

Abstract
Aberrant cell surface glycosylation is prevalent in tumor cells, and there is ample evidence that glycans have functional roles in carcinogenesis. Nonetheless, many molecular details remain unclear. Tumor cells frequently exhibit increased α2–6 sialylation on N-glycans, a modification that is added by the ST6Gal-I sialyltransferase, and emerging evidence suggests that ST6Gal-I–mediated sialylation promotes the survival of tumor cells exposed to various cell stressors. Here we report that ST6Gal-I protects cancer cells from hypoxic stress. It is well known that hypoxia-inducible factor 1α (HIF-1α) is stabilized in hypoxic cells, and, in turn, HIF-1α directs the transcription of genes important for cell survival. To investigate a putative role for ST6Gal-I in the hypoxic response, we examined HIF-1α accumulation in ovarian and pancreatic cancer cells in ST6Gal-I overexpression or knockdown experiments. We found that ST6Gal-I activity augmented HIF-1α accumulation in cells grown in a hypoxic environment or treated with two chemical hypoxia mimetics, deferoxamine and dimethyloxalylglycine. Correspondingly, hypoxic cells with high ST6Gal-I expression had increased mRNA levels of HIF-1α transcriptional targets, including the glucose transporter genes GLUT1 and GLUT3 and the glycolytic enzyme gene PDHK1. Interestingly, high ST6Gal-I–expressing cells also had an increased pool of HIF-1α mRNA, suggesting that ST6Gal-I may influence HIF-1α expression. Finally, cells grown in hypoxia for several weeks displayed enriched ST6Gal-I expression, consistent with a pro-survival function. Taken together, these findings unravel a glycosylation-dependent mechanism that facilitates tumor cell adaptation to a hypoxic milieu.
Keywords: sialyltransferase, glycosylation, hypoxia, cancer stem cells, hypoxia-inducible factor (HIF), anoxia, cell stress, cell surface glycosylation, glycan, ST6Gal-I
Introduction
The tumor microenvironment plays a prominent role in regulating cancer cell behavior. One major microenvironmental factor that influences tumor cell phenotype is oxygen availability. Hypoxic areas within tumors are well-established niches for stem-like cancer cells, referred to as cancer stem cells (CSCs).2 Furthermore, exposing tumor cells to low oxygen tension promotes CSC characteristics, suggesting that hypoxia actively reprograms cells to acquire stem-like properties. Under conditions of oxygen deprivation, a variety of cell signaling pathways become activated. Among these, the hypoxia-inducible factor (HIF) family of transcription factors serves as a principal mediator of the cell response to hypoxia. HIF proteins, such as HIF-1α, are constitutively synthesized but rapidly degraded under normoxic conditions. When oxygen is replete, HIF-1α is hydroxylated by prolyl hydroxylase enzymes, an event that targets HIF-1α for ubiquitination and proteolytic degradation. In low oxygen tension, the lack of HIF-1α hydroxylation stabilizes the protein, allowing HIF-1α to bind HIF-1β, and translocate into the nucleus to direct the transcription of genes that enable tumor cell survival within the hypoxic milieu. In particular, HIF-1α up-regulates angiogenic genes that stimulate tumor vascularization as well as metabolic genes that facilitate anaerobic glycolysis (1, 2). In addition, HIF-dependent transcriptional activity is a key contributor to the CSC phenotype (3–7).
Deciphering the molecular mechanisms that foster tumor cell adaptation to hypoxia represents an exceptionally active area of investigation; however, the role of the tumor glycome in this process has received limited attention. Glycans lie at the interface between the extracellular environment and intracellular signaling events that dictate cell response to the external milieu. Cell surface receptors are extensively decorated with glycoconjugates, and these function to modify the activity of the cognate proteins that carry these structures. For example, sialic acid, a negatively charged sugar, can markedly alter receptor conformation, oligomerization, and/or surface retention, depending on the specific protein carrier. As a consequence, receptor glycosylation tunes receptor-driven signaling cascades that control tumor cell behavior.
Changes in the composition of receptor glycans on malignant cells are well-documented. There is a distinct subset of glycan structures commonly up-regulated in cancer, including sialyl Lewis antigens, complex β1,6-branched N-glycans, and truncated O-glycans (e.g. Tn and sialyl Tn) (8–12). Another prevalent tumor-associated alteration is an increase in the amount of α2–6–linked sialic acid added to N-glycans, a modification elaborated by the ST6Gal-I sialyltransferase (13–17). ST6Gal-I is up-regulated in numerous types of epithelial cancers, and high ST6Gal-I expression is associated with a poor patient prognosis in breast, ovarian, colon, and pancreatic adenocarcinoma (18–22). Recent studies suggest that a central cornerstone of ST6Gal-I's pro-tumorigenic function is its role in conferring a CSC phenotype (18). ST6Gal-I expression correlates with established CSC markers such ALDH1 and CD133 (19), and ST6Gal-I activity imparts hallmark CSC characteristics, including tumor spheroid growth, cell invasiveness, tumor-initiating potential, and resistance to cytotoxic stimuli, including chemotherapeutic drugs (18, 23–31). ST6Gal-I also promotes epithelial-to-mesenchymal transition (32).
In view of evidence suggesting that ST6Gal-I endows tumor cells with CSC-like properties, we investigated whether ST6Gal-I is involved in the cellular response to hypoxia. ST6Gal-I was overexpressed or knocked down in various pancreatic and ovarian cancer cell lines, and the hypoxia-induced activation of HIF-1α signaling was examined. We report that cells with high ST6Gal-I expression have elevated levels of HIF-1α protein upon exposure to either chemical hypoxia mimetics or incubation in a hypoxia chamber. Moreover, long-term cell growth in hypoxia selects for a population with enhanced ST6Gal-I expression. Consistent with HIF-1α enrichment, high ST6Gal-I expressers exhibit increased transcription of HIF-1α target genes, including the glucose transporters GLUT1 and GLUT3 and the glycolytic enzyme pyruvate dehydrogenase kinase 1 (PDHK1). Finally, hypoxic cells with high ST6Gal-I expression have increased levels of HIF-1α mRNA, thus poising the cell for greater HIF-1α accumulation upon protein stabilization. These collective results point to a novel role for tumor cell glycans in aiding tumor cell survival within hypoxic microenvironments.
Results
ST6Gal-I regulates HIF-1α accumulation after hypoxia chemical mimetic treatment
To investigate the role of ST6Gal-I in hypoxia signaling, we modulated ST6Gal-I expression in two ovarian cancer lines, OV4 and PA-1, and one pancreatic cancer line, MiaPaCa-2. ST6Gal-I was stably overexpressed in the OV4 line (Fig. 1A), which is one of the few cancer lines that lacks detectable ST6Gal-I protein. As a control, OV4 cells were stably transduced with an empty lentiviral vector (EV). Contrarily, ST6Gal-I was stably knocked down in the MiaPaCa-2 and PA-1 cell lines, both of which have high endogenous ST6Gal-I expression (Fig. 1, B and C). To confirm that manipulation of ST6Gal-I expression correlated with changes in surface sialylation, cells were stained with the SNA lectin, which specifically recognizes α2–6–linked sialic acids, and evaluated by flow cytometry. ST6Gal-I OE in OV4 cells led to an increase in SNA reactivity (Fig. 1D), whereas SNA staining was diminished by ST6Gal-I KD in MiaPaCa-2 and PA-1 cells (Fig. 1, E and F).
Figure 1.
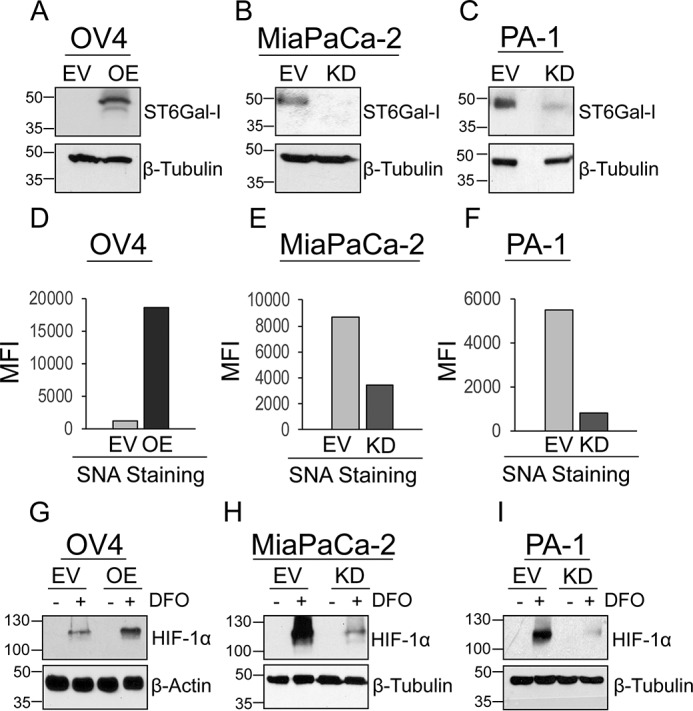
Cells with high ST6Gal-I expression have increased accumulation of HIF-1α after treatment with DFO. A, OV4 cells were stably transduced with a lentivirus encoding ST6Gal-I, and ST6Gal-I OE was verified by immunoblotting. Control cells were generated by stable transduction of an EV. B, MiaPaCa-2 cells were stably transduced with ST6Gal-I–targeting shRNA, and ST6Gal-I KD was confirmed by immunoblotting. C, PA-1 cells were stably transduced with shRNA for ST6Gal-I, and ST6Gal-I KD was verified by immunoblotting. D–F, OV4 (D), MiaPaCa-2 (E), and PA-1 cells (F) were stained with SNA-DyLIght649 to detect surface α2–6 sialic acids and evaluated by flow cytometry. The graphs depict mean fluorescence intensity (MFI). G, OV4 EV and OE cells were cultured with the hypoxia mimetic DFO for 12 h, and then whole-cell lysates were immunoblotted for HIF-1α. H and I, MiaPaCa-2 EV and KD cells (H) and PA-1 EV and KD cells (I) were cultured with DFO for 24 h and probed for HIF-1α.
Cells with variant ST6Gal-I expression were cultured in the presence or absence of the chemical hypoxia mimetic desferoxamine (DFO), an iron chelator that prevents cells from binding oxygen. In the OV4 line, greater accumulation of HIF-1α was observed in DFO-treated OE cells compared with EV cells (Fig. 1G). No detectable HIF-1α protein expression was apparent in the absence of DFO treatment, consistent with the extensive literature indicating that HIF-1α is constitutively degraded under normoxic conditions (1, 33–35). In MiaPaCa-2 and PA-1 cells, KD of ST6Gal-I caused a reduction in DFO-dependent HIF-1α expression (Fig. 1, H and I).
The role of ST6Gal-I in HIF-1α accumulation was also examined using a second chemical hypoxia mimetic, dimethyloxalylglycine (DMOG). DMOG is a specific inhibitor of prolyl hydroxylase, an enzyme that hydroxylates HIF-1α to target it for proteasomal degradation. Cells were treated with DMOG for 24 h and then immunoblotted for HIF-1α. As shown in Fig. 2A, HIF-1α was more abundant in DMOG-treated OV4 OE cells versus EV cells, whereas ST6Gal-I KD in MiaPaCa-2 cells diminished DMOG-induced HIF-1α levels (Fig. 2B). Interestingly, striking differences in cell morphology were apparent in DMOG-treated cells (Fig. 2C). DMOG treatment of OV4 EV cells (which lack ST6Gal-I) caused cell rounding and detachment from the plate, indicative of cell death. Notably, ST6Gal-I OE in OV4 cells had a strong protective effect against DMOG toxicity. Consistent with these results, DMOG did not alter the morphology of MiaPaCa-2 EV cells (with high endogenous ST6Gal-I), whereas substantial DMOG-induced cell detachment was apparent in ST6Gal-I KD cells. These results are in line with emerging literature highlighting ST6Gal-I as a potent tumor cell survival factor (18, 36–38).
Figure 2.
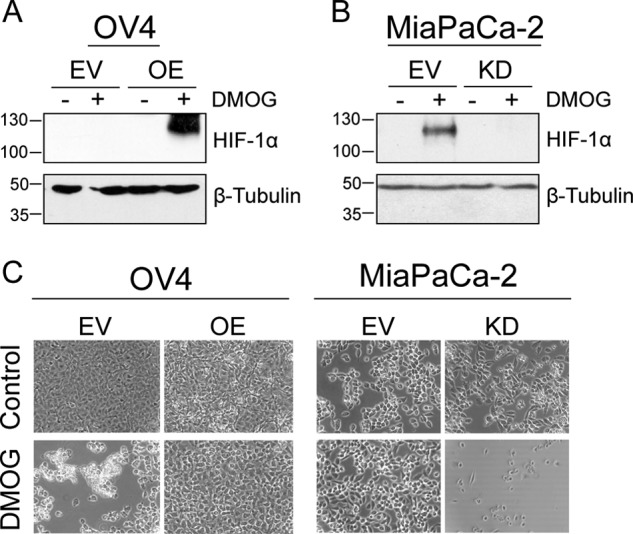
Cells with high ST6Gal-I expression have increased HIF-1α accumulation after treatment with DMOG. A, OV4 EV and OE cells were cultured with the hypoxia mimetic DMOG for 24 h, and then cell lysates were immunoblotted for HIF-1α. B, MiaPaCa-2 EV and KD cells were cultured with DMOG, and lysates were probed for HIF-1α. C, OV4 and MiaPaCa-2 cell models were imaged 24 h after treatment with DMOG.
ST6Gal-I activity enhances HIF-1α accumulation in physiological hypoxia
We next examined the contribution of ST6Gal-I to HIF-1α accumulation in cells exposed to physiological hypoxia. OV4 cells were grown for 24 h under either standard culture conditions (normoxia) or in a 2% oxygen chamber (hypoxia). After culture in hypoxia, OV4 OE cells exhibited increased levels of HIF-1α compared with EV cells (Fig. 3A). No detectable HIF-1α protein was observed in cells grown in normoxia, as expected. For the PA-1 line, cells were cultured in 0.5% O2, as these cells appeared to be less sensitive to hypoxia than OV4 cells. Compared with PA-1 EV cells, ST6Gal-I KD cells had reduced levels of HIF-1α protein at both 12 and 24 h after hypoxic growth (Fig. 3B). We also overexpressed ST6Gal-I in the PA-1 line and found that ST6Gal-I OE cells cultured in hypoxia had enriched HIF-1α at 24 h relative to EV cells (Fig. 3B). As an additional control, PA-1 cells were transduced with a nontargeting shRNA control sequence (ShC). After incubation in hypoxia, ShC cells displayed substantially greater HIF-1α abundance than KD cells (Fig. 3C). HIF-1α accumulation was next examined in the MiaPaCa-2 line. As shown in Fig. 3D, MiaPaCa-2 KD cells had lower levels of HIF1α than EV cells at all time points following exposure to hypoxia, whereas no detectable HIF-1α was observed in normoxic populations (positive control, generated from EV cells exposed to hypoxia for 24 h). Correspondingly, HIF-1α protein was reduced in hypoxic MiaPaCa-2 KD cells compared with MiaPaCa-2 cells transduced with ShC (Fig. 3E). To further verify that ST6Gal-I activity contributes to HIF-1α protein levels, an independent ST6Gal-I KD cell line was generated using a ST6Gal-I–targeting shRNA sequence different from that used to engineer KD cells (referred to as KD2). As with MiaPaCa-2 KD cells, hypoxic KD2 cells had diminished HIF-1α protein relative to the EV population (Fig. 3F). The combined results in Figs. 1–3 provide strong evidence that ST6Gal-I activity regulates the hypoxia-dependent accumulation of HIF-1α.
Figure 3.
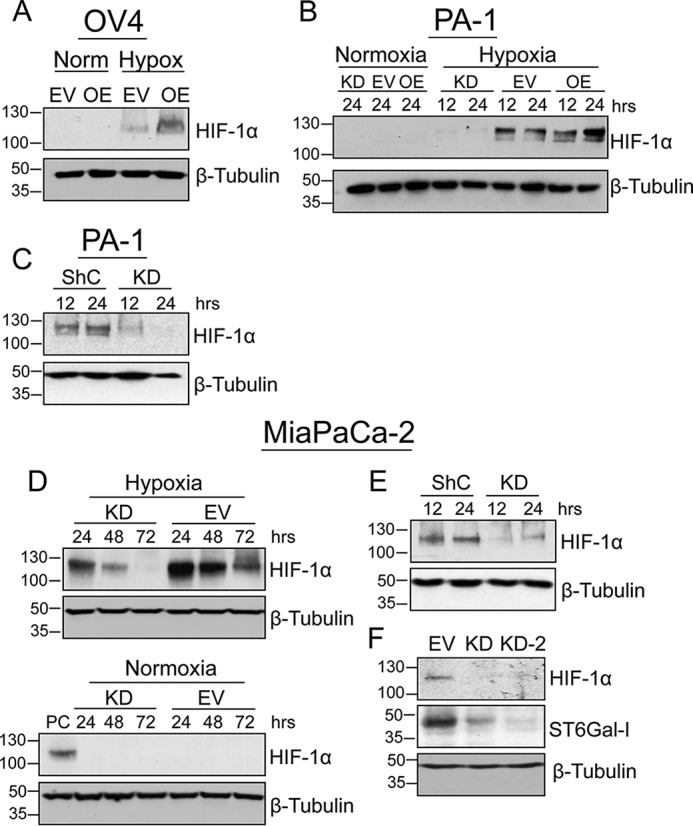
ST6Gal-I activity promotes HIF-1α accumulation in cells exposed to physiologic hypoxia. A, OV4 EV and OE cells were cultured in either normoxia (Norm) or hypoxia (Hypox) for 24 h, and then cell lysates were immunoblotted for HIF-1α. B, PA-1 cells were cultured for 12 or 24 h in hypoxia or, alternatively, for 24 h in normoxia. EV, KD, and OE cell lysates were probed for HIF-1α. C, PA-1 ShC and KD cells were cultured in hypoxia for 12 or 24 h and immunoblotted for HIF-1α. D, HIF-1α immunoblots of MiaPaCa-2 EV and KD cells cultured in hypoxia or normoxia for 24, 48, and 72 h. In the blot for normoxia (bottom panel), a positive control (PC) sample was loaded onto the gel to provide a detectable HIF-1α signal. The positive control lysate was from EV cells exposed to hypoxia for 24 h. E, MiaPaCa-2 ShC and KD cells were cultured in hypoxia for 12 or 24 h and then blotted for HIF-1α. F, MiaPaCa-2 cells transduced with two different shRNA constructs (KD and KD-2) were cultured in hypoxia for 24 h and immunoblotted for HIF-1α. Lysates were also probed for ST6Gal-I to confirm knockdown.
ST6Gal-I activity provides a proliferative advantage in hypoxic environments
Previous studies by our group have demonstrated that exposure of cells to cytotoxic stimuli or cell stressors exerts selective pressure, leading to the expansion of clonal variants with high ST6Gal-I expression (18, 19, 23, 24, 36, 37). To determine whether this was true in the context of hypoxia, we cultured parental MiaPaCa-2 cells in a hypoxia chamber for 6 weeks to generate a population capable of proliferating under chronic hypoxia (“hypoxia-adapted”). We then probed for ST6Gal-I expression and found that the hypoxia-adapted population had increased levels of ST6Gal-I relative to the initial parental population (Fig. 4A). To determine whether ST6Gal-I provides a growth advantage in hypoxia, we examined the rate of proliferation for MiaPaCa-2 EV and KD cultures exposed to hypoxia for 24, 48, or 72 h (Fig. 4B). At both 48 and 72 h, a significantly greater number of cells was apparent in EV cells compared with KD cells. Fig. 4C shows representative images for MiaPaCa-2 cells after culture in hypoxia for 72 h. These findings suggest that ST6Gal-I activity fosters the maintenance of proliferative potential in cells exposed to a hypoxic microenvironment.
Figure 4.
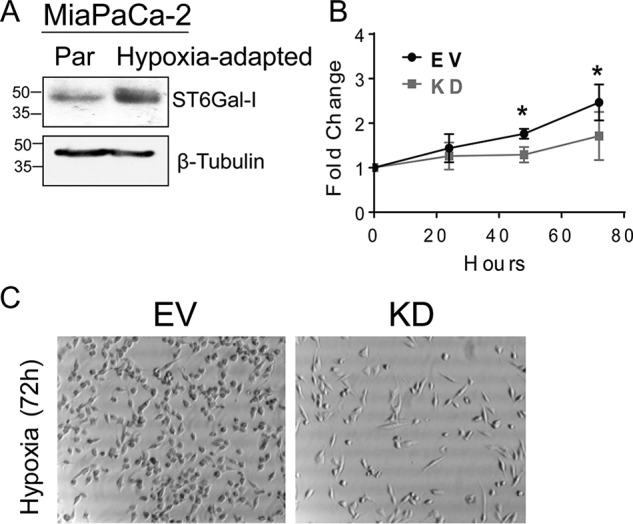
High ST6Gal-I expression provides a growth advantage. A, MiaPaCa-2 parental cells were cultured in hypoxia for 6 weeks to develop a hypoxia-adapted population. The original parental (Par) and hypoxia-adapted populations were immunoblotted for ST6Gal-I. B, MiaPaCa-2 EV and KD cells were seeded at equal density and allowed to adhere overnight in standard normoxic conditions. Cultures were then placed in hypoxia for 24, 48, or 72 h, and cell number was quantified by crystal violet staining. Values for hypoxic cultures were normalized to the overnight normoxic cultures. Values represent means and S.D. for three independent experiments. *, p < 0.05. C, representative images are shown for MiaPaCa-2 EV and KD cells grown in hypoxia for 72 h.
Cells with high ST6Gal-I expression display increased transcription of HIF target genes
Given that ST6Gal-I promoted hypoxia-induced HIF-1α accumulation (Figs. 1–3), we examined known targets of HIF-1α transcription by RT-qPCR. Under hypoxic conditions, OV4 cells with ST6Gal-I OE exhibited significantly greater transcription of GLUT1, GLUT3, and PDHK1 compared with EV cells (Fig. 5, A–C). Similarly, VEGFα mRNA levels were higher in OE cells versus EV cells; however, in this case, OE cells had increased levels of VEGFα under both hypoxic and normoxic conditions (Fig. 5D). In the MiaPaCa-2 cell line, ST6Gal-I KD cells grown in hypoxia displayed significantly reduced transcription of GLUT1, GLUT3, and PDHK1 compared with hypoxic ShC cells (Fig. 6, A–C). Interestingly, for GLUT3, ST6Gal-I KD also reduced its transcription in normoxia. However, in contrast to the OV4 cell model, ST6Gal-I activity appeared to have no significant effect on VEGFα expression in MiaPaCa-2 cells (Fig. 6D).
Figure 5.

ST6Gal-I overexpression in OV4 cells promotes the hypoxia-induced transcription of HIF-1α target genes. A–D, OV4 EV and OE cells were grown in normoxia or hypoxia for 12 or 24 h, and then RT-qPCR was used to measure the expression of GLUT1 (A), GLUT3 (B), PDHK1 (C), and VEGFα (D). Graphs depict means and S.D. from at least three independent experiments. *, p < 0.05.
Figure 6.

ST6Gal-I knockdown in MiaPaCa-2 cells decreases the hypoxia-induced transcription of HIF-1α target genes. A–D, MiaPaCa-2 ShC and KD cells were grown in normoxia or hypoxia for 12 or 24 h, and then RT-qPCR was used to measure the expression of GLUT1 (A), GLUT3 (B), PDHK1 (C), and VEGFα (D). Graphs depict means and S.D.s from at least three independent experiments. *, p < 0.05.
ST6Gal-I activity promotes the expression of HIF-1α under hypoxic conditions
Because ST6Gal-I sialylates proteins destined for the plasma membrane, we hypothesized that ST6Gal-I activity may affect HIF-1α expression. There is extensive literature suggesting that cell surface receptors primarily regulate HIF-1α biosynthesis rather than protein stabilization. Accordingly, we examined the effect of ST6Gal-I activity on HIF-1α mRNA levels. Fig. 7A shows that OV4 OE cells have higher HIF-1α mRNA expression than EV cells after culture in hypoxia for 24 h. In the MiaPaCa-2 line (Fig. 7B), ST6Gal-I KD cells had reduced expression of HIF-1α mRNA compared with ShC control cells under hypoxic conditions. These studies suggest that ST6Gal-I activity may contribute to HIF-1α accumulation by increasing HIF-1α mRNA pools.
Figure 7.

ST6Gal activity increases HIF-1α mRNA levels. A and B, OV4 (A) and MiaPaCa-2 (B) cells were cultured in normoxia or hypoxia for 12 or 24 h, and then HIF-1α mRNA expression was analyzed by RT-qPCR. C and D, levels of HIF-2α mRNA (C) or Oct4 mRNA (D) were evaluated by RT-qPCR in OV4 EV and OE cells grown in normoxia or hypoxia for 12 or 24 h. Graphs depict means and S.D. from at least three independent experiments. *, p < 0.05.
We also quantified the expression of HIF-2α. Like HIF-1α, HIF-2α is induced by hypoxia; however there is growing literature suggesting that HIF-2α is particularly associated with stem-like cancer cells (6). Moreover, although many of the transcriptional targets for HIF-1α and HIF-2α overlap, there are some distinct targets for these two factors. For instance, HIF-2α, but not HIF-1α, activates the transcription of Oct4 (39). Oct4 is a master transcription factor that maintains “stemness” in both normal stem cells and CSCs. Intriguingly, overexpression of ST6Gal-I in OV4 cells coordinately increased the mRNA levels of HIF-2α and Oct4 under both normoxic and hypoxic conditions (Fig. 7, C and D). These data are in agreement with our prior studies indicating that forced expression of ST6Gal-I in OV4 cells reprograms cells to adopt a CSC phenotype (18). However, unlike the OV4 model, MiaPaCa-2 cells did not exhibit significant differences in HIF-2α and Oct4 expression (data not shown). Further studies will be needed to dissect the role of ST6Gal-I in regulating the HIF-2α/Oct4 axis in CSCs.
Discussion
Areas of low oxygen tension are common in rapidly growing tumors because of insufficient vascularization (1). Tumor cells within these regions respond by activating the HIF pathway, which induces the expression of genes that drive angiogenesis and anaerobic metabolism. These processes are crucial for tumor cell survival. Not surprisingly, HIF signaling is often dysregulated in cancer cells. As an example, the von Hippel–Lindau enzyme, which ubiquitinates HIF-1α to target it for proteasomal degradation, is frequently inactivated in various cancers, leading to HIF-1α stabilization (40). HIF-1α expression is also up-regulated by oncogenes such as ras, Her-2/neu, and v-src (33, 41). High levels of HIF-1α protein and tissue hypoxia in tumors are correlated with poor patient outcomes as well as tumor resistance to chemo- and radiotherapy (1, 42–46). Suppression of HIF-1α expression or activity, via RNAi or dominant negative approaches, restores cancer cell responses to chemotherapeutic agents and sensitizes cells to apoptosis induced by oxygen and glucose deprivation (47–49). For these reasons, there is a compelling need to define the HIF-directed molecular events that protect tumor cells from hypoxic stress.
In this investigation, we describe a new glycosylation-dependent mechanism that stimulates HIF-1α signaling to foster hypoxia adaptation. Using multiple cell models with ST6Gal-I overexpression or knockdown, we show that ST6Gal-I activity augments hypoxia-induced HIF-1α accumulation and increases the transcription of HIF-1α target genes. In addition, cells with high ST6Gal-I expression have elevated levels of HIF-1α mRNA, which may position the cell for greater HIF-1α accumulation upon hypoxia-induced HIF-1α stabilization. Also important is that long-term culture of cells in low oxygen tension selects for a population with enriched ST6Gal-I expression, substantiating a pro-survival function for ST6Gal-I in hypoxic cells.
In tandem with regulating hypoxic response, HIFs, including HIF-1α and HIF-2α, endow tumor cells with stemlike features (3, 6, 7). As with ST6Gal-I, HIF-1α positively regulates CSC behaviors, including self-renewal, cell invasiveness, and epithelial-to-mesenchymal transition (50, 51). Moreover, HIFs are major drivers of a metabolic switch toward glycolysis. CSCs have greatly increased glucose uptake relative to more differentiated tumor cells, and CSCs are reportedly dependent on glycolysis for cell growth (50). Significantly, cells with high ST6Gal-I levels have enhanced expression of GLUT1 and GLUT3. Both of these glucose transporters play a seminal role in CSC survival (52, 53), and up-regulated GLUT3 is particularly associated with CSCs (53). HIF-2α also contributes to a CSC phenotype by activating the transcription of well-known stem cell genes such as Oct4. In the OV4 ovarian cancer model, ST6Gal-I overexpression increased the levels of HIF-2α and Oct4 mRNA under both normoxic and hypoxic conditions. These data are consistent with prior work indicating that ST6Gal-I overexpression reprograms cells into a CSC phenotype (18). Likewise, ST6Gal-I activity is important for maintaining the stemness of induced pluripotent stem cells (54).
Further investigation will be needed to fully understand the mechanism by which ST6Gal-I activity potentiates HIF-1α signaling. We speculate that ST6Gal-I is more likely to influence HIF-1α biosynthesis than HIF-1α protein stabilization, given that ST6Gal-I sialylates proteins destined for the plasma membrane (but not cytosolic proteins). Robust literature suggests that cell surface receptors mainly contribute to HIF-1α abundance through increasing HIF-1α transcription or translation (55–58). As one example, the epidermal growth factor receptor signals through phosphatidylinositol 3-kinase/Akt to activate mTOR and stimulate HIF-1α translation (59). On the other hand, the TNFR1/NF-κB axis enhances HIF-1α transcription (60, 61), and notably, NF-κB binds directly to the HIF-1α promoter (62). Our results highlighting elevated HIF-1α mRNA in cells with high ST6Gal-I expression hint at a possible role for the TNFR1/NF-κB pathway in ST6Gal-I–dependent HIF-1α accumulation. Previously we determined that TNFR1 is α2–6–sialylated by ST6Gal-I and that TNFR1 sialylation abrogates apoptotic signaling (37, 63). TNFR1 sialylation has been also shown to block receptor internalization, which, in turn, promotes long-term activation of NF-κB (37). Additionally, ST6Gal-I sustains NF-κB activation in cancer cells deprived of serum growth factors, thereby preventing exit from the cell cycle and prolonging cell viability (36).
Regardless of the mechanism by which ST6Gal-I contributes to HIF-1α accumulation, this study provides a conceptual advance by elucidating a novel role for a tumor-associated glycosyltransferase in facilitating hypoxia adaptation. Despite decades of evidence that tumor glycans contribute to a malignant cell phenotype, the mechanistic role of receptor glycans in modulating cell signaling remains a markedly underinvestigated area of cancer research. Accruing literature suggests that ST6Gal-I acts a master regulatory molecule to confer CSC properties that enable tumor cells to survive a plethora of cytotoxic assaults, including oxygen deprivation.
Experimental procedures
Cell culture
MiaPaCa-2 and PA-1 cells were obtained from the ATCC, whereas OV4 cells were a gift from Dr. Timothy Eberlein (Harvard University). For routine propagation, cell lines were cultured in Dulbecco's modified Eagle's medium (MiaPaCa-2 and PA-1) or Dulbecco's modified Eagle's medium/F12 (DME/F12) (OV4) medium, supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) antibiotics/antimycotics (GE Healthcare/Hyclone). For hypoxia mimetic studies, cells were grown in 1% FBS with either DFO or DMOG (both from Sigma) added to the medium at final concentrations of 150 μm and 1 mm, respectively (64, 65). Cells were harvested 24 h after DFO or DMOG treatment. For physiologic hypoxia studies, cells cultured in 1% FBS were placed in a hypoxia chamber for varying times. Hypoxia chambers were set up with varying levels of oxygen tension depending on the cell line. OV4 cells were typically cultured in 2% O2, whereas MiaPaCa-2 and PA-1 cells were normally grown in 0.5% O2.
Generation of cell lines with variant ST6Gal-I expression
Stable polyclonal cell lines with ST6Gal-I OE were created utilizing a lentivirus encoding the ST6Gal-I gene (Genecopoeia). Stable KD cells were generated using a lentivirus harboring shRNA against ST6Gal-I. The vectors for KD and KD-2 cells were both from Sigma (KD vector TRCN00000035432, sequence CCGGCGTGTGCTACTACTACCAGAACTCGAGTTCTGGTAGTAGTAGCACACGTTTTTG; KD-2 vector TCRN0000035433, sequence CCGGGCGCTTCCTCAAAGACAGTTTCTCGAGAAACTGTCTTTGAGGAAGCGCTTTTTG). We also generated control cell lines using an EV (Sigma). Alternatively, cells were stably transduced with ShC (Sigma, catalog no. SHC002V). Stably transduced cells were isolated by puromycin selection. Overexpression or knockdown of ST6Gal-I was verified by immunoblotting using an anti-ST6Gal-I goat polyclonal antibody, as described in more detail below. To confirm that changes in ST6Gal-I expression were correlated with alterations in cell surface α2–6 sialylation, cells were stained with the SNA lectin, which specifically binds α2–6 sialic acids. Cells were incubated for 40 min at 4 °C with a 1:200 dilution of SNA conjugated to DyLight649 (EY Laboratories, DY649-6802-1). Mean fluorescence intensity values were determined by flow cytometry.
Development of hypoxia-adapted cells
MiaPaCa-2 parental cells were cultured in 10% Dulbecco's modified Eagle's medium in a hypoxia chamber containing 2% O2 for 6 weeks. During this interval, cells were only removed from the chamber to allow passaging to prevent confluency. After 6 weeks of hypoxic culture, the hypoxia-adapted cells or initial parental MiaPaCa-2 cells were immunoblotted for ST6Gal-I.
Proliferation assay
MiaPaCa-2 EV and KD cells were seeded at equal densities in multiple plates and allowed to adhere overnight under standard normoxic conditions. After the overnight incubation, one set of plates was stained with crystal violet to obtain a baseline value for cell number. The remaining plates were placed into hypoxic culture (0.5% O2) for 24, 48, or 72 h. Subsequently, cells were fixed with 4% paraformaldehyde and then stained with a 0.5% (w/v) crystal violet solution (24). To quantify cell number, the crystal violet–stained cultures were solubilized using a 10% acetic acid solution, and solution absorbance was measured on a Biotek plate reader at 590 nm. Absorbance values for the hypoxic cultures were normalized to the baseline values (overnight culture in normoxia). Three independent experiments were performed, and differences in cell proliferation were assessed by a Student's t test.
Immunoblotting
Cells cultured with either hypoxia mimetics or in physiologic hypoxia were lysed in radioimmune precipitation assay buffer containing protease and phosphatase inhibitors (Thermo). Protein concentrations were quantified by BCA (Pierce). Samples were resolved by SDS-PAGE and transferred onto polyvinylidene difluoride membranes. Membranes were blocked by incubation in 5% nonfat dry milk dissolved in Tris-buffered saline containing 0.1% Tween 20. Membranes were then incubated with primary antibodies against ST6Gal-I (goat polyclonal, R&D Systems, catalog no. AF5924, lot no. CDSF0114101) or HIF-1α (Cell Signaling Technology, catalog no. 14179S, lot no. 1). Protein loading was confirmed using either anti-actin (Abcam, catalog no. ab20272, lot no. GR201277) or anti-β tubulin (Abcam, catalog no. ab21058, lot no. GR284232). Secondary antibodies conjugated to horseradish peroxidase were incubated with membranes, and protein was detected by enhanced chemiluminescence.
RT-qPCR
RNA was extracted utilizing the Ambion RNA extraction kit (Life Technologies) following the manufacturer's instructions. Complementary DNA was synthesized utilizing M-MLV reverse transcriptase (Promega). Preparation for RT-qPCR samples was implemented using TaqMan Fast Advanced Master Mix (Thermo). Primers for the following gene targets were purchased from Applied Biosystems (Thermo): GLUT1 (catalog no. 4331182, assay ID Hs00892681_m1), GLUT3 (catalog no. 4331182, assay ID Hs00359840_m1), PDHK1 (catalog no. 4331182, assay ID Hs01561847_m1), VEGFα (catalog no. 4331182, assay ID Hs00900055_m1), HIF-1α (catalog no. 4331182, assay ID Hs00153153_m1), HIF-2α (catalog no. 4331182, assay ID Hs01026149_m1), and Oct 4 (catalog no. 4331182, assay ID Hs00999632_g1). Data were normalized to expression of RPLPO (Thermo, catalog no. 4333761F, lot no. 1604105). Significance was determined as p < 0.05 using a Student's t test from at least three independent experiments, with each experiment performed in triplicate.
Author contributions
R. B. J., A. B. H., and S. L. B. conceptualization; R. B. J., K. A. D., and S. L. B. data curation; R. B. J., K. A. D., A. B. H., and S. L. B. formal analysis; R. B. J., A. B. H., and S. L. B. methodology; R. B. J. and S. L. B. writing-original draft; A. B. H. and S. L. B. supervision; S. L. B. funding acquisition; S. L. B. project administration; S. L. B. writing-review and editing.
Acknowledgments
We thank the University of Alabama at Birmingham Flow Cytometry Core Facility (P30AR048311 and P30AI027767) for assistance.
Note added in proof
In the version of this article that was published as a Paper in Press on February 23, 2018, images in Fig. 2C were inadvertently duplicated. This error has now been corrected.
This work was supported by National Institutes of Health Grant R01 GM111093 (to S. L. B.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- CSC
- cancer stem cell
- HIF
- hypoxia-inducible factor
- EV
- empty vector
- OE
- overexpression
- KD
- knockdown
- DFO
- desferoxamine
- DMOG
- dimethyloxalylglycine
- VEGF
- vascular endothelial growth factor
- FBS
- fetal bovine serum
- shRNA
- short hairpin RNA
- RT-qPCR
- reverse transcription quantitative PCR
- SNA
- Sambucus nigra agglutinin.
References
- 1. Semenza G. L. (2014) Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol. 9, 47–71 10.1146/annurev-pathol-012513-104720 [DOI] [PubMed] [Google Scholar]
- 2. Kim J. W., Tchernyshyov I., Semenza G. L., and Dang C. V. (2006) HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 3, 177–185 10.1016/j.cmet.2006.02.002 [DOI] [PubMed] [Google Scholar]
- 3. Qin J., Liu Y., Lu Y., Liu M., Li M., Li J., and Wu L. (2017) Hypoxia-inducible factor 1 α promotes cancer stem cells-like properties in human ovarian cancer cells by upregulating SIRT1 expression. Sci. Rep. 7, 10592 10.1038/s41598-017-09244-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang Y., Liu Y., Malek S. N., Zheng P., and Liu Y. (2011) Targeting HIF1α eliminates cancer stem cells in hematological malignancies. Cell Stem Cell 8, 399–411 10.1016/j.stem.2011.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mohyeldin A., Garzón-Muvdi T., and Quiñones-Hinojosa A. (2010) Oxygen in stem cell biology: a critical component of the stem cell niche. Cell Stem Cell 7, 150–161 10.1016/j.stem.2010.07.007 [DOI] [PubMed] [Google Scholar]
- 6. Heddleston J. M., Li Z., Lathia J. D., Bao S., Hjelmeland A. B., and Rich J. N. (2010) Hypoxia inducible factors in cancer stem cells. Br. J. Cancer 102, 789–795 10.1038/sj.bjc.6605551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Heddleston J. M., Li Z., McLendon R. E., Hjelmeland A. B., and Rich J. N. (2009) The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 8, 3274–3284 10.4161/cc.8.20.9701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Munkley J., and Elliott D. J. (2016) Hallmarks of glycosylation in cancer. Oncotarget 7, 35478–35489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stowell S. R., Ju T., and Cummings R. D. (2015) Protein glycosylation in cancer. Annu. Rev. Pathol. 10, 473–510 10.1146/annurev-pathol-012414-040438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pinho S. S., and Reis C. A. (2015) Glycosylation in cancer: mechanisms and clinical implications. Nat. Rev. Cancer 15, 540–555 10.1038/nrc3982 [DOI] [PubMed] [Google Scholar]
- 11. Taniguchi N., and Kizuka Y. (2015) Glycans and cancer: role of N-glycans in cancer biomarker, progression and metastasis, and therapeutics. Adv. Cancer Res. 126, 11–51 10.1016/bs.acr.2014.11.001 [DOI] [PubMed] [Google Scholar]
- 12. Varki A., Kannagi R., Toole B., and Stanley P. (2015) in Essentials of Glycobiology (Varki A., Cummings R. D., Esko J. D., Stanley P., Hart G. W., Aebi M., Darvill A. G., Kinoshita T., Packer N. H., Prestegard J. H., Schnaar R. L., and Seeberger P. H., eds.) 3rd ed., pp. 597–609, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 13. Schultz M. J., Swindall A. F., and Bellis S. L. (2012) Regulation of the metastatic cell phenotype by sialylated glycans. Cancer Metastasis Rev. 31, 501–518 10.1007/s10555-012-9359-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lu J., and Gu J. (2015) Significance of β-galactoside α2,6 sialyltransferase 1 in cancers. Molecules 20, 7509–7527 10.3390/molecules20057509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dall'Olio F., Malagolini N., Trinchera M., and Chiricolo M. (2014) Sialosignaling: sialyltransferases as engines of self-fueling loops in cancer progression. Biochim. Biophys. Acta 1840, 2752–2764 10.1016/j.bbagen.2014.06.006 [DOI] [PubMed] [Google Scholar]
- 16. Bhide G. P., and Colley K. J. (2017) Sialylation of N-glycans: mechanism, cellular compartmentalization and function. Histochem. Cell Biol. 147, 149–174 10.1007/s00418-016-1520-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Harduin-Lepers A., Krzewinski-Recchi M. A., Colomb F., Foulquier F., Groux-Degroote S., and Delannoy P. (2012) Sialyltransferases functions in cancers. Front. Biosci. 4, 499–515 [DOI] [PubMed] [Google Scholar]
- 18. Schultz M. J., Holdbrooks A. T., Chakraborty A., Grizzle W. E., Landen C. N., Buchsbaum D. J., Conner M. G., Arend R. C., Yoon K. J., Klug C. A., Bullard D. C., Kesterson R. A., Oliver P. G., O'Connor A. K., Yoder B. K., and Bellis S. L. (2016) The tumor-associated glycosyltransferase ST6Gal-I regulates stem cell transcription factors and confers a cancer stem cell phenotype. Cancer Res. 76, 3978–3988 10.1158/1538-7445.AM2016-3978,10.1158/0008-5472.CAN-15-2834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Swindall A. F., Londoño-Joshi A. I., Schultz M. J., Fineberg N., Buchsbaum D. J., and Bellis S. L. (2013) ST6Gal-I protein expression is upregulated in human epithelial tumors and correlates with stem cell markers in normal tissues and colon cancer cell lines. Cancer Res. 73, 2368–2378 10.1158/0008-5472.CAN-12-3424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lise M., Belluco C., Perera S. P., Patel R., Thomas P., and Ganguly A. (2000) Clinical correlations of α2,6-sialyltransferase expression in colorectal cancer patients. Hybridoma 19, 281–286 10.1089/027245700429828 [DOI] [PubMed] [Google Scholar]
- 21. Recchi M. A., Hebbar M., Hornez L., Harduin-Lepers A., Peyrat J. P., and Delannoy P. (1998) Multiplex reverse transcription polymerase chain reaction assessment of sialyltransferase expression in human breast cancer. Cancer Res. 58, 4066–4070 [PubMed] [Google Scholar]
- 22. Hsieh C. C., Shyr Y. M., Liao W. Y., Chen T. H., Wang S. E., Lu P. C., Lin P. Y., Chen Y. B., Mao W. Y., Han H. Y., Hsiao M., Yang W. B., Li W. S., Sher Y. P., and Shen C. N. (2017) Elevation of β-galactoside α2,6-sialyltransferase 1 in a fructose-responsive manner promotes pancreatic cancer metastasis. Oncotarget 8, 7691–7709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schultz M. J., Swindall A. F., Wright J. W., Sztul E. S., Landen C. N., and Bellis S. L. (2013) ST6Gal-I sialyltransferase confers cisplatin resistance in ovarian tumor cells. J. Ovarian Res. 6, 25 10.1186/1757-2215-6-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chakraborty A., Dorsett K. A., Trummell H. Q., Yang E. S., Oliver P. G., Bonner J. A., Buchsbaum D. J., and Bellis S. L. (2018) ST6Gal-I sialyltransferase promotes chemoresistance in pancreatic ductal adenocarcinoma by abrogating gemcitabine-mediated DNA damage. J. Biol. Chem. 293, 984–994 10.1074/jbc.M117.808584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Britain C. M., Holdbrooks A. T., Anderson J. C., Willey C. D., and Bellis S. L. (2018) Sialylation of EGFR by the ST6Gal-I sialyltransferase promotes EGFR activation and resistance to gefitinib-mediated cell death. J. Ovarian Res. 11, 12 10.1186/s13048-018-0385-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Christie D. R., Shaikh F. M., Lucas J. A. 4th, Lucas J. A. 3rd, and Bellis S. L. (2008) ST6Gal-I expression in ovarian cancer cells promotes an invasive phenotype by altering integrin glycosylation and function. J. Ovarian Res. 1, 3 10.1186/1757-2215-1-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shaikh F. M., Seales E. C., Clem W. C., Hennessy K. M., Zhuo Y., and Bellis S. L. (2008) Tumor cell migration and invasion are regulated by expression of variant integrin glycoforms. Exp. Cell Res. 314, 2941–2950 10.1016/j.yexcr.2008.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lin S., Kemmner W., Grigull S., and Schlag P. M. (2002) Cell surface α 2,6 sialylation affects adhesion of breast carcinoma cells. Exp. Cell Res. 276, 101–110 10.1006/excr.2002.5521 [DOI] [PubMed] [Google Scholar]
- 29. Zhu Y., Srivatana U., Ullah A., Gagneja H., Berenson C. S., and Lance P. (2001) Suppression of a sialyltransferase by antisense DNA reduces invasiveness of human colon cancer cells in vitro. Biochim. Biophys. Acta 1536, 148–160 10.1016/S0925-4439(01)00044-8 [DOI] [PubMed] [Google Scholar]
- 30. Chen X., Wang L., Zhao Y., Yuan S., Wu Q., Zhu X., Niang B., Wang S., and Zhang J. (2016) ST6Gal-I modulates docetaxel sensitivity in human hepatocarcinoma cells via the p38 MAPK/caspase pathway. Oncotarget 7, 51955–51964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Park J. J., Yi J. Y., Jin Y. B., Lee Y. J., Lee J. S., Lee Y. S., Ko Y. G., and Lee M. (2012) Sialylation of epidermal growth factor receptor regulates receptor activity and chemosensitivity to gefitinib in colon cancer cells. Biochem. Pharmacol. 83, 849–857 10.1016/j.bcp.2012.01.007 [DOI] [PubMed] [Google Scholar]
- 32. Lu J., Isaji T., Im S., Fukuda T., Hashii N., Takakura D., Kawasaki N., and Gu J. (2014) β-Galactoside α2,6-sialyltranferase 1 promotes transforming growth factor-β-mediated epithelial-mesenchymal transition. J. Biol. Chem. 289, 34627–34641 10.1074/jbc.M114.593392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Semenza G. (2002) Signal transduction to hypoxia-inducible factor 1. Biochem. Pharmacol. 64, 993–998 10.1016/S0006-2952(02)01168-1 [DOI] [PubMed] [Google Scholar]
- 34. Gordan J. D., and Simon M. C. (2007) Hypoxia-inducible factors: central regulators of the tumor phenotype. Curr. Opin. Genet. Dev. 17, 71–77 10.1016/j.gde.2006.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bárdos J. I., and Ashcroft M. (2005) Negative and positive regulation of HIF-1: a complex network. Biochim. Biophys. Acta 1755, 107–120 [DOI] [PubMed] [Google Scholar]
- 36. Britain C. M., Dorsett K. A., and Bellis S. L. (2017) The glycosyltransferase ST6Gal-I protects tumor cells against serum growth factor withdrawal by enhancing survival signaling and proliferative potential. J. Biol. Chem. 292, 4663–4673 10.1074/jbc.M116.763862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Holdbrooks A. T., Britain C. M., and Bellis S. L. (2018) ST6Gal-I sialyltransferase promotes tumor necrosis factor (TNF)-mediated cancer cell survival via sialylation of the TNF receptor 1 (TNFR1) death receptor. J. Biol. Chem. 293, 1610–1622 10.1074/jbc.M117.801480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Swindall A. F., and Bellis S. L. (2011) Sialylation of the Fas death receptor by ST6Gal-I provides protection against Fas-mediated apoptosis in colon carcinoma cells. J. Biol. Chem. 286, 22982–22990 10.1074/jbc.M110.211375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Covello K. L., Kehler J., Yu H., Gordan J. D., Arsham A. M., Hu C. J., Labosky P. A., Simon M. C., and Keith B. (2006) HIF-2α regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 20, 557–570 10.1101/gad.1399906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Maxwell P. H. (2005) The HIF pathway in cancer. Semin. Cell Dev. Biol. 16, 523–530 10.1016/j.semcdb.2005.03.001 [DOI] [PubMed] [Google Scholar]
- 41. Jiang B. H., Agani F., Passaniti A., and Semenza G. L. (1997) V-SRC induces expression of hypoxia-inducible factor 1 (HIF-1) and transcription of genes encoding vascular endothelial growth factor and enolase 1: involvement of HIF-1 in tumor progression. Cancer Res. 57, 5328–5335 [PubMed] [Google Scholar]
- 42. Hoffmann A. C., Mori R., Vallbohmer D., Brabender J., Klein E., Drebber U., Baldus S. E., Cooc J., Azuma M., Metzger R., Hoelscher A. H., Danenberg K. D., Prenzel K. L., and Danenberg P. V. (2008) High expression of HIF1a is a predictor of clinical outcome in patients with pancreatic ductal adenocarcinomas and correlated to PDGFA, VEGF, and bFGF. Neoplasia 10, 674–679 10.1593/neo.08292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chang Q., Jurisica I., Do T., and Hedley D. W. (2011) Hypoxia predicts aggressive growth and spontaneous metastasis formation from orthotopically grown primary xenografts of human pancreatic cancer. Cancer Res. 71, 3110–3120 10.1158/0008-5472.CAN-10-4049 [DOI] [PubMed] [Google Scholar]
- 44. Dhani N., Fyles A., Hedley D., and Milosevic M. (2015) The clinical significance of hypoxia in human cancers. Semin. Nucl. Med. 45, 110–121 10.1053/j.semnuclmed.2014.11.002 [DOI] [PubMed] [Google Scholar]
- 45. Sowa T., Menju T., Chen-Yoshikawa T. F., Takahashi K., Nishikawa S., Nakanishi T., Shikuma K., Motoyama H., Hijiya K., Aoyama A., Sato T., Sonobe M., Harada H., and Date H. (2017) Hypoxia-inducible factor 1 promotes chemoresistance of lung cancer by inducing carbonic anhydrase IX expression. Cancer Med. 6, 288–297 10.1002/cam4.991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sullivan R., Paré G. C., Frederiksen L. J., Semenza G. L., and Graham C. H. (2008) Hypoxia-induced resistance to anticancer drugs is associated with decreased senescence and requires hypoxia-inducible factor-1 activity. Mol. Cancer Ther. 7, 1961–1973 10.1158/1535-7163.MCT-08-0198 [DOI] [PubMed] [Google Scholar]
- 47. Li S., Wei Q., Li Q., Zhang B., and Xiao Q. (2015) Down-regulating HIF-1α by lentivirus-mediated shRNA for therapy of triple negative breast cancer. Cancer Biol. Ther. 16, 866–875 10.1080/15384047.2015.1040958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen J., Zhao S., Nakada K., Kuge Y., Tamaki N., Okada F., Wang J., Shindo M., Higashino F., Takeda K., Asaka M., Katoh H., Sugiyama T., Hosokawa M., and Kobayashi M. (2003) Dominant-negative hypoxia-inducible factor-1 α reduces tumorigenicity of pancreatic cancer cells through the suppression of glucose metabolism. Am. J. Pathol. 162, 1283–1291 10.1016/S0002-9440(10)63924-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hao J., Song X., Song B., Liu Y., Wei L., Wang X., and Yu J. (2008) Effects of lentivirus-mediated HIF-1α knockdown on hypoxia-related cisplatin resistance and their dependence on p53 status in fibrosarcoma cells. Cancer Gene Ther. 15, 449–455 10.1038/cgt.2008.4 [DOI] [PubMed] [Google Scholar]
- 50. Carnero A., and Lleonart M. (2016) The hypoxic microenvironment: a determinant of cancer stem cell evolution. BioEssays 38, S65–S74 10.1002/bies.201670911 [DOI] [PubMed] [Google Scholar]
- 51. Méndez O., Zavadil J., Esencay M., Lukyanov Y., Santovasi D., Wang S. C., Newcomb E. W., and Zagzag D. (2010) Knock down of HIF-1α in glioma cells reduces migration in vitro and invasion in vivo and impairs their ability to form tumor spheres. Mol. Cancer 9, 133 10.1186/1476-4598-9-133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shibuya K., Okada M., Suzuki S., Seino M., Seino S., Takeda H., and Kitanaka C. (2015) Targeting the facilitative glucose transporter GLUT1 inhibits the self-renewal and tumor-initiating capacity of cancer stem cells. Oncotarget 6, 651–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Flavahan W. A., Wu Q., Hitomi M., Rahim N., Kim Y., Sloan A. E., Weil R. J., Nakano I., Sarkaria J. N., Stringer B. W., Day B. W., Li M., Lathia J. D., Rich J. N., and Hjelmeland A. B. (2013) Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat. Neurosci. 16, 1373–1382 10.1038/nn.3510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang Y. C., Stein J. W., Lynch C. L., Tran H. T., Lee C. Y., Coleman R., Hatch A., Antontsev V. G., Chy H. S., O'Brien C. M., Murthy S. K., Laslett A. L., Peterson S. E., and Loring J. F. (2015) Glycosyltransferase ST6GAL1 contributes to the regulation of pluripotency in human pluripotent stem cells. Sci. Rep. 5, 13317 10.1038/srep13317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chun Y. S., Kim M. S., and Park J. W. (2002) Oxygen-dependent and -independent regulation of HIF-1α. J. Korean Med. Sci. 17, 581–588 10.3346/jkms.2002.17.5.581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Masoud G. N., and Li W. (2015) HIF-1α pathway: role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 5, 378–389 10.1016/j.apsb.2015.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tavare A. N., Perry N. J., Benzonana L. L., Takata M., and Ma D. (2012) Cancer recurrence after surgery: direct and indirect effects of anesthetic agents. Int. J. Cancer 130, 1237–1250 10.1002/ijc.26448 [DOI] [PubMed] [Google Scholar]
- 58. Laughner E., Taghavi P., Chiles K., Mahon P. C., and Semenza G. L. (2001) HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1α (HIF-1α) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol. Cell. Biol. 21, 3995–4004 10.1128/MCB.21.12.3995-4004.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Semenza G. L. (2003) Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 3, 721–732 10.1038/nrc1187 [DOI] [PubMed] [Google Scholar]
- 60. Jung Y., Isaacs J. S., Lee S., Trepel J., Liu Z. G., and Neckers L. (2003) Hypoxia-inducible factor induction by tumour necrosis factor in normoxic cells requires receptor-interacting protein-dependent nuclear factor κB activation. Biochem. J. 370, 1011–1017 10.1042/bj20021279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Görlach A., and Bonello S. (2008) The cross-talk between NF-κB and HIF-1: further evidence for a significant liaison. Biochem. J. 412, e17–e19 10.1042/BJ20080920 [DOI] [PubMed] [Google Scholar]
- 62. van Uden P., Kenneth N. S., and Rocha S. (2008) Regulation of hypoxia-inducible factor-1α by NF-κB. Biochem. J. 412, 477–484 10.1042/BJ20080476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Liu Z., Swindall A. F., Kesterson R. A., Schoeb T. R., Bullard D. C., and Bellis S. L. (2011) ST6Gal-I regulates macrophage apoptosis via α2–6 sialylation of the TNFR1 death receptor. J. Biol. Chem. 286, 39654–39662 10.1074/jbc.M111.276063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lim C. S., Kiriakidis S., Paleolog E. M., and Davies A. H. (2012) Increased activation of the hypoxia-inducible factor pathway in varicose veins. J. Vasc. Surg. 55, 1427–1439 10.1016/j.jvs.2011.10.111 [DOI] [PubMed] [Google Scholar]
- 65. Peyssonnaux C., Zinkernagel A. S., Schuepbach R. A., Rankin E., Vaulont S., Haase V. H., Nizet V., and Johnson R. S. (2007) Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). J. Clin. Invest. 117, 1926–1932 10.1172/JCI31370 [DOI] [PMC free article] [PubMed] [Google Scholar]