

Abstract
Furan‐ and indole‐derived boronate complexes react with alkyl iodides under radical (photoredox) or polar (SN2) conditions to generate three‐component alkylation products with high efficiency and complete stereospecificity. The methodology allows the incorporation of versatile functional groups such as nitriles, ketones, esters, sulfones, and amides, providing rapid access to complex chiral heteroaromatic molecules in enantioenriched form. Interestingly, while indolyl boronate complexes react directly with alkyl halides in a polar pathway, furyl boronates require photoredox catalysis. Careful mechanistic analysis revealed that the boronate complex not only serves as a substrate in the reaction but also acts as a reductive quencher for the excited state of the photocatalyst.
Keywords: alkylation, boron, heterocycles, metallate rearrangement, photoredox catalysis
Heteroaromatic compounds are ubiquitous in natural products and bioactive compounds and play a central role in organic chemistry. In particular, the furan ring is a versatile moiety for organic synthesis, readily undergoing oxidations, acid‐catalyzed rearrangements and cycloaddition reactions,1 while the indole ring is one of the most common motifs in alkaloids and drug candidates.2 Our group recently introduced an enantiospecific sp3–sp2 coupling between aromatic rings and chiral boronic esters 1 to access a variety of enantioenriched monofunctionalized aromatic molecules (e.g. 3, Scheme 1 a).3 Furthermore, inspired by the intriguing reactivity of unsaturated boronate complexes,4 we showed that nucleophilic furyl‐derived boronate complexes 2 react with electrophiles in an enantiospecific three‐component coupling reaction (leading to 4, Scheme 1 a).5 Key to the success of this process was the use of highly activated electrophiles, such as the Umemoto trifleoromethylating agent6 or carbocationic species. Interestingly, the trifluoromethylation reaction was found to proceed through a self‐initiated radical chain mechanism. Independently, Studer and our group have shown that electrophilic radicals add to vinyl boronate complexes 5 (Scheme 1 b).7 The resulting α‐boronate radicals 7 undergo facile single electron oxidation to trigger 1,2‐migration and form boronic ester adducts 8.
Scheme 1.
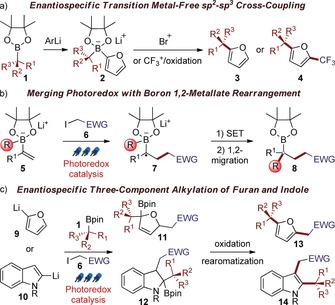
a) Enantiospecific cross‐coupling and three‐component trifluoromethylation of furans. b) Photoredox‐mediated three‐component alkylation of vinyl boronic esters. c) Planned strategy: photoredox‐mediated enantiospecific three‐component coupling of furan and indole. SET: single electron transfer, EWG: electron‐withdrawing group, pin: pinacolato.
We envisioned extending this merger of photoredox catalysis8 with boron 1,2‐metallate rearrangements to aromatic boronate systems (Scheme 1 c).9, 10 Photoredox radical‐mediated reaction of alkyl halides 6 11 with boronate complexes derived from chiral boronic esters 1 and aryllithiums 9 and 10 should lead to dearomatized intermediates 11 and 12. As 1,2‐migration is well‐known to be a stereospecific process12 the stereochemistry within boronic ester 1 should be conserved in the process. In situ oxidation/rearomatization of 11 and 12 would then provide chiral aromatic compounds 13 and 14 in enantioenriched form. Such a transformation would constitute a novel enantiospecific three‐component alkylation protocol, providing valuable routes to complex chiral heteroaromatic structures. Herein, we describe the successful development of this new methodology, which takes advantage of the versatility and mild conditions of photoredox catalysis to introduce a suite of synthetically valuable functional groups.
Attracted by the versatility of nitriles in organic synthesis,13 we commenced our studies by investigating the reaction of furan with iodoacetonitrile (6 a) (Table 1).14 Boronate complex 2 a was formed by addition of cyclohexyl boronic acid pinacol ester (1 a) to 2‐furyllithium, generated by lithiation of furan with n‐butyllithium in THF. Solutions of iodoacetonitrile (6 a) and a photocatalyst were then added and the mixture irradiated with blue LEDs for 1 h. Gratifyingly, in a preliminary experiment carried out using acetonitrile/THF as solvent (entry 1), we found that the reaction proceeded smoothly to give the desired dearomatized intermediate 11 a in 50 % yield using Ru(bpy)3 2+ as a photocatalyst. A solvent screen led to the identification of a mixture of DMF/THF (2:1) as optimal, giving 11 a in 73 % yield (entry 4). The use of the more reducing photocatalyst Ir(ppy)3 gave similar results (entry 5). In contrast to the photochemical three‐component alkylation of vinyl boronate complexes,7a we observed that the use of a photocatalyst was crucial for obtaining high yields in this reaction (compare entries 4 and 6). A control experiment carried out in the dark did not give any product (entry 7), showing the photochemical nature of this transformation. Intermediate 11 a could be oxidized by addition of iodine and potassium acetate15 to the reaction vessel to give the desired three‐component alkylation product 13 a in 71 % isolated yield (entry 4).
Table 1.
Reaction optimization.
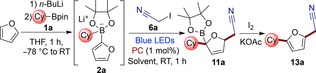
| |||
|---|---|---|---|
| Entry[a] | Solvent[b] | Photocatalyst [PC] | Yield of 11 a [%][c] |
| 1 | CH3CN/THF | Ru(bpy)3Cl2⋅6 H2O | 50 |
| 2 | DMSO/THF | Ru(bpy)3Cl2⋅6 H2O | 66 |
| 3 | DMI/THF | Ru(bpy)3Cl2⋅6 H2O | 70 |
| 4 | DMF/THF | Ru(bpy)3Cl2⋅6 H2O | 73 (71) |
| 5 | DMF/THF | Ir(ppy)3 | 71 |
| 6[d] | DMF/THF | – | 26 |
| 7[e] | DMF/THF | Ru(bpy)3Cl2⋅6 H2O | 0 |
[a] All the reactions were carried out using 1.2 equiv of furan, 1.15 equiv of n‐butyllithium, 1.0 equiv of 1 a and 1.5 equiv of iodoacetonitrile 6 a on a 0.2 mmol scale. [b] Mixture of solvents are intended solvent/THF 2:1. DMI: 1,3‐dimethyl‐2‐imidazolidinone. [c] Yield measured through NMR analysis of the crude mixture using dibromomethane as an internal standard. Intermediate 11 a was obtained as a 1:1 mixture of diastereoisomers. Number in parenthesis is the isolated yield of compound 13 a after oxidation and chromatographic purification. [d] Photochemical step time: 2 hours. [e] Reaction carried out in the dark.
Having identified the optimum conditions, we explored the scope of our photochemical transformation (Scheme 2). A wide range of synthetically versatile functional groups could be efficiently introduced by varying the alkyl iodide radical precursor. Iodoacetophenones proved to be good substrates, leading to the corresponding ketones in good yields (13 b and 13 c). Ethyl iodoacetate and iodoacetamide provided ester 13 d and unprotected amide 13 e. Iodomethyl phenylsulfone led to sulfone 13 g in 64 % yield.16, 17 In addition, perfluoroalkyl chains could be connected to the furan scaffold (13 f) starting from readily available perfluoroalkyl iodides. The employment of tertiary and primary boronic esters led to products 13 h and 13 i in good yields, demonstrating that the process tolerates a wide spectrum of steric demand. Furthermore, starting from enantioenriched chiral boronic esters, compounds 13 j and 13 k were obtained with complete stereospecificity.
Scheme 2.
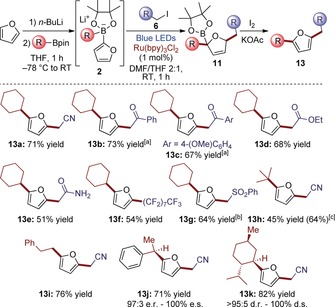
Scope of the enantiospecific three‐component alkylation of furan. All the yields refer to isolated product after chromatographic purification. [a] Intermediate oxidation conditions: NaClO (aq.), DMF, −20 °C. [b] 4 equiv of alkyl iodide were used. [c] Volatile product, number in parenthesis is the NMR yield using CH2Br2 as internal standard.
Attracted by the importance of the indole ring in synthetic and medicinal chemistry, we sought to extend our enantiospecific three‐component alkylation reaction to N‐methyl indole (Scheme 3). Pleasingly, the reaction of boronate 15 with iodoacetonitrile (6 a) gave the desired product 14 a in 70 % yield. However, in control experiments we found that neither light nor the photocatalyst were required for the transformation. Furthermore, addition of the radical inhibitor 1,1‐diphenylethylene7a had no effect on the reaction outcome (see Supporting Information–3.5 for details), ruling out a possible electron‐transfer initiated radical chain process5 and supporting a polar SN2‐like pathway. The nucleophilic reactivity of indole‐derived borates generated from difficult‐to‐handle trialkyl boranes has been described,18 however the difficulties in accessing enantioenriched chiral boranes has prevented the use of this methodology for the synthesis of chiral compounds in enantioenriched form. Surprisingly, reactions of boronates derived from stable boronic esters are rare with only a single report very recently disclosed by Ready et al. (with π‐allyl palladium complexes).19
Scheme 3.
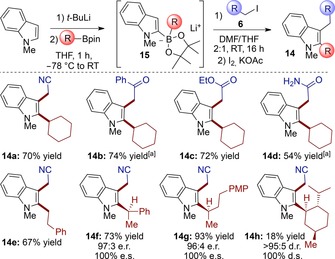
Scope of the enantiospecific three‐component alkylation of indole. All the yields refer to the isolated product after chromatographic purification. [a] Intermediate oxidation conditions: H2O2(aq.)/NaOH(aq.), 0 °C, DMF/THF 2:1.
Through our simple protocol, indole‐derived boronates 15 could be alkylated to introduce a diverse range of functional groups, including nitriles, ketones, esters and unprotected amides, providing functionalized indoles 14 a–d in good yields. Various other alkyl boronic esters were also applied to the coupling reaction, including primary (14 e) and enantioenriched chiral secondary (14 f and 14 g) examples, all proceeding in high yields and with complete stereospecificity. However, using the bulky menthyl boronic ester provided the desired product 14 h in only 18 % yield (albeit with excellent stereospecificity) showing that steric hindrance has an impact in this reaction. Finally, a simple control experiment showed that N‐methylindole does not undergo Friedel–Crafts alkylation with iodoacetonitrile 6 a under our reaction conditions (see Supporting Information–3.5 for details), thus highlighting the importance of the boronate σ‐donation to the indolyl π‐system for the nucleophilicity of 15. Indeed, Mayr has shown that a BF3K moiety (which is not as electron‐donating as RBpinLi) at the 2 position increases the nucleophilicity of N‐Boc indole by >105.20
To glean insights into the mechanism of the furan three‐component coupling reaction, selected spectroscopic and electrochemical studies were carried out. Quantum yield measurements gave a value of Φ=27.8 (see Supporting Information–3.4 for details), suggesting a radical chain pathway to be operative.21 Fluorescence quenching analysis revealed that boronate complex 2 a was an effective quencher of the excited state photocatalyst, whereas iodoacetonitrile (6 a) was ineffective (see Supporting Information–3.2 for details). Based on these results, we propose the mechanism depicted in Scheme 4. The highly reducing RuI is generated by single electron transfer (SET) from a sacrificial amount of boronate complex 2 a (E p/2=+0.26 V vs. SCE, Figure S5) to the excited RuII catalyst (E 0[RuII*/I]=+0.77 V vs. SCE).22, 23 Since one electron oxidation of analogous boronate complexes has been shown to lead to the generation of alkyl radicals through C−B bond fragmentation,24 this reductive quenching phenomenon is expected to lead to by‐product 16, which was indeed observed in the crude reaction mixtures. Once formed, the electron‐rich RuI species (E 0[RuI/II]=−1.33 V vs. SCE) undergoes single electron transfer with iodoacetonitrile (6 a, E p/2=−1.24 V vs. SCE, Figure S6) leading to the formation of the reactive electrophilic radical 17 and the regeneration of the RuII catalyst. Radical 17 then adds to the furyl system of 2 a generating radical anion 18. The electron‐rich radical anion is expected to undergo SET with another molecule of iodoacetonitrile (6 a),7, 25 forming radical 17 and a zwitterionic species (not shown), which undergoes 1,2‐migration to release intermediate 11 a.26, 27 In this process, the excited RuII* photocatalyst operates a smart initiation,28 with the RuI species being regenerated by reductive quenching of RuII* either by intermediate 18 or by another molecule of boronate 2 a. Considering the high bimolecular quenching rate constant found for boronate 2 a (k q=1.98×109 m −1 s−1, close to the diffusion limit, see Supporting Information–3.2 for details) and the expected low concentration of reactive intermediate 18 in solution, we believe that the second option is more likely. In this scenario a sacrificial amount of boronate 2 a is required, rather like a “tax” that has to be paid, to sustain the radical chain and balance undesired termination events.
Scheme 4.
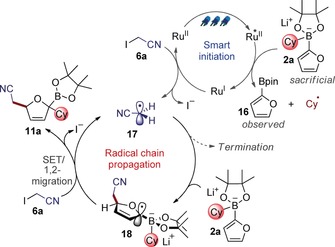
Proposed reaction mechanism for the three‐component alkylation of furan.
In conclusion, we have developed a novel stereospecific three‐component alkylation reaction of furans and indoles with boronic esters and electron‐deficient alkyl iodides. Mechanistically, the more electron‐rich indole boronates are sufficiently nucleophilic to react directly with alkyl iodides through a polar pathway. Conversely, alkylation of the less reactive furyl boronates proceeded through a radical pathway induced by photoredox catalysis. Careful mechanistic analysis showed that the furyl boronate complex 2 plays a dual role, acting both as sacrificial reductive quencher for RuII* (giving the reductant RuI) and as reactant for the three‐component alkylation reaction.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the University of Bristol, the ERC (670668) and the EPSRC (EP/I038071/1) for financial support. M.S. thanks the EU for H‐2020 Marie Skłodowska‐Curie Fellowship (grant no. 744242). R.S. thanks the Swiss National Science Foundation fellowship program (P2EZP2 175145).
M. Silvi, R. Schrof, A. Noble, V. K. Aggarwal, Chem. Eur. J. 2018, 24, 4279.
References
- 1.
- 1a. Eicher T., Hauptmann S., Speicher A., The Chemistry of Heterocycles, Wiley, Weinheim, 2012, pp. 61–80; [Google Scholar]
- 1b. Merino P., Tejero T., Delso J. I., Matute R., Curr. Org. Chem. 2007, 11, 1076–1091; [Google Scholar]
- 1c. Piutti C., Quartieri F., Molecules 2013, 18, 12290–12312; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1d. Piancatelli G., Scettri A., Barbadoro S., Tetrahedron Lett. 1976, 17, 3555–3558; [Google Scholar]
- 1e. Kappe C. O., Murphree S. S., Padwa A., Tetrahedron 1997, 53, 14179–14233. [Google Scholar]
- 2.For selected reviews, see:
- 2a. Gribble G., Indole Ring Synthesis: from Natural Products to Drug Discovery, Wiley, Chichester, 2016, pp. 1–38; [Google Scholar]
- 2b. Netz N., Opatz T., Mar. Drugs 2015, 13, 4814–4914; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2c. Homer J. A., Sperry J., J. Nat. Prod. 2017, 80, 2178–2187; [DOI] [PubMed] [Google Scholar]
- 2d. Sravanthi T. V., Manju S. L., Eur. J. Pharm. Sci. 2016, 91, 1–10. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Bonet A., Odachowski M., Leonori D., Essafi S., Aggarwal V. K., Nat. Chem. 2014, 6, 584–589; [DOI] [PubMed] [Google Scholar]
- 3b. Llaveria J., Leonori D., Aggarwal V. K., J. Am. Chem. Soc. 2015, 137, 10958–10961; [DOI] [PubMed] [Google Scholar]
- 3c. Odachowski M., Bonet A., Essafi S., Conti-Ramsden P., Harvey J. N., Leonori D., Aggarwal V. K., J. Am. Chem. Soc. 2016, 138, 9521–9532; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3d. Ganesh V., Odachowski M., Aggarwal V. K., Angew. Chem. Int. Ed. 2017, 56, 9752–9756; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 9884–9888. [Google Scholar]
- 4.
- 4a. Armstrong R. J., Aggarwal V. K., Synthesis 2017, 49, 3323–3336; [Google Scholar]
- 4b. Zhang L., Lovinger G. J., Edelstein E. K., Szymaniak A. A., Chierchia M. P., Morken J. P., Science 2016, 351, 70–74; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4c. Lovinger G. J., Aparece M. D., Morken J. P., J. Am. Chem. Soc. 2017, 139, 3153–3160; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4d. Lovinger G. J., Morken J. P., J. Am. Chem. Soc. 2017, 139, 17293–17296; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4e. Armstrong R. J., Sandford C., Garcia-Ruiz C., Aggarwal V. K., Chem. Commun. 2017, 53, 4922–4925. [DOI] [PubMed] [Google Scholar]
- 5. Wang Y., Noble A., Sandford C., Aggarwal V. K., Angew. Chem. Int. Ed. 2017, 56, 1810–1814; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1836–1840. [Google Scholar]
- 6. Umemoto T., Ishihara S., Tetrahedron Lett. 1990, 31, 3579–3582. [Google Scholar]
- 7.
- 7a. Silvi M., Sandford C., Aggarwal V. K., J. Am. Chem. Soc. 2017, 139, 5736–5739; [DOI] [PubMed] [Google Scholar]
- 7b. Kischkewitz M., Okamoto K., Mück-Lichtenfeld C., Studer A., Science 2017, 355, 936–938. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Prier C. K., Rankic D. A., MacMillan D. W. C., Chem. Rev. 2013, 113, 5322–5363; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Shaw M. H., Twilton J., MacMillan D. W. C., J. Org. Chem. 2016, 81, 6898–6926; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8c. Staveness D., Bosque I., Stephenson C. R. J., Acc. Chem. Res. 2016, 49, 2295–2306; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8d. Goddard J.-P., Ollivier C., Fensterbank L., Acc. Chem. Res. 2016, 49, 1924–1936. [DOI] [PubMed] [Google Scholar]
- 9.For selected reviews about the reactivity of aromatic rings with radicals, see:
- 9a. Studer A., Bossart M. in Radicals in Organic Synthesis, Vol. 2 (Eds.: P. Renaud, M. P. Sibi), Wiley, Weinheim, 2001, pp. 62–80; [Google Scholar]
- 9b. Studer A., Angew. Chem. Int. Ed. 2012, 51, 8950–8958; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 9082–9090; [Google Scholar]
- 9c. Gurry M., Aldabbagh F., Org. Biomol. Chem. 2016, 14, 3849–3862; [DOI] [PubMed] [Google Scholar]
- 9d. Tauber J., Imbri D., Opatz T., Molecules 2014, 19, 16190–16222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.For seminal examples of photoredox-mediated reactions of five-membered heteroaromatics, see:
- 10a. Nagib D. A., MacMillan D. W. C., Nature 2011, 480, 224–228; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Furst L., Matsuura B. S., Narayanam J. M. R., Tucker J. W., Stephenson C. R. J., Org. Lett. 2010, 12, 3104–3107. [DOI] [PubMed] [Google Scholar]
- 11.For representative photoredox-induced reactions involving reduction of alkyl halides, see:
- 11a. Nicewicz D. A., MacMillan D. W. C., Science 2008, 322, 77–80; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Narayanam J. M. R., Tucker J. W., Stephenson C. R. J., J. Am. Chem. Soc. 2009, 131, 8756–8757; [DOI] [PubMed] [Google Scholar]
- 11c. Nguyen J. D., Tucker J. W., Konieczynska M. D., Stephenson C. R. J., J. Am. Chem. Soc. 2011, 133, 4160–4163; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11d. Wallentin C.-J., Nguyen J. D., Finkbeiner P., Stephenson C. R. J., J. Am. Chem. Soc. 2012, 134, 8875–8884. [DOI] [PubMed] [Google Scholar]
- 12.For a review, see: Sandford C., Aggarwal V. K., Chem. Commun. 2017, 53, 5481–5494. [DOI] [PubMed] [Google Scholar]
- 13. Schaefer F. C. in PATAI′S Chemistry of Functional Groups, The Cyano Group (Ed.: Z. Rappoport), Wiley, Belfast, 1970, pp. 239–305. [Google Scholar]
- 14. Welin E. R., Warkentin A. A., Conrad J. C., MacMillan D. W. C., Angew. Chem. Int. Ed. 2015, 54, 9668–9672; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 9804–9808. [Google Scholar]
- 15.An extensive screen of different bases revealed that potassium acetate [instead of potassium carbonate (see Ref. 5)] led to a significant improvement in oxidation yield. With other substrates we found that NaClO (aq.), in DMF was required. See the Supporting Information for details.
- 16.For selected examples of the use of halomethyl sulfones as radical precursors, see:
- 16a. Renaud P., Schubert S., Angew. Chem. Int. Ed. Engl. 1990, 29, 433–434; [Google Scholar]; Angew. Chem. 1990, 102, 416–417; [Google Scholar]
- 16b. Masnyk M., Tetrahedron Lett. 1991, 32, 3259–3262; [Google Scholar]
- 16c. Liu F., Li P., J. Org. Chem. 2016, 81, 6972–6979; [DOI] [PubMed] [Google Scholar]
- 16d. Filippini G., Silvi M., Melchiorre P., Angew. Chem. Int. Ed. 2017, 56, 4447–4451; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 4518–4522. [Google Scholar]
- 17.Sulfones open significant opportunities for further C−C bond formations, see: Simpkins N. S., Sulphones in Organic Synthesis Pergamon, Exeter: 1993. [Google Scholar]
- 18.
- 18a. Levy A. B., Tetrahedron Lett. 1979, 20, 4021–4024; [Google Scholar]
- 18b. Ishikura M., Terashima M., J. Chem. Soc. Chem. Commun. 1991, 1219–1221; [Google Scholar]
- 18c. Ishikura M., Kato H., Ohnuki N., Chem. Commun. 2002, 220–221; [DOI] [PubMed] [Google Scholar]
- 18d. Ishikura M., Kato H., Tetrahedron 2002, 58, 9827–9838; [Google Scholar]
- 18e. Ishikura M., Ida W., Yanada K., Tetrahedron 2006, 62, 1015–1024; [Google Scholar]
- 18f. Vaultier M., Carboni B. in Comprehensive Organometallic Chemistry II, Vol. 11 (Ed.: A. McKillop), Pergamon, Oxford, 1995, p. 229. [Google Scholar]
- 19. Panda S., Ready J. M., J. Am. Chem. Soc. 2017, 139, 6038–6041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.
- 20a. Berionni G., Morozova V., Heininger M., Mayer P., Knochel P., Mayr H., J. Am. Chem. Soc. 2013, 135, 6317–6324; [DOI] [PubMed] [Google Scholar]
- 20b. Berionni G., Leonov A. I., Mayer P., Ofial A. R., Mayr H., Angew. Chem. Int. Ed. 2015, 54, 2780–2783; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 2820–2824. [Google Scholar]
- 21. Cismesia M. A., Yoon T. P., Chem. Sci. 2015, 6, 5426–5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bock C. R., Connor J. A., Gutierrez A. R., Meyer T. J., Whitten D. G., Sullivan B. P., Nagle J. K., J. Am. Chem. Soc. 1979, 101, 4815–4824. [Google Scholar]
- 23.For an analogous sacrificial role of a reaction intermediate in photoredox, see Ref. [11a].
- 24. Rasappan R., Aggarwal V. K., Nat. Chem. 2014, 6, 810–814. [DOI] [PubMed] [Google Scholar]
- 25.For a review about electron catalysis, see: Studer A., Curran D. P., Nat. Chem. 2014, 6, 765–773. [DOI] [PubMed] [Google Scholar]
- 26.SET and 1,2-migration is likely to be a concerted process. For a DFT study on the reaction of furyl systems with Br+-type electrophiles, see Ref. [3c].
- 27.At this stage, an atom-transfer mechanism cannot be ruled out. In this scenario, radical anion 18 abstracts an I atom from another molecule of iodoacetonitrile, generating radical 17 and an α-iodo boronate complex, which is expected to undergo 1,2-migration rapidly to afford product 11 a.
- 28. Studer A., Curran D. P., Angew. Chem. Int. Ed. 2016, 55, 58–102; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 58–106. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary