

Abstract
The pathogen Vibrio cholerae is the causative agent of cholera. Emergence of antibiotic-resistant V. cholerae strains is increasing, but the underlying mechanisms remain unclear. Herein, we report that the stringent response regulator and stress alarmone guanosine tetra- and pentaphosphate ((p)ppGpp) significantly contributes to antibiotic tolerance in V. cholerae. We found that N16961, a pandemic V. cholerae strain, and its isogenic (p)ppGpp-overexpressing mutant ΔrelAΔspoT are both more antibiotic-resistant than (p)ppGpp0 (ΔrelAΔrelVΔspoT) and ΔdksA mutants, which cannot produce or utilize (p)ppGpp, respectively. We also found that additional disruption of the aconitase B–encoding and tricarboxylic acid (TCA) cycle gene acnB in the (p)ppGpp0 mutant increases its antibiotic tolerance. Moreover, expression of TCA cycle genes, including acnB, was increased in (p)ppGpp0, but not in the antibiotic-resistant ΔrelAΔspoT mutant, suggesting that (p)ppGpp suppresses TCA cycle activity, thereby entailing antibiotic resistance. Importantly, when grown anaerobically or incubated with an iron chelator, the (p)ppGpp0 mutant became antibiotic-tolerant, suggesting that reactive oxygen species (ROS) are involved in antibiotic-mediated bacterial killing. Consistent with that hypothesis, tetracycline treatment markedly increased ROS production in the antibiotic-susceptible mutants. Interestingly, expression of the Fe(III) ABC transporter substrate–binding protein FbpA was increased 10-fold in (p)ppGpp0, and fbpA gene deletion restored viability of tetracycline-exposed (p)ppGpp0 cells. Of note, FbpA expression was repressed in the (p)ppGpp-accumulating mutant, resulting in a reduction of intracellular free iron, required for the ROS-generating Fenton reaction. Our results indicate that (p)ppGpp-mediated suppression of central metabolism and iron uptake reduces antibiotic-induced oxidative stress in V. cholerae.
Keywords: bacterial metabolism, bacterial pathogenesis, antibiotic resistance, oxidative stress, stress response, antibiotic tolerance, iron uptake, oxidative stress, stringent response, Vibrio cholerae, cholera, guanosine tetra- and pentaphosphate, alarmone, stress signaling, ROS
Introduction
Cholera, the epidemic acute diarrheal disease caused by Vibrio cholerae, occurs in many developing countries that have poor sanitation (1). The toxigenic strains express various pathogenic factors, including toxin-co-regulated pilus and cholera toxin (CT)3 to acquire the host environmental niche where it survives in the acidic gastric conditions and enters the small intestine (1–5). These virulence factors permit substantial fluid transport from epithelial cells to lumen, which leads to severe watery diarrhea. Thus, treatments mainly used for cholera patients are oral or intravenous hydration therapy that are effective for reducing stool output (6). According to the cholera treatment guidelines from the World Health Organization and the Centers for Disease Control and Prevention, antibiotic treatment in conjunction with an oral rehydration solution (ORS) is recommended for patients who have severe symptoms or are seriously dehydrated and continue to pass a large amount of stool (7). By evaluating many antibiotics, those that are effective at (i) reducing stool output, (ii) reducing the duration of diarrhea, and (iii) inducing bacterial shedding have been selected for treating cholera patients (8–12). Antibiotic treatment in conjunction with ORS allows patients to recover more rapidly compared with ORS treatment alone. Tetracycline is the most effective antibiotic to reduce the cholera morbidity (8). However, doxycycline, a proxy for tetracycline, is currently the first-line drug of choice for cholera treatment due to its easy administration and low dosage requirement compared with tetracycline (13). Alternative drugs for treatment include chloramphenicol, erythromycin, azithromycin, and furazolidone. This provides drug treatment flexibility dependent on infected regions or antibiotic resistance rates. These antibiotics are only administered in combination with rehydration therapy and are not permitted to be used for the prophylaxis of cholera infection to prevent the induction of antibiotic resistance (14, 15). Recently, most isolates of V. cholerae O1 serotype from patient stools showed resistance to antibiotics that are commonly used for cholera treatment (16–20), but with no clear underlying mechanisms.
In our previous studies, we revealed that stringent response (SR) regulates V. cholerae viability and virulence by providing the bacterium with increased fitness in unfavorable environments (21, 22). SR is characterized as one of the global regulatory systems in bacteria, which is activated by a variety of growth-inhibiting stresses (23). SR induces rapid adaptation under various stress conditions via (p)ppGpp accumulation (24). (p)ppGpp, known as a stress alarmone, forms a complex with RNA polymerase and induces profound reprogramming of global gene expression, which leads to growth arrest (25–27). In most Gram-negative bacteria, (p)ppGpp production is regulated by two enzymes, RelA and SpoT (24, 28). RelA, a monofunctional synthetase, recognizes the uncharged tRNA at the A site of a ribosome, and it starts to synthesize (p)ppGpp. SpoT is the bifunctional enzyme that has both synthetase and hydrolase domains but shows mostly strong hydrolysis and weak synthetase activities. In the case of V. cholerae, it has an additional novel (p)ppGpp synthetase called RelV, which loses its N-terminal hydrolase domain (29, 30). Because this enteric pathogen encounters the host-derived immune system during infection, human intestinal environments have the potential to influence nutrient availability to and the viability of V. cholerae (1, 31).
Interestingly, recent studies have reported that SR-mediated transcriptional switching impacts bacterial physiology and increases antibiotic resistance (32–34). For example, (p)ppGpp accumulation raises penicillin resistance by inhibiting peptidoglycan synthesis in both Gram-positive and -negative bacteria (35–38). This is not limited to β-lactam antibiotics, and the resistance to other antimicrobials is also linked to (p)ppGpp accumulation (39, 40). It has been suggested that SR-mediated growth defects reduce antibiotic sensitivity, but this hypothesis is not fully understood. Recently, some studies have suggested that decreased levels of superoxide dismutase and catalase activities in SR mutants are associated with susceptibility to multiclasses of antibiotics (41, 42). This report described the famous mechanistic model that different bactericidal antibiotics, regardless of their primary drug target, had a common pathway that generated deleterious ROS (43). According to this concept, ROS formation following antibiotic treatment enhances antibiotic lethality as well as the interaction with their traditional targets. Importantly, this ROS stress is derived from alterations of bacterial physiology, including hyperactivation of central metabolism and cellular respiration and disruption of iron homeostasis, by disrupting specific drug targets (43–45). It might seem strange that antibiotic treatment induces the overflow of metabolism, but to support this idea, many reports have described the transcriptomic and proteomic response to the bactericidal antibiotics (43, 44). Therefore, we hypothesized that the potential capacity for reducing oxidative stress is essential for bacteria to survive during antibiotic treatment.
In this study, we showed that (p)ppGpp accumulation induced antibiotic resistance in V. cholerae by suppressing its central metabolism. We investigated the molecular basis of the specific physiology in (p)ppGpp-null V. cholerae that restored their viability against antibiotics when the acnB gene was totally abolished. Additionally, we showed that the (p)ppGpp-nonproducing V. cholerae mutant carried a higher level of intracellular free iron, the crucial source of ROS production. This report provides a novel insight into the stepwise regulation of SR that contributes to defend against oxidative stress following antibiotic treatment.
Results
(p)ppGpp-accumulated V. cholerae exhibits antibiotic tolerance
SR regulates bacterial responses to overcome unfavorable growth conditions in V. cholerae. To explore whether V. cholerae develops antibiotic tolerance in an SR-dependent manner, we measured survival rates of a range of SR-related mutant strains of V. cholerae. Front-line drugs for cholera treatment, tetracycline (Tc), erythromycin (Em), and chloramphenicol (Cp), were used for the bacterial treatments. To elucidate whether antibiotic tolerance was specifically induced by (p)ppGpp accumulation, we first chemically induced (p)ppGpp overproduction in N16961, a seventh pandemic O1 V. cholerae strain. When N16961 was treated for 2 h with serine hydroxamate (SHX), a serine analogue that activates SR by mimicking amino acid starvation (41), a marked increase in (p)ppGpp production was induced (Fig. 1A). Although ppGpp and pppGpp were not clearly distinguished in our TLC assay, SHX-treated N16961 produced dramatically abundant (p)ppGpp compared with untreated controls (Fig. 1A). To assess the effect of (p)ppGpp accumulation on bacterial survival under antibiotic stress, we treated N16961 grown in Luria–Bertani medium (LB) or LB + SHX with varying concentrations of Tc, Em, or Cp for 4 h. Loss of viability was detected in LB-grown N16961. Complete killing was observed after 4 h of treatment with 10 μg/ml Tc (Fig. 1B), 60 μg/ml Em (Fig. 1C), and 25 μg/ml Cp (Fig. 1D). SHX-treated bacterial cells, however, were tolerant to the same antibiotic treatment and maintained their viability under conditions lethal to the control groups (Fig. 1, B–D). Although the extent to which these changes occurred was similar in all instances, bacterial viability was least affected in the Tc-treated group, with ∼106 cfu/ml recovered even after treatment with the 20 μg/ml concentration (Fig. 1B). These results suggest that a clear mechanistic link probably exists between SR and antibiotic tolerance in V. cholerae.
Figure 1.

Effects of (p)ppGpp on viability of V. cholerae strains against clinically used antibiotics. A, detection of (p)ppGpp accumulation pattern in wildtype N16961 by TLC. Wildtype N16961 was divided into two groups and grown in LB supplemented with or without SHX in the presence of [32P]orthophosphate for 2 h. Cellular extracts were prepared and analyzed in TLC. ΔrelAΔspoT, (p)ppGpp0, and ΔdksA mutants were also grown with [32P]orthophosphate and processed for the TLC assay. B–J, bacterial viabilities under various clinically used antibiotics were measured in each growth condition. Wildtype N16961 and various (p)ppGpp-associated mutant strains were inoculated in LB and aerobically grown to each growth stage. Aliquots of each culture were resuspended in several concentrations of antibiotic-containing LB broth, sampled at 4 h post-inoculation, and 10-fold serially diluted to assess the number of cfu.
To further elucidate the effect of intracellular (p)ppGpp accumulation on bacterial susceptibility to antibiotics in V. cholerae, we tested isogenic ΔrelAΔspoT double and ΔrelAΔrelVΔspoT (termed (p)ppGpp0) triple mutant strains. The ΔrelAΔspoT mutant showed highly elevated levels of (p)ppGpp production due to (i) the action of RelV, an additional enzyme involved in (p)ppGpp biosynthesis and (ii) the lack of SpoT that hydrolyzes (p)ppGpp (21). The (p)ppGpp0 mutant, which lacked all enzymes involved in (p)ppGpp metabolism, was found to produce no (p)ppGpp (21). The level of (p)ppGpp produced in the ΔrelAΔspoT mutant was similar to that in the SHX-treated N16961 cells, whereas no (p)ppGpp was produced in the (p)ppGpp0 triple mutant (Fig. 1A). We also included a ΔdksA mutant, defective in DksA, that binds to RNA polymerase to facilitate the function of (p)ppGpp during SR (46). The capability of the ΔdksA mutant to produce (p)ppGpp was not affected, as shown in Fig. 1A. Because (p)ppGpp levels are known to accumulate as bacterial cells enter the stationary phase (28), we also examined how each bacterial strain, harvested at either the exponential or stationary phase, responded to antibiotic stresses.
When exponential-phase cells were treated with antibiotics, (p)ppGpp0 and ΔdksA mutant strains as well as the WT strain N16961 invariably lost their viability. The ΔrelAΔspoT mutant, which accumulates intracellular (p)ppGpp, was found to be the only strain that maintained viability (Fig. 1, E–G). In contrast, bacterial strains harvested at the stationary phase displayed marked differences from those grown in the exponential phase. Whereas the ΔrelAΔspoT mutant continued to be antibiotic-tolerant, WT N16961 was also found to be tolerant to the same antibiotic treatment (Fig. 1, H–J). Furthermore, these two strains developed tolerance to significantly higher concentrations of antibiotics. Their viabilities were not affected even in the presence of 50 μg/ml Tc, 600 μg/ml Em, or 500 μg/ml Cp, respectively.
To further verify the role of (p)ppGpp in conferring antibiotic tolerance in V. cholerae, we tested the antibiotic-susceptible (p)ppGpp0 mutant harboring pRelVBAD, a plasmid that can express the relV gene via an arabinose-inducible promoter. When the relV gene was expressed, the mutant cells became tolerant to Tc treatment (Fig. S1). Restored tolerance was observed in cells harvested at either growth phase (Fig. S1, A and B). Such a restoration was not detected in the mutant transformed with a control plasmid. Together, these results demonstrated that the ability to produce and utilize (p)ppGpp, a SR regulator, was critically important in affecting V. cholerae susceptibility to antibiotics.
Mutations in the acnB gene resulted in increased tolerance to antibiotic stresses
To provide a mechanistic insight into the increased antibiotic susceptibility of the (p)ppGpp0 mutant, we sought to identify additional mutations that rendered the mutant tolerant to antibiotic treatments. To achieve this goal, we constructed a transposon (Tn) insertion library of the (p)ppGpp0 mutant and looked for mutants that survived Tc treatment. Experimental procedures are described in Fig. 2A. After three successive subcultures in Tc-containing media, we recovered seven mutants that exhibited unencumbered growth. Subsequent analysis indicated that these mutants harbored Tn insertions in the acnB gene encoding a TCA cycle enzyme, aconitase B (Fig. 2B). Importantly, Tn insertion was found to occur at different locations of the gene in each of these mutants (Fig. 2B), suggesting that independent mutational events resulted in an identical consequence. We then introduced an in-frame deletion of the acnB gene into N16961 and (p)ppGpp0 mutants and measured bacterial responses to the antibiotic treatments. Notably, the viability of bacterial cells, when harvested at the exponential phase, was dramatically increased in both ΔacnB and the quadruple (p)ppGpp0ΔacnB mutants compared with their parental strains (Fig. 2, C–E). cfu of up to ∼106/ml were observed following Tc (Fig. 2C) and Cp (Fig. 2E) treatments, whereas ∼104/ml was recovered after treatment with Em (Fig. 2D). Bacterial cells remained viable even in the presence of the highest concentrations of antibiotics. When the (p)ppGpp0ΔacnB quadruple mutant, harvested at the stationary phase, was treated with antibiotics, higher levels of bacterial survival were detected (Fig. 2, F–H).
Figure 2.

Screening of Tn-insertion mutant strains, derived from the (p)ppGpp0 strain, which recovered viability under antibiotic treatment. A, Tn-insertion mutants, derived from the (p)ppGpp0 mutant, that survived in the presence of a lethal concentration of tetracycline (50 μg/ml) were selected. Tetracycline was treated three times every 4 h to the entire recruited culture. B, a genetic map of the acnB gene (VC0604) in V. cholerae. Arrowheads indicate the positions of Tn insertions. C–H, bacterial viability of acnB-deletion mutants under tetracycline (C and F), erythromycin (D and G), and chloramphenicol (E and H) treatments. Aliquots of each culture were resuspended in several concentrations of antibiotic-containing LB broth, sampled at 4 h post-inoculation, and serially diluted for cfu counting.
Importantly, when the plasmid-born acnB gene was expressed by the arabinose-inducible promoter, bacterial strains became susceptible to Tc treatments (Fig. S2). Consistent with previous results, sharper decreases in bacterial viability were detected when using exponential-phase cells (Fig. S2, A and C). Together, these results demonstrated that deletion of the acnB gene conferred a survival advantage to V. cholerae in the presence of antibiotic stresses.
Intracellular (p)ppGpp levels inversely regulate TCA cycle activity, which affects bacterial growth and cell morphology
Aconitase B, encoded by acnB, is the enzyme that catalyzes the interconversion of citrate and isocitrate in the TCA cycle. To examine whether the bacterial TCA cycle was regulated depending on the intracellular concentration of (p)ppGpp, we monitored transcript levels of genes encoding TCA cycle enzymes by RNA-sequencing analysis. First, acnB gene expression was ∼3-fold higher in the (p)ppGpp0 mutant than in the N16961 or in the (p)ppGpp-accumulating ΔrelAΔspoT mutant (Fig. 3A). It is of note that N16961 also possesses the acnA gene, which encodes a phylogenetically distinct aconitase. However, its expression was not detected in all three strains, indicating that aconitase B is probably the major enzyme in the TCA cycle (Fig. 3A). Expressions of mdh, gltA, icd, sucA, sucC, and sucD genes were also noticeably increased in the (p)ppGpp0 mutant, whereas their expressions were decreased (albeit to varying degrees) in the ΔrelAΔspoT mutant (Fig. 3A). Although clear (p)ppGpp dependences were not observed in transcriptional regulation of the sdhA, sdhC, sdhD, frdB, and ttdA genes (Fig. 3A), our RNA-sequencing results strongly suggest that TCA cycle activity is inversely regulated by intracellular (p)ppGpp concentrations.
Figure 3.

Effects of (p)ppGpp on central metabolism and growth of V. cholerae strains. A, RNA-sequencing analysis of TCA cycle enzymes dependent on (p)ppGpp accumulation. RNA samples extracted from three independent bacterial cultures were pooled together for the analysis. Transcripts were extracted at 16 h from bacterial cells grown under LB. Each bar represents the RPKM value of the transcript. B, bacterial cells were inoculated in LB and grown in aerobic conditions for 10 h. A600 values were measured every 1 h. C, scanning electron microscope analysis of N16961 and various (p)ppGpp-associated mutant strains grown under identical conditions as seen in Fig. 1. The plots show the distribution of diameter and length of each strain. The solid horizontal lines represent the geometric mean values. *, p < 0.001.
We then asked whether the (p)ppGpp-dependent regulation of TCA cycle gene expression was reflected in bacterial growth. When wildtype N16961 was grown aerobically in LB, A600 values were reached at ∼1.38 at 4 h post-inoculation and gradually increased up to ∼3.5 for the rest of the experimental period (Fig. 3B). Consistent with a previous finding (21), bacterial growth was significantly retarded in the ΔrelAΔspoT mutant, which accumulates (p)ppGpp. Final A600 values were ∼2.0. In contrast, the (p)ppGpp0 mutant exhibited faster growth compared with N16961, and final A600 was reached at ∼4.5 (Fig. 3B). Similarly, the ΔdksA mutant grew more vigorously, compared with N16961, further confirming that the ΔdksA mutant shared growth-associated phenotypes with the (p)ppGpp0 mutant (Fig. 3B). To reveal the effect of acnB gene deletion on bacterial growth, we also monitored the growth of ΔacnB and (p)ppGpp0ΔacnB mutants. Both mutants exhibited a significantly retarded growth during the early growth stage. For the first 3 h, their growth was comparable with that of the ΔrelAΔspoT mutant (Fig. 3B). Importantly, the robust growth phenotype of the (p)ppGpp0 mutant disappeared when the acnB gene was additionally disrupted, further suggesting that derepressed aerobic growth of the (p)ppGpp0 mutant was associated with increased activity of the acnB gene product (Fig. 3B).
Elevated antibiotic resistance is observed when bacterial cells exhibit a persister phenotype that is often accompanied by cell shape changes (48–50). To address this possibility, we analyzed digitized images of various bacterial strains that showed distinct antibiotic susceptibility. N16961 cells, harvested at the stationary phase and thus more resistant to antibiotics, were shorter and thinner than those harvested at the exponential phase (Fig. 3C and Fig. S3). Antibiotic-tolerant ΔrelAΔspoT mutant and SHX-treated N16961 cells looked thinner and shorter, regardless of when they were harvested (Fig. 3C and Fig. S3). In contrast, antibiotic-susceptible (p)ppGpp0 and ΔdksA mutants maintained their regular curve-shaped morphotype even at the stationary phase (Fig. 3C and Fig. S3). These results clearly suggest that bacterial cells with reduced size are probably more resistant to antibiotic treatment. Of particular interest is that disruption of the acnB gene also resulted in shorter and thinner morphotypes, a phenotype observed under (p)ppGpp-accumulating conditions (Fig. 3C and Fig. S3). Together, these results demonstrated that (i) cell shape changes could be induced upon intracellular (p)ppGpp accumulation or metabolic alterations by acnB gene mutation and (ii) such changes are closely related to bacterial responses to antibiotic treatment.
Other TCA cycle mutants also exhibit increased antibiotic tolerance
To see whether other TCA cycle mutants also exhibit phenotypes similar to that of the acnB mutant, we additionally constructed in-frame deletion mutants of TCA cycle enzymes (Δicd, ΔsucDC, and Δmdh) in both wildtype N16961 and (p)ppGpp0 mutant background and monitored their responses to the Tc treatment. As shown in Fig. 4, TCA cycle mutants were also found to be resistant to Tc. The Δicd mutant showed the most similar resistance to the ΔacnB mutant, whereas the remaining two mutants (ΔsucDC and Δmdh) showed lower resistance (Fig. 4, A and C). Importantly, when each of these mutations was introduced in the antibiotic-sensitive (p)ppGpp0 mutant, antibiotic resistance was restored (Fig. 4, B and D). Compared with the (p)ppGpp0 mutant, (p)ppGpp0ΔacnB and (p)ppGpp0Δicd mutants maintained their viabilities at ∼108 cells/ml following 4-h Tc treatment at 50 μg/ml. The (p)ppGpp0ΔsucDC and (p)ppGpp0Δmdh survived at ∼106 cells/ml. Taken together, these results suggest that disruptions of the TCA cycle function due to the mutation of either the acnB gene or other constituting genes affected antibiotic resistance in V. cholerae. Our results also show that ablation of the acnB or icd gene, whose products mediate early steps of the TCA cycle, produced a more profound effect on bacterial responses to antibiotic stresses.
Figure 4.

Effects of TCA cycle gene mutations on V. cholerae antibiotic tolerance. Bacterial viabilities under various Tc concentrations were measured. Each mutation indicated below in different colors was introduced in N16961 (A and C) and the (p)ppGpp0 mutant (B and D). Bacterial strains were inoculated in LB and aerobically grown to exponential (A and B) or stationary phase (C and D). Experimental conditions were identical to those described in Fig. 1, E and H.
Antibiotic-mediated bacterial killing occurs only in the presence of molecular oxygen and iron
Our results so far showed that (i) expression of TCA cycle genes, such as acnB, icd, and mdh, was highly induced in the antibiotic-susceptible (p)ppGpp0 mutant, (ii) antibiotic-mediated bacterial killing was reduced in each of these TCA cycle mutants, and (iii) when (p)ppGpp was accumulated, bacterial cells shrank and became antibiotic-tolerant. These findings led us to hypothesize that (p)ppGpp accumulation results in metabolic slowdown, rendering bacterial cells unresponsive to antibiotics. To explore whether active metabolism, possibly fueled by aerobic respiration, is necessary for antibiotic-mediated bacterial killing, we compared bacterial responses under aerobic versus anaerobic environments. Exponential-phase N16961 cells, which were found to be antibiotic-sensitive, maintained their viability when further grown for 4 h anaerobically with Tc (Fig. 5A). Anerobiosis also provided an additional protective effect (>10-fold) on stationary-phase N16961 cells against 50 μg/ml Tc (Fig. 5D). Likewise, only marginal viability loss was observed upon treatment with Tc in two antibiotic-sensitive mutants, (p)ppGpp0 (Fig. 5, B and E) and ΔdksA (Fig. 5, C and F), under strict anaerobic environments. These results demonstrated that antibiotic-mediated bacterial killing occurred only during aerobic growth.
Figure 5.

Recovery of tetracycline resistance of (p)ppGpp-deficient strains under anaerobic and iron-deplete conditions. A–F, tetracycline resistance of N16961 (circles), (p)ppGpp0 mutant (triangles), and ΔdksA mutant (squares) strains under anaerobic conditions. Wildtype N16961 and various (p)ppGpp-associated mutant strains were inoculated in LB and aerobically grown to each growth stage. Aliquots of each culture were resuspended in several concentrations of antibiotic-containing LB broth and incubated for 4 h in anaerobic conditions. Each aliquot was sampled and 10-fold serially diluted to assess the number of cfu. G–L, tetracycline resistance of N16961 and (p)ppGpp-deficient mutant strains under iron-depleted conditions. Aliquots of each culture were resuspended in several concentrations of tetracycline-containing LB supplemented with 2,2′-dipyridyl, sampled at 4 h post-inoculation, and serially diluted for cfu counting.
Hydroxyl radical (OH•), the most bactericidal ROS, was reported to be produced during antibiotic treatments in large quantities (43). Because its production is catalyzed by the iron-mediated Fenton reaction (51, 52), we next examined how bacterial responses were altered under iron-depleted conditions. To this end, bacterial strains were treated with Tc in the presence of 2,2′-dipyridyl, an iron chelator (43). As shown in Fig. 5 (G–L), bacterial strains remained viable under iron-deficient conditions after being treated with as high as 50 μg/ml Tc for 4 h (Fig. 5, J–L). Again, two antibiotic-vulnerable mutants and exponential-phase N16961 cells were completely killed by the same treatment in iron-sufficient LB. We then examined whether bacterial killing was alleviated by co-treatment with N-acetylcysteine (NAC), an ROS scavenger. Bacterial survival was substantially restored when antibiotic-sensitive (p)ppGpp0 and ΔdksA mutants were co-treated with 5 mm NAC (Fig. 6, B and C). As expected, NAC treatment exerted no protective effects on stationary-phase N16961 cells already resistant to Tc treatment (Fig. 6A). Together, these results indicated that (i) iron availability determined bacterial survival during antibiotic stress and (ii) ROS, produced aerobically during antibiotic treatment, were responsible for bacterial killing.
Figure 6.

Effects of an ROS scavenger and additional catalase production on bacterial tetracycline resistance. A–C, tetracycline resistance of N16961 and (p)ppGpp-deficient mutant strains under ROS-scavenger treatment. Aliquots of each culture were resuspended in several concentrations of tetracycline-containing LB supplemented with 5 mm NAC, respectively, sampled at 4 h post-inoculation, and serially diluted for cfu counting. D, bacterial viability of (p)ppGpp-deficient mutants and eKatE-producing N16961 (N16961::pVIK112+eKatE) under the tetracycline treatment.
Consistent with these results, post-antibiotic ROS production was indeed increased in two antibiotic-sensitive mutants (Fig. 7). When treated with Tc for 1 h, ROS-specific fluorescent signals increased ∼100- and ∼1,000-fold in (p)ppGpp0 and ΔdksA mutant, respectively. In contrast, fluorescent signals increased only ∼10-fold in stationary-phase N16961 cells and (p)ppGpp-accumulating ΔrelAΔspoT mutant (Fig. 7). It is of particular importance that post-antibiotic ROS production was not detected in the ΔacnB single and (p)ppGpp0ΔacnB quadruple mutant (Fig. 7), thereby further demonstrating that the recovered antibiotic resistance by acnB gene deletion is associated with the lack of ROS production.
Figure 7.
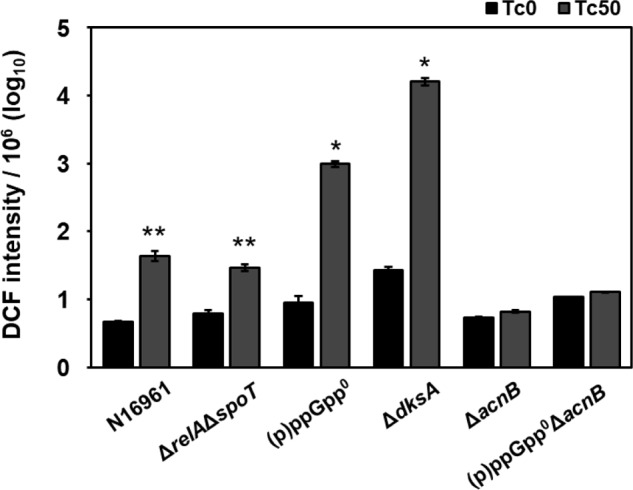
Antibiotic-induced ROS production in (p)ppGpp-deficient mutant strains. Bacterial strains indicated at the bottom were treated with 50 μg/ml Tc for 1 h (gray bars) or left untreated (black bars). Following treatment, bacterial cells were stained with 50 μm DCF for 30 min to measure intracellular ROS levels. DCF intensity was normalized with bacterial cfu and is displayed in logarithmic scale. *, p < 0.0001 versus values of untreated groups. **, p < 0.001 versus values of untreated groups. Error bars, S.D.
Recently, we reported a recombinant N16961 strain that harbored an eKatE gene encoding a robust catalase (53). The eKatE gene is derived from a commensal Escherichia coli strain, and this strain was found to be resistant to 2 mm H2O2 concentration. This strain, when harvested at the exponential phase, developed clear resistance to Tc treatment. Whereas the control N16961 strain perished completely during the same treatment, ∼104 cells remained viable (Fig. 6D). This result further supports our finding that ROS resistance can help with antibiotic tolerance.
Larger amounts of intracellular free iron are present in SR-negative mutants
Next, we sought to further elucidate mechanisms by which (p)ppGpp regulates iron-dependent ROS production during aerobic antibiotic treatment. Table 1 lists the top five genes, expression of which was highly induced in the antibiotic-susceptible (p)ppGpp0 mutant, as normalized with that of each counterpart in the antibiotic-tolerant ΔrelAΔspoT mutant. Among these are genes encoding heme transport protein (hutA), enterobactin receptor protein (irgA), and periplasmic Fe(III) ABC transporter substrate–binding protein (fbpA). Because these proteins are involved in iron acquisition, these RNA-sequencing data strongly suggest that the iron uptake system may be inversely regulated by intracellular (p)ppGpp levels. Consistent with this notion, production of FbpA protein was markedly increased in two SR-negative mutants, whereas its production was not detected in the (p)ppGpp-accumulating ΔrelAΔspoT mutant (Fig. 8A).
Table 1.
List of genes showing the expression significantly increased in (p)ppGpp0 compared with ΔrelAΔspoT mutant
| Gene no. | ΔrelAΔspoT | (p)ppGpp0 | (p)ppGpp0/ΔrelAΔspoT | Product |
|---|---|---|---|---|
| VCA0819 | 129.41 | 2612.98 | 20.19 | Co-chaperonin GroES |
| VC2664 | 217.95 | 3203.18 | 14.70 | Molecular chaperone GroEL |
| VCA0576 | 134.48 | 1804.06 | 13.42 | Heme transport protein HutA |
| VC0475 | 101.92 | 1343.82 | 13.19 | Enterobactin receptor protein IrgA |
| VC0608 | 219.09 | 2653.88 | 12.11 | Fe(III) ABC transporter substrate–binding protein FbpA |
Figure 8.

Effects of fbpA gene deletion on tetracycline resistance and the levels of intracellular free iron in V. cholerae. A, SDS-PAGE analysis of periplasmic fractions in wildtype and (p)ppGpp-associated mutants. Periplasmic fraction was extracted at 16 h from bacterial cells grown under LB and loaded onto SDS-PAGE. B, the levels of intracellular unincorporated iron in N16961 and (p)ppGpp-deficient mutant and fbpA-deletion mutant strains were measured by whole-cell electron paramagnetic resonance (EPR) analysis. The EPR peaks represent the content of total free iron converted to ferric form. C–H, bacterial viability of the (p)ppGpp-deficient mutant and fbpA-deletion mutants under tetracycline treatment.
To understand the role of FbpA in iron uptake and antibiotic tolerance, we introduced an in-frame deletion of the fbpA gene in the N16961, (p)ppGpp0, and ΔdksA mutants (Fig. S4). Bacterial growth was not affected by fbpA gene deletion (data not shown). When we measured intracellular free iron concentrations by whole-cell EPR spectrometry, iron-specific signals were invariably decreased in the N16961, (p)ppGpp0, or ΔdksA strains when the fbpA gene was inactivated (Fig. 8B). The (p)ppGpp0ΔfbpA quadruple and ΔdksAΔfbpA double mutants contained ∼2-fold and ∼3.3-fold decreased levels of free iron, respectively, when compared with their parental mutants (Fig. 8B). These results showed that FbpA protein played an important role in iron uptake in V. cholerae. It is of particular interest that substantially increased amounts of free iron were detected in two antibiotic-susceptible mutants, as compared with N16961 (Fig. 8B). Together, these results clearly demonstrated that the iron import system was suppressed by (p)ppGpp accumulation, and this down-regulation could contribute to reducing the level of intracellular free iron, the crucial source of hydroxyl radical (OH•) formation.
In line with these findings, bacterial survival under Tc treatment was significantly increased in ΔfbpA mutants (Fig. 8, C–H). When exponential-phase cells were used for treatment, bacterial viability of up to ∼104-fold was recovered in ΔfbpA, (p)ppGpp0ΔfbpA, and ΔdksAΔfbpA mutants compared with each of their background strains (Fig. 8, C, E, and G). Moreover, (p)ppGpp0ΔfbpA and ΔdksAΔfbpA mutants, harvested at the stationary stage, also exhibited Tc resistance (Fig. 8, F and H).
Discussion
Cholera is characterized by CT-induced profuse watery diarrhea. Rapid loss of body fluids often leads to fatal dehydration (54–56). Although up to 80% of cholera patients can be treated with oral rehydration therapy, annual deaths up to 120,000 are reported (81). Mortality includes victims who fail to receive immediate interventions and young patients with immature stomach function (82). These cases apparently need prompt antibiotic treatment to reduce the volume of diarrhea and kill the causative pathogen, V. cholerae. Treating cholera patients with antibiotics, however, has been a challenge due to the increased emergence of antibiotic-tolerant V. cholerae strains (16–20, 57).
In this study, we proposed that SR, a conserved bacterial stress response mechanism, regulates antibiotic tolerance in V. cholerae via mechanisms that eventually suppress ROS production. SR controls metabolic activity and intracellular iron level, both of which affect bacterial growth and thereby antibiotic-induced ROS generation. Our successful demonstration on this important issue was made possible by the availability of the ΔrelAΔspoT double mutant that spontaneously accumulates intracellular (p)ppGpp. Whereas (p)ppGpp-null phenotypes have been well-documented in various bacterial species (21, 41, 58, 59), cellular phenotypes induced by natural (p)ppGpp accumulation have not been clearly described. The (p)ppGpp-accumulating ΔrelAΔspoT mutant (i) metabolized slowly, (ii) exhibited a smaller cell-size phenotype, and (iii) produced significantly decreased levels of FbpA protein involved in iron acquisition. All of these phenotypes were invariably reversed in (p)ppGpp0 and ΔdksA mutants, determined to be antibiotic-susceptible. Among these phenotypes, FbpA-mediated iron metabolism provided us with a clue that helped us to precisely elucidate the role of ROS in antibiotic-mediated bacterial killing.
V. cholerae is an enteric pathogen that is transmitted through the fecal-oral route. The toxigenic V. cholerae is able to rapidly spread through bacterial shedding by evoking deadly diarrhea. It passes through the esophagus and comes into contact with the acid environment of the stomach, to which V. cholerae is known to be susceptible. Moreover, V. cholerae colonizes the small intestine, where it must compete with diverse species of commensal microbes for nutrients (60). To overcome such unfavorable conditions, it is highly likely that SR is activated in V. cholerae inside the host intestine. Consistent with this idea, our previous work (21) demonstrated that (i) SR-defective mutants are incapable of colonizing mouse intestine and (ii) the (p)ppGpp-accumulating ΔrelAΔspoT mutant produces a markedly increased level of CT. We therefore proposed that strains that survive and shed out to the environments may have active SR and thereby be more able to infect subsequent hosts and overcome antibiotic stresses. To further validate this hypothesis, it will be important to compare SR activity status in pandemic versus non-virulent environmental strains.
Our results demonstrated that (p)ppGpp, when accumulated, down-regulated expression of many TCA cycle genes, including acnB. Not surprisingly, bacterial metabolism and growth were reported to be suppressed by SR in diverse bacterial species (25, 61, 62). Studies also suggested that (p)ppGpp-mediated regulation participated in bacterial persister formations by mechanisms that involved toxin-antitoxin modules (63, 64). Bacterial persisters are transient phenotypic variants that are stochastically induced within a subset of cells in a given population (64). Persister cells, in general, are more tolerant of antibiotic treatments (65). Ronayne et al. (48) showed that E. coli cells undergo cell elongation during the acquisition of a persister phenotype. In contrast, V. cholerae cells, incubated for a long time in nutrient-poor media, were shown to be smaller in size when compared with normal state cells (50), suggesting that the potential persister-like phenotype may be achieved in a distinct manner in V. cholerae. Our results showed that the (p)ppGpp-accumulating ΔrelAΔspoT mutant and SHX-treated N16961 cells were thinner and shorter. Interestingly, reduced cell size was also observed in the ΔacnB single mutant. These results indicated that cell shape change and a resultant increase in antibiotic tolerance ensued from (p)ppGpp accumulation or acnB gene deletion in V. cholerae and presented a new question as to which of these two cellular processes was the primary cause of the phenotype. Because a similar cell-size reduction was also detected in the (p)ppGpp0ΔacnB mutant cells, we postulate that acnB gene deletion is a necessary and sufficient condition for the phenotype. Likewise, cell size reduction in the ΔrelAΔspoT mutant was probably induced by suppressed aconitase activity, not directly by (p)ppGpp accumulation. Cell biological features need to be further explored in terms of how inactive metabolism can lead to changes in cell shape.
Periplasmic Fe(III) ABC transporter substrate–binding protein, encoded by fbpA, was highly produced in antibiotic-susceptible (p)ppGpp0 and ΔdksA mutants. More importantly, EPR analysis clearly showed that intracellular free iron was concomitantly increased in these two mutants. Like other bacterial species, V. cholerae requires iron for growth and possesses a variety of iron uptake systems (66). The iron acquisition systems in V. cholerae involve synthesis, secretion, and uptake of a range of siderophore molecules, such as vibriobactin (67), enterobactin (68, 69), and ferrichrome (67, 70). In addition, V. cholerae possesses feo and fbp gene clusters encoding systems for the acquisition of ferrous and ferric iron, respectively (66). The Fbp system is a periplasmic binding protein–dependent ABC transport system and consists of three genes, fbpA (VC0608), fbpB (VC0609), and fbpC (VC0610). The fbpB and fbpC encode membrane-spanning proteins forming a pore across the cell membrane and ATP binding proteins, respectively. The fbpA encodes a substrate-binding protein that carries ferric iron to the membrane-spanning proteins. Studies indicate that the potential Fur box (ferric uptake regulator–binding motif) exists in the promoter region of the fbpABC operon (66, 71), suggesting that expression of the operon is highly induced under iron-deficient conditions. However, in our experiments, bacterial strains were grown in LB, considered to be a nutrient-rich medium. It was of particular interest to us that the Fur protein is inactivated by the presence of ROS (72). ROS oxidizes Fe2+, a co-factor of Fur, converting active Fur into an inactive apo-form. Therefore, one possible explanation for the antibiotic-mediated bacterial killing in the antibiotic-susceptible (p)ppGpp0 and ΔdksA mutants would be that ROS produced during derepressed aerobic growth stimulated FbpA-mediated iron uptake, which in turn amplified further ROS production. In support of this idea, the ΔrelAΔspoT mutant that grew very slowly produced undetectable levels of FbpA protein. Furthermore, we also found that FbpA production was reduced in the ΔacnB mutant that exhibited a slow growth phenotype (data not shown).
In our experiments, the first-line drugs for treating cholera patients, tetracycline, erythromycin, and chloramphenicol, were used. We found that SR-deficient mutants of V. cholerae completely lost their viability as the antibiotic concentration increased. Interestingly, however, the antibiotics that turned out to be very effective at killing V. cholerae strains are commonly known as bacteriostatic. Although it is still debatable how to classify antibiotics as bacteriostatic or bactericidal based on their in vitro test results, recent studies described their differences based on their effects on bacterial metabolism. Lobritz et al. (44) found that bacteriostatic and bactericidal have opposing effects on bacterial respiration in E. coli and Staphylococcus aureus. They demonstrated that bacteriostatic antibiotics, such as tetracycline, chloramphenicol, and erythromycin, decelerate cellular respiration, whereas bactericidal antibiotics accelerate basal respiration and lead to the production of ROS as a by-product. On the other hand, we found that bacteriostatic antibiotics killed bacteria and produced deleterious ROS. Interestingly, in another study, the antibiotics that did not trigger SOS responses in E. coli caused SOS responses in V. cholerae (73). SOS stress responses were activated when exogenous and endogenous triggers provoked DNA damage, and some antibiotics act as exogenous triggers that stimulate ROS production, the crucial weapon of DNA disruption. Aminoglycosides, tetracycline, and chloramphenicol induce SOS responses in V. cholerae, unlike E. coli, and this result suggests the possibility that these antibiotics have more deleterious effects on V. cholerae. Taken together, our results suggest that effects of bacteriostatic and bactericidal antibiotics on bacteria vary from species to species.
In conclusion, our results revealed that bacterial SR regulates antibiotic tolerance by modulating ROS production. Central metabolism and iron transport systems are subject to (p)ppGpp-mediated regulation (summarized in Fig. 9). When (p)ppGpp is accumulated, the TCA cycle is down-regulated to slow down bacterial growth. At the same time, FbpA-mediated iron uptake is also suppressed in (p)ppGpp-accumulating cells. These dual cellular events both contribute to physiological changes resulting in metabolic slowdown and therefore antibiotic-tolerant states. Bacterial SR has been a target to be inhibited. Chemical compounds that can suppress (p)ppGpp production, such as relacin (74) and iMAC (75), can potentially be used as antibiotic adjuvants. We anticipate that experimental data provided in the current study will stimulate future investigations that eventually help us come up with better strategies to combat bacterial infections, including one by the deadly enteric pathogen V. cholerae.
Figure 9.
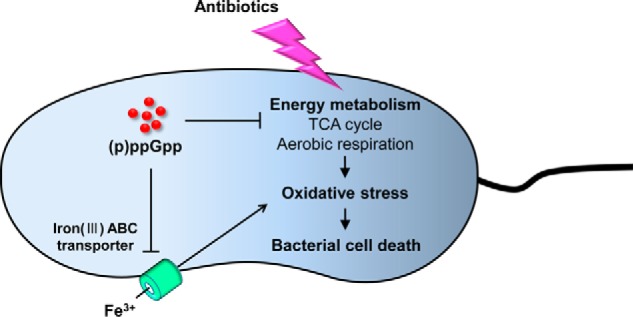
Summary of (p)ppGpp-mediated regulation of antibiotic resistance in V. cholerae. Antibiotic treatment stimulates ROS production by hyperactivating bacterial central metabolism. The released ROS leads to damage of intracellular DNA, proteins, and lipids, which results in cell death. However, (p)ppGpp negatively regulates TCA cycle and aerobic respiration. The down-regulation of aerobic respiration can reduce oxidative stress and eventually prevent cell death. The (p)ppGpp can maximize this effect by restricting free iron uptake from the iron-transporting system.
Experimental procedures
Bacterial strains and growth conditions
All of the bacterial strains and plasmids used in this study are listed in Table 2. Bacterial cultures were grown in LB (1% (w/v) tryptone, 0.5% (w/v) yeast extract, and 1% (w/v) sodium chloride) at 37 °C, and antibiotics were used at the following concentrations: streptomycin (Duchefa), 200 μg/ml; ampicillin (Sigma), 50 μg/ml; kanamycin (Duchefa), 50 μg/ml. All bacterial single colonies on LB plates were picked and inoculated in LB broth for precultures and grown overnight. Precultures were diluted 100-fold in fresh LB broth for subculture and incubated at 37 °C and 220 rpm. The incubation time was dependent on experimental procedures.
Table 2.
Bacterial strains and plasmids used in this study
| Strains and plasmids | Relevant characteristic | Source |
|---|---|---|
| V. cholerae strains | ||
| N16961 | Wildtype, O1 serogroup, biotype El Tor | Laboratory collection |
| ΔacnB | N16961, acnB deleted | This study |
| ΔdksA | N16961, dksA deleted | This study |
| Δicd | N16961, icd deleted | This study |
| ΔsucDC | N16961, sucDC deleted | This study |
| Δmdh | N16961, mdh deleted | This study |
| ΔfbpA | N16961, fbpA deleted | This study |
| ΔdksAΔfbpA | N16961, dksA and fbpA deleted | This study |
| ΔrelAΔspoT | N16961, relA and spoT deleted | Ref. 21 |
| (p)ppGpp0 | N16961, relA, relV, and spoT deleted | Ref. 21 |
| (p)ppGpp0ΔacnB | N16961, relA, relV, spoT, and acnB deleted | This study |
| (p)ppGpp0Δicd | N16961, relA, relV, spoT, and icd deleted | This study |
| (p)ppGpp0ΔsucDC | N16961, relA, relV, spoT, and sucDC deleted | This study |
| (p)ppGpp0Δmdh | N16961, relA, relV, spoT, and mdh deleted | This study |
| (p)ppGpp0ΔfbpA | N16961, relA, relV, spoT, and fbpA deleted | This study |
| N16961::pVIK112+eKatE | Ref. 53 | |
| E. coli strains | ||
| SM10/λpir | Kmr thi-1 thr leu tonA lacY supE recA::RP4–2-Tc::Mu pir+, for conjugal transfer | Laboratory collection |
| Plasmids | ||
| pBAD24 | Ampr, cloning vector | Laboratory collection |
| pCVD442 | sacB suicide vector from plasmid pUM24 | Laboratory collection |
| pTnKGL3 | Suicide vector bearing TnKGL3, Cmr, Kmr | Laboratory collection |
Antibiotic tolerance assay
All antibiotic tolerance assays in this study were performed as described previously with a few modifications (41). For bacterial drug experiments, we used tetracycline (Sigma), erythromycin (Sigma), chloramphenicol (Sigma).
Exponential-phase bacteria
Bacteria from precultures were diluted 100-fold in LB broth and grown shaking at 37 °C until the growth reached A600 0.5. 5 mm serine hydroxamate (Sigma) was added to some of the cultures after 1 h of growth and further incubated for 1 h. Aliquots were then resuspended in serial diluted antibiotic-LB to A600 0.05 and incubated statically for 4 h. The cfu were measured by serial dilution of individual aliquots on LB plates for statistical testing.
Stationary-phase bacteria
Overnight subcultures (16 h) were resuspended in serial diluted antibiotic-LB aliquots to A600 0.3 and statically incubated for 4 h. The survival rate of individual aliquots was also measured by viable cell counting.
Determination of intracellular (p)ppGpp concentration by TLC
Intracellular ppGpp concentration was measured as described previously with a few modifications (22). To detect intracellular (p)ppGpp, bacterial cells were grown aerobically with 100 μCi/ml [32P]orthophosphate (PerkinElmer Life Sciences) at each growth phase. The bacterial cell cultures were extracted with cold 10 mm Tris-HCl buffer (pH 8.0). After centrifugation to remove the cell supernatant, cell pellets were resuspended with cold 10 mm Tris-HCl buffer and 19 m formic acid and then freeze-thawed for three cycles. After centrifugation to remove cell debris, cell supernatants were spotted on PEI cellulose F TLC plates (Merck). The TLC plates were developed in 1.5 m KH2PO4 buffer (pH 3.4) in a humidified chamber and imaged with autoradiography.
Construction of in-frame deletion mutants
V. cholerae mutants were created by allele replacement, as described previously (21, 76). To induce mutation, 500-bp flanking sequences located at both ends of the ORF were amplified by PCR with the primers listed in Table 3. The primers used to amplify each flanking region were carried by restriction enzyme sites that were located in multiple cloning sites of pCVD442 suicide vectors. The purified forward flanking sequences were ligated in pCVD442 vector with T4 ligase, and extracted vectors were transformed to heat-shock competent cells, SM10/λpir strains. The transformed cells were selected on LB plates containing 100 μg/ml ampicillin, and cloning vectors were purified from single survival colonies, following recombination of another flanking sequence going through the same steps to transformation. The SM10/λpir strains with cloned vectors and V. cholerae recipient strains were mixed at a ratio of 3:1 onto LB plates and incubated for 6 h at 37 °C. The mixed pool was suspended in fresh LB broth and spread on LB plates containing 200 μg/ml streptomycin or 100 μg/ml ampicillin for the first step in allelic exchange. After overnight culture, for plasmid excision from the chromosome by second cross-over, single colonies were selected and streaked on LB plates with 8% sucrose and 200 μg/ml streptomycin without NaCl. Screening of in-frame deletion sites for each colony was preceded by PCR to identify the desired allele with primers that contained flanking sequences and ORF.
Table 3.
Primers used in this study
| Gene name | Directiona | Primer sequence (5′–3′)a |
|---|---|---|
| Cloning | ||
| ΔacnB left | Forward | GCAAGCATGCAAACCTCGTTTACCGTTACC |
| ΔacnB left | Reverse | CTCTGAGCTCGACTTTTTCCTCTCATTGCG |
| ΔacnB right | Forward | TTGCGAGCTCGCAGAGTGATTGAATCCTCT |
| ΔacnB right | Reverse | GCGACCCGGGATTTTGAATAAAGCTTTGCC |
| Δicd left | Forward | TATTGCATGCTGGCTTAAAGTGTCATAAGG |
| Δicd left | Reverse | GTCTAAACTAGAGAACTTTCCCTATCTGTTCT |
| Δicd right | Forward | TAGGGAAAGTTCTCTAGTTTAGACACCAAAAC |
| Δicd right | Reverse | ATGTGCATGCATACGTTCGTCCTATTGACT |
| ΔsucDC left | Forward | TATTGCATGCGTGATTTGCTCGCGATTAGC |
| ΔsucDC left | Reverse | TGGAACAACACATCTACCGCGATTACTTACTC |
| ΔsucDC right | Forward | AATCGCGGTAGATGTGTTGTTCCATTTGTTTA |
| ΔsucDC right | Reverse | ATCTGAGCTCATACCTTGAGTTTGGCGCAA |
| Δmdh left | Forward | AGTTGCATGCGCGATACTTTGGATTGGTTG |
| Δmdh left | Reverse | ATCGATTGTCGACGTAAATCTCCTTGAGAGTA |
| Δmdh right | Forward | AGGAGATTTACGTCGACAATCGATTCAAGCAT |
| Δmdh right | Reverse | ATTAGAGCTCTGAACCAATCACTAGCGCCG |
| ΔfbpA left | Forward | TGCTGCATGCTTTTAGTGTGTAAAACCACT |
| ΔfbpA left | Reverse | CGGCGAGCTCTGTATTATAGGAATGTTCAA |
| ΔfbpA right | Forward | TCGCGAGCTCGTAAAATCAGGGGTATAACG |
| ΔfbpA right | Reverse | CGCGCCCGGGATACACATAAGGATAAAGTA |
| Arbitrary PCR | ||
| KGL3-Mar1, 1 round | Forward | GGGAATCATTTGAAGGTTGGT |
| Arb1, 1 round | Reverse | GGCCACGCGTCGACTAGTACNNNNNNNNNNGATAT |
| Arb6, 1 round | Reverse | GGCCACGCGTCGACTAGTACNNNNNNNNNNACGCC |
| KGL3-Mar2, 2 round | Forward | TAGCGACGCCATCTATGTGTC |
| Arb2, 2 round | Reverse | GGCCACGCGTCGACTAGTAC |
a Restriction enzyme recognition sequences are underlined.
Construction of a random Tn-insertion mutant library
A Tn-inserted mutant library was constructed by using the mariner-based Tn, TnKGL3, which contains a kanamycin resistance marker. Tn mutagenesis of ΔrelAΔrelVΔspoT (i.e. (p)ppGpp0) mutant was performed with SM10/λpir, which carries the TnKGL3. SM10/λpir strains and the V. cholerae recipient strain were mixed at a ratio of 2:1 onto LB plates and incubated for 6 h at 37 °C. The mixed pool was suspended with fresh LB broth and spread on LB plates containing 200 μg/ml streptomycin, 100 μg/ml kanamycin and incubated overnight. The survival bacterial pool was harvested in LB broth and diluted in LB aliquots containing 50 μg/ml tetracycline to A600 0.3 and statically incubated for 4 h. The whole bacterial cells of each aliquot were recruited for centrifugation and resuspended again in LB aliquots containing 50 μg/ml tetracycline and incubated for 4 h. These procedures were repeated three times, and final aliquots were partially spread on LB plates containing streptomycin and kanamycin. The surviving bacterial colonies were collected into a library, and the Tn-insertion site for each mutant was determined by arbitrary PCR. The first round of arbitrary PCR was performed by the transposon TnKGL3-specific primer Mar1 and two random primers, Arb1 and Arb6. The following second round of PCR proceeded with Mar2 and Arb2, by using the first-round PCR products for templates. The PCR products were sequenced using the primer Mar2. The Tn-inserted locations were identified by comparison with the public database of the V. cholerae genome sequence.
Scanning electron microscopy analysis
Characterization of bacterial cell morphology and size were visualized with scanning electron microscopy, following procedures described previously (77). Briefly, for the sample preparation, bacterial cell cultures were fixed with PBS containing 2% glutaraldehyde and 0.1% paraformaldehyde for 2 h and stained with 1% OsO4. Samples were then coated with gold by an ion sputter (IB-3, Eiko, Japan) and examined with a scanning electron microscope (FE SEM S-800, Hitachi, Japan) at an acceleration voltage of 20 kV. Images were processed with ESCAN 4000 software (Bummi Universe Co., Ltd., Seoul, Korea). For measuring the cell length and diameter, more than 100 straight-lined cells were randomly chosen from the digitized scanning electron microscopy images, and the distance between the two ends was automatically calculated.
RNA-sequencing analysis
Bacterial cultures grown in LB were harvested at 16 h post-inoculation. Aliquots of each culture (n = 3) were pooled together in one tube for RNA analysis. To extract high-quality bacterial RNA, an RNeasy Protect kit (Qiagen) was used with an RNeasy minikit (Qiagen) following the manufacturer's protocol. The quantity and quality of total RNA were evaluated using RNA electropherograms (Agilent 2100 Bioanalyzer, Agilent Technologies, Waldbroon, Germany) and by assessing the RNA integrity number. From each sample with an RNA integrity number value greater than 8.0, 8 μg of the total RNA was used as a starting material and treated with the MICROBExpressTM mRNA enrichment kit (Invitrogen). The resulting mRNA samples were processed for the sequencing libraries using Illumina mRNA-Seq sample preparation kit (Illumina, San Diego, CA) following the manufacturer's protocols. One lane per sample was used for sequencing with the Illumina Genome Analyzer IIx (Illumina) to generate nondirectional, single-ended, 36-base pair reads. Quality-filtered reads were mapped to the reference genome sequences (NCBI Bio-Project accession number PRJNA57623, identification number 57623) using CLRNASeq version 0.80 (Chunlab, Seoul, Korea). Relative transcript abundance was computed by counting the RPKM (78).
ROS measurement
Chemical hydrolysis of 2′,7′-dichlorofluorescin diacetate was performed following procedures described elsewhere (79). Briefly, 0.5 ml of 5 mm 2′,7′-dichlorofluorescin diacetate, dissolved in 100% ethanol, was reacted with 2 ml of 0.1 n NaOH at room temperature for 30 min. The reaction was stopped by adding 7.5 ml of 100 mm PBS, giving a final DCF concentration of 50 μm. Bacterial suspensions prepared from stationary-phase cultures were treated with 50 μg/ml tetracycline for 1 h. After treatment, the suspensions were centrifuged at 13,000 rpm for 3 min, and cell-free supernatants were removed. Cell pellets were resuspended with 50 μm DCF solution and incubated for 30 min. Then the DCF intensity, which is indicative of the intracellular ROS level, was measured with a Victor X4 plate reader (PerkinElmer Life Sciences).
SDS-PAGE analysis and protein identification
Preparation of periplasmic fractions followed procedures described previously (80). V. cholerae strains were grown anaerobically in LB for 16 h. Cell pellets were resuspended with PBS containing 250 μg/ml polymyxin B and incubated for 15 min at 4 °C. After incubation, the mixtures were centrifuged at 13,000 rpm for 20 min at 4 °C, and the supernatant was used for separation of periplasmic proteins. Protein was quantified by the method of Bradford, and 5 μg of proteins were separated by 12% SDS-PAGE. The SDS-polyacrylamide gel fractions were submitted to Yonsei Proteome Research Center for protein identification.
EPR analysis
Intracellular free iron levels were measured as described previously with a few modifications (47). Bacterial cells were grown in LB and harvested at 16 h post-inoculation. A bacterial cell pellet was resuspended in 5 ml of prewarmed fresh LB broth that contained 10 mm diethylentriaminepentaacetic acid, pH 7.0, and 20 mm desferrioxamine (pH 8.0). Diethylentriaminepentaacetic acid blocks further iron import, whereas desferrioxamine diffuses into cells and binds unincorporated iron in an EPR-visible ferric form. The concentrated cells were incubated at 37 °C for 15 min in a shaking incubator. The cells were washed with ice-cold 20 mm Tris-Cl (pH 7.4) twice. Cells were then resuspended in 200 μl of ice-cold 10% glycerol, 20 mm Tris-Cl (pH 7.4). The cell suspension (200 μl) then was transferred into an EPR tube and frozen in liquid nitrogen. Ferric sulfate standards were mixed with desferrioxamine and prepared in the same Tris buffer containing glycerol. The spectrometer settings were as follows: microwave power, 1 milliwatt; microwave frequency, 9.64 GHz; modulation amplitude, 10 Gauss at 100 KHz; temperature, 15 K.
Statistical analysis
The data are expressed as the means ± S.D. Unpaired Student's t tests (two-tailed, unequal variance) were used to analyze the differences between experimental groups. p values < 0.05 were considered statistically significant. All experiments were repeated for reproducibility.
Author contributions
H. Y. K., Y. T. O., and S. S. Y. conceptualization; H. Y. K. and Y. T. O. investigation; H. Y. K. and S. S. Y. writing-original draft; J. G. and K.-M. L. data curation; S. S. Y. supervision.
Supplementary Material
This work was supported by National Research Foundation (NRF) of Korea Grants 2017R1A2A2A05019987, 2015M3C9A2054024, 2014R1A1A2056139, 2014R1A4A1008625, and 2017M3A9F3041233, funded by the Korean government. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S4.
- CT
- cholera toxin
- (p)ppGpp
- guanosine tetra- and pentaphosphate
- TCA
- tricarboxylic acid
- ROS
- reactive oxygen species
- SR
- stringent response
- ABC
- ATP-binding cassette
- ORS
- oral rehydration solution
- Tc
- tetracycline
- Em
- erythromycin
- Cp
- chloramphenicol
- SHX
- serine hydroxamate
- LB
- Luria–Bertani medium
- cfu
- colony-forming unit(s)
- RPKM
- reads per kilobase per million mapped
- NAC
- N-acetylcysteine
- OH•
- hydroxyl radical
- Tn
- transposon.
References
- 1. Faruque S. M., Albert M. J., and Mekalanos J. J. (1998) Epidemiology, genetics, and ecology of toxigenic Vibrio cholerae. Microbiol. Mol. Biol. Rev. 62, 1301–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Matson J. S., Withey J. H., and DiRita V. J. (2007) Regulatory networks controlling Vibrio cholerae virulence gene expression. Infect. Immun. 75, 5542–5549 10.1128/IAI.01094-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Thelin K. H., and Taylor R. K. (1996) Toxin-coregulated pilus, but not mannose-sensitive hemagglutinin, is required for colonization by Vibrio cholerae O1 El Tor biotype and O139 strains. Infect. Immun. 64, 2853–2856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Childers B. M., and Klose K. E. (2007) Regulation of virulence in Vibrio cholerae: the ToxR regulon. Future Microbiol. 2, 335–344 10.2217/17460913.2.3.335 [DOI] [PubMed] [Google Scholar]
- 5. Krebs S. J., and Taylor R. K. (2011) Protection and attachment of Vibrio cholerae mediated by the toxin-coregulated pilus in the infant mouse model. J. Bacteriol. 193, 5260–5270 10.1128/JB.00378-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Patra F. C., Sack D. A., Islam A., Alam A. N., and Mazumder R. N. (1989) Oral rehydration formula containing alanine and glucose for treatment of diarrhoea: a controlled trial. BMJ 298, 1353–1356 10.1136/bmj.298.6684.1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Leibovici-Weissman Ya., Neuberger A., Bitterman R., Sinclair D., Salam M. A., and Paul M. (2014) Antimicrobial drugs for treating cholera. Cochrane Database Syst. Rev. CD008625 10.1002/14651858.CD008625.pub2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Greenough W. B. 3rd, Rosenberg I. S., Gordon R. S. Jr., Davies B. I., and Benenson A. S. (1964) Tetracycline in the treatment of cholera. Lancet 1, 355–357 [DOI] [PubMed] [Google Scholar]
- 9. Lindenbaum J., Greenough W. B., and Islam M. R. (1967) Antibiotic therapy of cholera. Bull. World Health Organ. 36, 871–883 [PMC free article] [PubMed] [Google Scholar]
- 10. Rahaman M. M., Majid M. A., Alam A. K. M. J., and Islam M. R. (1976) Effects of doxycycline in actively purging cholera patients: a double-blind clinical trial. Antimicrob. Agents Chemother. 10, 610–612 10.1128/AAC.10.4.610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Roy S. K., Islam A., Ali R., Islam K. E., Khan R. A., Ara S. H., Saifuddin N. M., and Fuchs G. J. (1998) A randomized clinical trial to compare the efficacy of erythromycin, ampicillin and tetracycline for the treatment of cholera in children. Trans. R. Soc. Trop. Med. Hyg. 92, 460–462 10.1016/S0035-9203(98)91094-X [DOI] [PubMed] [Google Scholar]
- 12. Kaushik J. S., Gupta P., Faridi M. M. A., and Das S. (2010) Single dose azithromycin versus ciprofloxacin for cholera in children: a randomized controlled trial. Indian Pediatr. 47, 309–315 [DOI] [PubMed] [Google Scholar]
- 13. De S., Chaudhuri A., Dutta P., Dutta D., De S. P., and Pal S. C. (1976) Doxycycline in the treatment of cholera. Bull. World Health Organ. 54, 177–179 [PMC free article] [PubMed] [Google Scholar]
- 14. Weber J. T., Mintz E. D., Cañizares R., Semiglia A., Gomez I., Sempértegui R., Dávila A., Greene K. D., Puhr N. D., and Cameron D. N. (1994) Epidemic cholera in Ecuador: multidrug-resistance and transmission by water and seafood. Epidemiol. Infect. 112, 1–11 10.1017/S0950268800057368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Towner K. J., Pearson N. J., Mhalu F. S., and O'Grady F. (1980) Resistance to antimicrobial agents of Vibrio cholerae El Tor strains isolated during the fourth cholera epidemic in the United Republic of Tanzania. Bull. World Health Organ. 58, 747–751 [PMC free article] [PubMed] [Google Scholar]
- 16. Wang R., Lou J., Liu J., Zhang L., Li J., and Kan B. (2012) Antibiotic resistance of Vibrio cholerae O1 El Tor strains from the seventh pandemic in China, 1961–2010. Int. J. Antimicrob. Agents 40, 361–364 10.1016/j.ijantimicag.2012.06.010 [DOI] [PubMed] [Google Scholar]
- 17. Jesudason M. (2006) Change in serotype and appearance of tetracycline resistance in V. cholerae O1 in Vellore, South India. Indian J. Med. Microbiol. 24, 152–153 10.4103/0255-0857.25224 [DOI] [PubMed] [Google Scholar]
- 18. Chomvarin C., Johura F.-T., Mannan S. B., Jumroenjit W., Kanoktippornchai B., Tangkanakul W., Tantisuwichwong N., Huttayananont S., Watanabe H., Hasan N. A., Huq A., Cravioto A., Colwell R. R., and Alam M. (2013) Drug response and genetic properties of Vibrio cholerae associated with endemic cholera in north-eastern Thailand, 2003–2011. J. Med. Microbiol. 62, 599–609 10.1099/jmm.0.053801-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mandal J., Dinoop K. P., and Parija S. C. (2012) Increasing antimicrobial resistance of Vibrio cholerae OI biotype EI Tor strains isolated in a tertiary-care centre in India. J. Health Popul. Nutr. 30, 12–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Klontz E. H., Das S. K., Ahmed D., Ahmed S., Chisti M. J., Malek M. A., Faruque A. S., and Klontz K. C. (2014) Long-term comparison of antibiotic resistance in Vibrio cholerae O1 and Shigella species between urban and rural Bangladesh. Clin. Infect. Dis. 58, e133–e136 10.1093/cid/ciu040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Oh Y. T., Park Y., Yoon M. Y., Bari W., Go J., Min K. B., Raskin D. M., Lee K.-M., and Yoon S. S. (2014) Cholera toxin production during anaerobic trimethylamine N-oxide respiration is mediated by stringent response in Vibrio cholerae. J. Biol. Chem. 289, 13232–13242 10.1074/jbc.M113.540088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Oh Y. T., Lee K.-M., Bari W., Raskin D. M., and Yoon S. S. (2015) (p)ppGpp, a small nucleotide regulator, directs the metabolic fate of glucose in Vibrio cholerae. J. Biol. Chem. 290, 13178–13190 10.1074/jbc.M115.640466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Magnusson L. U., Farewell A., and Nyström T. (2005) ppGpp: a global regulator in Escherichia coli. Trends Microbiol. 13, 236–242 [DOI] [PubMed] [Google Scholar]
- 24. Hauryliuk V., Atkinson G. C., Murakami K. S., Tenson T., and Gerdes K. (2015) Recent functional insights into the role of (p)ppGpp in bacterial physiology. Nat. Rev. Microbiol. 13, 298–309 10.1038/nrmicro3448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Potrykus K., and Cashel M. (2008) (p) ppGpp: still magical? Annu. Rev. Microbiol. 62, 35–51 10.1146/annurev.micro.62.081307.162903 [DOI] [PubMed] [Google Scholar]
- 26. Dalebroux Z. D., and Swanson M. S. (2012) ppGpp: magic beyond RNA polymerase. Nat. Rev. Microbiol. 10, 203–212 10.1038/nrmicro2720 [DOI] [PubMed] [Google Scholar]
- 27. Gaca A. O., Colomer-Winter C., and Lemos J. A. (2015) Many means to a common end: the intricacies of (p)ppGpp metabolism and its control of bacterial homeostasis. J. Bacteriol. 197, 1146–1156 10.1128/JB.02577-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dalebroux Z. D., Svensson S. L., Gaynor E. C., and Swanson M. S. (2010) ppGpp conjures bacterial virulence. Microbiol. Mol. Biol. Rev. 74, 171–199 10.1128/MMBR.00046-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Das B., Pal R. R., Bag S., and Bhadra R. K. (2009) Stringent response in Vibrio cholerae: genetic analysis of spoT gene function and identification of a novel (p)ppGpp synthetase gene. Mol. Microbiol. 72, 380–398 10.1111/j.1365-2958.2009.06653.x [DOI] [PubMed] [Google Scholar]
- 30. Dasgupta S., Basu P., Pal R. R., Bag S., and Bhadra R. K. (2014) Genetic and mutational characterization of the small alarmone synthetase gene relV of Vibrio cholerae. Microbiology 160, 1855–1866 10.1099/mic.0.079319-0 [DOI] [PubMed] [Google Scholar]
- 31. Sikora A. E. (2013) Proteins Secreted via the type II secretion system: smart strategies of Vibrio cholerae to maintain fitness in different ecological niches. PLoS Pathog. 9, e1003126 10.1371/journal.ppat.1003126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Aedo S., and Tomasz A. (2016) Role of the stringent stress response in the antibiotic resistance phenotype of methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 60, 2311–2317 10.1128/AAC.02697-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim C., Mwangi M., Chung M., Milheirço C., de Lencastre H., and Tomasz A. (2013) The mechanism of heterogeneous β-lactam resistance in MRSA: key role of the stringent stress response. PLoS One 8, e82814 10.1371/journal.pone.0082814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Abranches J., Martinez A. R., Kajfasz J. K., Chávez V., Garsin D. A., and Lemos J. A. (2009) The molecular alarmone (p)ppGpp mediates stress responses, vancomycin tolerance, and virulence in Enterococcus faecalis. J. Bacteriol. 191, 2248–2256 10.1128/JB.01726-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wu J., Long Q., and Xie J. (2010) (p)ppGpp and drug resistance. J. Cell. Physiol. 224, 300–304 10.1002/jcp.22158 [DOI] [PubMed] [Google Scholar]
- 36. Ishiguro E. E., and Ramey W. D. (1980) Inhibition of in vitro peptidoglycan biosynthesis in Escherichia coli by guanosine 5′-diphosphate 3′-diphosphate. Can. J. Microbiol. 26, 1514–1518 10.1139/m80-253 [DOI] [PubMed] [Google Scholar]
- 37. Rodionov D. G., and Ishiguro E. E. (1995) Direct correlation between overproduction of guanosine 3′,5′-bispyrophosphate (ppGpp) and penicillin tolerance in Escherichia coli. J. Bacteriol. 177, 4224–4229 10.1128/jb.177.15.4224-4229.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Heath R. J., Jackowski S., and Rock C. O. (1994) Guanosine tetraphosphate inhibition of fatty acid and phospholipid synthesis in Escherichia coli is relieved by overexpression of glycerol-3-phosphate acyltransferase (plsB). J. Biol. Chem. 269, 26584–26590 [PubMed] [Google Scholar]
- 39. Greenway D. L. A., and England R. R. (1999) The intrinsic resistance of Escherichia coli to various antimicrobial agents requires ppGpp and σs. Lett. Appl. Microbiol. 29, 323–326 10.1046/j.1472-765X.1999.00642.x [DOI] [PubMed] [Google Scholar]
- 40. Hesketh A., Hill C., Mokhtar J., Novotna G., Tran N., Bibb M., and Hong H.-J. (2011) Genome-wide dynamics of a bacterial response to antibiotics that target the cell envelope. BMC Genomics 12, 226 10.1186/1471-2164-12-226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nguyen D., Joshi-Datar A., Lepine F., Bauerle E., Olakanmi O., Beer K., McKay G., Siehnel R., Schafhauser J., Wang Y., Britigan B. E., and Singh P. K. (2011) Active starvation responses mediate antibiotic tolerance in biofilms and nutrient-limited bacteria. Science 334, 982–986 10.1126/science.1211037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Khakimova M., Ahlgren H. G., Harrison J. J., English A. M., and Nguyen D. (2013) The stringent response controls catalases in Pseudomonas aeruginosa and is required for hydrogen peroxide and antibiotic tolerance. J. Bacteriol. 195, 2011–2020 10.1128/JB.02061-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kohanski M. A., Dwyer D. J., Hayete B., Lawrence C. A., and Collins J. J. (2007) A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130, 797–810 [DOI] [PubMed] [Google Scholar]
- 44. Lobritz M. A., Belenky P., Porter C. B. M., Gutierrez A., Yang J. H., Schwarz E. G., Dwyer D. J., Khalil A. S., and Collins J. J. (2015) Antibiotic efficacy is linked to bacterial cellular respiration. Proc. Natl. Acad. Sci. U.S.A. 112, 8173–8180 10.1073/pnas.1509743112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dwyer D. J., Belenky P. A., Yang J. H., MacDonald I. C., Martell J. D., Takahashi N., Chan C. T. Y., Lobritz M. A., Braff D., Schwarz E. G., Ye J. D., Pati M., Vercruysse M., Ralifo P. S., Allison K. R., et al. (2014) Antibiotics induce redox-related physiological alterations as part of their lethality. Proc. Natl. Acad. Sci. U.S.A. 111, E2100–E2109 10.1073/pnas.1401876111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pal R. R., Bag S., Dasgupta S., Das B., and Bhadra R. K. (2012) Functional characterization of the stringent response regulatory gene dksA of Vibrio cholerae and its role in modulation of virulence phenotypes. J. Bacteriol. 194, 5638–5648 10.1128/JB.00518-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jang S., and Imlay J. A. (2010) Hydrogen peroxide inactivates the Escherichia coli Isc iron-sulfur assembly system, and OxyR induces the Suf system to compensate. Mol. Microbiol. 78, 1448–1467 10.1111/j.1365-2958.2010.07418.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ronayne E. A., Wan Y. C. S., Boudreau B. A., Landick R., and Cox M. M. (2016) P1 Ref endonuclease: a molecular mechanism for phage-enhanced antibiotic lethality. PLOS Genet. 12, e1005797 10.1371/journal.pgen.1005797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang X., Kim Y., Ma Q., Hong S. H., Pokusaeva K., Sturino J. M., and Wood T. K. (2010) Cryptic prophages help bacteria cope with adverse environments. Nat. Commun. 1, 147 10.1038/ncomms1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jubair M., Morris J. G. Jr., and Ali A. (2012) Survival of Vibrio cholerae in nutrient-poor environments is associated with a novel “persister” phenotype. PLoS One 7, e45187 10.1371/journal.pone.0045187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Imlay J. A., Chin S. M., and Linn S. (1988) Toxic DNA damage by hydrogen peroxide through the Fenton reaction in vivo and in vitro. Science 240, 640–642 10.1126/science.2834821 [DOI] [PubMed] [Google Scholar]
- 52. Jang S., and Imlay J. A. (2007) Micromolar intracellular hydrogen peroxide disrupts metabolism by damaging iron-sulfur enzymes. J. Biol. Chem. 282, 929–937 10.1074/jbc.M607646200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yoon M. Y., Min K. B., Lee K.-M., Yoon Y., Kim Y., Oh Y. T., Lee K., Chun J., Kim B.-Y., Yoon S.-H., Lee I., Kim C. Y., and Yoon S. S. (2016) A single gene of a commensal microbe affects host susceptibility to enteric infection. Nat. Commun. 7, 11606 10.1038/ncomms11606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Brandt K. G., Castro Antunes M. M., and Silva G. A. (2015) Acute diarrhea: evidence-based management. J. Pediatr. 91, S36–S43 10.1016/j.jped.2015.06.002 [DOI] [PubMed] [Google Scholar]
- 55. Das J. K., Ali A., Salam R. A., and Bhutta Z. A. (2013) Antibiotics for the treatment of cholera, shigella and cryptosporidium in children. BMC Public Health 13, S10 10.1186/1471-2458-13-S3-S10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Leibovici-Weissman Y., Neuberger A., Bitterman R., Sinclair D., Salam M. A., and Paul M. (2014) Antimicrobial drugs for treating cholera. Cochrane Database Syst. Rev. CD008625 10.1002/14651858.CD008625.pub2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mwansa J. C. L., Mwaba J., Lukwesa C., Bhuiyan N. A., Ansaruzzaman M., Ramamurthy T., Alam M., and Balakrish Nair G. (2007) Multiply antibiotic-resistant Vibrio cholerae O1 biotype El Tor strains emerge during cholera outbreaks in Zambia. Epidemiol. Infect. 135, 847–853 10.1017/S0950268806007254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sugisaki K., Hanawa T., Yonezawa H., Osaki T., Fukutomi T., Kawakami H., Yamamoto T., and Kamiya S. (2013) Role of (p)ppGpp in biofilm formation and expression of filamentous structures in Bordetella pertussis. Microbiology 159, 1379–1389 10.1099/mic.0.066597-0 [DOI] [PubMed] [Google Scholar]
- 59. Jiang M., Sullivan S. M., Wout P. K., and Maddock J. R. (2007) G-protein control of the ribosome-associated stress response protein SpoT. J. Bacteriol. 189, 6140–6147 10.1128/JB.00315-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yoon M. Y., Lee K., and Yoon S. S. (2014) Protective role of gut commensal microbes against intestinal infections. J. Microbiol. 52, 983–989 10.1007/s12275-014-4655-2 [DOI] [PubMed] [Google Scholar]
- 61. Potrykus K., Murphy H., Philippe N., and Cashel M. (2011) ppGpp is the major source of growth rate control in E. coli. Environ. Microbiol. 13, 563–575 10.1111/j.1462-2920.2010.02357.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gaca A. O., Kajfasz J. K., Miller J. H., Liu K., Wang J. D., Abranches J., and Lemos J. A. (2013) Basal levels of (p)ppGpp in Enterococcus faecalis: the magic beyond the stringent response. mBio 4, e00646–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Maisonneuve E., Castro-Camargo M., and Gerdes K. (2013) (p)ppGpp controls bacterial persistence by stochastic induction of toxin-antitoxin activity. Cell 154, 1140–1150 10.1016/j.cell.2013.07.048 [DOI] [PubMed] [Google Scholar]
- 64. Maisonneuve E., and Gerdes K. (2014) Molecular mechanisms underlying bacterial persisters. Cell 157, 539–548 10.1016/j.cell.2014.02.050 [DOI] [PubMed] [Google Scholar]
- 65. Lewis K. (2010) Persister cells. Annu. Rev. Microbiol. 64, 357–372 10.1146/annurev.micro.112408.134306 [DOI] [PubMed] [Google Scholar]
- 66. Wyckoff E. E., Mey A. R., Leimbach A., Fisher C. F., and Payne S. M. (2006) Characterization of ferric and ferrous iron transport systems in Vibrio cholerae. J. Bacteriol. 188, 6515–6523 10.1128/JB.00626-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Griffiths G. L., Sigel S. P., Payne S. M., and Neilands J. B. (1984) Vibriobactin, a siderophore from Vibrio cholerae. J. Biol. Chem. 259, 383–385 [PubMed] [Google Scholar]
- 68. Mey A. R., Wyckoff E. E., Oglesby A. G., Rab E., Taylor R. K., and Payne S. M. (2002) Identification of the Vibrio cholerae enterobactin receptors VctA and IrgA: IrgA is not required for virulence. Infect. Immun. 70, 3419–3426 10.1128/IAI.70.7.3419-3426.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wyckoff E. E., Valle A.-M., Smith S. L., and Payne S. M. (1999) A multifunctional ATP-binding cassette transporter system from Vibrio cholerae transports vibriobactin and enterobactin. J. Bacteriol. 181, 7588–7596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Rogers M. B., Sexton J. A., DeCastro G. J., and Calderwood S. B. (2000) Identification of an operon required for ferrichrome iron utilization in Vibrio cholerae. J. Bacteriol. 182, 2350–2353 10.1128/JB.182.8.2350-2353.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Davies B. W., Bogard R. W., and Mekalanos J. J. (2011) Mapping the regulon of Vibrio cholerae ferric uptake regulator expands its known network of gene regulation. Proc. Natl. Acad. Sci. U.S.A. 108, 12467–12472 10.1073/pnas.1107894108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Brynildsen M. P., and Liao J. C. (2009) An integrated network approach identifies the isobutanol response network of Escherichia coli. Mol. Syst. Biol. 5, 277 10.1038/msb.2009.34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Baharoglu Z., and Mazel D. (2011) Vibrio cholerae triggers SOS and mutagenesis in response to a wide range of antibiotics: a route towards multiresistance. Antimicrob. Agents Chemother. 55, 2438–2441 10.1128/AAC.01549-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wexselblatt E., Oppenheimer-Shaanan Y., Kaspy I., London N., Schueler-Furman O., Yavin E., Glaser G., Katzhendler J., and Ben-Yehuda S. (2012) Relacin, a novel antibacterial agent targeting the stringent response. PLoS Pathog. 8, e1002925 10.1371/journal.ppat.1002925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Oh Y. T., Kim H. Y., Kim E. J., Go J., Hwang W., Kim H. R., Kim D. W., and Yoon S. S. (2016) Selective and efficient elimination of Vibrio cholerae with a chemical modulator that targets glucose metabolism. Front. Cell. Infect. Microbiol. 6, 156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Philippe N., Alcaraz J.-P., Coursange E., Geiselmann J., and Schneider D. (2004) Improvement of pCVD442, a suicide plasmid for gene allele exchange in bacteria. Plasmid 51, 246–255 10.1016/j.plasmid.2004.02.003 [DOI] [PubMed] [Google Scholar]
- 77. Yoon M. Y., Lee K.-M., Park Y., Yoon S. S., and Kaushal D. (2011) Contribution of cell elongation to the biofilm formation of Pseudomonas aeruginosa during anaerobic respiration. PLoS One 6, e16105 10.1371/journal.pone.0016105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Fonseca N. A., Marioni J., and Brazma A. (2014) RNA-Seq gene profiling: a systematic empirical comparison. PLoS One 9, e107026 10.1371/journal.pone.0107026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Possel H., Noack H., Augustin W., Keilhoff G., and Wolf G. (1997) 2,7-Dihydrodichlorofluorescein diacetate as a fluorescent marker for peroxynitrite formation. FEBS Lett. 416, 175–178 10.1016/S0014-5793(97)01197-6 [DOI] [PubMed] [Google Scholar]
- 80. Lee K.-M., Park Y., Bari W., Yoon M. Y., Go J., Kim S. C., Lee H-I., and Yoon S. S. (2012) Activation of cholera toxin production by anaerobic respiration of trimethylamine N-oxide in Vibrio cholerae. J. Biol. Chem. 287, 39742–39752 10.1074/jbc.M112.394932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. World Health Organization (2014) Cholera, 2013. Wkly. Epidemiol. Rec. 89, 345–355 [PubMed] [Google Scholar]
- 82. World Health Organization (2012) Cholera, 2011. Releve epidemiologique hebdomadaire 87, 289–304 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.