

Abstract
This study examines the effect of co‐administration of antimicrobial peptides and the synthetic glycolipid FP7, which is active in inhibiting inflammatory cytokine production caused by TLR4 activation and signaling. The co‐administration of two lipopolysaccharide (LPS)‐neutralizing peptides (a cecropin A–melittin hybrid peptide and a human cathelicidin) enhances by an order of magnitude the potency of FP7 in blocking the TLR4 signal. Interestingly, this is not an additional effect of LPS neutralization by peptides, because it also occurs if cells are stimulated by the plant lectin phytohemagglutinin, a non‐LPS TLR4 agonist. Our data suggest a dual mechanism of action for the peptides, not exclusively based on LPS binding and neutralization, but also on a direct effect on the LPS‐binding proteins of the TLR4 receptor complex. NMR experiments in solution show that peptide addition changes the aggregation state of FP7, promoting the formation of larger micelles. These results suggest a relationship between the aggregation state of lipid A‐like ligands and the type and intensity of the TLR4 response.
Keywords: aggregation, antimicrobial peptides, FP7, small-molecule antagonists, toll-like receptor 4
Introduction
Toll‐like receptors (TLRs) belong to the family of pattern recognition receptors and participate in immune surveillance by detecting pathogen‐associated molecular patterns (PAMPs).1, 2 Upon recognition of PAMPs, TLRs recruit adapter molecules and initiate a wide range of reactions leading to both innate and adaptive immune responses. Toll‐like receptor 4 (TLR4) is expressed at the surface of innate immune cells (e.g., macrophages, dendritic cells) and specifically recognizes bacterial endotoxins, for example, lipopolysaccharide (LPS) or lipooligosaccharide (LOS),3 the main molecular components of the outer leaflet of the outer membrane of Gram‐negative bacterial cell walls. Lipid A, the membrane‐anchoring moiety of LPS, is the biologically active part of LPS and LOS.4 The current view of the molecular mechanism of TLR4 activation by endotoxins (LPS or LOS) is based on the serial interactions of increasing affinity of a single LPS molecule with LPS‐binding proteins: lipopolysaccharide‐binding protein (LBP), cluster of differentiation 14 (CD14) and finally with myeloid differentiation factor 2 (MD‐2),5 inducing the dimerization of TLR4 by forming the complex (TLR4–MD‐2–lipid A)2 that in turn triggers intracellular Toll/interleukin‐1 receptor (TIR) domain interactions and myddosome assembly.6 Myeloid differentiation primary response gene 88 (MyD88)‐dependent intracellular signaling of the membrane complex (TLR4–MD‐2–lipid A)2 and/or, after its endosome internalization, TIR‐domain‐containing adapter‐inducing interferon‐β (TRIF)‐dependent signaling, lead to the production of, respectively, inflammatory cytokines and interferon‐β. LPS and LOS are amphiphilic molecules with typically low sub‐micromolar/nanomolar critical micelle concentration (CMC) values in an aqueous environment, and hence have a tendency to aggregate in concentration ranges relevant for biological responses. CMC values between 1.3 and 1.6 μm have been measured by fluorescence correlation microscopy for Escherichia coli LPS.7 However, as for every amphiphile, monomers are also present in a dynamic equilibrium depending on the actual concentration. The issue of whether large or small aggregates, or monomers, are the biologically active units of endotoxins has been thoroughly debated in the literature.8, 9, 10 The first steps of LPS recognition and binding by LBP, albumin and CD14 are probably influenced by the size and 3D shape of LPS aggregates, whereas the formation of supramolecular complexes between single LPS molecules and receptors (CD14 and MD‐2) is the basis of sensitive and selective endotoxin molecular recognition by the (TLR4–MD‐2–lipid A)2 active complex.5, 6, 11, 12 Modulation of the TLR4 signal by agonists and antagonists is an innovative approach to developing either immunostimulating agents, for example, vaccine adjuvants, or therapeutics that target inflammatory pathologies due to excessive TLR4 activation by bacterial PAMPs and endogenous damage‐associated molecular patterns (DAMPs).13, 14
Two strategies are usually adopted to interfere with TLR4 activation and signaling with small molecules: 1) LPS‐binding molecules that prevent the interaction of LPS with receptors, and 2) inhibition of activated (TLR4–MD‐2–LPS)2 complex formation by molecules directly competing with LPS for the binding to MD‐2 and CD14 receptors. Whereas the first strategy is mainly used to block TLR4 stimulation by LPS in sepsis and septic shock, the second one could be, in principle, applied to block a wider array of pathologies deriving from TLR4 activation by DAMPs.
In the first approach, positively charged antimicrobial peptides (AMPs) are known to bind to and neutralize LPS and interact with endotoxin.15 The prototypic AMP is polymyxin B,16 a cationic, small, cyclic lipopeptide. Further examples are cecropins,17 magainins,18 proline–arginine‐rich peptides,19 tachyplesin,20 defensins,21 and others.22, 23, 24 The structures of many of these peptides are known and include turn/loop, helix or β‐sheet motifs. Neutralization of LPS by AMPs involves a strong exothermic coulombic interaction between the two species, with ensuing fluidization of LPS acyl chains, and a drastic change in LPS aggregate type from cubic into multilamellar and an increase in aggregate sizes; together these effects bring about the inhibition of the binding of LBP and other mammalian proteins to the endotoxin.25
The second approach to block the LPS–TLR4 signal is based on molecules that directly compete with LPS or other ligands for the binding of MD‐2 and CD14 co‐receptors. Several small molecules with potent TLR4 antagonist activity are known, such as synthetic phosphorylated disaccharides mimicking lipid A (Eisai's eritoran being the most famous),26 synthetic monosaccharides,27, 28, 29 or natural and synthetic compounds with structures unrelated to lipid A.14
We recently synthesized FP7 (Figure 1), a diphosphorylated glucosamine monosaccharide bearing two myristic (C14) chains linked to the C‐2 and C‐3 positions, which was shown to be active as a TLR4 antagonist in cells and in animal models.30, 31, 32 Similarly to disaccharide‐based lipid A mimetics that block TLR4, such as eritoran, FP7 binds to MD‐2 by inserting its fatty acid C14 linear chains into the receptor's binding cavity.30 We were interested in investigating whether cationic peptides that interact with anionic LPS aggregates could also interact with the anionic monosaccharide FP7 and thus modulate its TLR4 antagonist activity. To this end, we decided to combine FP7 with cecropin A–melittin (CA–M) hybrids, a class of AMPs in which the cationic N terminus of cecropin A (CA) is fused to the hydrophobic N terminus of melittin (M; AMPs 1–5), and with LL‐37, a human cathelicidin that possesses a variety of activities including endotoxin neutralization (AMP 6).33, 34, 35 Specifically, we screened six synthetic CA–M hybrids (Table 1), specifically CA(1–8)M(1–18)36 (AMP 1), CA(1–7)M(2–9)37 (AMP 2), [K6(Me3)]CA(1–7)M(2–9)38 (AMP 3), N α‐Oct‐CA(1–7)M(2–9)39 (AMP 4), CA(1–7)M(5–9)37 (AMP 5), and LL‐3740 (AMP 6), for their effects on TLR4 activation if added alone to cells or in combination with the TLR4 antagonist FP7.
Figure 1.
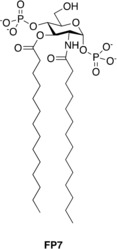
Molecular formula of synthetic glycolipid FP7.
Table 1.
Primary structures of the peptides used in this study.
| Entry | Common name | Sequence | Ref. |
|---|---|---|---|
| AMP 1 | CA(1–8)M(1–18) | KWKLFKKIGIGAVLKVLTTGLPALIS‐amide | 36 |
| AMP 2 | CA(1–7)M(2–9) | KWKLFKKIGAVLKVL‐amide | 37 |
| AMP 3 | [K6(Me3)]CA(1–7)M(2–9) | KWKLFK(Me3)KIGAVLKVL‐amide | 38 |
| AMP 4 | N α‐Oct‐CA(1–7)M(2–9) | octanoyl‐KWKLFKKIGAVLKVL‐amide | 39 |
| AMP 5 | CA(1–7)M(5–9) | KWKLFKKVLKVL‐amide | 37 |
| AMP 6 | LL‐37 | LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES | 40 |
Results and Discussion
AMPs potentiation of FP7 antagonist activity on LPS–TLR4 signaling in HEK‐Blue hTLR4 cells
The effect of FP7/AMP co‐administration was initially investigated in HEK‐Blue hTLR4 cells. These cells are HEK293 cells stably transfected with human TLR4, MD‐2, and CD14 genes. In addition, these cells possess a reporter gene encoding a secreted embryonic alkaline phosphatase (SEAP), which is produced upon activation of NF‐κB. LPS binding triggers in sequence TLR4 dimerization, myddosome formation and NF‐κB activation, leading to SEAP production and secretion. The activity of compound FP7 as TLR4 antagonist was confirmed, exhibiting dose‐dependent inhibition of LPS‐stimulated TLR4 activation with a calculated IC50 of 2.5 μm (Figure 2 and Table 2).30 Interestingly, AMPs administered alone did not show any antagonist effect on the same cell line at the concentration range used (Figure S1 in the Supporting Information). FP7 was than co‐administered (1:1 stoichiometric ratio) with AMPs 1–6. In the concentration range (0 to 10 μm) tested, AMPs 2–5 weakly enhanced FP7 antagonist activity (co‐administration IC50 was around 1.1–1.5 μm, Table 2), whereas AMPs 1 and 6 showed a stronger effect (IC50 0.56 and 0.18 μm, respectively; Figure 2 and Table 2). To exclude the possibility that the activity increase was due to a cytotoxic effect, all co‐administrations were assessed by the MTT test, and showed no or very low toxicity up to the highest concentration tested (10 μm; Figure S2). CA(1–8)M(1–18) (AMP 1) and LL‐37 (AMP 6) most efficiently improve the TLR4 antagonist activity of FP7 in HEK‐Blue hTLR4 cells.
Figure 2.
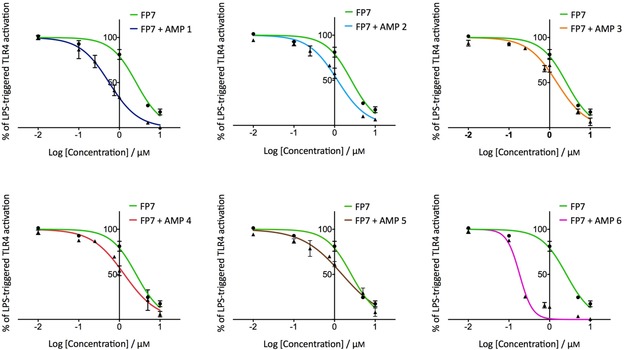
Dose‐dependent inhibition of the LPS‐stimulated TLR4 signal in HEK‐Blue hTLR4 cells by FP7/AMP co‐administration. HEK‐Blue hTLR4 cells were pre‐treated with increasing concentrations of FP7 and FP7/AMP mixtures and stimulated with LPS (100 ng mL−1) after 30 min. Data were normalized to stimulation with LPS alone. Concentration–effect data were fitted to a sigmoidal four‐parameter logistic equation to determine IC50 values. Data points represent the mean percentage ± standard error of the mean (SEM) of at least three independent experiments. Table 2 summarizes the IC50 values for the inhibition of LPS‐ and PHA‐stimulated TLR4 signal in HEK‐Blue hTLR4 cells.
Table 2.
Effects of FP7/AMP administration on the LPS‐ and PHA lectin‐stimulated TLR4 signal in HEK‐Blue hTLR4 cells.
| Treatment | IC50 [μm] | |
|---|---|---|
| LPS | PHA | |
| FP7 | 2.5 | 1.21 |
| FP7+AMP 1 | 0.56 | – |
| FP7+AMP 2 | 1.18 | – |
| FP7+AMP 3 | 1.51 | – |
| FP7+AMP 4 | 1.32 | – |
| FP7+AMP 5 | 1.54 | – |
| FP7+AMP 6 | 0.18 | 0.14 |
We then tested the activity of AMPs 1–6 as TLR4 antagonists in HEK‐Blue hTLR4 cells. Peptides showed no or weak activity in preventing LPS‐triggered TLR4 activation. AMP 1 and AMP 6 showed IC50 values of 21.9 and 23.1 μm, respectively, whereas AMPs 2–5 were found to be inactive in neutralizing the LPS stimulus. An IC50 value of approximately 0.3 μm has been reported for LL‐37 (AMP 6) in limulus amebocyte lysate (LAL) and whole blood (WB) assays.34 The discrepancy in the IC50 values we determined from HEK‐Blue hTLR4 cell experiments and that previously reported using LAL or WB assays is about two orders of magnitude. This can be explained by experimental differences, such as incubation times (30 min pre‐incubation with peptide for LAL and WB assays, no pre‐incubation in our experiments).
LL‐37 (AMP 6) potentiation of FP7 antagonist activity in lectin‐stimulated cells
Considering that several AMPs are known to interact with high affinity with LPS to promote its neutralization,33, 35, 41 we next investigated if the potentiation of FP7 antagonist activity by AMPs was due exclusively to a neutralizing effect on the endotoxin. If so, the additive effect would be lost by stimulating cells with a TLR4 agonist different from LPS. The phytohemagglutinin (PHA) lectins PHA‐L and PHA‐P induce TLR4‐dependent NF‐κB activation in a dose‐dependent manner, with a lower potency than LPS.42 In our test of the activity of PHA‐P as a TLR4 agonist (Figure 3 A), we aimed to ensure that the TLR4 activity of the lectin was not due to LPS contamination, thus we treated cells with either PHA‐P alone or in the presence of the LPS sequestrant polymyxin B, obtaining similar activation values (Figure 3 B). Next we used PHA‐P‐stimulated HEK‐Blue hTLR4 cells to evaluate the antagonist activity of FP7 in the presence of LL‐37 (AMP 6). As expected, treatment with FP7 inhibited TLR4 activation in a dose‐dependent manner, confirming that the compound interferes with receptor–ligand recognition (Figure 3 C). Interestingly, on the same PHA‐P‐activated cells the potentiation of FP7 antagonism by LL‐37 co‐administration was maintained (Figure 3 C), suggesting that the enhancement of FP7 activity is at least in part independent from a LPS‐neutralizing effect.
Figure 3.
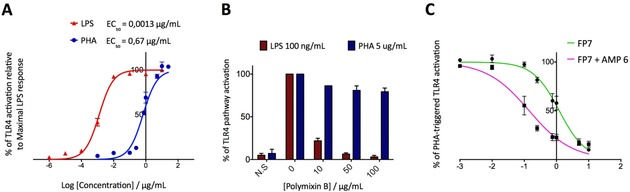
A) Dose‐dependent PHA‐ and LPS‐stimulated TLR4 activation in HEK‐Blue hTLR4 cells. HEK‐Blue hTLR4 cells were stimulated with increasing concentrations of LPS and PHA lectin and SEAP levels in the medium were quantified after 16 h. The percentages of TLR4 activation are relative to the maximal LPS response. B) HEK‐Blue hTLR4 cells were stimulated with LPS (100 ng mL−1) and PHA lectin (5 μg mL−1) in the absence or presence of increasing concentrations of polymyxin B. C) Dose‐dependent inhibition of PHA‐stimulated TLR4 activation by FP7 and FP7/AMP 6. Cells were treated with increasing concentrations of compounds and stimulated with PHA‐P (5 μg mL−1). The results represent data normalized with the positive control (PHA‐P alone). Concentration–effect data were fitted to a sigmoidal four‐parameter logistic equation to determine IC50 values and represent the mean of percentage±SEM of at least three independent experiments. The IC50 values are shown in Table 1.
LL‐37 (AMP 6) potentiation of FP7 antagonist activity in human peripheral blood mononuclear cells
We investigated whether the capacity of the most potent peptide, LL‐37 (AMP 6), to enhance FP7 antagonist activity occurs also in human monocytes that naturally express TLR4. For this purpose, human peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats, pre‐incubated with increasing concentrations (0.1–10 μm) of FP7 or FP7/AMP 6 mix and stimulated with LPS (100 ng mL−1) after 30 min. We evaluated the production of the NF‐κB‐dependent pro‐inflammatory cytokine interleukin‐1β (IL‐1β) as a readout for TLR4 pathway activation. As expected, FP7 was able to decrease the production of IL‐1β in a dose‐dependent manner, halving the amount of cytokine released if administered at a concentration of 5 μm. The addition of AMP 6 to FP7 produces a much more powerful inhibitory response, inhibiting the production of interleukin at a lower dose of 1 μm (Figure 4).
Figure 4.
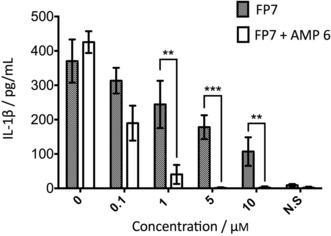
LL‐37 (AMP 6) potentiation of FP7 antagonist activity in human PBMCs. PBMCs isolated from buffy coats were pre‐incubated with FP7 or FP7/AMP 6 mix for 30 min and then stimulated with LPS (100 ng mL−1). IL‐1β production was quantified after incubation overnight. Data represent the mean±SEM (**P<0.01, ***P<0.001) of at least three independent experiments.
NMR analysis of glycolipid–peptide interactions
The additive effect of AMP 6 on FP7 activity in lectin‐stimulated cells suggests that a direct interaction between peptides and glycolipid might have an effect on TLR4 antagonism. The peptide–glycolipid interaction was studied by NMR for AMP 1 and AMP 6, by analyzing the perturbations observed on characteristic NMR parameters (e.g., chemical shifts, line widths, and signal intensities) of either binding partner. The titration of AMP 1 and AMP 6 with FP7 (Figures S3 and S11, respectively) revealed the broadening of the peptide NMR resonance signals upon addition of FP7, mainly a clear perturbation of the signals of the lateral chains of the hydrophobic amino acids. The experimentally observed reductions in intensity (Figures S4 and S12), due to specific line broadening of these signals, probably arise from the changes in the transverse relaxation times of these signals, a clear indication of binding between these two AMPs and FP7. The diffusion‐ordered spectroscopy (DOSY) spectra of AMP 1 and AMP 6 showed a strikingly low diffusion coefficient, much different from its low/medium molecular weight, indicating peptide aggregation (see Figures S5 and S13). This experimental evidence was also confirmed by using TEM negative staining analysis (Figures S6 and S14). Indeed, filament‐like shapes were observed for the peptides alone. Interestingly, the diffusion coefficient of the both AMPs remained unaltered in the presence of FP7 (Figures S5 and S13). Thus, the interaction of FP7 with these two AMPs (in excess), does not show a large effect on the average size of the AMP aggregates.
The process was also monitored by measuring the NMR signals of FP7 protons upon addition of AMP 1 (Figure 5 A‐I) and AMP 6 (Figure 5 B‐I). In both cases, the dramatic reduction of the intensity of the NMR signals corresponding to aliphatic moieties allowed us to conclude that the interaction with AMPs 1 and 6 involved the lipid chains (Figure 5 A‐I and B–I for AMP 1 and AMP 6, respectively). Three alternative hypotheses could explain these data. The peptide could act as linker between different FP7 aggregates (Figure S7) and/or deform the FP7 micelle (Figure S8). Alternatively, the peptide could participate in the formation of large aggregates (Figure S9), behaving as a large molecule, as earlier described for MD‐2.30
Figure 5.
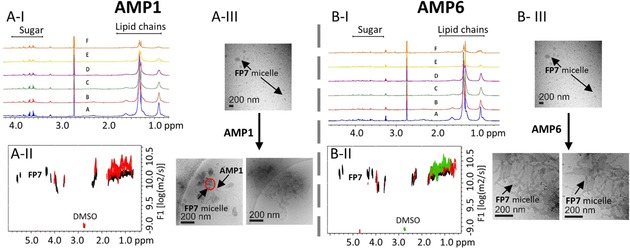
A‐I) 1H NMR titration of FP7 with AMP 1. A: FP7 alone (500 μm); B: +10 μm AMP 1; C: +30 μm AMP 1; D: +50 μm AMP 1; E: +90 μm AMP 1; F: +170 μm AMP 1. A‐II) DOSY spectrum; black: FP7 (500 μm); red: FP7 (500 μm) +AMP 1 (30 μm). A‐III) Cryo‐TEM of FP7 alone (2.5 mg mL−1) and after addition of AMP 1 (80 μm), with a nominal magnification of 30 000× (0.36 nm per pixel). B‐I) 1H NMR titration of FP7 with AMP 6. A: FP7 alone (500 μm); B: +10 μm AMP 6; C: +20 μm AMP 6; D: +30 μm AMP 6; E: +50 μm AMP 6; F: +90 μm AMP 6. B‐II) DOSY spectrum; black: FP7 (500 μm) alone; red: FP7 (500 μm) with AMP 6 (10 μm); green: FP7 (500 μm) with AMP 6 (50 μm). B‐III) Cryo‐TEM of FP7 (2.5 mg mL−1) and after addition of AMP 6 (400 μm), nominal magnification of 30 000× (0.36 nm per pixel). The samples were prepared in 10 % DMSO in PBS (100 mm, pH 5.5). Two cryo‐TEM images taken after addition of AMPs are shown to demonstrate that the same structures are present in different grid locations.
This model is in agreement with the DOSY observations (Figures S5 and S13) described above. Indeed, the DOSY experiment of FP7 in the presence of AMPs (Figure 5 A‐IIand B‐II for AMP 1 and AMP 6, respectively) showed clear perturbations of the diffusion coefficient measured for FP7 alone. In the case of AMP 1, for substoichiometric ratios of the peptide ([AMP 1]/[FP7]=0.06), the decrease in the diffusion coefficient of FP7 is evident (Table 3). AMP 6 causes a decrease in the diffusion coefficient of FP7, although the observed perturbation is smaller than that in the presence of AMP 1 (Table 3).
Table 3.
Diffusion coefficient values estimated for FP7 (500 μm) from DOSY NMR experiments.
| Compound(s) | D [m2 s−1] |
|---|---|
| FP7 | 6.31×10−11 |
| [AMP 1]/[FP7]=0.06 | 3.16×10−11 |
| [AMP 6]/[FP7]=0.02 | 5.01×10−11 |
| [AMP 6]/[FP7]=0.1 | 3.98×10−11 |
This effect could, in principle, be due to changes either in the size or shape (Figures S7 and S8) of the lipid. Thus, we used TEM with negative staining analysis (Figures S10 and S14) and cryo‐microscopy (Figure 4 A‐III and B‐III) to obtain the required morphological information. It was possible to observe that the presence of AMP 1 induced formation of aggregates between different FP7 micelles, thus supporting the change in size. The peptide is linking various FP7 micelles, displaying peanut‐shaped structures. This suggests the presence of fusion events (indicated with the red circle in Figure 4 A‐III and Figure S10). In contrast, the TEM analysis of FP7 in the presence of AMP 6 showed a dramatic change in the shape of the micelles, from spheres to cylinders. Long entangled cylindrical micelles are displayed in the cryo‐TEM image (Figure 5 B‐III) and in the negative staining analysis (Figure S14, right).
Conclusions
Compound FP7 has been rationally designed to be a MD‐2 ligand and shown to inhibit TLR4 signal in cells in the low micromolar range.30 The mechanism of action is based on direct competition with LPS for interacting with the MD‐2 binding pocket.30 We recently observed that FP7 targets selectively TLR4 and not TLR2, and that it is able to block the TLR4 signal activated by microbial PAMPs and also by endogenous DAMPs such as HMGB1 protein.32 The potent and selective TLR4 antagonist activity and its lack of toxicity make FP7 a good lead for therapeutic development against pathologies generated by PAMP–TLR4 and DAMP–TLR4 signaling. There is a pressing need for new antimicrobial drugs that can neutralize bacterial products such as endotoxins. In some pathologies such as inflammatory bowel disease (IBD; Crohn's disease and ulcerative colitis), inflammation induces damage of the intestinal epithelium, which becomes more permeable to bacteria.43 With a view to studying the synergistic combination of TLR modulators with antibacterial drugs for these and other inflammatory and autoimmune pathologies, we examined the combination of a potent TLR4 antagonist with various AMPs and shown that co‐administration of FP7 with cationic AMPs potentiates the antagonist action of FP7 on cells expressing hTLR4 and murine cells. Out of the six AMPs studied, AMP 1 and AMP 6 present the most pronounced effect. A solution structure for CA(1–8)M(1–18) (AMP 1) is not available. Nevertheless, a six‐residue‐shorter version, CA(1–8)M(1–12), in aqueous solution (in the presence of structure‐inducing trifluoroethanol)44 has been shown to adopt a major helical structure, with three and one helix turns in the melittin and cecropin A moieties, respectively, separated by a flexible hinge (residues Gly–Ile–Gly). These features have been postulated to be required for membrane disruption against both prokaryotic and eukaryotic cells. On the other hand, a high quality structure of human cathelicidin LL‐37 (AMP 6) in SDS micelles has been determined by NMR spectroscopy using a 13C,15N‐labeled version.45 In the SDS micelles, the peptide adopted a curved amphipathic helix‐bend‐helix motif spanning most of its length (residues 2–31), a pattern not unlike that of CA(1–8)M(1–12) discussed above. Several mechanisms can underlie the potentiating action of AMPs. A direct neutralizing effect on LPS by AMP interaction cannot completely explain the result, which is still observed if cells are stimulated by the plant lectin PHA instead of LPS. Three possible mechanisms are: 1) direct binding of AMPs to CD14 and/or MD‐2 receptors, with concomitant LPS displacement; 2) the aggregation state of FP7 is affected by AMPs; 3) an allosteric action of AMPs that stabilizes the inactive FP7–MD‐2–TLR4 complex.
The amphiphilic glycolipid FP7 has a CMC of approximately 9 μm,30 the same order of magnitude as that calculated for E. coli LPS (between 1.3 and 1.6 μm).7 In the concentration range of our cell experiments (0.1–10 μm), FP7 is therefore in equilibrium between aggregate and monomeric species.
Both NMR spectroscopy and TEM experiments clearly show an effect of both AMP 1 or AMP 6 on the FP7 aggregation state. NMR spectroscopy shows that addition of either AMP to FP7 causes the formation of larger aggregates, as revealed by the reduction in the intensities of signals corresponding to the FP7 aliphatic chain protons and the decrease of the FP7 diffusion coefficient in DOSY. Cryo‐TEM images support these data and clearly show that, upon peptide addition, FP7 micellar aggregates (at 500 μm concentration) undergo a change in size and 3D shape from spheres to rod‐like cylinders. NMR experiments provide a valuable indication on the ability of these AMPs to affect FP7 aggregation state in an aqueous environment. However, because they have been performed at a concentration two orders of magnitude higher than that at which FP7 is biologically active, they might suggest a similar behavior of the peptides on FP7 under biological conditions but do not allow a definitive conclusion on this. A more detailed physicochemical characterization of FP7–AMP co‐aggregates is in progress.
In pathologies in which inflammation is exacerbated by bacterial infection, the combined use of anti‐TLR (anti‐inflammatory) small molecules with AMPs, as discussed here for the TLR4 antagonist FP7 and peptides CA(1–8)M(1–18) and LL‐37, might become a valuable and innovative therapeutic approach. Moreover, the capacity of AMP peptides to potentiate the TLR4 antagonist action also in the absence of LPS opens the way to new mechanisms of antagonism based on the direct binding of AMPs to MD‐2 or formation of AMP–antagonist co‐aggregates or allosteric modulation of TLR4 by AMPs.
Experimental Section
NMR spectroscopy
All NMR experiments were recorded on a Bruker Avance III 800 MHz spectrometer equipped with a TCI cryoprobe or a Bruker Avance III 600 MHz spectrometer equipped with a TBI probe. The 1H NMR resonances of the peptides (AMPs 1 and 6) were characterized through 2D TOCSY (75 ms mixing time) and 2D NOESY experiments (300 ms mixing time). The concentration of the compounds was fixed at 500 μm (AMP 6) and 300 μm (AMP 1) in perdeuterated PBS (100 μm) in H2O/D2O (90:10) with 10 % DMSO at an uncorrected pH meter reading of 5.5, at 293 K. The resonance of 2,2,3,3‐tetradeutero‐3‐trimethylsilylpropionic acid was used as a chemical shift reference in the 1H NMR experiments (δ=0 ppm). Peak lists for the 2D TOCSY and 2D NOESY spectra were generated by interactive peak picking using the CARA software (R. Keller, Computer‐aided Resonance Assignment Tutorial CARA, Cantina Verlag, Goldau, Switzerland, 2004). The DOSY spectra of FP7 were recorded at 310 K with the tdDOSYccbp.2D pulse sequence by acquisition of 256 scans, with a diffusion time of 300 ms, a gradient length of 2 ms, and a gradient ramp from 5 % to 95 % in 16 linear steps. Additions of the peptides to the solution were then made and new DOSY spectra were recorded up to molar ratios of [AMP 1]/[FP7]=0.06 and [AMP 6]/[FP7]=0.1.
The DOSY spectra of the isolated AMP 1 and AMP 6 peptides as blanks were recorded at 310 K with the tdDOSYccbp.2D pulse sequence by acquisition of 128 scans, with a diffusion time of 250 ms, a gradient length of 1.5 ms, and a gradient ramp from 5 % to 95 % in 16 linear steps. Additions of FP7 to the solution were then made and new DOSY spectra were recorded up to molar ratios of [FP7]/[AMP 1]=0.667 and [FP7]/[AMP 6]=0.667. FP7 samples were prepared by diluting the stock solution of FP7 (50 mm in DMSO) with PBS buffer (100 mm pH 5.5), with a final DMSO content of 10 %. Peptide samples were prepared by dissolving the solid molecules in DMSO (20 mm stock solution).
Transmission electron microscopy
All samples were prepared with in H2O containing 10 % DMSO. Samples for negative staining were applied to glow‐discharged carbon‐coated copper grids and stained with 2 % (w/v) NanoVan (methylamine vanadate), Nanoprobes. Digital micrographs were taken at room temperature in low‐dose radiation mode on a Jeol transmission electron microscope (JEM‐1230) operated at 100 kV and equipped with an Orius SC1000 (4008×2672 pixels) cooled slow‐scan CCD camera (GATAN, UK). For cryo‐microscopy studies the samples were vitrified on Quantifoil 2/2 grids, using a Vitrobot (FEI) and were analyzed at liquid nitrogen temperature with a transmission electron microscope operated at 200 kV in low‐dose conditions. The samples were applied to glow‐discharged carbon‐coated copper grids and stained with 2 % (w/v) NanoVan. Micrographs were taken at a low radiation dose on a JEM‐2200FS/CR transmission electron microscope (JEOL, Japan), equipped with an UltraScan 4000 SP (4008×4008 pixels) cooled slow‐scan CCD camera (GATAN, UK).
Peptides and biochemicals
Six AMPs were produced in high purity (>95 % by analytical HPLC) by Fmoc solid‐phase synthesis methods, as reported (see Table 1). The HPLC‐purified materials had the expected composition as determined by electrospray or MALDI‐TOF mass spectrometry. E. coli LPS (O55:B5) and PHA‐P lectin (from Phaseolus vulgaris) were supplied from Sigma–Aldrich. Polymyxin B was purchased from InvivoGen.
HEK‐Blue hTLR4 cell assay
HEK‐Blue hTLR4 cells were purchased from InvivoGen and cultured according to the manufacturer's instructions in DMEM (Euroclone) supplemented with 10 % FBS, glutamine (2 mm), antibiotics (penicillin/streptomycin) and 1× HEK‐Blue Selection (InvivoGen). Cells were detached from the plate using a cell scraper, mixed 1:1 with trypan blue solution and counted. Cells were then diluted in complete DMEM with FBS (10 %) and glutamine (2 mm), plated in a 96‐well plate (3×104 cells per well) and incubated overnight (37 °C, 5 % CO2, 95 % humidity). Supernatants were removed and replaced with premixed DMEM without FBS (190 μL per well) and test compound solution (different concentrations, 10 μL per well). After 30 min cells were stimulated with LPS (100 ng mL−1) and incubated for 16 h, as above. Supernatants were then collected and incubated with p‐nitrophenyl phosphate solution (0.8 mm). Absorbance was monitored at 405 nm. All curves are representative of at least three independent experiments.
Human peripheral blood mononuclear cells
PBMCs were isolated by density gradient centrifugation (Lympholyte‐H, Cedarlane Labs) from buffy coats. In brief, buffy coats were diluted 1:1 with phosphate‐buffered saline (PBS), and layered on Lympholyte‐H for density gradient centrifugation according to the manufacturer's instructions. PBMCs were harvested from the interface and washed in PBS. The isolated cells were counted, checked for viability using 0.1 % trypan blue and resuspended in RPMI 1640 supplemented with 10 % fetal bovine serum (FBS), penicillin (100 U mL−1) and streptomycin (100 U mL−1). Cells were plated in 96‐well U‐bottomed multiwell culture plates (Euroclone), pre‐incubated with increasing concentrations of FP7 and FP7/AMP 6 mix (0.1, 1, 5, 10 μm) and stimulated with smooth lipopolysaccharide (S‐LPS; E. coli 055:B5; Sigma; 100 ng mL−1) after 30 min. Each patient who took part in the study gave written informed consent, and the study protocol was approved by the local ethics committees (agreement between Ospedale Niguarda Cà Granda and Università degli Studi di Milano‐Bicocca for the supply of buffy coats for research use).
MTT cell viability assay
HEK‐Blue hTLR4 cells were seeded in a 96‐well multiwell plate at the density of 3×104 cells per well. After overnight incubation, the test compounds were added at different concentrations to each well and the plate was incubated for 16 h. PBS was included as an internal control. The supernatant of each well was then collected and replaced with of DMEM (90 μL) and MTT solution (5 mg mL−1 in PBS, 10 μL). The plate was incubated for 2–4 h at 37 °C, 5 % CO2, 95 % humidity. Formazan crystals were dissolved by adding HCl in 2‐propanol (0.1 n, 100 μL per well) and the absorbance was measured at 570 nm (LT‐4000 Microplate Reader, Labtech). The results were normalized to untreated cells (PBS) and expressed as the mean percentage±SEM of three independent experiments.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This study was financially supported by the H2020‐MSC‐ETN‐642157 project TOLLerant. The Italian Ministry for Foreign Affairs and International Cooperation (MAECI) is acknowledged. Work at Pompeu Fabra University was supported by MINECO (grants SAF2011‐24899, AGL2014‐52395‐C2‐2‐R to D.A., CTQ2015‐64597‐C2‐1‐P and MINECO‐Severo Ochoa Excellence Acreditation 2017–2021 (SEV‐2016‐0644) to J.J.‐B.) with FEDER funds, and by Generalitat de Catalunya (2014SGR692).
F. A. Facchini, H. Coelho, S. E. Sestito, S. Delgado, A. Minotti, D. Andreu, J. Jiménez-Barbero, F. Peri, ChemMedChem 2018, 13, 280.
Contributor Information
Dr. Fabio A. Facchini, Email: f.facchini16@gmail.com
Prof. Francesco Peri, Email: francesco.peri@unimib.it.
References
- 1. Akira S., Takeda K., Nat. Rev. Immunol. 2004, 4, 499–511. [DOI] [PubMed] [Google Scholar]
- 2. Miyake K., Semin. Immunol. 2007, 19, 3–10. [DOI] [PubMed] [Google Scholar]
- 3. Poltorak A., He X., Smirnova I., Liu M.-Y., Huffel C. V., Du X., Birdwell D., Alejos E., Silva M., Galanos C., Freudenberg M., Ricciardi-Castagnoli P., Layton B., Beutler B., Science 1998, 282, 2085. [DOI] [PubMed] [Google Scholar]
- 4. Molinaro A., Holst O., Di Lorenzo F., Callaghan M., Nurisso A., D′Errico G., Zamyatina A., Peri F., Berisio R., Jerala R., Jiménez-Barbero J., Silipo A., Martín-Santamaría S., Chem. Eur. J. 2015, 21, 500–519. [DOI] [PubMed] [Google Scholar]
- 5. Teghanemt A., Widstrom R. L., Gioannini T. L., Weiss J. P., J. Biol. Chem. 2008, 283, 21881–21889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Park B. S., Song D. H., Kim H. M., Choi B.-S., Lee H., Lee J.-O., Nature 2009, 458, 1191–1195. [DOI] [PubMed] [Google Scholar]
- 7. Yu L., Tan M., Ho B., Ding J. L., Wohland T., Anal. Chim. Acta 2006, 556, 216–225. [DOI] [PubMed] [Google Scholar]
- 8. Schromm A. B., Howe J., Ulmer A. J., Wiesmüller K.-H., Seyberth T., Jung G., Rössle M., Koch M. H. J., Gutsmann T., Brandenburg K., J. Biol. Chem. 2007, 282, 11030–11037. [DOI] [PubMed] [Google Scholar]
- 9. Mueller M., Lindner B., Kusumoto S., Fukase K., Schromm A. B., Seydel U., J. Biol. Chem. 2004, 279, 26307–26313. [DOI] [PubMed] [Google Scholar]
- 10. Gutsmann T., Schromm A. B., Brandenburg K., Int. J. Med. Microbiol. 2007, 297, 341–352. [DOI] [PubMed] [Google Scholar]
- 11. Jerala R., Int. J. Med. Microbiol. 2007, 297, 353–363. [DOI] [PubMed] [Google Scholar]
- 12. Gioannini T. L., Teghanemt A., Zhang D., Coussens N. P., Dockstader W., Ramaswamy S., Weiss J. P., Proc. Natl. Acad. Sci. USA 2004, 101, 4186–4191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Peri F., Piazza M., Biotechnol. Adv. 2012, 30, 251–260. [DOI] [PubMed] [Google Scholar]
- 14. Peri F., Calabrese V., J. Med. Chem. 2014, 57, 3612–3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Andreu D., Rivas L., Pept. Sci. 1998, 47, 415–433. [DOI] [PubMed] [Google Scholar]
- 16. Rifkind D., J. Infect. Dis. 1967, 117, 433–438. [DOI] [PubMed] [Google Scholar]
- 17. Lee J.-Y., Boman A., Chuanxin S., Andersson M., Jörnvall H., Mutt V., Boman H. G., Proc. Natl. Acad. Sci. USA 1989, 86, 9159–9162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zasloff M., Martin B., Chen H.-C., Proc. Natl. Acad. Sci. USA 1988, 85, 910–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Agerberth B., Lee J.-Y., Bergman T., Carlquist M., Boman H. G., Mutt V., Jörnvall H., Eur. J. Biochem. 1991, 202, 849–854. [DOI] [PubMed] [Google Scholar]
- 20. Kawano K., Yoneya T., Miyata T., Yoshikawa K., Tokunaga F., Terada Y., Iwanaga S., J. Biol. Chem. 1990, 265, 15365–15367. [PubMed] [Google Scholar]
- 21. Lehrer R. I., Ganz T., Selsted M. E., Cell 1991, 64, 229–230. [DOI] [PubMed] [Google Scholar]
- 22. Mayo K. H., Haseman J., Young H. C., Mayo J. W., Biochem. J. 2000, 349, 717–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Japelj B., Pristovšek P., Majerle A., Jerala R., J. Biol. Chem. 2005, 280, 16955–16961. [DOI] [PubMed] [Google Scholar]
- 24. Andrä J., Lohner K., Blondelle S. E., Jerala R., Moriyon I., Koch M. H. J., Garidel P., Brandenburg K., Biochem. J. 2005, 385, 135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kaconis Y., Kowalski I., Howe J., Brauser A., Richter W., Razquin-Olazarán I., Iñigo-Pestaña M., Garidel P., Rössle M., de Tejada G. M., Gutsmann T., Brandenburg K., Biophys. J. 2011, 100, 2652–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim H. M., Park B. S., Kim J.-I., Kim S. E., Lee J., Oh S. C., Enkhbayar P., Matsushima N., Lee H., Yoo O. J., Lee J.-O., Cell 2007, 130, 906–917. [DOI] [PubMed] [Google Scholar]
- 27. Matsuura M., Kiso M., Hasegawa A., Infect. Immun. 1999, 67, 6286–6292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Funatogawa K., Matsuura M., Nakano M., Kiso M., Hasegawa A., Infect. Immun. 1998, 66, 5792–5798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tamai R., Asai Y., Hashimoto M., Fukase K., Kusumoto S., Ishida H., Kiso M., Ogawa T., Immunology 2003, 110, 66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cighetti R., Ciaramelli C., Sestito S. E., Zanoni I., Kubik L., Arda-Freire A., Calabrese V., Granucci F., Jerala R., Martin-Santamaria S., Jiménez-Barbero J., Peri F., ChemBioChem 2014, 15, 250–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. De Paola M., Sestito S. E., Mariani A., Memo C., Fanelli R., Freschi M., Bendotti C., Calabrese V., Peri F., Pharmacol. Res. 2016, 103, 180–187. [DOI] [PubMed] [Google Scholar]
- 32. Perrin-Cocon L., Aublin-Gex A., Sestito S. E., Shirey K. A., Patel M. C., Andre P., Blanco J. C., Vogel S. N., Peri F., Lotteau V., Sci. Rep. 2017, 7, 40791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Scott A., Weldon S., Buchanan P. J., Schock B., Ernst R. K., McAuley D. F., Tunney M. M., Irwin C. R., Elborn J. S., Taggart C. C., PLoS One 2011, 6, e26525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nell M. J., Tjabringa G. S., Wafelman A. R., Verrijk R., Hiemstra P. S., Drijfhout J. W., Grote J. J., Peptides 2006, 27, 649–660. [DOI] [PubMed] [Google Scholar]
- 35. Rosenfeld Y., Papo N., Shai Y., J. Biol. Chem. 2006, 281, 1636–1643. [DOI] [PubMed] [Google Scholar]
- 36. Wade D., Boman A., Wahlin B., Drain C. M., Andreu D., Boman H. G., Merrifield R. B., Proc. Natl. Acad. Sci. USA 1990, 87, 4761–4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Andreu D., Ubach J., Boman A., Wahlin B., Wade D., Merrifield R. B., Boman H. G., FEBS Lett. 1992, 296, 190–194. [DOI] [PubMed] [Google Scholar]
- 38. Fernández-Reyes M., Díaz D., de la Torre B. G., Cabrales-Rico A., Vallès-Miret M., Jiménez-Barbero J., Andreu D., Rivas L., J. Med. Chem. 2010, 53, 5587–5596. [DOI] [PubMed] [Google Scholar]
- 39. Chicharro C., Granata C., Lozano R., Andreu D., Rivas L., Antimicrob. Agents Chemother. 2001, 45, 2441–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gudmundsson G. H., Magnusson K. P., Chowdhary B. P., Johansson M., Andersson L., Boman H. G., Proc. Natl. Acad. Sci. USA 1995, 92, 7085–7089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wu J., Mu L., Zhuang L., Han Y., Liu T., Li J., Yang Y., Yang H., Wei L., Parasites Vectors 2015, 8, 561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Unitt J., Hornigold D., Biochem. Pharmacol. 2011, 81, 1324–1328. [DOI] [PubMed] [Google Scholar]
- 43. Spadoni I., Zagato E., Bertocchi A., Paolinelli R., Hot E., Di Sabatino A., Caprioli F., Bottiglieri L., Oldani A., Viale G., Penna G., Dejana E., Rescigno M., Science 2015, 350, 830. [DOI] [PubMed] [Google Scholar]
- 44. Oh D., Shin S. Y., Kang J. H., Hahm K.-S., Kim K. L., Kim Y., J. Pept. Res. 1999, 53, 578–589. [DOI] [PubMed] [Google Scholar]
- 45. Wang G., J. Biol. Chem. 2008, 283, 32637–32643. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary