

Abstract
S-Adenosyl-l-homocysteine hydrolase (AdoHcy hydrolase; Sah1 in yeast/AHCY in mammals) degrades AdoHcy, a by-product and strong product inhibitor of S-adenosyl-l-methionine (AdoMet)-dependent methylation reactions, to adenosine and homocysteine (Hcy). This reaction is reversible, so any elevation of Hcy levels, such as in hyperhomocysteinemia (HHcy), drives the formation of AdoHcy, with detrimental consequences for cellular methylation reactions. HHcy, a pathological condition linked to cardiovascular and neurological disorders, as well as fatty liver among others, is associated with a deregulation of lipid metabolism. Here, we developed a yeast model of HHcy to identify mechanisms that dysregulate lipid metabolism. Hcy supplementation to wildtype cells up-regulated cellular fatty acid and triacylglycerol content and induced a shift in fatty acid composition, similar to changes observed in mutants lacking Sah1. Expression of the irreversible bacterial pathway for AdoHcy degradation in yeast allowed us to dissect the impact of AdoHcy accumulation on lipid metabolism from the impact of elevated Hcy. Expression of this pathway fully suppressed the growth deficit of sah1 mutants as well as the deregulation of lipid metabolism in both the sah1 mutant and Hcy-exposed wildtype, showing that AdoHcy accumulation mediates the deregulation of lipid metabolism in response to elevated Hcy in yeast. Furthermore, Hcy supplementation in yeast led to increased resistance to cerulenin, an inhibitor of fatty acid synthase, as well as to a concomitant decline of condensing enzymes involved in very long-chain fatty acid synthesis, in line with the observed shift in fatty acid content and composition.
Keywords: fatty acid, homocysteine, lipid, Saccharomyces cerevisiae, triacylglycerol, S-adenosyl-L-homocysteine, S-adenosyl-L-homocysteine hydrolase, hyperhomocysteinemia
Introduction
Methylation plays a pivotal role in cellular metabolism. These reactions, catalyzed by more than 200 different methyltransferases in mammals (1), depend on S-adenosyl-l-methionine (AdoMet)2 as a methyl donor and generate S-adenosyl-l-homocysteine (AdoHcy) as a by-product (2). AdoHcy is a strong product inhibitor of AdoMet-dependent methyltransferases (2). S-Adenosyl-l-homocysteine hydrolase (Sah1 in yeast/AHCY in mammals) is the sole enzyme responsible for the degradation of AdoHcy to adenosine and homocysteine (Hcy) (Fig. 1A) (2). Deletion of the locus overlapping the AHCY gene in mice leads to embryonic lethality (3). Deletion of the SAH1 gene in yeast results in drastically reduced growth (4). Growth of the sah1 mutants is fully abolished by additional deletion of the MET25 gene (or any other gene) in the “sulfur assimilation pathway” (4). This pathway, which is absent in mammals, allows yeast to synthesize Hcy from sulfate in the growth medium. Hcy synthesized in the course of methylation metabolism or by the sulfur assimilation pathway can be remethylated to methionine, which can be further activated to AdoMet, or converted to cysteine, a precursor of glutathione (Fig. 1A) (5). Thus, Sah1/AHCY not only plays an important role in regulating methylation metabolism by degrading AdoHcy, but in mammals it also constitutes the sole pathway for the synthesis of Hcy, which is typically absent from the diet (5).
Figure 1.
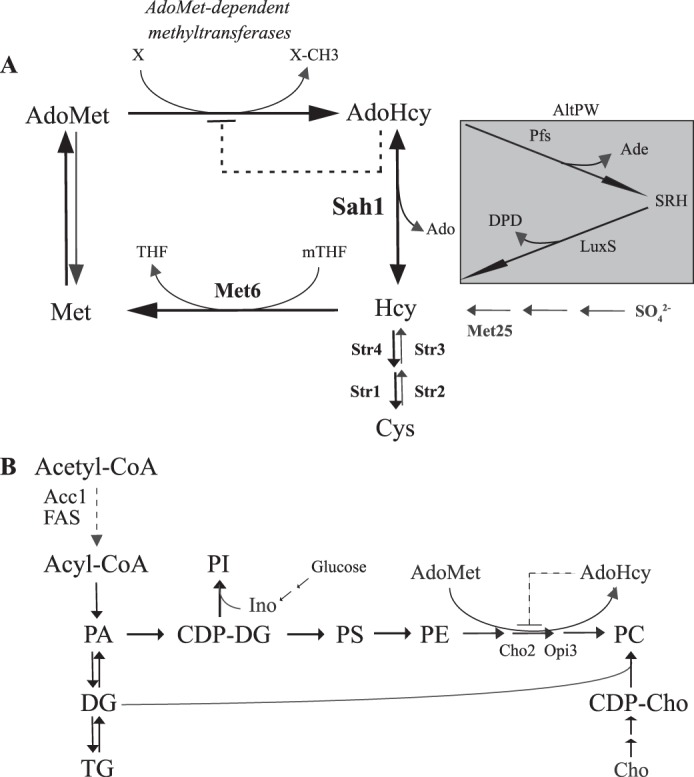
Methylation and glycerolipid metabolism in yeast. A, Sah1 catalyzes the reversible hydrolysis of AdoHcy to Hcy and adenosine. B, an accumulation of AdoHcy inhibits PC synthesis from PE by the three-step AdoMet-dependent methylation pathway. The alternative bacterial pathway for AdoHcy catabolism (AltPW) is shown in the gray box in A.
A pathological condition in humans termed hyperhomocysteinemia (HHcy) is characterized by elevated levels of Hcy in the serum (6). HHcy is linked to a number of disorders of modern society, including cardiovascular and neurological diseases, fatty liver, insulin resistance, and cancer (6–9). Notably, HHcy is present in 5–10% of the general population (10) and up to 30% of the elderly (>65 years) (11); however, mechanisms linking Hcy accumulation to disease phenotypes are unclear. Because the Sah1/AHCY-catalyzed reaction is reversible and the equilibrium of the reaction in vivo strongly favors the anabolic direction (4, 12–14), any elevation of Hcy levels, such as in HHcy, drives the formation of AdoHcy, which was shown to be a more sensitive marker of HHcy-associated pathology than Hcy (15, 16) .
Elevated Hcy and/or AdoHcy are associated with triacylglycerol (TG) accumulation in liver, endothelial, and smooth muscle cells (7, 17–19), adipocyte dysfunction (20, 21), and overall loss of fat mass (22), indicating a major impact on cellular lipid homeostasis in mammals. Furthermore, both elevated Hcy and AdoHcy lead to a depletion of polyunsaturated fatty acids in liver and plasma phospholipids (23–26). Some of these phenotypes are consistent with an inhibition of phospholipid (PL) methylation, which is also associated with TG accumulation in the liver and polyunsaturated fatty acid depletion (27–29). In mammals, about 50% of total AdoMet is used for the de novo synthesis of the major membrane PL, phosphatidylcholine (PC), by the three-step AdoMet-dependent methylation of phosphatidylethanolamine (PE) (Fig. 1B) (30, 31). PL methyltransferases that catalyze PC synthesis by the methylation pathway in mammals as well as in yeast are particularly sensitive to AdoHcy in vitro (32, 33).
Sah1/AHCY (EC 3.3.1.1) is an exceptionally well-conserved enzyme that exhibits 70% sequence identity at the protein level between human and yeast orthologs (34). Because degradation of AdoHcy is a universal regulator of methylation metabolism, some organisms, including Escherichia coli, that lack Sah1/AHCY orthologs, have evolved an alternative irreversible two-step pathway for AdoHcy catabolism that involves S-adenosylhomocysteine nucleosidase (Pfs, EC 3.2.2.9) and S-ribosylhomocysteine lyase (LuxS, EC 4.4.1.21) (35) (Fig. 1A). This pathway is referred to as AltPW (alternative pathway) throughout this study. We have previously shown that Sah1 is regulated at the transcriptional level in coordination with PL biosynthetic genes; SAH1 expression is repressed in the presence and derepressed in the absence of lipid precursors, inositol and choline (36). Similarly to Hcy supplementation, down-regulation of SAH1 expression leads to AdoHcy and TG accumulation in yeast in the absence of inositol and choline (4). Here, we dissected AdoHcy- from Hcy-evoked effects by expressing the alternative irreversible bacterial pathway for AdoHcy degradation in wildtype yeast and sah1 mutants. The AltPW indeed fully suppressed the growth defect of the sah1 mutant, providing clear evidence that the poor growth of the sah1 mutant is linked to AdoHcy accumulation. In addition, the AltPW suppressed Hcy-induced lipid alterations, including TG accumulation, increased FA content, and altered FA profiles, showing that indeed AdoHcy accumulation is the key trigger of the deregulation of lipid metabolism in response to Hcy in yeast. This deregulation appears to involve the deregulation of the enzymes involved in FA synthesis, FA synthase (FAS) as well as condensing enzymes of FA elongase complex and FA desaturase, by AdoHcy, contributing to the complex alterations in cellular lipid homeostasis.
Results
The growth defects of the sah1 single and sah1met25 double mutants are of different nature
Sah1/AHCY offers a single way to dissipate the potent product inhibitor AdoHcy in yeast and mammals and also provides Hcy for cysteine synthesis and/or remethylation to methionine. The yeast sah1 mutant is viable, displaying markedly impaired growth; additional disruption of the sulfur assimilation pathway in the sah1 mutant leads, however, to inviability of the resulting double mutant, unless it is cultivated in the presence of Hcy (4) (Fig. 2). Because Hcy can be used for cysteine/glutathione or methionine/AdoMet synthesis, we wanted to investigate blockage of which pathway is responsible for the inviability of the sah1met25 double mutant. Supplementation of 1 mm Hcy suppressed the growth defect of the sah1met25 but not of the sah1 mutant (Fig. 2). In contrast, the addition of 1 mm methionine to the standard growth medium (which already contains ∼0.1 mm methionine) did not improve growth of either the sah1met25 or sah1 mutant (Fig. 2). This indicates that inviability of the sah1met25 double mutant is due to a block in Hcy synthesis, presumably leading to glutathione depletion and oxidative stress induction. In contrast, the growth defect of the sah1 mutant is instead due to AdoHcy accumulation (Fig. 2).
Figure 2.
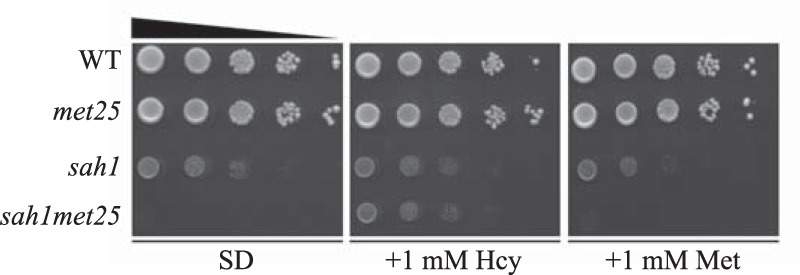
The growth defect of the yeast sah1met25 but not sah1 mutant is rescued by Hcy supplementation. Wildtype yeast and the met25, sah1, and sah1met25 mutants were analyzed for growth on SD medium containing or not containing 1 mm Hcy and 1 mm Met. Images were taken after 2 days of growth at 30 °C.
Wildtype cells exposed to Hcy show similar alterations in lipid composition as the sah1 mutant
We have previously shown that down-regulation of SAH1 expression in yeast leads to a concomitant increase of AdoHcy and TG content in the absence of inositol and choline (4). To simulate conditions of HHcy in our yeast model, wildtype cells were supplemented with 1 mm Hcy and analyzed for lipid composition. In accordance with our previous observations, Hcy-supplemented wildtype yeast cultivated in the absence of inositol and choline (−I/−C) exhibited elevated TG levels similarly to the sah1 mutant grown in the absence of Hcy (Figs. 3A and 4A). Most notably, both Hcy-supplemented wildtype and the sah1 mutant displayed elevated total FA content by some 30–60%, respectively (Figs. 3B and 4B). This indicates that the altered glycerolipid profiles in Hcy-supplemented wildtype yeast and the sah1 mutant are not simply a result of the redirection of FA metabolic flux from PL toward TG due to inhibition of PE methylation and decreased PC synthesis via the CPD-choline pathway in the absence of choline (37) (Figs. 3A and 4A) but rather a result of an up-regulated de novo FA synthesis. Furthermore, analysis of total FA composition showed a selective increase in palmitoleic (C16:1) acid (Figs. 3B and 4B), which was also observed in glycerolipids and was most pronounced in PE and TG. PE32:2 was increased 2–4-fold, and TG48:3 was increased 2–5-fold in both Hcy-supplemented wildtype and the sah1 mutant, respectively, as compared with control cells (Figs. 3C and 4C). This shift in FA profiles suggests an overall up-regulated production of FAs but also a decreased FA elongation and an increased FA desaturation in response to Hcy in yeast (Figs. 3B and 4B). Of note, Hcy supplementation did not impair wildtype growth (Fig. 2). This suggests that the increase in AdoHcy through the bidirectionally functional Sah1 reaction in Hcy-supplemented wildtype yeast may not induce the same detrimental consequences on cellular methylation as a complete loss of Sah1 activity.
Figure 3.

Hcy-supplemented wildtype yeast accumulate FAs and exhibit altered FA composition in the main glycerolipids. A, total TG, PE, and PC levels. B, total FA levels and total FA composition. C, PE, PC, and TG species distribution. Logarithmic-phase wildtype cells were grown overnight in +I/+C, shifted to −I/−C containing or not containing 1 mm Hcy, and cultivated for 6 h before lipid extraction. TG, PE, PC, and FA were normalized to ISTDs. Data are mean ± S.D. (error bars) from three independent experiments (***, p < 0.001; **, p < 0.01; *, p < 0.05).
Figure 4.

The yeast sah1 mutant phenocopies the deregulation of lipid metabolism in Hcy-supplemented wildtype. A, total TG, PE, and PC levels. B, total FA levels and total FA composition. C, PE, PC, and TG species distribution. Logarithmic-phase wildtype and the sah1 mutant yeast cells were grown overnight in +I/+C, shifted to −I/−C, and cultivated for 6 h before lipid extraction. TG, PE, PC, and FA were normalized to ISTDs. Data are mean ± S.D. (error bars) from three independent experiments (***, p < 0.001; **, p < 0.01; *, p < 0.05).
Expression of the alternative irreversible pathway for AdoHcy catabolism (AltPW) functionally complements Sah1 deficiency in yeast
Hcy supplementation leads not only to Hcy but also to AdoHcy accumulation (4). Thus, we set out to dissect AdoHcy- and Hcy-triggered effects on lipid metabolism in yeast by expressing the AltPW (Fig. 1A) in wildtype and sah1 mutant cells. pfs and luxS genes encoding S-adenosylhomocysteine nucleosidase and S-ribosylhomocysteine lyase were cloned from E. coli wildtype cells into a yeast episomal plasmid as GST fusions, as described under “Experimental procedures.” Expression of both genes was under the control of the copper-inducible CUP1p promoter. As shown in Fig. 5A, both fusion proteins were expressed in yeast and yielded distinct bands with the corresponding sizes of 24 kDa for Pfs and 19 kDa for LuxS on a Western blot. Proteins were already detectable in the absence of additional copper supplementation, indicating that the traces of copper in the standard growth medium were already sufficient to drive Pfs and LuxS expression. The AltPW fully restored wildtype growth of the sah1 mutant, suggesting that the expressed enzymes indeed degraded AdoHcy. Consistent with the Western blot data shown in Fig. 5A, growth complementation of the sah1 mutant was already observed in the absence of copper supplementation (Fig. 5B), and the addition of 0.05 mm copper had no further effect on the growth. Because we proposed that growth inhibition of the sah1 mutant was due to a buildup of AdoHcy, we next measured the content of this metabolite both in wildtype and in the sah1 mutant transformed with the expression plasmid harboring the AltPW. As shown in Fig. 5C, wildtype cells transformed with the empty plasmid showed a massive accumulation of AdoHcy in the presence of Hcy in the growth medium, consistent with the reversal of the Sah1-catalyzed reaction. Expression of the AltPW completely abolished the AdoHcy buildup in wildtype cells in the presence of Hcy, further confirming that the AltPW was functional (Fig. 5C). In contrast, the sah1 mutant transformed with the empty plasmid displayed highly elevated AdoHcy content, as expected, in the absence of Hcy in the growth medium; the further addition of Hcy had no influence on the AdoHcy levels (Fig. 5C). Expression of the AltPW completely dissipated the AdoHcy buildup in the mutant (Fig. 5C). These data clearly show that (i) Hcy supplementation drives AdoHcy accumulation in wildtype cells, (ii) AdoHcy accumulation in the sah1 mutant is independent of Hcy supplementation to the medium, and (iii) functional expression of the unidirectional bacterial AltPW leads to complete degradation of AdoHcy that accumulates in wildtype in the presence of Hcy or in the sah1 mutant and also fully restores cell growth in the absence of Sah1.
Figure 5.

The AltPW functionally complements the yeast sah1 mutants. A, expression of N-terminally tagged GST-Pfs and GST-LuxS fusion proteins in yeast. Shown are heterozygous diploid sah1/SAH1 met25/MET25 transformants carrying the empty pYEX4T-1 or pYEX-4T-1 [Pfs/LuxS] plasmid containing E. coli pfs and luxS genes under the regulation of the CUP1p promoter. B, the AltPW suppresses the growth defect of the sah1 mutant. Wildtype yeast and the sah1 mutant carrying pYEX4T-1 or pYEX-4T-1 [Pfs/LuxS] plasmid were analyzed for growth on SD media with or without 0.05 mm CuSO4. Images were taken after 2 days of growth at 30 °C. C, the AltPW suppresses elevated AdoHcy levels in the sah1 mutant and in Hcy-supplemented wildtype yeast. Logarithmic-phase wildtype and the sah1 mutant yeast cells carrying pYEX4T-1 or pYEX-4T-1 [Pfs/LuxS] plasmid were cultivated in SD−Ura with or without 0.05 mm CuSO4 and 1 mm Hcy for 6 h before AdoHcy extraction. AdoHcy levels only for strains grown in the absence of additional CuSO4 supplementation are shown. AdoHcy was normalized to ISTD. Data are mean ± S.D. (error bars) from three independent experiments (***, p < 0.001).
AdoHcy is responsible for the deregulation of lipid metabolism in the sah1 mutant and Hcy-supplemented wildtype
Hcy supplementation to wildtype yeast and lack of Sah1 activity both induce major alterations in cellular lipid homeostasis, presumably through a common mechanism of AdoHcy accumulation. Because expression of the AltPW fully suppressed AdoHcy accumulation in both strain backgrounds, we next set out to analyze its impact on cellular lipid content. Because the sah1 mutant is an inositol-overproducing (opi) strain (4) and inositol may interfere with membrane lipid (38) and TG synthesis (39), we supplemented the culture medium with 10 μm inositol, which is present in standard yeast media (40). 10 μm inositol did not affect TG levels in the sah1 mutant and Hcy-supplemented wildtype expressing the empty plasmid, suggesting that inositol is not involved in the deregulation of lipid metabolism in response to Hcy (Figs. 3, 4, and 6). In contrast, expression of the AltPW in the sah1 mutant fully restored wildtype TG as well as FA content and composition (Fig. 6A and Fig. S1), showing that AdoHcy accumulation is indeed responsible for the deregulation of lipid metabolism in the sah1 mutant. Notably, PE levels were somewhat decreased and PC levels increased in the presence of the AltPW, indicating very efficient removal of cellular AdoHcy that may otherwise inhibit PE-to-PC methylation. Although Hcy-induced lipid alterations in wildtype cells were less pronounced, in accordance with lower AdoHcy levels (Fig. 5), expression of the AltPW in Hcy-supplemented wildtype suppressed an accumulation of TG, restored its wildtype composition, and decreased the levels of total FA and in particular C16:1 (Fig. 6B and Fig. S2), indicating that AdoHcy indeed is the key trigger of the deregulation of lipid metabolism in response to Hcy in yeast.
Figure 6.

Expression of the AltPW suppresses the deregulation of lipid metabolism in the sah1 mutant and Hcy-supplemented wildtype yeast. A, total TG, PE, and PC levels in the yeast sah1 mutant. B, total FA levels and total FA composition in the yeast sah1 mutant. C, total TG, PE, and PC levels in Hcy-supplemented wildtype yeast. D, total FA levels and total FA composition in Hcy-supplemented wildtype yeast. Logarithmic-phase wildtype and the sah1 mutant yeast cells expressing the AltPW or carrying the empty plasmid were cultivated in SD−Ura (containing 10 μm inositol), reinoculated into the same medium with or without 0.05 mm CuSO4, and cultivated for 6 h before lipid extraction. Wildtype yeast expressing the AltPW or carrying the empty plasmid were cultivated in the presence or absence of 1 mm Hcy only in the SD−Ura without additional CuSO4 supplementation. Data for wildtype yeast and the sah1 mutant grown in the presence of additional CuSO4 supplementation are not shown. TG, PE, PC, and FA were normalized to ISTDs. Data are mean ± S.D. (error bars) from three independent experiments (***, p < 0.001; **, p < 0.01; *, p < 0.05).
AdoHcy accumulation impacts FA synthesis at multiple levels
A hallmark of AdoHcy accumulation both in the sah1 mutant and in Hcy-supplemented wildtype was the increase in cellular FA and TG content and alterations in FA composition. Notably, these alterations reflect metabolic changes observed under conditions of HHcy in mammals (7, 8, 18, 19, 23–26). To obtain further insight into potential mechanisms underlying these alterations, we analyzed the sensitivity of Hcy-supplemented wildtype and the sah1 mutant to the FAS inhibitor, cerulenin, which provides a reliable readout for in vivo activity of this enzyme (41). In addition, abundance and subcellular localization of FAS subunits Fas1 and Fas2 as well as condensing enzymes Elo1, Elo2, and Elo3 of the FA elongation complex and Ole1, the sole FA desaturase in yeast, were determined by Western blotting and live cell fluorescence microscopy of C-terminal GFP fusions, respectively. Markedly increased resistance of Hcy-supplemented wildtype to cerulenin suggests increased FAS activity (Fig. 7). Although the sah1 mutant displayed overall reduced growth under all conditions, it did not appear to show any response to the inhibitor in the absence or presence of Hcy (Fig. 7). Because this mutant accumulates AdoHcy under all conditions, this finding suggests that cerulenin resistance is linked to AdoHcy content. In contrast, the cho2opi3 mutant, deficient in PL methylation without being impaired in cellular methylation due to AdoHcy accumulation, exhibited wildtype sensitivity to cerulenin in the absence or presence of Hcy (Fig. 7). This shows that cerulenin resistance of Hcy-supplemented wildtype and the sah1 mutant is not mediated by the inhibition of PL methylation. Furthermore, Hcy-supplemented wildtype and the sah1 mutant displayed cerulenin resistance in the presence of inositol and choline (complete YPD medium), further indicating that neither choline nor inositol mediates cerulenin resistance of Hcy-supplemented wildtype and the sah1 mutant.
Figure 7.
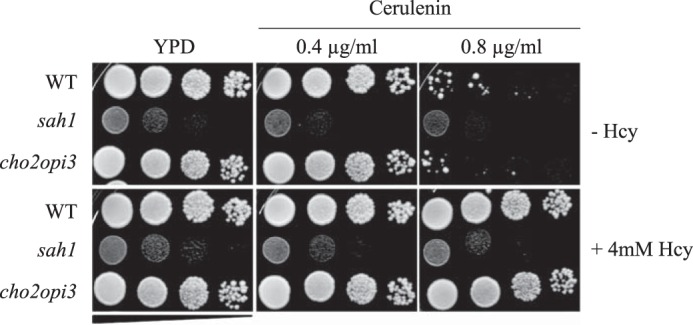
Hcy-supplemented wildtype yeast as well as the sah1 mutant are resistant to cerulenin. Wildtype yeast and the yeast sah1 and cho2opi3 mutants were analyzed for growth on YPD containing or not containing 0.4–0.6 μg/ml cerulenin and 4 mm Hcy, as indicated. Images were taken after 2 days of growth at 30 °C.
Although cerulenin resistance of wildtype cells grown in the presence of Hcy suggests an up-regulated FAS activity consistent with increased cellular FA content, this increase was not reflected in the amount of protein (Fig. 8, A and B), suggesting perhaps posttranslational modifications improving enzyme activity of an as yet unknown nature. Ole1 appears to be unaffected by the presence of Hcy (Fig. 8, A and B). In contrast, the abundance of Elo2- and Elo3-GFP fusion proteins was significantly attenuated in response to Hcy, as shown by Western blotting (Fig. 8A). Furthermore, analysis of subcellular localization of proteins involved in FA synthesis reveled that Elo2-GFP was significantly reduced at the cortical ER but not at the nuclear ER in response to Hcy (Fig. 8, B and C). The physiological relevance of this observation is currently unclear; however, the overall reduction of protein content indicates a reduced capacity of FA elongation in response to Hcy. This correlates with the observed changes in the FA chain length distribution in Hcy-supplemented wildtype and the sah1 mutant (Figs. 3 and 4). A slight increase in the abundance of Ole1 (Fig. 8A), which also correlates with increased C16:1 content (Figs. 3 and 4), suggests an up-regulation of FA desaturation in response to Hcy in yeast. However, live cell fluorescence microscopy showed neither increased protein levels nor changes in protein localization of Ole1-GFP (Fig. 8, A and B). Taken together, the up-regulated FA and TG content, the shift in FA composition of cellular lipids, and the deregulation of the enzymes involved in FA synthesis indicate that Hcy regulates lipid metabolism in yeast. Suppression of dysregulated lipid metabolism in Hcy-supplemented wildtype by the AltPW shows that the deregulation of lipid metabolism in response to Hcy in yeast is due to AdoHcy accumulation. The dysregulation of FA synthesis in response to Hcy and AdoHcy further indicates that lipid metabolic enzymes beyond PL methyltransferases are highly susceptible to elevated Hcy and an accumulation of the product inhibitor of AdoMet-dependent methylation, AdoHcy.
Figure 8.

Abundance and localization of GFP-tagged proteins involved in FA synthesis in yeast cultivated in the presence and absence of Hcy. A, Fas1, Fas2, Elo1, Elo2, Elo3, and Ole1-GFP fusion protein levels. B, subcellular localization of Fas1, Fas2, Elo1, Elo2, Elo3, and Ole1-GFP fusion proteins. Logarithmic-phase yeast strains expressing C-terminally tagged GFP fusion proteins Fas1, Fas2, Elo1, Elo2, Elo3, and Ole1-GFP (EUROSCARF) were cultivated overnight in YPD medium, washed, shifted to −I/−C containing or not containing 2 mm Hcy, and cultivated for 6 h before analysis of GFP fusion protein levels by Western blotting (A) and GFP fusion protein localization by confocal laser-scanning microscopy (B). C, quantitation of Elo2-GFP and Elo3-GPF nuclear ER- and cortical ER-labeled structures. For details, see “Experimental procedures.” Data are mean ± S.D. (error bars) from more than 150 cells (p = 0.0019).
Discussion
An accumulation of Hcy and AdoHcy is linked to a number of pathological consequences and numerous disorders of modern society (6, 42–44). Because both metabolites are physiologically connected through the Sah1/AHCY-catalyzed reaction, an elevation of Hcy levels may lead to similar pathological consequences as an elevation of AdoHcy levels (4, 12–14). Both an accumulation of Hcy and an accumulation of AdoHcy are linked to the deregulation of lipid metabolism in mammals as well as in yeast (4, 7, 8, 18, 19, 23–26), suggesting that underlying mechanisms are related.
Here, we developed a yeast model of HHcy to identify mechanisms that dysregulate lipid metabolism. Hcy supplementation to wildtype yeast, similarly to AdoHcy accumulation in the sah1 mutant, up-regulated cellular FA and TG content and induced a shift in FA composition (Figs. 3 and 4). Expression of the irreversible AltPW from E. coli enabled the dissection of AdoHcy- from Hcy-dependent effects and showed that indeed AdoHcy is the key trigger of the deregulation of lipid metabolism in response to Hcy in wildtype yeast (Fig. 6 and Figs. S1 and S2).
Defective PE methylation and concomitant inhibition of PC synthesis in the absence of choline may lead to a redirection of FA metabolic flux from membrane PL, particularly PC, toward storage TG. Indeed, mutants lacking the Cho2 and Opi3 PE- or PL-N-methyltransferases, respectively, similarly to Hcy-supplemented wildtype and the sah1 mutant, display reduced PC and elevated TG content in the absence of choline (4). TG accumulation in the mutants deficient in PL methylation, however, is suppressed by the addition of choline, which channels any buildup of the TG precursor diacylglycerol (DG) into the synthesis of PC via the CDP-choline pathway (4). In contrast, choline supplementation of the yeast mutant depleted of Sah1, while restoring PC content, could not suppress TG accumulation (4), suggesting that mechanisms leading to TG accumulation in yeast deficient in PL methylation and AdoHcy hydrolysis differ. In line with this, we observed that the yeast cho2opi3 mutant, which lacks both methyltransferases involved in PE-to-PC methylation, did not display similar resistance to cerulenin as Hcy-supplemented wildtype and the sah1 mutant (Fig. 7), further indicating that the deregulation of lipid metabolism in response to Hcy/AdoHcy goes beyond deficient PL methylation.
Resistance of the sah1 mutant and wildtype cells treated with Hcy to cerulenin (Fig. 7) suggests an up-regulation of FAS activity in response to Hcy/AdoHcy. Surprisingly, content of Fas1 and Fas2 subunits of FAS complex was unaltered, and no change in the cytosolic localization of FAS subunits in response to Hcy was observed (Fig. 8). In contrast, we observed alterations in protein levels of the condensing enzymes Elo2 and Elo3 of the FA elongase complex and Ole1, the sole FA desaturase, as well as a decrease of Elo2-GFP at the cortical ER in response to Hcy (Fig. 8, B and C). Whereas overall these alterations are consistent with the observed changes in FA profiles, the underlying molecular mechanisms affecting enzyme levels, activities, and/or localization in response to elevated Hcy are unclear. In mammals, FAS was reported to be phosphorylated (45) and acetylated (46), but there is no evidence available at present that protein methylation plays any role in regulating FAS activity in mammals or yeast. An involvement of methylation in the regulation of enzymatic activity was shown for protein phosphatase 2A; the C-terminal methylation of the Pph21/22 subunits of the protein phosphatase 2A regulates complex formation and enzymatic activity (47). However, whether the observed enzyme changes leading to deregulated lipid metabolism in response to Hcy/AdoHcy may be mediated by direct methylation defects or are a consequence of indirect effects (e.g. on (as yet unknown) kinases/phosphatases) still remains to be determined.
Experimental procedures
Strains, media, and growth conditions
Saccharomyces cerevisiae strains used in this study are congenic with BY4741, a derivative of S288C, and are listed in Table 1. Yeast strains expressing C-terminally tagged GFP full-length fusion proteins Fas1, Fas2, Elo1, Elo2, Elo3, and Ole1-GFP were obtained from EUROSCARF (48). Cells were grown at 30 °C in YPD medium containing 10 g/liter yeast extract, 20 g/liter peptone, and 20 g/liter glucose or in inositol- and choline-free synthetic minimal medium (−I/−C) containing 1.9 g/liter yeast nitrogen base without inositol, amino acids, and ammonium sulfate (Formedium), 5 g/liter ammonium sulfate, 20 g/liter glucose, supplemented with 20 mg/liter adenine, 20 mg/liter arginine, 20 mg/liter histidine, 60 mg/liter leucine, 230 mg/liter lysine, 20 mg/liter methionine, 20 mg/liter tryptophan, and 40 mg/liter uracil (49). Standard synthetic defined medium (SD), which contained 1.9 g/liter yeast nitrogen base without amino acids and ammonium sulfate (Formedium), 5 g/liter ammonium sulfate, 20 g/liter glucose, and amino acids as described above, differed from −I/−C only in that it contained 10 μm inositol. Inositol- and choline-containing medium (+I/+C) was the −I/−C medium containing 75 μm inositol and 1 mm choline. Sporulation medium contained 2.5 g/liter yeast extract, 1 g/liter glucose, and 10 g/liter potassium acetate. To select for the pYEX4T-1 (Clontech) plasmid, yeast transformants were grown on SD lacking uracil (SD−Ura). Induction of the CUP1p promoter was achieved by supplementation of media with 0.05 mm CuSO4. Media were solidified by the addition of 20 g/liter agar.
Table 1.
Strains used in this study
| Strain | Genotype | Source |
|---|---|---|
| WT | MAT a his3Δ1 leu2Δ0 ura3Δ0 | This study |
| met25 | MAT a his3Δ1 leu2Δ0 ura3Δ0 met15Δ0 | This study |
| WT pYEX4T-1 | MAT a his3Δ1 leu2Δ0 ura3Δ0 pYEX4T-1 | This study |
| WT pYEX4T-1 [Pfs/LuxS] | MAT a his3Δ1 leu2Δ0 ura3Δ0 pYEX4T-1 [Pfs/LuxS] | This study |
| sah1/SAH1 | MAT a/a his3Δ1/his3Δ1 leu2Δ0/leu2Δ0 ura3Δ0/ura3Δ0 LYS2/lys2Δ0 met25Δ0/MET25 sah1::kanMX4/SAH1 | Euroscarf |
| sah1 | MAT a his3Δ1 leu2Δ0 ura3Δ0 sah1::kanMX4 | This study |
| sah1 pYEX4T-1 | MAT a his3Δ1 leu2Δ0 ura3Δ0 sah1::kanMX4 pYEX4T-1 | This study |
| sah1 pYEX4T-1 [Pfs/LuxS] | MAT a his3Δ1 leu2Δ0 ura3Δ0 sah1::kanMX4 pYEX4T-1 [Pfs/LuxS] | This study |
| sah1met25 | MAT a his3Δ1 leu2Δ0 ura3Δ0 met15Δ0 sah1::kanMX4 | This study |
| cho2opi3 | MAT a his3Δ1 leu2Δ0 ura3Δ0 cho2::kanMX4 opi3::natMX4 | This study |
| Fas1-GFP | MAT a his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 Fas1-GFP-HIS3MX6 | Euroscarf |
| Fas2-GFP | MAT a his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 Fas2-GFP-HIS3MX6 | Eurascarf |
| Elo1-GFP | MAT a his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 Elo1-GFP-HIS3MX6 | Euroscarf |
| Elo2-GFP | MAT a his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 Elo2-GFP-HIS3MX6 | Euroscarf |
| Elo3-GFP | MAT a his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 Elo3-GFP-HIS3MX6 | Euroscarf |
| Ole1-GFP | MAT a his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 Ole1-GFP-HIS3MX6 | Euroscarf |
Cloning of genes encoding E. coli enzymes Pfs and LuxS
A 699-base pair fragment containing the pfs gene encoding S-adenosylhomocysteine nucleosidase (EC 3.2.2.9) was amplified from E. coli XL-1 Blue genomic DNA (StratageneTM) by PCR using 5′-TAGTAGGATCCATGAAAATCGGCATCATTGG-3′ and 5′-TGTATGAATTCTTAGCCATGTGCAAGTTTCTG-3′ forward and reverse primers, respectively, and Pwosuper yield DNA polymerase (Roche Applied Science). A 516-base pair fragment containing the luxS gene encoding S-ribosylhomocysteine lyase (EC 4.4.1.21) was amplified from E. coli K12 genomic DNA by PCR using 5′-TCTGAGAATTCATGCCGTTGTTAGATAGCTTCAC-3′ and 5′-TATTCTCGAGCTAGATGTGCAGTTCCTGCAACTT-3′ forward and reverse primers, respectively. The pfs fragment was cloned into the BamHI and EcoRI restriction sites of the multiple cloning site of the pYEX4T-1 vector yielding the plasmid pYEX4T-1 [Pfs]. The luxS fragment was cloned into the EcoRI and XhoI restrictions sites of the multiple cloning site of pYEX4T-1 yielding the plasmid pYEX4T-1 [LuxS]. For the construction of a vector containing both GST-tagged pfs and luxS genes, the whole expression cassette for the pfs gene was amplified from pYEX4T-1 [Pfs] by PCR using 5′-TATTTTGACGTCCCAAGCTTGCATGTCTTTTGCT-3′ and 5′-TAAATTGACGTCCATCGCGGTACCATGAATAGCTT-3′ primers and inserted into the AatII restriction site of the pYEX4T-1 [LuxS] plasmid. The plasmids pYEX4T-1 [Pfs], pYEX4T-1 [LuxS], and pYEX4T-1 [Pfs/LuxS] were verified for correct insertion by restriction analysis and by sequencing.
Expression of N-terminally GST-tagged E. coli enzymes Pfs and LuxS in yeast
Yeast wildtype and sah1 mutant cells were transformed by the method of Gietz et al. (50). Transformants were selected for uracil prototrophy on SD−Ura medium. To detect expression of the GST fusion proteins, transformants were inoculated from overnight SD−Ura cultures in 5 ml of SD−Ura containing or not 0.05 mm CuSO4 at A600 = 1. Cells were collected after cultures reached A600 = 10, and whole-yeast cell extracts were prepared by the method of Baerends et al. (51). Proteins were separated on 10% SDS-polyacrylamide gels and transferred to a nitrocellulose membrane (Bio-Rad). Goat anti-GST antibody (Sigma) followed by horseradish peroxidase-conjugated anti-goat IgG (Pierce) secondary antibody was used to detect GST fusion proteins. Chemiluminescent Western blotting substrate (Thermo Scientific) was used for detection.
AdoHcy quantification
AdoHcy was extracted by the method of Gellekink (52) with minor modifications. Yeast were grown overnight in SD−Ura, rediluted in the same medium to A600 = 0.03, grown for an additional 24 h, and reinoculated in the same medium to A600 = 0.3. After 6 h of growth, 100 A600 units were collected, stored at −70 °C, or processed immediately. For AdoHcy extraction, cells were resuspended in 5 ml of cold 0.091 m acetic acid and broken with 5-ml glass beads in a Merckenschlager homogenizer (Braun Biotech International) under CO2 cooling in the presence of 15 nmol of AdoHcy-d4 (CDN Isotopes) as an internal standard (ISTD). Glass beads were separated from the lysate by centrifugation and washed twice with 0.091 m cold acetic acid, and the supernatants were combined. AdoHcy and the internal standard were extracted from the cell lysates by solid-phase extraction on Bond Elut PBA columns (Varian Inc.) (52). Chromatographic separation of AdoHcy was performed using an AQUITY-UPLC® system (Waters) on a BEH-C18 column, 1.7-μm particle size, 3 × 150 mm (Waters). The gradient started at 96% solvent A (0.1% formic acid in water) and increased to 100% solvent B (methanol, 0.1% formic acid) over 9 min at a flow rate of 100 μl/min. AdoHcy was measured in positive ESI mode on a SYNAPTTM G1 qTOF HD mass spectrometer (Waters). The extracted ion chromatograms of m/z 385 (AdoHcy) and m/z 389 (AdoHcy-d4) were used for peak integration.
Lipid analysis
Yeast wildtype or the sah1 mutant cells were grown overnight in +I/+C, washed, inoculated to A600 = 0.3 in −I/−C containing or not 1 mm Hcy, and cultivated for 6 h. Afterward, 20 A600 units were collected, stored at −70 °C, or processed immediately. Yeast expressing the AltPW were cultivated as described under “AdoHcy quantification.” Cells were resuspended in 5 ml of chloroform/methanol (2:1, v/v) (CM), and 50 μl of 0.05 mg/ml lipid ISTD mix (53) were added. Cells were disrupted in the presence of glass beads in a Heidolph Multi Reax test tube shaker at 4 °C for 30 min. Lipids were extracted according to the Folch method (54). For analysis in positive ESI mode, dried total lipid extracts were dissolved in 1 ml of CM and diluted with 2-propanol (1:5, v/v). For analysis in negative ESI mode, dried total lipid extracts were dissolved in 1 ml of CM/2-propanol (1:10, v/v). Chromatographic separation was performed using an AQUITY-UPLC system equipped with a BEH-C18 column, 2.1 × 150 mm, 1.7 μm (Waters). A binary gradient was applied; solvent A consisted of water/methanol (1:1, v/v), and solvent B was 2-propanol. Both solvents contained phosphoric acid (8 μm), ammonium acetate (10 mm), and formic acid (0.1 volume %). The linear gradient started at 45% solvent B and increased to 100% solvent B within 32 min, and the flow rate was 150 μl/min. The column compartment was kept at 50 °C. A SYNAPTTM G1 qTOF HD mass spectrometer equipped with an ESI source was used for analysis. The following source parameters were used: capillary temperature, 100 °C; desolvatization temperature, 400 °C; N2 as nebulizer gas. The capillary voltage was 2.6 kV in positive and 2.1 kV in negative ionization mode. Leucine-enkephaline ([MH+]: m/z 556.2771 and [M−H−]: m/z 554.2615) was used as reference substance in the lock-spray. Data acquisition was done by MassLynx version 4.1 software (Waters). For lipid analysis, Lipid Data Analyzer version 1.6.2 software was used (55). Data were normalized (analyte/ISTD) for recovery, extraction, and ionization efficacy using ISTDs.
FA analysis
Dried lipid extracts were resuspended in 1 ml of CM. A 200-μl aliquot of the extract was transferred to a Pyrex glass tube, and 100 μl of ISTD (C17:0 FA, 250 μm in methanol) were added. FAs were transesterified using 2 ml of 2% HCl in methanol and 0.5 ml of toluene. After a 1-h incubation at 100 °C, 1 ml of ice-cold water was added, and methyl esters were extracted twice with 2 ml of hexane. The samples were dried under a stream of nitrogen, resuspended in 100 μl of hexane, and transferred to autosampler vials. GC-FID was performed using a Trace GC (Thermo Scientific). 1 μl of sample solution was injected (injector temperature 250 °C) and separated on a 25-m column (CP-FFAP CB, ID 0.32 mm, 0.3-μm film) using a temperature gradient from 110 to 300 °C. Data analysis was performed using XCalibur version 1.4 software (Thermo Scientific). GC-MS was performed using the Trace GC coupled to a DSQ single-stage quadrupole mass spectrometer (Thermo Scientific). 1 μl of sample solution was injected (injector temperature 250 °C) and separated on a 60-m column (CP-FFAP CB, ID 0.25 mm, 0.25 μm film) using a temperature gradient from 110 to 300 °C. Data analysis was performed using XCalibur version 1.4 software. Calibration for FA C16:0/1 and C18:0/1 was performed between 1 and 133 μg/ml, C17:0 FA was used as internal standard.
Western blotting
Yeast strains expressing chromosomally C-terminally tagged GFP fusion proteins Fas1, Fas2, Elo1, Elo2, Elo3, and Ole1-GFP were grown overnight in YPD, washed, inoculated at A600 = 0.25 in −I/−C in the presence or absence of 2 mm Hcy, and cultivated for an additional 6 h. 3 A600 units of logarithmically growing cells were harvested, and whole-yeast cell extracts were prepared by the method of Horvath and Riezman (56). Proteins were resolved on 10% SDS-polyacrylamide gels and blotted onto nitrocellulose membranes (Bio-Rad). Mouse monoclonal anti-GFP antibody (Roche Applied Science) followed by horseradish peroxidase–conjugated anti-mouse IgG (Pierce) secondary antibody was used to detect GFP fusion proteins. Rabbit polyclonal anti-GAPDH antibody (57) followed by horseradish peroxidase–conjugated anti-rabbit IgG (Pierce) secondary antibody was used to detect GAPDH, which served as loading control. Chemiluminescent Western blotting substrate (Thermo Scientific) was used for detection.
Confocal laser-scanning microscopy
Imaging was performed using a Leica SP5 confocal microscope (Leica Inc.) with spectral detection and a HCX PL APO ×63 numeric aperture 1.4 oil immersion objective. GFP was excited at 488 nm, and emission was detected between 500–550 nm. Fluorescence signal was detected using a hybrid photon detector. Fluorescence and transmission images were acquired simultaneously.
Image processing
Image processing and quantification of fluorescent protein signal were performed using the open-source software Fiji (58). Initially, image noise was reduced by Gaussian filtering (σ value 1). Ringlike nER structures typically showing increased signal compared with cER structures were extracted from the data sets using the autothreshold/triangle method. False-segmented structures located at the cER were manually removed from the binary data. Not entirely registered nER structures were closed using the “close” binary function. The mean gray-level intensity of segmented nER structures was computed. All segmented nER structures were selected using the “create selection” function, and selections were filled with the background color (black) using the “fill” function. Afterward, cER structures were extracted from the fluorescence data sets using the autothreshold/entropy method while excluding the selections previously created from the segmentation process. Again, the mean gray-level intensity of extracted cER structures was computed.
Statistics
Data are shown as the means ± S.D. Statistical significance between two groups was determined by unpaired Student's two-tailed t test. The following levels of statistical significance were used: *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Author contributions
M. V. constructed all strains used in this study; M. V., M. R., T. O. E., G. N. R., and O. T. performed lipid analysis; M. V. and M. R. performed plate tests; H. W. performed microscopy and image analysis; M. V. and M. R. performed Western blotting; S. S. and N. M. cloned and functionally expressed the AltPW in yeast; M. V. performed AdoHcy analysis; O. T. designed the study and wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Sepp D. Kohlwein for critical and helpful discussions and gratefully acknowledge excellent technical assistance by Brigitte Spreitzer and Gabriela Gogg-Fassolter. Rabbit polyclonal anti-GAPDH antibody was kindly provided by Günther Daum.
This work was supported by the Austrian Science Fund (FWF) Projects P24216 and P18094 (to O. T.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1 and S2.
- AdoMet
- S-adenosyl-l-methionine
- AHCY
- S-adenosyl-l-homocysteine hydrolase in mammals
- Sah1
- S-adenosyl-l-homocysteine hydrolase in yeast
- AdoHcy
- S-adenosyl-l-homocysteine
- Hcy
- homocysteine
- HHcy
- hyperhomocysteinemia
- FA
- fatty acid
- TG
- triacylglycerol
- PL
- phospholipid
- Pfs
- S-adenosylhomocysteine nucleosidase
- LuxS
- S-ribosylhomocysteine lyase
- AltPW
- alternative pathway for AdoHcy catabolism
- PC
- phosphatidylcholine
- PE
- phosphatidylethanolamine
- FAS
- FA synthase
- GST
- glutathione S-transferase
- GFP
- green fluorescent protein
- DG
- diacylglycerol
- ISTD
- internal standard
- CM
- chloroform/methanol
- SD
- synthetic defined
- ER
- endoplasmic reticulum
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase.
References
- 1. Petrossian T. C., and Clarke S. G. (2010) Uncovering the human methyltransferasome. Mol. Cell. Proteomics 10, M110.000976 10.1074/mcp.M110.000976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tehlivets O., Malanovic N., Visram M., Pavkov-Keller T., and Keller W. (2013) S-adenosyl-l-homocysteine hydrolase and methylation disorders: yeast as a model system. Biochim. Biophys. Acta 1832, 204–215 10.1016/j.bbadis.2012.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Miller M. W., Duhl D. M., Winkes B. M., Arredondo-Vega F., Saxon P. J., Wolff G. L., Epstein C. J., Hershfield M. S., and Barsh G. S. (1994) The mouse lethal nonagouti (a (x)) mutation deletes the S-adenosylhomocysteine hydrolase (Ahcy) gene. EMBO J. 13, 1806–1816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Malanovic N., Streith I., Wolinski H., Rechberger G., Kohlwein S. D., and Tehlivets O. (2008) S-adenosyl-l-homocysteine hydrolase, key enzyme of methylation metabolism, regulates phosphatidylcholine synthesis and triacylglycerol homeostasis in yeast: implications for homocysteine as a risk factor of atherosclerosis. J. Biol. Chem. 283, 23989–23999 10.1074/jbc.M800830200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Finkelstein J. D., and Martin J. J. (2000) Homocysteine. Int. J. Biochem. Cell Biol. 32, 385–389 10.1016/S1357-2725(99)00138-7 [DOI] [PubMed] [Google Scholar]
- 6. Schalinske K. L., and Smazal A. L. (2012) Homocysteine imbalance: a pathological metabolic marker. Adv. Nutr. 3, 755–762 10.3945/an.112.002758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Werstuck G. H., Lentz S. R., Dayal S., Hossain G. S., Sood S. K., Shi Y. Y., Zhou J., Maeda N., Krisans S. K., Malinow M. R., and Austin R. C. (2001) Homocysteine-induced endoplasmic reticulum stress causes dysregulation of the cholesterol and triglyceride biosynthetic pathways. J. Clin. Invest. 107, 1263–1273 10.1172/JCI11596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bravo E., Palleschi S., Aspichueta P., Buqué X., Rossi B., Cano A., Napolitano M., Ochoa B., and Botham K. M. (2011) High fat diet-induced non alcoholic fatty liver disease in rats is associated with hyperhomocysteinemia caused by down regulation of the transsulphuration pathway. Lipids Health Dis. 10, 60 10.1186/1476-511X-10-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pang Y., Li Y., Lv Y., Sun L., Zhang S., Li Y., Wang Y., Liu G., Xu M. J., Wang X., and Jiang C. (2016) Intermedin restores hyperhomocysteinemia-induced macrophage polarization and improves insulin resistance in mice. J. Biol. Chem. 291, 12336–12345 10.1074/jbc.M115.702654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Refsum H., Ueland P. M., Nygård O., and Vollset S. E. (1998) Homocysteine and cardiovascular disease. Annu. Rev. Med. 49, 31–62 10.1146/annurev.med.49.1.31 [DOI] [PubMed] [Google Scholar]
- 11. Selhub J., Jacques P. F., Wilson P. W., Rush D., and Rosenberg I. H. (1993) Vitamin status and intake as primary determinants of homocysteinemia in an elderly population. JAMA 270, 2693–2698 10.1001/jama.1993.03510220049033 [DOI] [PubMed] [Google Scholar]
- 12. Isa Y., Mishima T., Tsuge H., and Hayakawa T. (2006) Increase in S-adenosylhomocysteine content and its effect on the S-adenosylhomocysteine hydrolase activity under transient high plasma homocysteine levels in rats. J. Nutr. Sci. Vitaminol. 52, 479–482 10.3177/jnsv.52.479 [DOI] [PubMed] [Google Scholar]
- 13. Dayal S., Bottiglieri T., Arning E., Maeda N., Malinow M. R., Sigmund C. D., Heistad D. D., Faraci F. M., and Lentz S. R. (2001) Endothelial dysfunction and elevation of S-adenosylhomocysteine in cystathionine β-synthase-deficient mice. Circ. Res. 88, 1203–1209 10.1161/hh1101.092180 [DOI] [PubMed] [Google Scholar]
- 14. Chen Z., Karaplis A. C., Ackerman S. L., Pogribny I. P., Melnyk S., Lussier-Cacan S., Chen M. F., Pai A., John S. W., Smith R. S., Bottiglieri T., Bagley P., Selhub J., Rudnicki M. A., James S. J., and Rozen R. (2001) Mice deficient in methylenetetrahydrofolate reductase exhibit hyperhomocysteinemia and decreased methylation capacity, with neuropathology and aortic lipid deposition. Hum. Mol. Genet. 10, 433–443 10.1093/hmg/10.5.433 [DOI] [PubMed] [Google Scholar]
- 15. Kerins D. M., Koury M. J., Capdevila A., Rana S., and Wagner C. (2001) Plasma S-adenosylhomocysteine is a more sensitive indicator of cardiovascular disease than plasma homocysteine. Am. J. Clin. Nutr. 74, 723–729 10.1093/ajcn/74.6.723 [DOI] [PubMed] [Google Scholar]
- 16. Liu C., Wang Q., Guo H., Xia M., Yuan Q., Hu Y., Zhu H., Hou M., Ma J., Tang Z., and Ling W. (2008) Plasma S-adenosylhomocysteine is a better biomarker of atherosclerosis than homocysteine in apolipoprotein E-deficient mice fed high dietary methionine. J. Nutr. 138, 311–315 10.1093/jn/138.2.311 [DOI] [PubMed] [Google Scholar]
- 17. Matthews R. P., Lorent K., Mañoral-Mobias R., Huang Y., Gong W., Murray I. V., Blair I. A., and Pack M. (2009) TNFα-dependent hepatic steatosis and liver degeneration caused by mutation of zebrafish S-adenosylhomocysteine hydrolase. Development 136, 865–875 10.1242/dev.027565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kharbanda K. K., Mailliard M. E., Baldwin C. R., Beckenhauer H. C., Sorrell M. F., and Tuma D. J. (2007) Betaine attenuates alcoholic steatosis by restoring phosphatidylcholine generation via the phosphatidylethanolamine methyltransferase pathway. J. Hepatol. 46, 314–321 10.1016/j.jhep.2006.08.024 [DOI] [PubMed] [Google Scholar]
- 19. Medici V., Shibata N. M., Kharbanda K. K., LaSalle J. M., Woods R., Liu S., Engelberg J. A., Devaraj S., Török N. J., Jiang J. X., Havel P. J., Lönnerdal B., Kim K., and Halsted C. H. (2013) Wilson's disease: changes in methionine metabolism and inflammation affect global DNA methylation in early liver disease. Hepatology 57, 555–565 10.1002/hep.26047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang Z., Pini M., Yao T., Zhou Z., Sun C., Fantuzzi G., and Song Z. (2011) Homocysteine suppresses lipolysis in adipocytes by activating the AMPK pathway. Am. J. Physiol. Endocrinol. Metab. 301, E703–E712 10.1152/ajpendo.00050.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ngo S., Li X., O'Neill R., Bhoothpur C., Gluckman P., and Sheppard A. (2014) Elevated S-adenosylhomocysteine alters adipocyte functionality with corresponding changes in gene expression and associated epigenetic marks. Diabetes 63, 2273–2283 10.2337/db13-1640 [DOI] [PubMed] [Google Scholar]
- 22. Gupta S., and Kruger W. D. (2011) Cystathionine β-synthase deficiency causes fat loss in mice. PLoS One 6, e27598 10.1371/journal.pone.0027598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ikeda K., Kubo A., Akahoshi N., Yamada H., Miura N., Hishiki T., Nagahata Y., Matsuura T., Suematsu M., Taguchi R., and Ishii I. (2011) Triacylglycerol/phospholipid molecular species profiling of fatty livers and regenerated non-fatty livers in cystathionine β-synthase-deficient mice, an animal model for homocysteinemia/homocystinuria. Anal. Bioanal. Chem. 400, 1853–1863 10.1007/s00216-011-4703-2 [DOI] [PubMed] [Google Scholar]
- 24. Li D., Mann N. J., and Sinclair A. J. (2006) A significant inverse relationship between concentrations of plasma homocysteine and phospholipid docosahexaenoic acid in healthy male subjects. Lipids 41, 85–89 10.1007/s11745-006-5074-x [DOI] [PubMed] [Google Scholar]
- 25. Selley M. L. (2007) A metabolic link between S-adenosylhomocysteine and polyunsaturated fatty acid metabolism in Alzheimer's disease. Neurobiol. Aging 28, 1834–1839 10.1016/j.neurobiolaging.2006.08.003 [DOI] [PubMed] [Google Scholar]
- 26. Innis S. M., Davidson A. G., Chen A., Dyer R., Melnyk S., and James S. J. (2003) Increased plasma homocysteine and S-adenosylhomocysteine and decreased methionine is associated with altered phosphatidylcholine and phosphatidylethanolamine in cystic fibrosis. J. Pediatr. 143, 351–356 10.1067/S0022-3476(03)00326-3 [DOI] [PubMed] [Google Scholar]
- 27. Li Z., Agellon L. B., Allen T. M., Umeda M., Jewell L., Mason A., and Vance D. E. (2006) The ratio of phosphatidylcholine to phosphatidylethanolamine influences membrane integrity and steatohepatitis. Cell Metab. 3, 321–331 10.1016/j.cmet.2006.03.007 [DOI] [PubMed] [Google Scholar]
- 28. Jacobs R. L., Zhao Y., Koonen D. P., Sletten T., Su B., Lingrell S., Cao G., Peake D. A., Kuo M. S., Proctor S. D., Kennedy B. P., Dyck J. R., and Vance D. E. (2010) Impaired de novo choline synthesis explains why phosphatidylethanolamine N-methyltransferase-deficient mice are protected from diet-induced obesity. J. Biol. Chem. 285, 22403–22413 10.1074/jbc.M110.108514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Watkins S. M., Zhu X., and Zeisel S. H. (2003) Phosphatidylethanolamine-N-methyltransferase activity and dietary choline regulate liver-plasma lipid flux and essential fatty acid metabolism in mice. J. Nutr. 133, 3386–3391 10.1093/jn/133.11.3386 [DOI] [PubMed] [Google Scholar]
- 30. Noga A. A., Stead L. M., Zhao Y., Brosnan M. E., Brosnan J. T., and Vance D. E. (2003) Plasma homocysteine is regulated by phospholipid methylation. J. Biol. Chem. 278, 5952–5955 10.1074/jbc.M212194200 [DOI] [PubMed] [Google Scholar]
- 31. Stead L. M., Brosnan J. T., Brosnan M. E., Vance D. E., and Jacobs R. L. (2006) Is it time to reevaluate methyl balance in humans? Am. J. Clin. Nutr. 83, 5–10 10.1093/ajcn/83.1.5 [DOI] [PubMed] [Google Scholar]
- 32. Clarke S., and Banfield K. (2001) S-adenosylmethionine-dependent methyltransferases. In Homocysteine in Health and Disease (Carmel R., and Jackobsen D. W., eds) pp. 63–78, Cambridge University Press, Cambridge, UK [Google Scholar]
- 33. Gaynor P. M., and Carman G. M. (1990) Phosphatidylethanolamine methyltransferase and phospholipid methyltransferase activities from Saccharomyces cerevisiae: enzymological and kinetic properties. Biochim. Biophys. Acta 1045, 156–163 10.1016/0005-2760(90)90145-N [DOI] [PubMed] [Google Scholar]
- 34. Mushegian A. R., Garey J. R., Martin J., and Liu L. X. (1998) Large-scale taxonomic profiling of eukaryotic model organisms: a comparison of orthologous proteins encoded by the human, fly, nematode, and yeast genomes. Genome Res. 8, 590–598 10.1101/gr.8.6.590 [DOI] [PubMed] [Google Scholar]
- 35. Shimizu S., Shiozaki S., Ohshiro T., and Yamada H. (1984) Occurrence of S-adenosylhomocysteine hydrolase in prokaryote cells: characterization of the enzyme from Alcaligenes faecalis and role of the enzyme in the activated methyl cycle. Eur. J. Biochem. 141, 385–392 10.1111/j.1432-1033.1984.tb08203.x [DOI] [PubMed] [Google Scholar]
- 36. Tehlivets O., Hasslacher M., and Kohlwein S. D. (2004) S-Adenosyl-l-homocysteine hydrolase in yeast: key enzyme of methylation metabolism and coordinated regulation with phospholipid synthesis. FEBS Lett. 577, 501–506 10.1016/j.febslet.2004.10.057 [DOI] [PubMed] [Google Scholar]
- 37. Henry S. A., Kohlwein S. D., and Carman G. M. (2012) Metabolism and regulation of glycerolipids in the yeast Saccharomyces cerevisiae. Genetics 190, 317–349 10.1534/genetics.111.130286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gaspar M. L., Aregullin M. A., Jesch S. A., and Henry S. A. (2006) Inositol induces a profound alteration in the pattern and rate of synthesis and turnover of membrane lipids in Saccharomyces cerevisiae. J. Biol. Chem. 281, 22773–22785 10.1074/jbc.M603548200 [DOI] [PubMed] [Google Scholar]
- 39. Gaspar M. L., Hofbauer H. F., Kohlwein S. D., and Henry S. A. (2011) Coordination of storage lipid synthesis and membrane biogenesis: evidence for cross-talk between triacylglycerol metabolism and phosphatidylinositol synthesis. J. Biol. Chem. 286, 1696–1708 10.1074/jbc.M110.172296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Henry S. A., Gaspar M. L., and Jesch S. A. (2014) The response to inositol: regulation of glycerolipid metabolism and stress response signaling in yeast. Chem. Phys. Lipids 180, 23–43 10.1016/j.chemphyslip.2013.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kawaguchi A., Tomoda H., Nozoe S., Omura S., and Okuda S. (1982) Mechanism of action of cerulenin on fatty acid synthetase: effect of cerulenin on iodoacetamide-induced malonyl-CoA decarboxylase activity. J. Biochem. 92, 7–12 10.1093/oxfordjournals.jbchem.a133933 [DOI] [PubMed] [Google Scholar]
- 42. Baric I., Fumic K., Glenn B., Cuk M., Schulze A., Finkelstein J. D., James S. J., Mejaski-Bosnjak V., Pazanin L., Pogribny I. P., Rados M., Sarnavka V., Scukanec-Spoljar M., Allen R. H., Stabler S., et al. (2004) S-Adenosylhomocysteine hydrolase deficiency in a human: a genetic disorder of methionine metabolism. Proc. Natl. Acad. Sci. U.S.A. 101, 4234–4239 10.1073/pnas.0400658101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ohmori O., Hirano H., Ono T., Abe K., and Mita T. (1996) Down-regulation of S-adenosylhomocysteine hydrolase in the active methyl transfer system in the brain of genetically epileptic El mice. Neurochem. Res. 21, 1173–1180 10.1007/BF02532392 [DOI] [PubMed] [Google Scholar]
- 44. Leal J. F., Ferrer I., Blanco-Aparicio C., Hernández-Losa J., Ramón Y. C., Carnero A., and Lleonart M. E. (2008) S-Adenosylhomocysteine hydrolase downregulation contributes to tumorigenesis. Carcinogenesis 29, 2089–2095 10.1093/carcin/bgn198 [DOI] [PubMed] [Google Scholar]
- 45. Jin Q., Yuan L. X., Boulbes D., Baek J. M., Wang Y. N., Gomez-Cabello D., Hawke D. H., Yeung S. C., Lee M. H., Hortobagyi G. N., Hung M. C., and Esteva F. J. (2010) Fatty acid synthase phosphorylation: a novel therapeutic target in HER2-overexpressing breast cancer cells. Breast Cancer Res. 12, R96 10.1186/bcr2777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhao S., Xu W., Jiang W., Yu W., Lin Y., Zhang T., Yao J., Zhou L., Zeng Y., Li H., Li Y., Shi J., An W., Hancock S. M., He F., et al. (2010) Regulation of cellular metabolism by protein lysine acetylation. Science 327, 1000–1004 10.1126/science.1179689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wei H., Ashby D. G., Moreno C. S., Ogris E., Yeong F. M., Corbett A. H., and Pallas D. C. (2001) Carboxymethylation of the PP2A catalytic subunit in Saccharomyces cerevisiae is required for efficient interaction with the B-type subunits Cdc55p and Rts1p. J. Biol. Chem. 276, 1570–1577 10.1074/jbc.M008694200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Huh W. K., Falvo J. V., Gerke L. C., Carroll A. S., Howson R. W., Weissman J. S., and O'Shea E. K. (2003) Global analysis of protein localization in budding yeast. Nature 425, 686–691 10.1038/nature02026 [DOI] [PubMed] [Google Scholar]
- 49. Villa-García M. J., Choi M. S., Hinz F. I., Gaspar M. L., Jesch S. A., and Henry S. A. (2011) Genome-wide screen for inositol auxotrophy in Saccharomyces cerevisiae implicates lipid metabolism in stress response signaling. Mol. Genet. Genomics 285, 125–149 10.1007/s00438-010-0592-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gietz R. D., Schiestl R. H., Willems A. R., and Woods R. A. (1995) Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast 11, 355–360 10.1002/yea.320110408 [DOI] [PubMed] [Google Scholar]
- 51. Baerends R. J., Faber K. N., Kram A. M., Kiel J. A., van der Klei I. J., and Veenhuis M. (2000) A stretch of positively charged amino acids at the N terminus of Hansenula polymorpha Pex3p is involved in incorporation of the protein into the peroxisomal membrane. J. Biol. Chem. 275, 9986–9995 10.1074/jbc.275.14.9986 [DOI] [PubMed] [Google Scholar]
- 52. Gellekink H., van Oppenraaij-Emmerzaal D., van Rooij A., Struys E. A., den Heijer M., and Blom H. J. (2005) Stable-isotope dilution liquid chromatography-electrospray injection tandem mass spectrometry method for fast, selective measurement of S-adenosylmethionine and S-adenosylhomocysteine in plasma. Clin. Chem. 51, 1487–1492 10.1373/clinchem.2004.046995 [DOI] [PubMed] [Google Scholar]
- 53. Knittelfelder O. L., Weberhofer B. P., Eichmann T. O., Kohlwein S. D., and Rechberger G. N. (2014) A versatile ultra-high performance LC-MS method for lipid profiling. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 951, 119–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Folch J., Lees M., and Sloane Stanley G. H. (1957) A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 226, 497–509 [PubMed] [Google Scholar]
- 55. Hartler J., Trötzmüller M., Chitraju C., Spener F., Köfeler H. C., and Thallinger G. G. (2011) Lipid data analyzer: unattended identification and quantitation of lipids in LC-MS data. Bioinformatics 27, 572–577 10.1093/bioinformatics/btq699 [DOI] [PubMed] [Google Scholar]
- 56. Horvath A., and Riezman H. (1994) Rapid protein extraction from Saccharomyces cerevisiae. Yeast 10, 1305–1310 10.1002/yea.320101007 [DOI] [PubMed] [Google Scholar]
- 57. Schmidt C., Athenstaedt K., Koch B., Ploier B., and Daum G. (2013) Regulation of the yeast triacylglycerol lipase TGl3p by formation of nonpolar lipids. J. Biol. Chem. 288, 19939–19948 10.1074/jbc.M113.459610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J. Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., and Cardona A. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.