role for receptor phosphorylation in the ubiquitination process and implying signaling crosstalk between receptor phosphorylation and ubiquitination.
Figure 2.

PTHR degradation and ubiquitination. A, time course of PTHR ubiquitination induced by PTH. HEK-293S GnTI− cells stably expressing TAP(HA)-PTHR were transfected with Myc-ubiquitin. 48 h later, cells were serum-starved for 2 h and treated with 10 μm MG132 for 30 min, followed by a 5-min treatment with 5 μm staurosporine where indicated. 100 nm PTH(1–34) was added for 5, 15, 30, 45, or 60 min. Cells were harvested and lysed as described under “Experimental procedures,” incubated overnight with streptavidin-agarose beads at 4 °C. The eluted protein using SDS sample buffer was incubated at 37 °C for 30 min. A higher-percentage gel (12%) was run for a shorter time to produce sharper bands on the immunoblot. Anti-ubiquitin P4D1 and HA antibodies (Table 3) were used to detect ubiquitination and PTHR. β-Actin was used as a loading control. Molecular weights are indicated at the left. The illustrated immunoblots are representative of three replicates. Blocking kinase activity with staurosporine virtually abolished receptor ubiquitination (lane 1). B, time-dependent degradation of WT-PTHR and 0K-PTHR. HEK-293S GnTI− cells stably expressing TAP(HA)WT-PTHR or TAP(HA)0K-PTHR were transfected with Myc-ubiquitin. After 36 h, cells were seeded on 6-cm dishes and grown overnight at 37 °C, serum-starved for 2 h, and incubated with 10 μg/ml cycloheximide for 1 h. 100 nm PTH(1–34) was added for the indicated time. Protein lysates were resolved by SDS-PAGE. WT-PTHR and 0K-PTHR were detected with an anti-HA antibody. Molecular weights are indicated at the left. C, quantification of receptor degradation. WT-PTHR and 0K-PTHR abundance was calculated from the respective band intensity and was normalized to β-actin. Results are reported as means ± S.D. (error bars) (n = 3; ***, p < 0.001, two-way ANOVA).
Sites of PTHR ubiquitination
We next sought to identify intracellular Lys residues ubiquitinated upon PTH treatment. GnTI− cells stably expressing WT-PTHR or the various Lys-deficient mutant PTHRs were transfected with Myc-ubiquitin and treated with PTH(1–34) for 30 min. Similar amounts of receptor protein were pulled down by streptavidin beads and detected by immunoblotting (Fig. 3A). The characteristic smearing of ubiquitinated WT-PTHR was absent in 2K-PTHR and 0K-PTHR (Fig. 3A). In contrast, 3K-PTHR and CTK-PTHR constructs, where loop 3 Lys and C terminus Lys are present, displayed ubiquitination comparable that of WT-PTHR, suggesting that loop 3 and the carboxyl PTHR tail harbor the principal sites of Lys ubiquitination.
Figure 3.

Sites of PTHR ubiquitination. A, ubiquitination of WT-PTHR and the indicated 0K, 2K, 3K, and CTK-PTHR mutants. GnTI− cells stably expressing the specified PTHR were transfected with Myc-ubiquitin. After 48 h, cells were serum-starved for 2 h and treated with 10 μm MG132 for 30 min followed by a 30-min exposure to 100 nm PTH(1–34). Empty TAP-PTHR vector was transfected as a negative control. PTHR was isolated on streptavidin-agarose beads. The bound receptor was separated by SDS-PAGE and immunoblotted with antibodies specific to ubiquitin (P4D1) (top) and HA for receptor expression (middle). β-Actin was used as the loading control (bottom). Molecular weights are indicated at the right. Shown is a representative blot of three experiments. B and C, MS/MS spectra for the identified ubiquitinated peptides containing Lys388 or Lys484. The peptide sequences are shown at the top of the MS/MS spectra with ubiquitinated residues highlighted in red. Peak heights indicate the relative abundance of the corresponding fragmentation ions, with the annotation of the identified, matched N terminus–containing ions (b ion) in blue and C terminus–containing ions (y ions) in red. The spectra are representative of three independent MS/MS experiments. D, immunoblot detection of ubiquitination of loop 3 Lys mutants. Cells were transiently transfected with WT-PTHR or mutant PTHR and Myc-ubiquitin using Lipofectamine 3000. GnTI− cells transfected with Myc-ubiquitin (Myc) alone were used as a negative control. PTHR was isolated and detected by immunoblotting using antibodies directed against ubiquitin (P4D1) (top) and HA for PTHR (bottom). Molecular weights are indicated at the left. Experiments were repeated three times with similar results.
We applied mass spectrometry of purified receptor protein to identify Lys residues targeted for ubiquitination. After a 30-min challenge with PTH(1–34), Lys388 in the third loop and Lys484 in the C terminus exhibited the characteristic covalently linked di-Gly modifications (Fig. 3 B and C and Table 1) indicative of ubiquitination. Loop 3 harbors lysines at Lys388, Lys405, and Lys408. The latter two sites are not covered by trypsin digestion as needed for mass spectrometry. Therefore, to determine whether these residues in the Loop 3 construct are ubiquitinated, we separately reverted each individual mutated Arg to Lys (R388K, R405K, or R408K) and transiently transfected them together with Myc-ubiquitin in GnTI− cells. As shown in Fig. 3D, 0K-PTHR displayed no ubiquitination as expected, and R388K-PTHR showed a high-molecular weight ubiquitination signal similar to WT-PTHR, confirming the mass spectrometry results for this Lys residue. In contrast, neither R405K nor R408K exhibited detectable ubiquitination (Fig. 3D). Collectively, these results show that only Lys388 in the third loop and Lys484 in the intracellular PTHR C terminus are ubiquitinated upon PTH treatment.
Table 1.
PTHR Lys post-translational modifications
Overexpressed TAP-PTHR was purified as described under “Experimental procedures” and digested by trypsin (cleavage site: Arg or Lys). Xcorr values indicate how well the spectrum fits an ideal spectrum for the matched peptide. ppm, parts per million, indicates the deviation of the observed peptide mass with the calculated peptide sequence.
| Peptide | Modification | Charge state | Xcorr | Precursor mass error |
|---|---|---|---|---|
| ppm | ||||
| VLATKubiLR | Ubiquitinated Lys388 | +2 | 0.62 | −10.50 |
| WTLALDFKubiR | Ubiquitinated Lys484 | +3 | 2.02 | −0.84 |
β-Arrestin dependence of PTHR ubiquitination
β-Arrestins function as adaptors for ubiquitination of several GPCRs, such as the β2AR (31) and μ-opioid receptors (32). To discern a role for β-arrestins in PTHR ubiquitination, wildtype or β-arrestin1/2 double knockout MEF cells (33) were transfected with PTHR harboring an N-terminal tandem-affinity purification tag (TAP-PTHR) and Myc-ubiquitin, with or without FLAG-β-arrestin1 or FLAG-β-arrestin2. As shown in Fig. 4, PTH promotes PTHR ubiquitination in wildtype MEF cells. However, ubiquitination was undetectable in β-arrestin1/2 knockout MEF cells. Overexpressing FLAG-β-arrestin1 or FLAG-β-arrestin2 rescued PTH-induced receptor ubiquitination, indicating that PTHR ubiquitination is β-arrestin–dependent but has no discernable preference for β-arrestin1 or β-arrestin2 within the limit of detection by the β-arrestin1/2 antibody, which is more sensitive to β-arrestin1 than to β-arrestin2.
Figure 4.
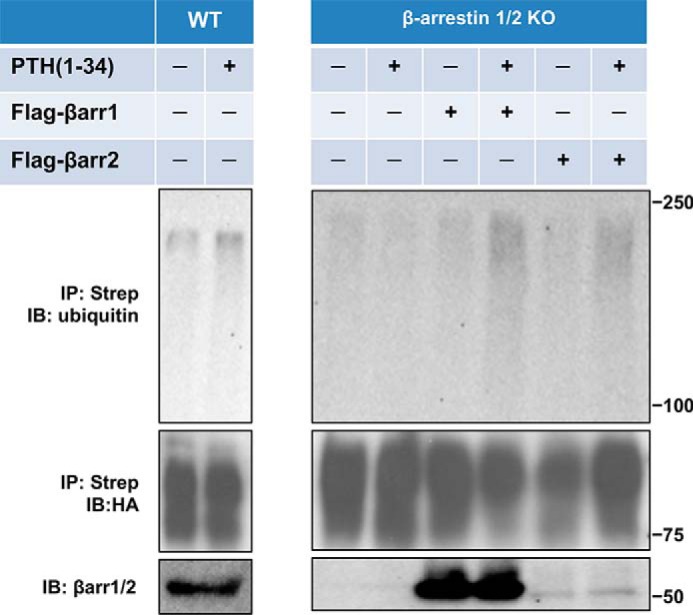
β-Arrestin-dependent PTHR ubiquitination. Wildtype or β-arrestin1/2 double KO MEF were transiently transfected with TAP(HA)-PTHR, Myc-ubiquitin, and FLAG-β-arrestin1 or FLAG-β-arrestin2 using Lipofectamine 3000. After 48 h, cells were serum-starved for 2 h and treated with 10 μm MG132 for 30 min followed by a 30-min challenge with 100 nm PTH(1–34). PTHR was isolated and detected by immunoblotting with antibodies against ubiquitin (P4D1) (top) and HA for receptor expression (middle). Expression of β-arrestin1 or -2 from cell lysates was detected with an anti-β-arrestin1/2 antiserum (bottom). The positions of molecular weight standards are marked at the right. Experiments performed in triplicate yielded similar results.
Following PTH stimulation, the PTHR is phosphorylated at Thr387 and Thr392, Ser489, Ser491, Ser492, and Ser493 (Fig. 5A and Table 2). These sites of Ser phosphorylation are consistent with a recent report (34). To determine which of these phosphorylation sites is involved in or responsible for β-arrestin recruitment, Thr at Thr387/Thr392 and Ser in the 489SGSSS493 cluster (Fig. 5B), alone or together, were mutated to Ala (Ala387/Ala392, 489AGAAA493). These constructs were then used to characterize the binding to β-arrestin1 or β-arrestin2. As shown in Fig. 5 (C and D), replacing Thr387/Thr392 decreased PTHR binding both to β-arrestin1 and β-arrestin2. In contrast, mutating the 489SGSSS493 motif failed to disrupt binding to β-arrestin1 or β-arrestin2. Combined mutation of Ala387/Ala392 with 489AGAAA49 had no greater effect than that of Ala387/Ala392 alone on binding to β-arrestin1 or β-arrestin2.
Figure 5.

β-Arrestin recruitment requires PTHR phosphorylation. A, representative MS/MS spectrum showing phosphorylated Ser489 (pS, red) in purified TAP(HA)-PTHR overexpressed in HEK-293S GnTI− cells. The peptide sequence corresponding to the MS/MS spectrum is shown at the top, with the phosphorylated residue highlighted in red. Peak heights indicate the relative abundance of the corresponding fragmentation ions. The identified matched N terminus–containing ions (b ion) in blue and C terminus–containing ions (y ions) in red are annotated. The experiment was repeated in triplicate with identical results. B, summary of constructs with their respective sequences for WT and mutant PTHR to define loop 3 phosphorylation sites. In the 387 construct, Thr387 and Thr392 were replaced by Ala (A). In the 489 construct, all Ser residues were changed to Ala. The 387/489 double mutant construct has all Thr and Ser residues in both regions replaced by Ala. C, representative immunoblots showing FLAG-β-arrestin1 (left) and FLAG-β-arrestin2 (right) from immunoprecipitated WT-, 387-, 489-, or 387/489-TAP(HA)-PTHR. After pulldown by streptavidin beads, protein samples were analyzed by Western blot with antibodies specific to β-arrestin1/2 (top) and HA for receptor expression (middle). Expression of β-arrestin1 or -2 in cell lysates is shown in the bottom panel. Molecular weights are indicated at the right. Note that some β-arrestin1/2 background was evident with empty vector (EV) despite HA-PTHR being undetectable. This background was subtracted during the subsequent quantification. Experiments were repeated three times with similar outcomes. D, quantitative summary of β-arrestin1 (top) or β-arrestin2 (bottom) binding to WT-PTHR of results shown in C. Data are means ± S.D. (error bars) of n = 3 independent observations for each group. Binding of β-arrestin to WT-PTHR was set as 1 (*, p < 0.05; **, p < 0.01 by ANOVA with post hoc analysis). E, effect of site-specific Thr/Ser mutation on PTHR ubiquitination. WT-PTHR or the indicated Ser/Thr mutant PTHR construct shown in B, together with Myc-ubiquitin, were transfected into HEK-293S GnTI− cells using Lipofectamine 3000. After 48 h, cells were serum-starved for 2 h and treated with 10 μm MG132 for 30 min, followed by a 30-min challenge with saline vehicle or 100 nm PTH(1–34). Ubiquitinated TAP-PTHR was pulled down with streptavidin beads and probed with an anti-ubiquitin antibody (P4D1, top) and HA for receptor expression (bottom). Molecular weight size markers are shown at the right. The 387 construct, where Thr387 and Thr392 were replaced by Ala, decreased but did not eliminate PTH-induced ubiquitination. In contrast, the 489 construct lacking all five Ser residues, did not affect receptor ubiquitination. The sample shown is illustrative of three experiments.
Table 2.
Identified phospho-Ser, and phospho-Thr

β-Arrestin binding to phosphorylated receptors initiates desensitization. As shown here for the PTHR, phosphorylation sites in the Thr387/Thr392 region are critical to β-arrestin recruitment (Fig. 5). Interestingly, ubiquitinated Lys388 is located within the 387TKLRET392 region. Overlap of phosphorylation and ubiquitination positions suggests that receptor phosphorylation and ubiquitination are at least correlated and probably interdependent. Indeed, abrogation of phosphorylation at positions Thr387/Thr392 dramatically decreased PTHR ubiquitination (Fig. 5E). However, preventing phosphorylation within the 489SGSSS493 cluster did not markedly interfere with PTHR binding to β-arrestin (Fig. 5, C and D) and did not affect receptor ubiquitination (Fig. 5E), suggesting that β-arrestin binding to the receptor site-specifically phosphorylated at 387TKLRET392 is required for subsequent PTHR ubiquitination.
Ubiquitinated PTHR differentially regulates PTHR-mediated MAPK signaling
We further inquired into the cellular and signaling consequences and biological function of ubiquitinated PTHR. Early evidence revealed that β-arrestins play an important role not only in GPCR desensitization (35) and trafficking (36), but also in G-protein–independent MAPK signaling (37). We reasoned that if β-arrestin-dependent receptor ubiquitination affects PTHR-mediated downstream MAPK signaling, then G-protein–independent signaling of WT-PTHR or 0K-PTHR stimulated by PTH(7–34) should be impaired. MAPK signaling pathways include ERK1 and ERK2 (ERK1/2), JNK, and p38 (38). Because β-arrestins can scaffold MAPKs (39, 40), we investigated the effects of β-arrestin–dependent PTHR ubiquitination on downstream MAPK signaling. The PDZ protein NHERF1 (Na+/H+ exchanger regulatory factor 1) can inhibit PTH-induced ERK signaling (24). Therefore, we used GnTI− cells, where endogenous NHERF1 expression is negligible (data not shown) and does not affect receptor trafficking (Fig. 1, C and E), to examine the influence of PTHR ubiquitination on downstream MAPK signaling. Further, to exclude the possibility of Gq/protein kinase C–induced ERK activation (41, 42), we used PTH(7–34), which triggers PTHR ubiquitination and MAPK signaling without activating PKC. Here, PTH(7–34) induced Lys48/Lys63-linked PTHR polyubiquitination in a time-dependent fashion (Fig. 6A). Lys48 ubiquitination is the dominant form of modification and targets PTHR for degradation (19). As shown here, polyubiquitination can also be Lys63-linked in the presence of either PTH(1–34) or PTH(7–34) (Fig. 6C). The observed discrete bands characteristic of Lys63-linked ubiquitination are lower than combined Lys63 plus Lys48 high-molecular-weight smears detected by the P4D1 ubiquitin antibody (Fig. 6A). This difference probably arises from the absence of Lys48-linked polyubiquitin. A similar pattern of antibody-dependent size differences has been described (44, 45). Lys63-linked ubiquitination has been implicated in a variety of cellular events, including ERK signal transduction (46).
Figure 6.
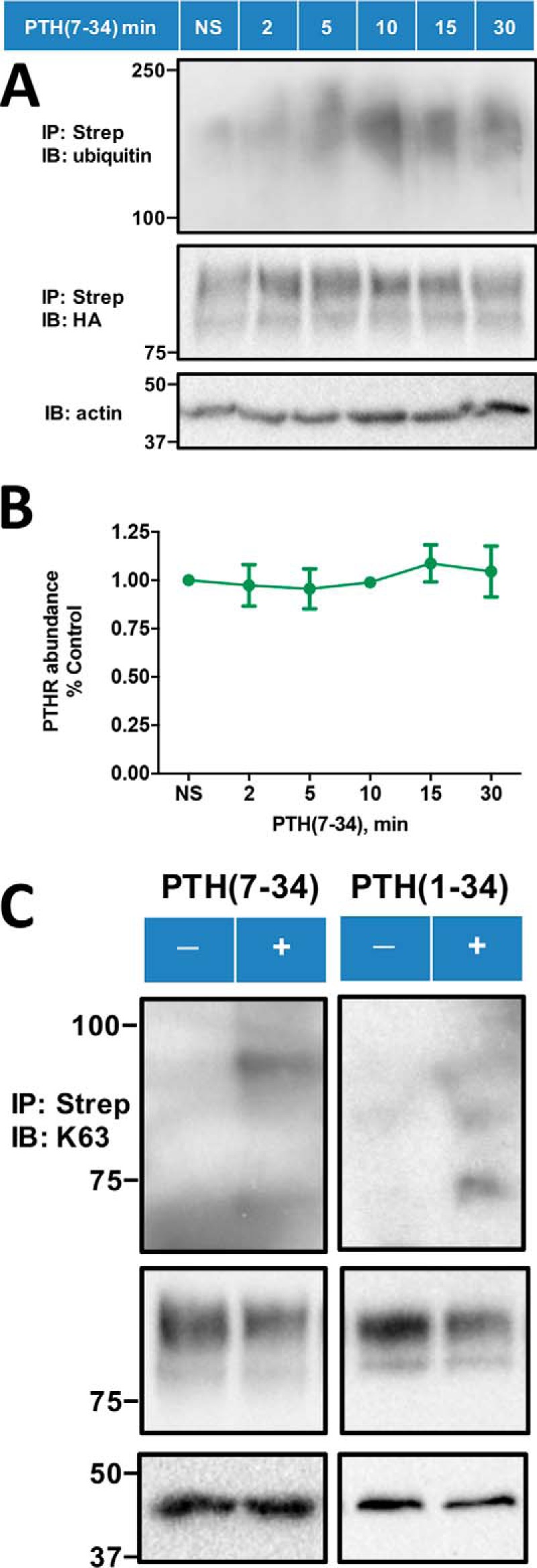
PTH(7–34)-induced receptor ubiquitination. A, PTH(7–34) stimulates receptor ubiquitination in a time-dependent manner. HEK-293S GnTI− cells stably expressing TAP(HA)-PTHR were transiently transfected with Myc-ubiquitin using Lipofectamine 3000. 48 h post-transfection, cells were serum-starved for 2 h and subsequently treated with 10 μm MG132 for 30 min and then challenged with 1 μm PTH(7–34) for the indicated time (0, 2, 5, 10, 15, or 30 min). PTHR was isolated on streptavidin-agarose beads. Ubiquitin was detected with a global anti-ubiquitin antibody that recognizes both Lys48 and Lys63 (P4D1) (top) and PTHR with an anti-HA antibody (middle). β-Actin was used as the loading control (bottom). Molecular weight standards are shown at the left. The illustrated blot is representative of three independent experiments. B, quantification of WT-PTHR expression upon PTH(7–34) treatment. The calculation was performed as described in the legend to Fig. 2C. Results are reported as means ± S.D. (error bars) (n = 3) and indicate that PTHR expression did not change for the first 30 min after challenge with PTH(7–34). C, PTH(7–34) and PTH(1–34) promoted specific Lys63-linked PTHR polyubiquitination. The experimental procedure was similar to that in A, where the protein sample was now detected with a Lys63 linkage-specific polyubiquitin antibody (Table 3) (top) and an anti-HA antibody for receptor expression (middle). β-Actin was used as the loading control (bottom). Positions of molecular weight standards are indicated at the left. Shown is a representative blot from three independent experiments.
MAPK signaling was analyzed from 0 to 30 min in the absence of proteasome inhibitor MG132 because PTHR ubiquitination (Fig. 6A) and degradation (Fig. 6B) were stable for the first 30 min following PTH(7–34). PTH(7–34) stimulated ERK1/2 phosphorylation in a time-dependent manner in cells expressing WT-PTHR. Phosphorylation of ERK1/2 reached a maximum at 5 min, after which the level declined (Fig. 7, A and C). In marked contrast, the 0K-PTHR exhibited a small early ERK1/2 increase at 2 min but a markedly lower peak at 5 min and a modestly delayed response at 10 min (Fig. 7, B and C).
Figure 7.

WT-PTHR and 0K-PTHR mediate distinct patterns of ERK signaling. HEK-293S GnTI− cells stably expressing WT-PTHR (A) or 0K-PTHR (B) were transfected with Myc-ubiquitin in 10-cm dishes. 36 h later, cells were split into six equal aliquots and incubated on 6-cm dishes for 16 h. Cells were serum-starved for 4 h and stimulated with 1 μm PTH(7–34) for the indicated times. 1 μm PMA for 10 min served as a positive control, and native GnTI− cells were transiently transfected with Myc-ubiquitin as a negative control. Cells were immediately washed and lysed as detailed under “Experimental procedures.” Total protein concentrations were measured with BCA. Western blot analysis using equal amounts of total protein from the corresponding cell lysates was performed to detect phosphorylated ERK1/2 (top) and total ERK1/2 (bottom). Positions of molecular weight standards are marked at the left. C, quantitative analysis of ERK signaling. Phospho-ERK signaling was normalized to that of total ERK at each time. Results are means ± S.D. (error bars) of three independent experiments (**, p < 0.01, two-way ANOVA).
p38 displayed a conspicuously different pattern of ubiquitination-sensitive activation compared with ERK1/2. Upon PTH(7–34) treatment, p38 signaling of WT-PTHR dramatically decreased at 5 min compared with 0K-PTHR (Fig. 8, A–C), where there was no measurable change. The substantial difference in p38 signaling between WT-PTHR and 0K-PTHR in response to PTH(7–34) suggested that PTH-induced ubiquitination of discrete Lys residues is required to inhibit p38 signaling. We used the PTHR mutants described earlier (Figs. 1A and 3D) to test this hypothesis. As shown in Fig. 8 (D and E), refined analysis of p38 signaling by the various PTHR Lys mutants revealed a pattern of differential responses. 3K-PTHR, CTK-PTHR, and 388K-PTHR exhibited decreased p38 signaling similar to WT-PTHR, whereas 2K-PTHR and 0K-PTHR were refractory to PTH(7–34) stimulation, thus implicating Lys388 and Lys484 as critical for PTH-induced receptor ubiquitination.
Figure 8.

p38 signaling by WT-PTHR and 0K-PTHR. Differential effects of PTH(7–34) on p38 signaling in WT-PTHR (A and C) and 0K-PTHR (B and C). Cells were treated and processed as in Fig. 7. WT-PTHR and 0K-PTHR cells were treated with 25 μg/ml anisomycin for 5 min as positive controls. HEK-293S GnTI− cells transiently transfected with Myc-ubiquitin served as negative controls. Molecular weight standards are indicated at the left. The signal for phospho-p38 was normalized to that of total p38 at each time point. Results are the average of n = 3 for each time point and group (***, p < 0.001, two-way ANOVA). D, inhibition of p38 signaling by the indicated PTHR mutants upon PTH(7–34) stimulation for 5 min. HEK-293S GnTI− cells stably expressing WT-, 0K-, 2K-, 3K-, or CTK-PTHR (transient expression for 388K-PTHR) were transfected with Myc-ubiquitin. 48 h post-transfection, cells were treated and processed as in A and B. Unstimulated WT-PTHR cells served as a control. Molecular weights are indicated at the left. E, quantitative analysis of p38 signaling shown in D. Results represent the mean ± S.D. (error bars) of three independent experiments (*, p < 0.05; **, p < 0.01 versus WT-PTHR, two-way ANOVA).
Compared with ERK (where 0K-PTHR signaling diminished) or p38 (where WT-PTHR signaling decreased), no significant difference of JNK signaling was found between WT-PTHR and 0K-PTHR upon PTH(7–34) treatment (data not shown). Together, these results provide direct evidence that ubiquitinated PTHR differentially and specifically regulates MAPK signaling of ERK1/2 and p38.
The differential response of β-arrestin-dependent MAPK signaling triggered by PTH(7–34) suggests that ubiquitination initiates distinct MAPK signaling signatures. MAPK signaling is importantly involved in cell proliferation and apoptosis (47). We therefore tested the effect of PTH on these biological processes in cells expressing WT-PTHR or 0K-PTHR. PTH(7–34) increased the rate of proliferation in cells expressing WT-PTHR but not in cells expressing 0K-PTHR (Fig. 9A). Enhanced proliferation was accompanied by reduced apoptosis in cells expressing WT-PTHR, whereas cells expressing 0K-PTHR exhibited increased apoptosis (Fig. 9B).
Figure 9.

Loss of receptor ubiquitination leads to reduced proliferation and increased apoptosis. In the presence of 1 μm PTH(7–34), HEK-293S GnTI− cells stably expressing WT-PTHR or 0K-PTHR were grown at 37 °C for 72 h. PTH(7–34) was replenished every 12 h. A, cell proliferation was evaluated at 2, 24, 48, and 72 h by ELISA with bromodeoxyuridine. B, apoptosis was assessed by an ELISA that detects histones H1, H2A, H2B, H3, and H4 and an anti-DNA POD antibody that binds to single- and double-stranded DNA. n = 3 in each group; data are means ± S.D. (error bars) (**, p < 0.01; ***, p < 0.001 versus 2 h; two-way ANOVA).
Discussion
Cyclical receptor desensitization and down-regulation protect cells against persistent agonist-induced overstimulation that can result in blunted cellular signaling and actions. GPCR down-regulation requires post-translational phosphorylation and ubiquitination (17, 26). Previous work from our laboratory showed that ubiquitination promotes ligand-biased PTHR sorting. PTH(1–34) activates coupled PTHR ubiquitination and deubiquitination, whereas PTH(7–34) primarily stimulates ubiquitination (19). As a result, PTH(1–34) stimulates PTHR internalization and recycling. In contrast, PTH(7–34) activates internalization and down-regulates PTHR. We here identified bipartite sites of Lys ubiquitination located within the third intracellular loop and in the C-terminal PTHR tail. β-Arrestins have been implicated in scaffolding of ubiquitin ligases (48). Notably, PTHR ubiquitination requires antecedent phosphorylation and β-arrestin recruitment and causes differential activation of MAPK signaling upon PTH treatment. Thus, a possible scenario is that phosphorylated receptors recruit β-arrestin, which subsequently forms a multicomponent complex that includes the E3 ubiquitin ligase that is responsible for PTHR ubiquitination, as in the case of β2AR (31) and V2R (49).
Ubiquitination commonly targets GPCRs for degradation in lysosomes or proteasomes (50, 51). Internalized PTHR are targeted to endosomes, as demonstrated by colocalization with EEA1 (52). PTHR then undergoes recycling or degradation (53). Post-translational ubiquitin modification also has been implicated in regulating receptor endocytosis and cell signaling (54). Early evidence established that internalization of the yeast GPCR Ste2 depends on auto- ubiquitination (55, 56). Internalization of most mammalian GPCRs, however, has proven to be independent of ubiquitination (57). Interestingly, upon agonist stimulation, PAR1 displays a hybrid pattern of ubiquitination-dependent and -independent internalization (58). The Lys-deficient 0K-PTHR mutant lacking all intracellular Lys and hence refractory to ubiquitination internalized and recycled comparably with WT-PTHR, implying that ubiquitination is not required for PTHR internalization or recycling and does not affect the rate of receptor endocytosis.
Several lines of investigation reveal an interplay between receptor phosphorylation and ubiquitination (59). The earliest evidence again came from yeast Ste2p, where phosphorylation of the cytoplasmic receptor tail facilitated ubiquitination of a vicinal Lys (60). This phenomenon was subsequently extended to β2AR, the prototype mammalian GPCR (31). Ligand-induced activation results in β2AR and PTHR phosphorylation by GRK2 (12, 61–63). Because isotype-specific GRK inhibitors are unavailable to determine whether phosphorylation affects PTHR ubiquitination, we used staurosporine, a Ser/Thr kinase inhibitor that at high concentrations nonspecifically blocks a broad gamut of protein kinases, including GRKs (64). The results show clearly that preventing PTHR phosphorylation virtually abolished receptor ubiquitination (Fig. 2A). Similar findings have been reported for the platelet-derived growth factor receptor-β (65). Consistent with this observation, we found that as opposed to kinase inhibition, blocking phosphorylation by site-specific Ala substitution at Thr387/Thr392, but not at Ser489–Ser493, reduced β-arrestin binding to PTHR by 80% (Fig. 5, C and D) with attendant reduction of PTHR ubiquitination (Fig. 5E). The partial reduction of ubiquitination by Thr mutation but not protein kinase inhibition implies the presence of additional phosphorylation sites besides Thr387/Thr392 that contribute to PTHR binding to β-arrestin. Indeed, a recent report showed that the cluster Ser501–Thr506 contributes to the interaction of PTHR with β-arrestin (34). The remaining input may stem from Thr387/Thr392, which was not characterized therein.
To determine the extent to which β-arrestin affects PTHR ubiquitination, wildtype or β-arrestin knockout MEF cells (33) were transfected with TAP(HA)-PTHR and Myc-ubiquitin. As anticipated, PTHR ubiquitination exhibited β-arrestin dependence, and transfection of MEF knockout cells with either β-arrestin1 or β-arrestin2 rescued PTHR ubiquitination, thus independently confirming the β-arrestin requirement for PTHR ubiquitination. Unexpectedly, however, β-arrestin1 and -2 exerted comparable receptor ubiquitination upon PTH stimulation. Of course, such equivalence may result from overexpression of the two isoforms.
β-Arrestin2 mediates β2AR ubiquitination by recruiting the ubiquitin E3 ligase Nedd4 to the activated receptor (31, 57). In addition to serving as an adapter protein to engage ubiquitin E3 ligase to the receptor (66), β-arrestins also activate MAPK signaling cascades, including ERK, JNK, and p38 pathways, independent of G protein activation (67). Receptor ubiquitination plays an important role in activating ERK signaling in receptor tyrosine kinases, such as the insulin-like growth factor 1 receptor, IGF-1R (68), and p38 signaling in G protein–coupled PAR1 (69). G protein-dependent or independent MAPK signaling has been associated with Lys63-linked GPCR ubiquitination (70). PTH(7–34) does not activate G protein-dependent protein kinase A or protein kinase C signaling (42), both of which are linked to MAPK signaling (71). As demonstrated here, however, PTH(7–34) elicits G protein-independent, β-arrestin–dependent MAPK signaling and PTHR Lys63-linked ubiquitination. We speculated that PTH(7–34)-activated, Lys63-linked ubiquitination leads to β-arrestin–dependent differential MAPK signaling. The results bear out this supposition and show that PTHR exhibits distinct profiles of MAPK responses. In the case of ERK1/2, PTH(7–34) increased time-dependent signaling for WT-PTHR, as observed before (6), and was markedly attenuated for the 0K-PTHR. A second model of MAPK signaling emerged for p38. Here, PTH(7–34) decreased WT-PTHR signaling, whereas 0K-PTHR remained constant. Further investigation showed that ubiquitination of 3K-PTHR, CTK-PTHR, 388K-PTHR, and WT-PTHR decreased p38 signaling, whereas ubiquitination-deficient 2K-PTHR and 0K-PTHR were unresponsive to PTH.
β-Arrestins not only scaffold receptors but also bind to ERK, p38, and JNK (67). Ubiquitination may promote different MAPK signaling patterns by virtue of β-arrestin assembling variable multicomponent macromolecular complexes for ERK, p38, and JNK signaling. A recent report showed that transforming growth factor-β–activated protein kinase-1–binding protein-2 (TAP2) is essential for PAR1 ubiquitination-mediated p38 MAPK activation (69).
We initially expected ubiquitination to affect PTHR internalization or recycling, which turned out not to be the case. The distinct requirement for phosphorylation, on the one hand, and striking biased effects on MAPK signaling of Lys-deficient forms of the PTHR, on the other, suggested that cell proliferation and apoptosis may rely on or be influenced by ubiquitination. Indeed, Lys-deficient 0K-PTHR ubiquitination displayed reduced cell proliferation and increased apoptosis. ERK signaling has been shown to increase cell proliferation (47), consistent with our observation that WT-PTHR exhibits greater ERK activity than 0K-PTHR upon PTH(7–34) treatment. Increased cell death for 0K-PTHR cells is probably associated with the loss of regulated p38 signaling (72). The biased PTHR signaling upon ubiquitination could be connected with dysfunctional regulation and disease. For instance, PTH(7–34) accumulates to high levels in end-stage kidney disease (5). Further, ubiquitin ligase RNF146 has been implicated in cleidocranial dysplasia, an autosomal dominant form of abnormal bone disease (73). Moreover, PTH-induced osteoblast proliferation requires direct regulation of the ubiquitin-specific-processing protease 2 gene, USP2 (74), which we demonstrated is required for PTHR deubiquitination (19).
In summary, the present report identifies dual sites of PTHR ubiquitination and illustrates how receptor ubiquitination differentially affects MAPK signaling and cell proliferation. The divergent MAPK signaling responses activated by PTH(7–34)-mediated ubiquitination could be potential targets for pharmacological intervention against diseases such as chronic kidney disease-mineral and bone disorder involving PTHR dysfunction. Additional work will be needed to ascertain the origin of the biased agonism by which ubiquitination triggers differential MAPK signaling. MAPK is a prospective therapeutic target (75–77). Our work suggests that novel compounds could potentially target specific elements of MAPK signaling of or signaling of PTHR-related skeletal disorders or chronic kidney disease (78–80).
Experimental procedures
Reagents and antibodies
Human PTH(1–34) ((Nle8,18,Tyr34)-PTH(1–34); henceforth, PTH) was purchased from Bachem (H-9110; Torrance, CA). Human PTH(7–34) ((Nle8,18,d-Trp12,Tyr34)-PTH(7–34) NH2) was prepared using standard peptide synthesis and purification methods (81, 82). MG132 was from AG Scientific (San Diego, CA). Blasticidin, Geneticin, and puromycin were obtained from Thermo Fisher Scientific. Antisera were acquired from the indicated sources and used at the dilutions summarized in Table 3. Staurosporine and all other reagents were of the highest available grade and purchased from Sigma.
Table 3.
Antibodies, clone, dilution, and source
| Antibody | Clone | Dilution | Source | Catalog no. |
|---|---|---|---|---|
| Anti-HA (16B12) (DyLight 488) | Mouse | 1:500 | Abcam | Ab117488 |
| Goat anti-mouse IgG secondary antibody, Alexa Fluor 594 | Goat | 1:500 | Thermo Fisher Scientific | A-11032 |
| Ubiquitin (P4D1) Lys63/Lys48 mouse mAb | Mouse | 1:1000 | Cell Signaling | 3936S |
| Anti-HA.11 epitope tag antibody | Mouse | 1:1000 | Biolegend | 901503 |
| Monoclonal anti-β-actin antibody | Mouse | 1:5000 | Sigma | A1978 |
| β-arrestin 1/2 rabbit mAb | Rabbit | 1:1000 | Cell Signaling | 4674S |
| Anti-FLAG antibody | Rabbit | 1:1000 | Sigma | F7425 |
| Lys63 linkage-specific polyubiquitin rabbit mAb | Rabbit | 1:500 | Cell Signaling | 12930 |
| Phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204) Rabbit mAb | Rabbit | 1:1000 | Cell Signaling | 9101S |
| p44/42 MAPK (ERK1/2) rabbit mAb | Rabbit | 1:1000 | Cell Signaling | 4695S |
| Phospho-p38 MAPK (Thr180/Tyr182) rabbit mAb | Rabbit | 1:1000 | Cell Signaling | 4511S |
| p38 MAPK antibody | Rabbit | 1:1000 | Cell Signaling | 9212S |
| Phospho-SAPK/JNK (Thr183/Tyr185) rabbit mAb | Rabbit | 1:1000 | Cell Signaling | 4668S |
| SAPK/JNK antibody | Rabbit | 1:1000 | Cell Signaling | 9252P |
DNA constructs
Human TAP-PTHR in pIRES-puro-SS-GLUE (a gift from Drs. Jean-Luc Parent (University of Sherbrooke) and Terence E. Hébert (McGill University, Montreal, Canada)) was generated by PCR. TAP contains calmodulin-binding protein, an HA epitope, a tobacco etch virus cleavage site, and streptavidin-binding protein tags (83) and was inserted at the N terminus of PTHR. Mutant PTHRs were engineered by changing Lys to Arg, Arg to Lys, or Ser/Thr to Ala using the QuikChange (Agilent) kit following the manufacturer's instructions. All constructs were confirmed by DNA sequencing.
Cell culture and transfection
Murine embryonic fibroblasts (MEF) (33) and HEK-293S GnTI− cells were grown at 37 °C in high-glucose Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS and 100 units/ml penicillin-streptomycin in a humidified atmosphere containing 5% CO2. Plasmid transfections were performed using FuGENE 6 (Promega), Effectene (Qiagen), or Lipofectamine 3000 (Thermo Fisher Scientific) according to the manufacturer's instructions, unless otherwise specified. For transient transfection, cells were treated as above, and experiments were performed 48 h thereafter. Stably transfected cells expressing epitope-tagged receptors were generated by selecting for neomycin or puromycin resistance using 500 μg/ml G418 or 1 μg/ml puromycin. Positive colonies were isolated, and clones were further selected based on protein expression levels as assessed by immunoblot.
cAMP
cAMP was measured by GloSensor (Promega). Briefly, HEK-293 cells stably expressing the GloSensor cAMP reporter were transiently transfected with WT-PTHR or the various Lys mutant PTHRs. 48 h later, cells were washed twice with PBS and resuspended in 2 ml of Na2HPO4 and β-glycerophosphate, CO2-independent medium (Thermo Fisher Scientific, 18045088). The stipulated 1.5 × 104 cells were transferred to a 96-well black-walled plate (Costar, 07-200-762) and pretreated with 1 mm luciferin (Biotium, Fremont, CA) in the dark at room temperature for 30 min. Bioluminescence was measured at 2-min intervals for 30 min using a Mithras LB 940 multimode microplate reader (Berthold) in the absence or presence of 100 nm PTH. Each treatment was performed in quadruplicate.
Immunostaining and confocal microscopy
HEK-293S GnTI− cells stably expressing TAP(HA)-WT-PTHR or TAP(HA)-0K-PTHR were plated on coverslips coated with poly-d-Lys. Cells were labeled for 1 h at room temperature with an anti-HA primary antibody directly conjugated to DyLight 488, which recognizes the HA tag in the N-terminal domain of PTHR. After washout, the cells were incubated with vehicle or 100 nm PTH(1–34) for 5 min at 37 °C. Cells were fixed in 3.6% formaldehyde for 15 min and quenched in 0.1 m glycine for 5 min. Nonspecific binding was blocked by incubation in 10% goat serum. Non-internalized PTHR remaining on the cell surface were detected by labeling with a goat anti-mouse IgG secondary antibody conjugated to Alexa 594 that recognizes the mouse IgG heavy and light chains of the primary antibody. Coverslips were mounted on glass slides with FluoromountTM (Diagnostic BioSystems, Pleasanton, CA). Single-plane confocal images were captured using a Nikon Ti-E microscope with A1 confocal unit and ×60/1.45 numeric aperture objective. Fluorescent proteins were excited using 488-nm (DyLight488) and 560-nm (Alexa594) lasers. Laser intensity and microscope gain settings were maintained for all image acquisitions. Image acquisition was performed with NIS Elements software, and analysis of fluorescence intensity was executed using ImageJ software (84). The fraction of internalized receptor was calculated by comparing the intensity of Alexa 594 staining in control and in cells treated with PTH.
PTHR biotinylation and receptor internalization and recycling
Stably transfected HEK-293S GnTI− cells expressing either WT-PTHR or 0K-PTHR were grown to ≥80% confluence on 10-cm dishes and serum-starved overnight. Cells were then chilled, washed with ice-cold PBS, and immediately incubated with 0.5 mg/ml disulfide-cleavable EZ-Link sulfo-NHS-S-S-biotin (Thermo Fisher Scientific, 21331) in a buffer containing 10 mm HEPES, pH 7.6, 154 mm NaCl, 3 mm KCl, 10 mm MgCl2, 0.1 mm CaCl2, and 10 mm glucose for 45 min at 4 °C. Unreacted biotin was quenched by ice-cold PBS supplemented with 100 mm glycine. Cells were subsequently washed twice with ice-cold PBS and incubated with prewarmed, serum-free DMEM containing 100 nm PTH(1–34) for 15 min to internalize the PTHR. Cells were then chilled on ice, and remaining cell-surface biotinylated receptors were scavenged by incubating at 4 °C with two changes of 2-mercaptoethanesulfonate stripping buffer (50 mm Tris-HCl, pH 8.6, 50 mm Na-2-mercaptoethanesulfonate, 100 mm NaCl, 1 nm MgCl2, and 0.1 mm CaCl2) for 20 min each. Cells were washed twice with ice-cold PBS and incubated with prewarmed DMEM at 37 °C for 0, 15, or 30 min to permit PTHR recycling. At each time point, the first dish of each pair was stripped as above to cleave newly appearing surface biotin from recycled PTHR. The remaining PTHRs detected by immunoblot from this fraction represent non-recycled receptors. The second dish without the additional round of stripping represents the total internalized receptor. Cells were washed with ice-cold PBS and lysed in 50 mm Tris-HCl, pH 8.0, 150 mm NaCl, and 0.5% DDM. After centrifugation at 14,000 rpm for 30 min, supernatants containing equal amounts of total protein were digested with tobacco etch virus protease overnight to remove streptavidin-binding peptide in the PTHR TAP tag, followed by incubation with streptavidin-agarose beads overnight to capture biotinylated proteins. Beads were extensively washed. Protein was eluted with SDS sample buffer, resolved by SDS-PAGE, and transferred to polyvinylidene difluoride membranes and detected by immunoblotting. The recycled receptor fraction was calculated as the difference between total internalized receptor and non-recycled receptor for each pair.
Purification of TAP-PTHR
GnTI− cells stably expressing PTHR were grown on two 15-cm tissue culture dishes and transferred to 200 ml of Gibco FreeStyle 293 expression medium (Invitrogen). After a 60-h incubation at 37 °C at 8% CO2, cells were serum-starved for 1 h and treated with 10 μm MG132 for a second hour and then challenged with 100 nm PTH for 30 min. Cells were harvested by centrifugation at 1,500 rpm for 3 min and washed with cold PBS. Cells were lysed in a buffer containing 50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 10 mm N-ethylmaleimide, 0.5% DDM, and Protease Inhibitor Mixture Set I (Calbiochem, 539131). After overnight incubation at 4 °C, cell extracts were clarified by centrifugation at 16,000 rpm for 45 min. The resulting supernatant was mixed with pre-equilibrated streptavidin-conjugated agarose beads in the lysis buffer and incubated at 4 °C for 4 h. Beads were extensively washed with a buffer containing 50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 10 mm N-ethylmaleimide, 2 mm CaCl2, and 0.05% DDM. TAP-PTHR was eluted with a buffer consisting of 50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 10 mm biotin, 2 mm CaCl2, and 0.05% DDM. The protein samples were further incubated with pre-equilibrated calmodulin-conjugated agarose beads in a buffer of 50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 2 mm CaCl2, and 0.05% DDM. Proteins were eluted with a buffer consisting of 50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 10 mm EGTA, and 0.05% DDM. Protein concentrations were measured using the Bradford assay (Bio-Rad) (85).
Detection of ubiquitinated receptors
For transiently expressed receptors, GnTI− cells were grown on 10-cm dishes and transiently transfected with 2 μg each of DNA encoding WT-PTHR or the indicated mutant PTHR and Myc-ubiquitin. GnTI− cells stably expressing WT-TAP(HA)-PTHR, 0K-TAP(HA)-PTHR, or the indicated mutant were transiently transfected with Myc-ubiquitin. After 48 h, cells were washed with cold PBS, serum-staved for 2 h, and incubated with 10 μm MG132 for 30 min. Cells were then stimulated with 100 nm PTH(1–34) for 30 min. Cells were harvested, lysed, and incubated with streptavidin affinity beads as described above. After extensive washing, protein was eluted in SDS sample buffer and incubated at 37 °C for 30 min. Following electrophoresis, proteins were electroblotted onto a polyvinylidene difluoride membrane and detected by immunoblotting. To reprobe with other antibodies, the membrane was stripped in a denaturation buffer (62.5 mm Tris-HCl, pH 6.8, 100 mm β-mercaptoethanol, and 2% SDS) for 30 min at 50 °C. Proteins were detected by ECL Western blotting (GE Healthcare), unless otherwise stated, and the signals were quantified with ImageJ software (84).
PTHR degradation
HEK-293S GnTI− cells stably expressing either WT-PTHR or 0K-PTHR were transfected with Myc-ubiquitin. 36 h later, cells were seeded at equal density on 6-cm dishes and grown overnight at 37 °C. Cells were serum-starved for 2 h and incubated with 10 μg/ml cycloheximide for 1 h to arrest newly synthesized receptor. Cells were subsequently incubated at 37 °C with or without 100 nm PTH(1–34) for the times indicated, washed with cold PBS, and lysed in a buffer containing 50 mm Tris-HCl, pH 8.0, 150 mm NaCl, and 0.5% DDM supplemented with protease inhibitors (Protease Inhibitor Mixture Set I). Cell lysates were collected and normalized based on total protein concentration measured by the Bradford method (85). Equivalent amounts of cell lysates were analyzed by immunoblotting.
In-gel digestion and mass spectrometry
Purified PTHR protein was resolved by 7% SDS-PAGE and stained with Coomassie Brilliant Blue. After destaining, the gel band containing receptor protein was excised and cut into small pieces. The gel was further destained overnight in 50% acetonitrile containing 25 mm NH4HCO3 and dehydrated in 100% acetonitrile. The in-gel protein was reduced with 10 mm DTT and alkylated by 55 mm iodoacetamide. The gel was washed with 25 mm NH4HCO3 and dehydrated with 100% acetonitrile. Sufficient trypsin was added to the dried gel pieces to perform overnight in-gel digestion at 37 °C. The digested peptides were desalted using Pierce C18 spin columns and eluted in a buffer containing 75% acetonitrile and 0.1% TFA. The eluates were lyophilized and reconstituted in 0.1% formic acid. 3 μl of the peptide solution was injected and analyzed by LC-MS/MS using an LTQ Orbitrap Velos mass spectrometer (Thermo Fisher Scientific) as described (20). Collected raw data were analyzed for both ubiquitination and phosphorylation using the Sequest algorithm (86). For ubiquitination, a dynamic modification of 114.0429 Da was used for Lys residues to match the di-Gly signature. For phosphorylation, a dynamic modification of 79.9663 Da was used for Ser, Thr, and Tyr residues.
Cell proliferation and apoptosis
A total of 2 × 104 stably transfected GnTI− cells expressing WT-PTHR or 0K-PTHR were seeded in triplicate on 96-well plates (Costar, 07-200-95) and cultured at 37 °C for the time indicated. Cell proliferation was evaluated by measuring bromodeoxyuridine incorporation using the cell proliferation ELISA kit (catalog no. 11647229001) from Roche Applied Science. Apoptosis was assessed with the ELISAPLUSTM cell death detection kit (Roche Applied Science, 11544675001).
Data analysis
Results were analyzed using Prism version 7 software (GraphPad, La Jolla, CA). Results represent the mean ± S.D. of n ≥ 3 independent experiments and were compared by analysis of variance with post hoc testing using the Bonferonni procedure. p values < 0.05 were considered statistically significant.
Author contributions
Q. Z. resources; Q. Z., K. X., H. L., L. S., J. C. M., and W. B. S. investigation; Q. Z., W. B. S., and A. B. methodology; Q. Z. and P. A. F. writing-original draft; Q. Z., W. B. S., and P. A. F. writing-review and editing; W. B. S. and P. A. F. formal analysis; P. A. F. conceptualization; P. A. F. supervision; P. A. F. funding acquisition; P. A. F. validation; P. A. F. project administration.
Acknowledgments
Dr. R. J. Lefkowitz kindly provided β-arrestin MEF cells. We thank Drs. Jean-Luc Parent and Terence Hébert for the gift of the plasmid pIRES-puro-SS-GLUE.
This work was supported by National Institutes of Health Grants R01 DK105811-A1 and R01 DK111427-A1 (to P. A. F.) and R01 HL136382 (to A. B.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- PTHR
- parathyroid hormone receptor
- PTH
- parathyroid hormone
- GPCR
- G protein–coupled receptor
- β2AR
- β2-adrenergic receptor
- PAR
- protease-activated receptor
- WT-PTHR
- wildtype PTHR
- GRK
- G protein–coupled receptor kinase
- GnTI−
- N-acetylglucosaminyltransferase–deficient HEK-293S cells
- MEF
- mouse embryonic fibroblast(s)
- ERK
- extracellular signal-regulated kinase
- JNK
- Jun N-terminal kinase
- MAPK
- mitogen-activated protein kinase
- TAP2
- transforming growth factor-β–activated protein kinase-1–binding protein-2
- TAP-PTHR
- tandem affinity purification–tagged PTHR
- DDM
- n-dodecyl-β-d-maltoside
- PTHrP
- PTH-related peptide
- HEK
- human embryonic kidney
- DMEM
- Dulbecco's modified Eagle's medium
- ANOVA
- analysis of variance
- mAb
- monoclonal antibody
- SAPK
- stress-activated protein kinase
- IP
- immunoprecipitation
- IB
- immunoblotting.
References
- 1. Maeda A., Okazaki M., Baron D. M., Dean T., Khatri A., Mahon M., Segawa H., Abou-Samra A. B., Jüppner H., Bloch K. D., Potts J. T. Jr., and Gardella T. J. (2013) Critical role of parathyroid hormone (PTH) receptor-1 phosphorylation in regulating acute responses to PTH. Proc. Natl. Acad. Sci. U.S.A. 110, 5864–5869 10.1073/pnas.1301674110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vilardaga J. P., Romero G., Friedman P. A., and Gardella T. J. (2011) Molecular basis of parathyroid hormone receptor signaling and trafficking: a family B GPCR paradigm. Cell Mol. Life Sci. 68, 1–13 10.1007/s00018-010-0465-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bohinc B. N., and Gesty-Palmer D. (2012) Biased agonism at the parathyroid hormone receptor: a demonstration of functional selectivity in bone metabolism. Mini Rev. Med. Chem. 12, 856–865 10.2174/138955712800959125 [DOI] [PubMed] [Google Scholar]
- 4. Gardella T. J., and Vilardaga J.-P. (2015) International Union of Basic and Clinical Pharmacology. XCIII. The parathyroid hormone receptors: family B G protein-coupled receptors. Pharmacol. Rev. 67, 310–337 10.1124/pr.114.009464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Friedman P. A., and Goodman W. G. (2006) PTH(1–84)/PTH(7–84): a balance of power. Am. J. Physiol. Renal Physiol. 290, F975–F984 10.1152/ajprenal.00336.2005 [DOI] [PubMed] [Google Scholar]
- 6. Gesty-Palmer D., Chen M., Reiter E., Ahn S., Nelson C. D., Wang S., Eckhardt A. E., Cowan C. L., Spurney R. F., Luttrell L. M., Lefkowitz R. J. (2006) Distinct β-arrestin- and G protein-dependent pathways for parathyroid hormone receptor stimulated ERK1/2 activation. J. Biol. Chem. 281, 10856–10864 10.1074/jbc.M513380200 [DOI] [PubMed] [Google Scholar]
- 7. Vilardaga J. P., Jean-Alphonse F. G., and Gardella T. J. (2014) Endosomal generation of cAMP in GPCR signaling. Nat. Chem. Biol. 10, 700–706 10.1038/nchembio.1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tsao P., Cao T., and von Zastrow M. (2001) Role of endocytosis in mediating downregulation of G-protein-coupled receptors. Trends Pharmacol. Sci. 22, 91–96 10.1016/S0165-6147(00)01620-5 [DOI] [PubMed] [Google Scholar]
- 9. Tawfeek H. A., Qian F., and Abou-Samra A. B. (2002) Phosphorylation of the receptor for PTH and PTHrP is required for internalization and regulates receptor signaling. Mol. Endocrinol. 16, 1–13 10.1210/mend.16.1.0760 [DOI] [PubMed] [Google Scholar]
- 10. Blind E., Bambino T., and Nissenson R. A. (1995) Agonist-stimulated phosphorylation of the G protein coupled receptor for parathyroid hormone (PTH) and PTH-related protein. Endocrinology 136, 4271–4277 10.1210/endo.136.10.7664644 [DOI] [PubMed] [Google Scholar]
- 11. Blind E., Bambino T., Huang Z., Bliziotes M., and Nissenson R. A. (1996) Phosphorylation of the cytoplasmic tail of the PTH/PTHrP receptor. J. Bone Miner. Res. 11, 578–586 [DOI] [PubMed] [Google Scholar]
- 12. Dicker F., Quitterer U., Winstel R., Honold K., and Lohse M. J. (1999) Phosphorylation-independent inhibition of parathyroid hormone receptor signaling by G protein-coupled receptor kinases. Proc. Natl. Acad. Sci. U.S.A. 96, 5476–5481 10.1073/pnas.96.10.5476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rajagopal S., and Shenoy S. K. (2018) GPCR desensitization: acute and prolonged phases. Cell. Signal. 41, 9–16 10.1016/j.cellsig.2017.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Skieterska K., Rondou P., and Van Craenenbroeck K. (2017) Regulation of G protein-coupled receptors by ubiquitination. Int. J. Mol. Sci. 18, E923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shenoy S. K., Barak L. S., Xiao K., Ahn S., Berthouze M., Shukla A. K., Luttrell L. M., and Lefkowitz R. J. (2007) Ubiquitination of β-arrestin links seven-transmembrane receptor endocytosis and ERK activation. J. Biol. Chem. 282, 29549–29562 10.1074/jbc.M700852200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sun L., and Chen Z. J. (2004) The novel functions of ubiquitination in signaling. Curr. Opin. Cell Biol. 16, 119–126 10.1016/j.ceb.2004.02.005 [DOI] [PubMed] [Google Scholar]
- 17. Alonso V., and Friedman P. A. (2013) Ubiquitination-regulated G protein-coupled receptor signaling and trafficking. Mol. Endocrinol. 27, 558–572 10.1210/me.2012-1404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Komander D., and Rape M. (2012) The ubiquitin code. Annu. Rev. Biochem. 81, 203–229 10.1146/annurev-biochem-060310-170328 [DOI] [PubMed] [Google Scholar]
- 19. Alonso V., Magyar C. E., Wang B., Bisello A., and Friedman P. A. (2011) Ubiquitination–deubiquitination balance dictates ligand-stimulated PTHR sorting. J. Bone Miner. Res. 26, 2923–2934 10.1002/jbmr.494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xiao K., and Shenoy S. K. (2011) β2-Adrenergic receptor lysosomal trafficking is regulated by ubiquitination of lysyl residues in two distinct receptor domains. J. Biol. Chem. 286, 12785–12795 10.1074/jbc.M110.203091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hislop J. N., Henry A. G., and von Zastrow M. (2011) Ubiquitination in the first cytoplasmic loop of μ-opioid receptors reveals a hierarchical mechanism of lysosomal down-regulation. J. Biol. Chem. 286, 40193–40204 10.1074/jbc.M111.288555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Binkowski B. F., Butler B. L., Stecha P. F., Eggers C. T., Otto P., Zimmerman K., Vidugiris G., Wood M. G., Encell L. P., Fan F., and Wood K. V. (2011) A luminescent biosensor with increased dynamic range for intracellular cAMP. ACS Chem. Biol. 6, 1193–1197 10.1021/cb200248h [DOI] [PubMed] [Google Scholar]
- 23. Iida-Klein A., Guo J., Xie L. Y., Jüppner H., Potts J. T. Jr., Kronenberg H. M., Bringhurst F. R., Abou-Samra A. B., and Segre G. V. (1995) Truncation of the carboxyl-terminal region of the rat parathyroid hormone (PTH)/PTH-related peptide receptor enhances PTH stimulation of adenylyl cyclase but not phospholipase C. J. Biol. Chem. 270, 8458–8465 10.1074/jbc.270.15.8458 [DOI] [PubMed] [Google Scholar]
- 24. Wang B., Yang Y., and Friedman P. A. (2008) Na/H exchange regulator factor 1, a novel Akt-associating protein, regulates extracellular signal-related signaling through a B-Raf-mediated pathway. Mol. Biol. Cell 19, 1637–1645 10.1091/mbc.E07-11-1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Marchese A., and Trejo J. (2013) Ubiquitin-dependent regulation of G protein-coupled receptor trafficking and signaling. Cell. Signal. 25, 707–716 10.1016/j.cellsig.2012.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kennedy J. E., and Marchese A. (2015) Regulation of GPCR trafficking by ubiquitin. Prog. Mol. Biol. Transl. Sci. 132, 15–38 10.1016/bs.pmbts.2015.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chaudhary S., Pak J. E., Gruswitz F., Sharma V., and Stroud R. M. (2012) Overexpressing human membrane proteins in stably transfected and clonal human embryonic kidney 293S cells. Nat. Protoc. 7, 453–466 10.1038/nprot.2011.453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bisello A., Greenberg Z., Behar V., Rosenblatt M., Suva L. J., and Chorev M. (1996) Role of glycosylation in expression and function of the human parathyroid hormone parathyroid hormone-related protein receptor. Biochemistry 35, 15890–15895 10.1021/bi962111+ [DOI] [PubMed] [Google Scholar]
- 29. Zhou A. T., Assil I., and Abou-Samra A. B. (2000) Role of asparagine-linked oligosaccharides in the function of the rat PTH/PTHrP receptor. Biochemistry 39, 6514–6520 10.1021/bi992706f [DOI] [PubMed] [Google Scholar]
- 30. Gan L., Alexander J. M., Wittelsberger A., Thomas B., and Rosenblatt M. (2006) Large-scale purification and characterization of human parathyroid hormone-1 receptor stably expressed in HEK293S GnTI− cells. Protein Expr. Purif. 47, 296–302 10.1016/j.pep.2005.11.004 [DOI] [PubMed] [Google Scholar]
- 31. Shenoy S. K., McDonald P. H., Kohout T. A., and Lefkowitz R. J. (2001) Regulation of receptor fate by ubiquitination of activated β2-adrenergic receptor and β-arrestin. Science 294, 1307–1313 10.1126/science.1063866 [DOI] [PubMed] [Google Scholar]
- 32. Groer C. E., Schmid C. L., Jaeger A. M., and Bohn L. M. (2011) Agonist-directed interactions with specific β-arrestins determine μ-opioid receptor trafficking, ubiquitination, and dephosphorylation. J. Biol. Chem. 286, 31731–31741 10.1074/jbc.M111.248310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kohout T. A., Lin F. S., Perry S. J., Conner D. A., and Lefkowitz R. J. (2001) β-Arrestin 1 and 2 differentially regulate heptahelical receptor signaling and trafficking. Proc. Natl. Acad. Sci. U.S.A. 98, 1601–1606 10.1073/pnas.98.4.1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zindel D., Engel S., Bottrill A. R., Pin J. P., Prézeau L., Tobin A. B., Bünemann M., Krasel C., and Butcher A. J. (2016) Identification of key phosphorylation sites in PTH1R which determine arrestin3 binding and fine tune receptor signaling. Biochem. J. 473, 4173–4192 10.1042/BCJ20160740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Benovic J. L., Kühn H., Weyand I., Codina J., Caron M. G., and Lefkowitz R. J. (1987) Functional desensitization of the isolated β-adrenergic receptor by the β-adrenergic receptor kinase: potential role of an analog of the retinal protein arrestin (48-kDa protein). Proc. Natl. Acad. Sci. U.S.A. 84, 8879–8882 10.1073/pnas.84.24.8879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kang D. S., Tian X., and Benovic J. L. (2013) β-Arrestins and G protein-coupled receptor trafficking. Methods Enzymol. 521, 91–108 10.1016/B978-0-12-391862-8.00005-3 [DOI] [PubMed] [Google Scholar]
- 37. Luttrell L. M., Ferguson S. S. G., Daaka Y., Miller W. E., Maudsley S., Della Rocca G. J., Lin F., Kawakatsu H., Owada K., Luttrell D. K., Caron M. G., and Lefkowitz R. J. (1999) β-arrestin-dependent formation of β2 adrenergic receptor Src protein kinase complexes. Science 283, 655–661 10.1126/science.283.5402.655 [DOI] [PubMed] [Google Scholar]
- 38. Johnson G. L., and Lapadat R. (2002) Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 298, 1911–1912 10.1126/science.1072682 [DOI] [PubMed] [Google Scholar]
- 39. McDonald P. H., Chow C. W., Miller W. E., Laporte S. A., Field M. E., Lin F. T., Davis R. J., and Lefkowitz R. J. (2000) β-Arrestin 2: a receptor-regulated MAPK scaffold for the activation of JNK3. Science 290, 1574–1577 10.1126/science.290.5496.1574 [DOI] [PubMed] [Google Scholar]
- 40. Luttrell L. M., and Lefkowitz R. J. (2002) The role of β-arrestins in the termination and transduction of G-protein-coupled receptor signals. J. Cell Sci. 115, 455–465 [DOI] [PubMed] [Google Scholar]
- 41. Friedman P. A., Gesek F. A., Morley P., Whitfield J. F., and Willick G. E. (1999) Cell-specific signaling and structure-activity relations of parathyroid hormone analogs in mouse kidney cells. Endocrinology 140, 301–309 10.1210/endo.140.1.6462 [DOI] [PubMed] [Google Scholar]
- 42. Sneddon W. B., Magyar C. E., Willick G. E., Syme C. A., Galbiati F., Bisello A., and Friedman P. A. (2004) Ligand-selective dissociation of activation and internalization of the parathyroid hormone receptor: conditional efficacy of PTH peptide fragments. Endocrinology 145, 2815–2823 10.1210/en.2003-1185 [DOI] [PubMed] [Google Scholar]
- 43. Omasits U., Ahrens C. H., Müller S., and Wollscheid B. (2014) Protter: interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics 30, 884–886 10.1093/bioinformatics/btt607 [DOI] [PubMed] [Google Scholar]
- 44. Newton K., Matsumoto M. L., Wertz I. E., Kirkpatrick D. S., Lill J. R., Tan J., Dugger D., Gordon N., Sidhu S. S., Fellouse F. A., Komuves L., French D. M., Ferrando R. E., Lam C., Compaan D., et al. (2008) Ubiquitin chain editing revealed by polyubiquitin linkage-specific antibodies. Cell 134, 668–678 10.1016/j.cell.2008.07.039 [DOI] [PubMed] [Google Scholar]
- 45. El Ayadi A., Stieren E. S., Barral J. M., and Boehning D. (2012) Ubiquilin-1 regulates amyloid precursor protein maturation and degradation by stimulating K63-linked polyubiquitination of lysine 688. Proc. Natl. Acad. Sci. U.S.A. 109, 13416–13421 10.1073/pnas.1206786109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Takeda A. N., Oberoi-Khanuja T. K., Glatz G., Schulenburg K., Scholz R. P., Carpy A., Macek B., Remenyi A., and Rajalingam K. (2014) Ubiquitin-dependent regulation of MEKK2/3-MEK5-ERK5 signaling module by XIAP and cIAP1. EMBO J. 33, 1784–1801 10.15252/embj.201487808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang W., and Liu H. T. (2002) MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 12, 9–18 10.1038/sj.cr.7290105 [DOI] [PubMed] [Google Scholar]
- 48. Shenoy S. K., and Lefkowitz R. J. (2011) β-arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol. Sci. 32, 521–533 10.1016/j.tips.2011.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Martin N. P., Lefkowitz R. J., and Shenoy S. K. (2003) Regulation of V2 vasopressin receptor degradation by agonist-promoted ubiquitination. J. Biol. Chem. 278, 45954–45959 10.1074/jbc.M308285200 [DOI] [PubMed] [Google Scholar]
- 50. Dores M. R., and Trejo J. (2014) Atypical regulation of G protein-coupled receptor intracellular trafficking by ubiquitination. Curr. Opin. Cell Biol. 27, 44–50 10.1016/j.ceb.2013.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dupré D. J., Chen Z., Le Gouill C., Thériault C., Parent J. L., Rola-Pleszczynski M., and Stankova J. (2003) Trafficking, ubiquitination, and down-regulation of the human platelet-activating factor receptor. J. Biol. Chem. 278, 48228–48235 10.1074/jbc.M304082200 [DOI] [PubMed] [Google Scholar]
- 52. Chan A. S. M., Clairfeuille T., Landao-Bassonga E., Kinna G., Ng P. Y., Loo L. S., Cheng T. S., Zheng M., Hong W., Teasdale R. D., Collins B. M., and Pavlos N. J. (2016) Sorting Nexin 27 couples PTHR trafficking to retromer for signal regulation in osteoblasts during bone growth. Mol. Biol. Cell 27, 1367–1382 10.1091/mbc.E15-12-0851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. McGarvey J. C., Xiao K., Bowman S. L., Mamonova T., Zhang Q., Bisello A., Sneddon W. B., Ardura J. A., Jean-Alphonse F., Vilardaga J. P., Puthenveedu M. A., and Friedman P. A. (2016) Actin-sorting nexin 27 (SNX27)-retromer complex mediates rapid parathyroid hormone receptor recycling. J. Biol. Chem. 291, 10986–11002 10.1074/jbc.M115.697045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mukhopadhyay D., and Riezman H. (2007) Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science 315, 201–205 10.1126/science.1127085 [DOI] [PubMed] [Google Scholar]
- 55. Hicke L., and Riezman H. (1996) Ubiquitination of a yeast plasma membrane receptor signals its ligand-stimulated endocytosis. Cell 84, 277–287 10.1016/S0092-8674(00)80982-4 [DOI] [PubMed] [Google Scholar]
- 56. Terrell J., Shih S., Dunn R., and Hicke L. (1998) A function for monoubiquitination in the internalization of a G protein-coupled receptor. Mol. Cell 1, 193–202 10.1016/S1097-2765(00)80020-9 [DOI] [PubMed] [Google Scholar]
- 57. Shenoy S. K., Xiao K., Venkataramanan V., Snyder P. M., Freedman N. J., and Weissman A. M. (2008) Nedd4 mediates agonist-dependent ubiquitination, lysosomal targeting, and degradation of the β2-adrenergic receptor. J. Biol. Chem. 283, 22166–22176 10.1074/jbc.M709668200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chen B., Dores M. R., Grimsey N., Canto I., Barker B. L., and Trejo J. (2011) Adaptor protein complex-2 (AP-2) and epsin-1 mediate protease-activated receptor-1 internalization via phosphorylation- and ubiquitination-dependent sorting signals. J. Biol. Chem. 286, 40760–40770 10.1074/jbc.M111.299776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nguyen L. K., Kolch W., and Kholodenko B. N. (2013) When ubiquitination meets phosphorylation: a systems biology perspective of EGFR/MAPK signalling. Cell Commun. Signal. 11, 52 10.1186/1478-811X-11-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hicke L., Zanolari B., and Riezman H. (1998) Cytoplasmic tail phosphorylation of the α-factor receptor is required for its ubiquitination and internalization. J. Cell Biol. 141, 349–358 10.1083/jcb.141.2.349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Premont R. T., Inglese J., and Lefkowitz R. J. (1995) Protein kinases that phosphorylate activated G protein-coupled receptors. FASEB J. 9, 175–182 10.1096/fasebj.9.2.7781920 [DOI] [PubMed] [Google Scholar]
- 62. Diviani D., Lattion A. L., Larbi N., Kunapuli P., Pronin A., Benovic J. L., and Cotecchia S. (1996) Effect of different G protein-coupled receptor kinases on phosphorylation and desensitization of the α1B-adrenergic receptor. J. Biol. Chem. 271, 5049–5058 10.1074/jbc.271.9.5049 [DOI] [PubMed] [Google Scholar]
- 63. Flannery P. J., and Spurney R. F. (2001) Domains of the parathyroid hormone (PTH) receptor required for regulation by G protein-coupled receptor kinases (GRKs). Biochem. Pharmacol. 62, 1047–1058 10.1016/S0006-2952(01)00749-3 [DOI] [PubMed] [Google Scholar]
- 64. Arttamangkul S., Lau E. K., Lu H. W., and Williams J. T. (2012) Desensitization and trafficking of μ-opioid receptors in locus ceruleus neurons: modulation by kinases. Mol. Pharmacol. 81, 348–355 10.1124/mol.111.076208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Freedman N. J., Kim L. K., Murray J. P., Exum S. T., Brian L., Wu J. H., and Peppel K. (2002) Phosphorylation of the platelet-derived growth factor receptor-β and epidermal growth factor receptor by G protein-coupled receptor kinase-2: mechanisms for selectivity of desensitization. J. Biol. Chem. 277, 48261–48269 10.1074/jbc.M204431200 [DOI] [PubMed] [Google Scholar]
- 66. Drake M. T., Shenoy S. K., and Lefkowitz R. J. (2006) Trafficking of G protein-coupled receptors. Circ. Res. 99, 570–582 10.1161/01.RES.0000242563.47507.ce [DOI] [PubMed] [Google Scholar]
- 67. DeWire S. M., Ahn S., Lefkowitz R. J., and Shenoy S. K. (2007) β-Arrestins and cell signaling. Annu. Rev. Physiol. 69, 483–510 10.1146/annurev.physiol.69.022405.154749 [DOI] [PubMed] [Google Scholar]
- 68. Sehat B., Andersson S., Vasilcanu R., Girnita L., and Larsson O. (2007) Role of ubiquitination in IGF-1 receptor signaling and degradation. PLoS One 2, e340 10.1371/journal.pone.0000340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Grimsey N. J., Aguilar B., Smith T. H., Le P., Soohoo A. L., Puthenveedu M. A., Nizet V., and Trejo J. (2015) Ubiquitin plays an atypical role in GPCR-induced p38 MAP kinase activation on endosomes. J. Cell Biol. 210, 1117–1131 10.1083/jcb.201504007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Shenoy S. K. (2007) Seven-transmembrane receptors and ubiquitination. Circ. Res. 100, 1142–1154 10.1161/01.RES.0000261939.88744.5a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Goldsmith Z. G., and Dhanasekaran D. N. (2007) G protein regulation of MAPK networks. Oncogene 26, 3122–3142 10.1038/sj.onc.1210407 [DOI] [PubMed] [Google Scholar]
- 72. Xia Z., Dickens M., Raingeaud J., Davis R. J., and Greenberg M. E. (1995) Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 270, 1326–1331 10.1126/science.270.5240.1326 [DOI] [PubMed] [Google Scholar]
- 73. Matsumoto Y., La Rose J., Lim M., Adissu H. A., Law N., Mao X., Cong F., Mera P., Karsenty G., Goltzman D., Changoor A., Zhang L., Stajkowski M., Grynpas M. D., Bergmann C., and Rottapel R. (2017) Ubiquitin ligase RNF146 coordinates bone dynamics and energy metabolism. J. Clin. Invest. 127, 2612–2625 10.1172/JCI92233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Shirakawa J., Harada H., Noda M., and Ezura Y. (2016) PTH-induced osteoblast proliferation requires upregulation of the ubiquitin-specific peptidase 2 (Usp2) expression. Calcif. Tissue Int. 98, 306–315 10.1007/s00223-015-0083-5 [DOI] [PubMed] [Google Scholar]
- 75. Ravingerová T., Barancík M., and Strnisková M. (2003) Mitogen-activated protein kinases: a new therapeutic target in cardiac pathology. Mol. Cell. Biochem. 247, 127–138 10.1023/A:1024119224033 [DOI] [PubMed] [Google Scholar]
- 76. Cossa G., Gatti L., Cassinelli G., Lanzi C., Zaffaroni N., and Perego P. (2013) Modulation of sensitivity to antitumor agents by targeting the MAPK survival pathway. Curr. Pharm. Des. 19, 883–894 10.2174/138161213804547187 [DOI] [PubMed] [Google Scholar]
- 77. Cheng Y., Zhang G., and Li G. (2013) Targeting MAPK pathway in melanoma therapy. Cancer Metastasis Rev. 32, 567–584 10.1007/s10555-013-9433-9 [DOI] [PubMed] [Google Scholar]
- 78. Marie P. J. (2012) Signaling pathways affecting skeletal health. Curr. Osteoporos. Rep. 10, 190–198 10.1007/s11914-012-0109-0 [DOI] [PubMed] [Google Scholar]
- 79. Guo Y., Yuan W., Wang L., Shang M., and Peng Y. (2011) Parathyroid hormone-potentiated connective tissue growth factor expression in human renal proximal tubular cells through activating the MAPK and NF-κB signalling pathways. Nephrol. Dial. Transplant. 26, 839–847 10.1093/ndt/gfq521 [DOI] [PubMed] [Google Scholar]
- 80. Guo Y. S., Yuan W. J., Zhang A. P., Ding Y. H., and Wang Y. X. (2010) Parathyroid hormone-mitogen-activated protein kinase axis exerts fibrogenic effect of connective tissue growth factor on human renal proximal tubular cells. Chin. Med. J. 123, 3671–3676 [PubMed] [Google Scholar]
- 81. Ferrari S. L., Behar V., Chorev M., Rosenblatt M., and Bisello A. (1999) Endocytosis of ligand-human parathyroid hormone receptor 1 complexes is protein kinase C-dependent and involves β-arrestin2: real-time monitoring by fluorescence microscopy. J. Biol. Chem. 274, 29968–29975 10.1074/jbc.274.42.29968 [DOI] [PubMed] [Google Scholar]
- 82. Syme C. A., Friedman P. A., and Bisello A. (2005) Parathyroid hormone receptor trafficking contributes to the activation of extracellular signal-regulated kinases but is not required for regulation of cAMP signaling. J. Biol. Chem. 280, 11281–11288 10.1074/jbc.M413393200 [DOI] [PubMed] [Google Scholar]
- 83. Roy S. J., Glazkova I., Fréchette L., Iorio-Morin C., Binda C., Pétrin D., Trieu P., Robitaille M., Angers S., Hébert T. E., and Parent J. L. (2013) Novel, gel-free proteomics approach identifies RNF5 and JAMP as modulators of GPCR stability. Mol. Endocrinol. 27, 1245–1266 10.1210/me.2013-1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Schneider C. A., Rasband W. S., and Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nature Methods 9, 671–675 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 10.1016/0003-2697(76)90527-3 [DOI] [PubMed] [Google Scholar]
- 86. Yates J. R. 3rd, Eng J. K., McCormack A. L., and Schieltz D. (1995) Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal. Chem. 67, 1426–1436 10.1021/ac00104a020 [DOI] [PubMed] [Google Scholar]