

Summary
Background
Hepatitis B virus (HBV) infection is common with major clinical consequences. In Asian Americans, the HBsAg carrier rate ranges from 2% to 16% which approximates the rates from their countries of origin. Similarly, HBV is the most important cause of cirrhosis, hepatocellular carcinoma (HCC) and liver related deaths in HBsAg positive Asians worldwide.
Aim
To generate recommendations for the management of Asian Americans infected with HBV.
Methods
These guidelines are based on relevant data derived from medical reports on HBV from Asian countries as well as from studies in the HBsAg positive Asian Americans. The guidelines herein differ from other recommendations in the treatment of both HBeAg positive and negative chronic hepatitis B (CHB), in the approach to HCC surveillance, and in the management of HBV in pregnant women.
Results
Asian American patients, HBeAg positive or negative, with HBV DNA levels >2000 IU/mL (>104 copies/mL) and ALT values above normal are candidates for anti‐viral therapy. HBeAg negative patients with HBV DNA >2000 IU/mL and normal ALT levels but who have either serum albumin <3.5 g/dL or platelet count <130 000 mm3, basal core promoter (BCP) mutations, or who have first‐degree relatives with HCC should be offered treatment. Patients with cirrhosis and detectable HBV DNA must receive life‐long anti‐viral therapy. Indications for treatment include pregnant women with high viraemia, coinfected patients, and those requiring immunosuppressive therapy. In HBsAg positive patients with risk factors, life‐long surveillance for HCC with alpha‐fetoprotein (AFP) testing and abdominal ultrasound examination at 6‐month intervals is required. In CHB patients receiving HCC treatments, repeat imaging with contrast CT scan or MRI at 3‐month intervals is strongly recommended. These guidelines have been assigned to a Class (reflecting benefit vs. risk) and a Level (assessing strength or certainty) of evidence.
Conclusions
Application of the recommendations made based on a review of the relevant literature and the opinion of a panel of Asian American physicians with expertise in HBV treatment will inform physicians and improve patient outcomes.
1. INTRODUCTION
Hepatitis B virus infection is common and clinically consequential worldwide. The prevalence of HBV carriers globally ranges from 240 million to 300 million persons.1, 2 An estimated 75% of these infected individuals reside in the Asia‐Pacific region wherein HBV is responsible for up to one‐third of all cases of cirrhosis and over 50% of HCC cases. HBV infection is a major public health problem in the USA, especially among ethnic minorities. The prevalence of HBsAg positive persons in the USA is estimated to be 2.2 million persons. Of these, 1.32 million are foreign born with 58% immigrating from high or intermediate endemic areas of Asia.3 At present, immigrants account for 95% of newly diagnosed HBV infections in the USA.4 As a result, Asian Americans are the ethnic group with the highest prevalence of HBV, and have the highest rates of HCC incidence and mortality in this country. A recent study in 400 HBsAg positive Asian American patients showed that during a mean 84‐month follow‐up, 67 of 71 (94%) deaths were from either liver‐related complications or HCC.5 Recent reports indicate that both screening for HBV in the Asian American population and linkage to health care have been suboptimal.6, 7 Despite the availability of potent anti‐viral medications, improving the morbidity and mortality rates associated with HBV in Asian Americans require greater emphasis on HBV education among health care providers.
2. METHODS
In April 2016, a panel of Asian American physicians with expertise in the management of HBV convened in Pasadena, CA. Each member was pre‐assigned a section and instructed to review the relevant medical literature specifically aimed at Asian and Asian Americans with hepatitis B infection. During the meeting, each topic was presented and subsequently, the manuscript was compiled and edited (MJT). The manuscript was reviewed by the authors and a consensus on management of Hepatitis B in Asian Americans was finalised.
Unique for Asians and Asian Americans with CHB include maternal to child transmission (MTCT), the long period of chronicity beginning in the immune tolerant stage with high viraemia, presence of genotypes B or C, and the high rate of progression to liver related complications and HCC. More recently, disparities in HCC survival was reported in a subgroup of first generation Asian immigrants to the USA, specifically due to paucity of medical care, lack of hepatitis B screening and treatments, and late stage of HCC presentation.8 Thus, the aim of this manuscript is to review the natural history, virology and clinical characteristics as it pertains to the approach, management and treatment of Asian Americans with chronic hepatitis B. It is anticipated that the information herein will assist physicians in the day to day management of CHB in the Asian American population.
3. QUALITY OF EVIDENCE
This document was intended to provide recommendations regarding the management of CHB infection in adult Asian Americans. Recommendations set forth herein have been assigned to a Class (reflecting benefit vs risk) and a Level (assessing strength or certainty) of evidence according to the grading system set forth in the 2015 update of the American College of Cardiology/American Heart Association Clinical Practice Guideline Recommendation Classification System (Data S1).9
4. EPIDEMIOLOGY, NATURAL HISTORY, CLINICAL STAGES AND PROGRESSION OF DISEASE IN ASIANS
4.1. Prevalence of HBsAg
Hepatitis B virus carrier rates in Asia have been reported to be as high as 20% in the male population of Guang Xi Province and up to 27.5% in the Fujian Province of China.10 HBV carrier rates ranging from 10% to 15% have been reported in Hong Kong, Shanghai, Taiwan and Southeast Asia.11 According to the results of a recent study from China, HBV carrier rates have fallen to 7.2% in regions where HBV vaccination programmes have been implemented.12 The report also indicated that the carrier rate in children <5 years of age had decreased from 9.7% in 1992 to 1% in 2006, thus preventing an estimated 16‐20 million HBV carriers through HBV vaccination of infants and young children. A similar study in Taiwan showed a decrease in the HBV carrier rate in children from 10% in 1984 to 0.5% in 2009 as well as in the childhood HCC rate after implementation of HBV vaccination of infants at birth.13 However, in other parts of Asia, the HBsAg carrier rates remain high, particularly in countries in which HBV vaccine programmes have not yet been implemented. In these endemic areas, 80% of patients with chronic hepatitis, 92% with cirrhosis, and 80% with HCC are HBsAg positive.14, 15, 16, 17, 18, 19
Notably, the HBsAg carrier rates among Asian Americans are similar to rates reported in their countries of origin, especially in first generation immigrants to the USA3, 12, 19, 20 Table 1 shows a comparison of HBsAg prevalence rates derived from screening programmes in various cities in the USA, to a study which derived the HBsAg carrier rates by estimating the number of foreign born persons in the USA and the prevalence of HBsAg carriers reported from their native countries.3
Table 1.
The prevalence of HBsAg in Asian Americans
| Country of origin | Screening in the USA | Derived from prevalence in country of origin |
|---|---|---|
| China | 5.6%‐12.3% | 12.3% |
| Vietnam | 3.2%‐13.6% | 12.8% |
| Philippines | 5.5%‐7.4% | 7.4% |
| South Korea | 5.3% | 5.3% |
| Laos | 2.3%‐16.4% | 11.6% |
| Burma | 9.4%‐12.4% | 11.6% |
| Cambodian | 10.3% | 13.6% |
| Thailand | 3.9%‐6.1% | 12.5% |
| Malaysia | 8.8% | 5.6% |
| India | 1.5% | 3.2% |
4.2. Routes of infection
In high and intermediate HBV endemic areas (HBsAg prevalence rates of ≥8% and 2%‐7% respectively) in Asia, HBV infection takes place from mother to child during birth. Up to 90% of infants born to HBeAg positive mothers with high viraemia become chronically infected if immunoprophylaxis is not administered at birth. In low endemic areas (HBsAg prevalence rates <2%), exposure to HBV occurs primarily among adolescents and adults, in whom the carrier rate following acute infection is <5%. In Asian adults, sexual contact may account for up to 10% of all HBV carriers. HBV transmission also occurs at high rates among injection drug users in Asia.21
4.3. The phases of CHB infection
In Asian patients, the natural course of CHB infection can be divided into five distinct phases which include the immune tolerant, immune activation, low replication phase, reactivation and the clinical resolution phase (Figure 1). A description of the five phases is provided in Data S2.
Figure 1.
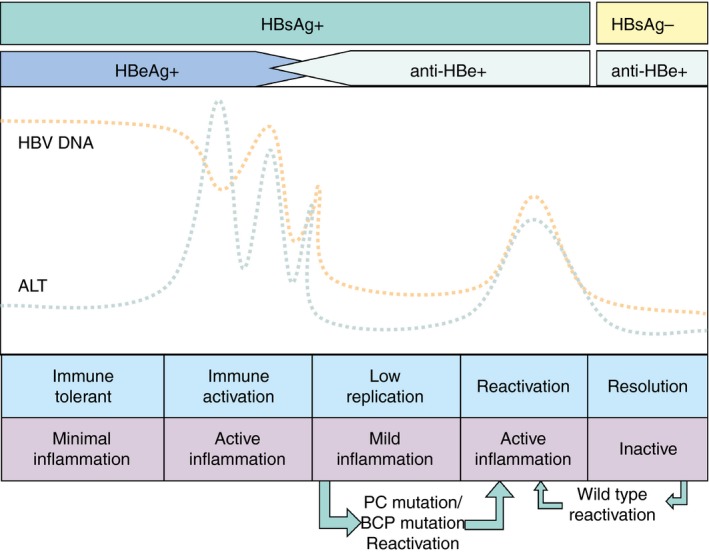
The five phases of chronic hepatitis B infection
4.4. The clinical stages of CHB infection
Based on HBeAg status, as well as ALT and HBV DNA levels at presentation, patients may be classified into one of several clinical stages of HBV infection (Figure 2).22 During subsequent visits, changes in the patient's virological and biochemical parameters may clarify the patient's clinical status, especially regarding the evaluation of disease progression and assessment of the need for anti‐viral therapy. The six clinical stages include: HBeAg positive immune tolerant, chronic hepatitis and cirrhosis patients, and the HBeAg negative low replication carrier, chronic hepatitis and cirrhosis patients. These clinical stages are further characterised in Table S1.
Figure 2.
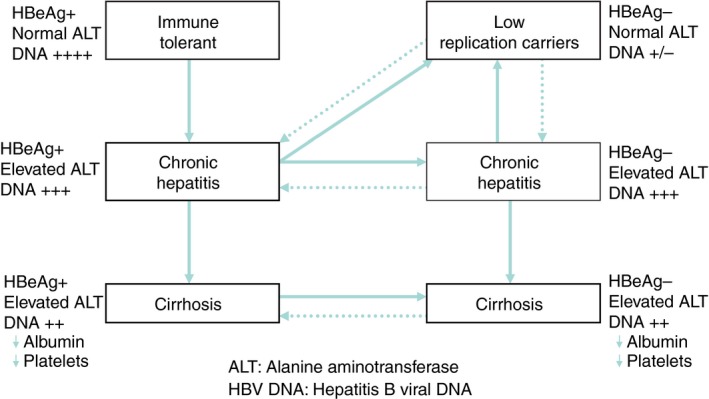
The clinical stages of hepatitis B
4.5. Predictors of disease progression
The annual rates of progression from chronic hepatitis to cirrhosis, decompensated cirrhosis, HCC and death are shown on Figure S1 and described in Data S3. Serologic predictors of disease progression in CHB patients include HBeAg, HBV genotype, HBV DNA titres, BCP mutants and PC mutants. A description of each of these factors are shown in Table S2. Tests for HBV genotype, BCP mutants and PC mutants are readily available at commercial laboratories in the USA. In addition, serum AFP levels are elevated in patients with acute and chronic liver diseases in the absence of HCC, especially in the presence of ongoing necroinflammation.23, 24 Elevation of AFP levels during ALT flares was detected in CHB‐infected patients who eventually developed cirrhosis, and thus is another parameter to use when evaluating patients for anti‐viral therapy.25
4.6. Family history of HCC
In a recent study, of 413 Asian American HBsAg positive patients, 173 with HCC and 240 without HCC were sub grouped into those with or without a family history of HCC and analysed for risk factors associated with HCC development.25 Forty‐four of 173 (25.4%) HCC patients had 82 other blood relatives with HCC. Of these, 69 (84.1%) were first degree relatives. In a multivariate logistic regression model, a family history of HCC was an independent factor associated with HCC development (OR = 2.58, P = 0.048). In addition, in a large population‐based cohort study of 22 472 participants from Taiwan, 374 cases of incident HCC were identified during 362 268 person‐years of follow‐up.26 The multivariate‐adjusted hazard ratio for HCC for HBsAg positive individuals with family history of HCC was 32.33 (P < 0.001), confirming a synergistic effect of family history and HBV on incidence of HCC.
5. SURVEILLANCE FOR HCC IN ASIAN PATIENTS WITH CHB INFECTION
Patients with CHB infection have a > 100‐fold increased risk for the development of HCC compared with those without HBV infection.27, 28 The annual incidence of HCC in low replication carriers is estimated to be 0.5% and increases to 3%‐10% in patients with cirrhosis.29 The REVEAL study from Taiwan by Chen et al demonstrated a strong correlation between increasing HBV DNA levels beginning at >2000 IU/mL (>104 copies/mL) and risk for HCC.27, 30 Another report on 820 patients with CHB infection from Hong Kong showed that male gender, increasing age, higher HBV DNA levels, presence of BCP mutations, and cirrhosis were independent risk factors for HCC.31 In a predominantly Asian American population, risk factors for HCC included older age, cirrhosis, PC mutations, BCP mutations, and high serum AFP levels.32, 33, 34
A randomised controlled trial for HCC screening in >18 000 subjects in Shanghai, China showed a 37% reduction in HCC‐related mortality among patients detected by surveillance.35 Another recent study in Asian Americans with HBV showed that surveillance identified HCC patients with smaller tumour burdens and better preserved liver function who were more likely to receive curative therapies which prolonged survival.36
Surveillance for HCC in all HBsAg positive patients should include both AFP testing and abdominal ultrasound examination (Figure 3). However, lesion detection rates by ultrasound may be compromised by body habitus, fatty liver and cirrhosis.37 Therefore, cross sectional imaging with contrast enhanced computed tomography (CT) or magnetic resonance imaging (MRI) should be considered in a surveillance programme whenever the image quality of an ultrasound is suboptimal (Figure S2). HBsAg positive patients at high risk for HCC development include those with more advanced stage of liver disease, ie, chronic hepatitis, cirrhosis and those with blood relatives with HCC.25, 38 Patients with low‐to‐moderate risk include low replication carriers and immune tolerant patients. In addition, since patients who clear HBsAg may still develop HCC, surveillance should be continued, especially if cirrhosis is present at the time of HBsAg loss.39 The current recommendation for HCC surveillance is every 6‐12 months in immune tolerant and low replication carriers, and every 6 months in chronic hepatitis and cirrhosis patients (Figure 4). In patients with persistently elevated AFP levels with undetectable lesions by ultrasound examination and in patients receiving HCC treatment, surveillance should include contrast CT scan or MRI at 3‐month intervals, which is based on the average HCC doubling time of 4.3 months.40
Figure 3.
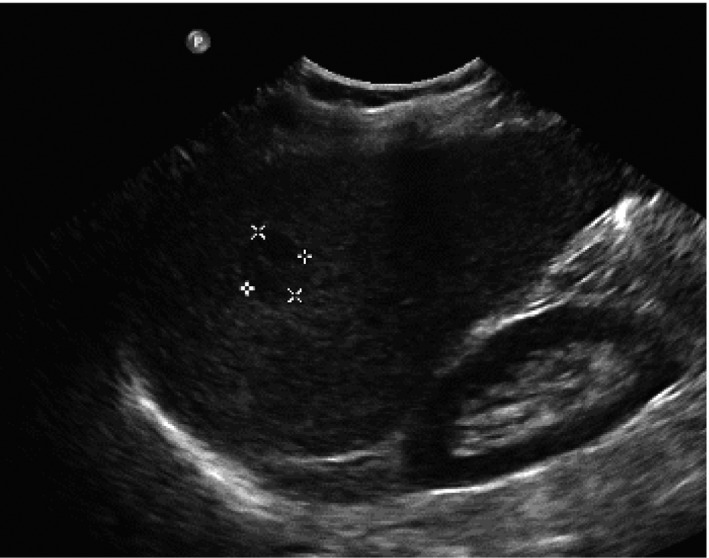
Ultrasound detection of a 3 cm HCC
Figure 4.
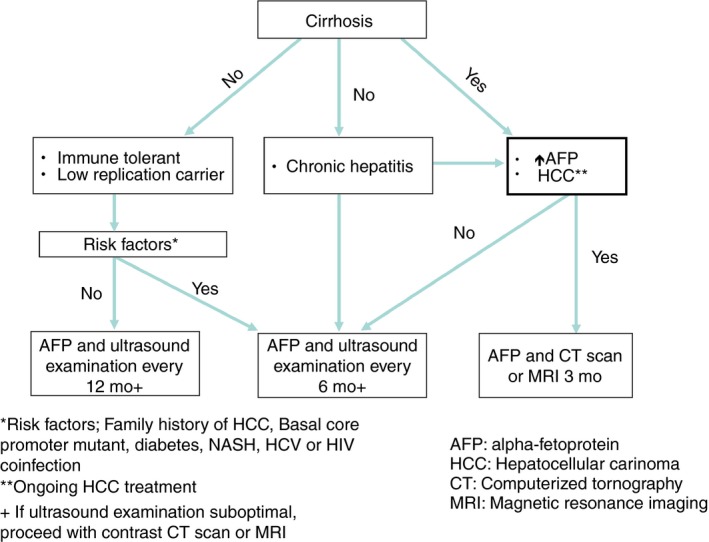
HCC surveillance intervals in Asian patients with hepatitis B
Although debate remains regarding the optimal age to begin HCC surveillance, it is probably not warranted among paediatric patients because of the low risk of HCC. The benefit of surveillance in HBsAg positive males aged <40 years and females aged <50 years has not been determined. However, up to 20% of HCC cases in Asians may be in this younger age range. If clinically active chronic hepatitis infection, cirrhosis or other risk factors for HCC are present, then surveillance for HCC should be instituted.36, 41, 42
Further management guidelines for liver lesions detected by surveillance are presented in Data S4. Confirmed cases of HCC must be referred to a multidisciplinary centre consisting of hepatologists, surgeons, interventional radiologists and oncologists who specialise in the treatments of this malignancy (Table S3).43 We recommend that during the initial workup of a new HBsAg positive patient, a baseline abdominal ultrasound and AFP measurement should be obtained. Thereafter, routine HCC surveillance with the above tests must be performed at the recommended intervals.
6. TREATMENT OF CHB INFECTION IN ASIAN AMERICANS: PROPOSED CANDIDATES FOR TREATMENT BASED ON CLINICAL STAGE
The goal of anti‐viral therapy in patients infected with HBV is to prevent progression from chronic hepatitis to cirrhosis, end‐stage liver disease, and development of HCC. The eligibility for anti‐viral therapy in recently published guidelines is based on HBeAg status, ALT values, and levels of HBV DNA in the setting of either chronic hepatitis or cirrhosis.44, 45, 46, 47
6.1. The significance of ALT
Elevated serum ALT levels are indicators of hepatic necroinflammatory activity. Because studies have shown that an elevated pre‐treatment ALT level was a significant predictor of HBeAg loss or seroconversion to treatments with interferon,48, 49 pegylated‐interferon alfa‐2a,50 lamivudine (LAM),51, 52 and adefovir (ADV),53 all of the published guidelines recommend anti‐viral therapy for patients with CHB infection who have elevated ALT levels. However, a significant proportion of CHB‐infected patients with normal to minimally increased ALT levels may have significant inflammation and fibrosis on liver biopsy.54, 55, 56, 57 Also, a meta‐analysis demonstrated that approximately one in five patients with persistently normal ALT had fibrosis scores of F2 or higher.58 Finally, a study from Hong Kong which included 3,233 CHB patients followed up for over 180 months were stratified by ALT level at presentation and during follow‐up (<0.5 × ULN, 0.5‐1 × ULN, 1‐2 × ULN, 2‐6 × ULN and >6 × ULN) and observed for development of decompensated cirrhosis or HCC.59 The highest risk of complications was associated with ALT levels 1–2 × ULN, followed by lesser risks in patients with ALT levels 2–6 × ULN, and 0.5–1 × ULN.
6.2. The significance of HBV DNA
The advent of PCR‐based assays with improved sensitivity heralded the era of serum HBV DNA as a marker of disease progression. HBeAg positive patients and many HBeAg negative patients have HBV DNA levels below 105 copies/mL.60 From 2001 through 2006, several studies noted the development of HCC in cirrhotic and noncirrhotic patients with CHB infection with HBV DNA levels as low as 3.7 log10 copies/mL.5, 61, 62, 63 In 2006, Chen et al published a study of 3653 HBsAg positive patients followed up for a mean of 11.4 years which showed that the incidence of HCC increased with serum HBV DNA levels at study entry in a dose‐related fashion.27 In a parallel publication, Iloeje and colleagues demonstrated a similar relationship between serum HBV DNA and the development of cirrhosis.64 In both studies, there was an increased risk of progression to cirrhosis and development of HCC at HBV DNA levels above 104 copies/mL. Since then, additional studies have confirmed a serum HBV DNA level >4 log10 copies/mL as predictive of an increased risk for HCC development.65, 66
6.3. Criteria from current guidelines and new era for a more comprehensive treatment strategy
Several recently published guidelines and an expert consensus algorithm focusing on the treatment of CHB infection are summarised in Table S4. The difference in selection of HBV patients for anti‐viral therapy in these published guidelines are discussed in Data S5. In a long term, natural history study of predominantly Asian CHB‐infected patients in the USA by Tong et al, the treatment criteria designated in the established treatment guidelines were applied to a cohort of 369 HBsAg positive patients followed up for a mean of 84 months (7 years).67 Using the threshold, HBV DNA and ALT values stated in the published documents, only 20%‐60% of patients who developed HCC and 27%‐70% who died from or were transplanted for non‐HCC‐related liver complications would have been identified initially as anti‐viral treatment candidates. Previously, Tong et al reported on a cohort of 400 CHB‐infected patients in the USA followed up for a mean of 83.6 months.5 The results of a multivariate analysis showed that older age (OR = 26.51, P = 0.007), PC mutants (OR = 4.23, P = 0.02), and BCP mutants (OR = 2.93, P = 0.02) were independent predictors for HCC. Additional studies on PC and BCP mutants are discussed in Data S6.
Recently, Chen et al showed that lower serum albumin levels was an independent risk factor for HCC development.27 In the aforementioned study by Tong et al,67 the addition of a platelet count <130 000 mm3 and serum albumin value <3.5 g/dL to the criteria established by the published treatment guidelines increased the detection rate of patients eligible for treatment to 89%‐100% of patients who died of non‐HCC liver‐related causes and 96%‐100% of patients who developed HCC.
6.4. Recommendations for treatment in Asian American patients with chronic hepatitis B
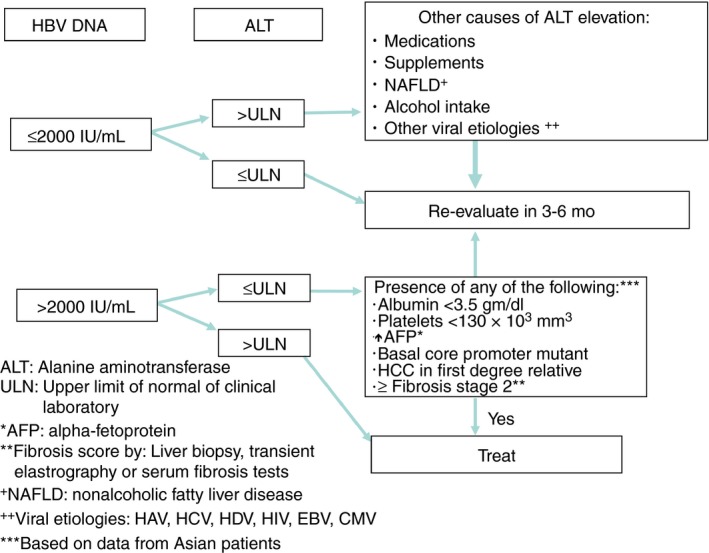
At present, anti‐viral treatment is not recommended for patients who are in the immune tolerant stage or in low replication carriers. Our guidelines are illustrated in a new algorithm, which includes additional laboratory parameters to better identify candidates for treatment to prevent disease progression (Table 2 and Figure 5). The algorithm specifies a quantitative HBV DNA threshold of 2000 IU/mL as the initial determination to consider treatment in both HBeAg positive and HBeAg negative patients, primarily based on data from the REVEAL‐HBV study from Taiwan indicating 104 copies/mL (2000 IU/mL) as the threshold HBV DNA level predictive of future cirrhosis and HCC development.27 In patients with an HBV DNA quantitation above this threshold level, determination of serum ALT is the next decision point for treatment. Patients with an ALT value > ULN in the physician's local laboratory in conjunction with an elevated serum HBV DNA would be eligible for consideration of anti‐viral therapy.
Table 2.
Recommendations for anti‐viral therapy in HBeAg positive and HBeAg negative chronic hepatitis B patients
| Clinical stage | HBeAg | HBV DNA | Comments | Quality of evidence |
|---|---|---|---|---|
| Immune tolerant | + | >2000 IU/mL | Insufficient evidence to warrant treatment | IIb C‐EO |
| Low replication carrier | − | <2000 IU/mL | Insufficient evidence to warrant treatment | IIb C‐EO |
| Chronic hepatitisa | +/− | >2000 IU/mL | See algorithm | IA |
| Cirrhosis | +/− | Detectableb | Treat | IA |
| Decompensated cirrhosis | +/− | Detectableb | Treat | I B‐NR |
| Hepatocellular carcinoma | +/− | Detectableb | Treat | IIb C‐EO |
EO, expert opinion; NR, Non randomised studies.
Differs from APASL and AASLD guidelines.
Option to treat if HBV DNA undetectable.
Figure 5.
Treatment algorithm for HBsAg positive patients with HBeAg positive/negative chronic hepatitis B
ALT values of 30 U/L for men and 19 U/L for women were established as the ULN in healthy patients with the lowest risk for subclinical liver disease68 and are used as the ULN determination in the AASLD guidelines and US algorithm. However, in real‐world practice, ALT values often exceed these “healthy” values due to underlying factors including steatosis, dyslipidemia, obesity and medications unrelated to CHB infection; therefore, these ALT values cannot be reliably interpreted to reflect inflammation due solely to HBV infection. Moreover, since “normal” ranges in clinical laboratories are determined by the patient population served by that laboratory in addition to nonstandardised assays for ALT measurement, these values will inherently vary between laboratories. The inclusion criteria used for patients treated with nucleoside/nucleotide analog anti‐viral therapy in several phase 3 clinical trials were based on > ULN ALT values derived from different clinical laboratories. Thus, the panel recommends ALT > ULN as the threshold value for treatment based on data from Hong Kong showing that significant inflammation and fibrosis were noted in patients with ALT 1‐1.5 × ULN.69
In patients with an elevated serum HBV DNA above threshold level but serum ALT ≤ ULN, further determination is necessary before initiating anti‐viral therapy. The next step is to assess the patient's serum albumin and platelet count. If the serum albumin is <3.5 g/dL or the platelet count is <130 000 mm3, anti‐viral therapy can be initiated. However, if serum albumin and platelet counts are persistently above threshold values, then testing to determine the presence of the BCP mutation is recommended. Testing for this mutation should be considered even in patients who are HBeAg positive, since these mutations may exist at baseline and if present, then anti‐viral therapy can be initiated. If laboratory testing is unremarkable for hypoalbuminemia, thrombocytopenia, or BCP mutations, then we recommend grading and staging of liver disease with liver biopsy or assessment by noninvasive methods such as transient elastography or serum markers for fibrosis. The presence of F2 fibrosis or higher in conjunction with inflammation would be an indication to start anti‐viral therapy. In the absence of active liver inflammation and advanced fibrosis, anti‐viral therapy can be deferred; however, the patient should be monitored at least twice annually or more frequently if indicated. Other risk factors for disease progression include first‐degree relatives with HCC.70 In addition, patients with elevated AFP levels during flares of hepatitis B (without HCC) may be more prone to have liver disease progression.25 The presence of any of these risk factors indicate that anti‐viral therapy should be initiated.
For patients with HBV DNA levels <2000 IU/mL and ALT >ULN, search for other causes of ALT elevation (ie, medications, NASH, excessive alcohol consumption, other viral infections, etc.). In patients with HBV DNA levels <2000 IU/mL and normal ALT values, continual evaluation should be repeated at 3‐6 month intervals.
Recent publications report that successful NA therapies reduces necroinflammation, reverses fibrosis and prevents or reverses clinical decompensation in chronic hepatitis and cirrhosis patients.71, 72 The benefit of reducing liver complications was demonstrated in a prospective trial of LAM in Asian patients with compensated cirrhosis.73 Benefit was primarily observed in patients with sustained viral suppression; however, less significant benefit was noted in patients who developed LAM resistance. Other studies showed that treatment with anti‐viral agents with low resistance profiles significantly improved clinical outcomes in patients with decompensated cirrhosis.71, 74 Furthermore, a recent report indicated that the risk of HCC is significantly increased in patients with HBV‐related cirrhosis with low viral loads.75 Based on these findings, patients with cirrhosis who have any detectable level of HBV DNA should receive lifelong anti‐viral therapy regardless of HBeAg status or ALT values. Patients with compensated cirrhosis with undetectable HBV DNA should have tests for ALT and HBV DNA every 3 months. Patients with decompensated cirrhosis should be started on anti‐viral therapy and immediately referred to a liver transplant centre.
7. ANTI‐VIRAL THERAPY
7.1. Current treatments for HBV infection
There are currently seven drugs licensed by the FDA for the treatment of CHB infection, including standard interferon alfa‐2b, LAM, ADV, pegylated interferon alfa‐2a, telbivudine, entecavir, tenofovir disoproxil fumarate (TDF), and tenofovir alafenamide fumarate (TAF), which represents a prodrug of the currently approved formulation of tenofovir. Interferon alfa‐2b is no longer used in the USA for the treatment of HBV infection. LAM, ADV, and telbivudine are considered second line agents and a summary of their effectiveness in HBV treatment is described in Data S7. The preferred or first‐line agents for the treatment of CHB infection include entecavir, TDF, and pegylated interferon alfa‐2a,46 although the latter is uncommonly used in the USA. TAF was recently approved for use in the USA in 2016 and also may be considered as a first line treatment for CHB. In this section, we review key data governing the efficacy of approved agents with regard to clinical endpoints including normalisation of ALT, suppression of HBV DNA to undetectable levels, HBeAg seroconversion, and HBsAg seroconversion. A summary of the four current agents for anti‐viral treatment in Asian CHB patients is shown in Table 3.
Table 3.
Virological, biochemical and histological responses to anti‐viral agents in treatment naive patients with CHB
| Asian patients | Trial endpoints | First line agents | ||||
|---|---|---|---|---|---|---|
| Pegylated interferon alfa 2a (up to 45 weeks) | Entecavir (up to 240 weeks) | Tenofovir (up to 192 weeks) | Tenofovir alafenamide (up to 48 weeks) | |||
| HBeAg(−) | HBV DNA | Year 1 undetectable HBV DNA | 19% | 93% | 82% (ITT) | 94% |
| Long‐term undetectable DNA | N/A | 98.3% | 97% | |||
| ALT | Year 1 ALT normalisation | 59% | 76% | 72% | 83% | |
| Long‐term ALT normalisation | N/A | 85.7% | 86% (combined with e‐pos patients) @ 192 weeks | |||
| HBsAg loss / sero‐conversion | Year 1 HBsAg sero‐conversion | 0/0% | 0/0% | 0%/0% | ||
| Long‐term HBsAg loss/sero‐conversion | 0/0% | 0/0% | ||||
| Histological Improvement | Year 1 histological improvement | N/A | 68% | 77% | ||
| Long‐term histological improvement | N/A | 100% | N/A | |||
| HBeAg(+) | HBV DNA | Year 1 undetectable HBV DNA | 14% @ 24 wk post Rx | 69% (<300 c/mL) | 85% (ITT) (<400 copies/mL) | 64% (<29 IU/mL) |
| Long‐term undetectable HBV DNA | N/A | 95% | 97% | |||
| ALT | Year 1 ALT normalisation | 41% @ 24 wk post Rx | 63% | 72% | 72% | |
| Long‐term ALT normalisation | N/A | 76% | 86% | |||
| HBeAg loss/seroconversion | Year 1 HBeAg loss/seroconversion | 31%@24 wk post Rx | 16/16% | 17/17% | 14/10% | |
| Long‐term HBeAg loss/sero‐conversion | 41% @ 48 wk post Rx | 40/18% | 35/26% | |||
| HBsAg loss/seroconversion | Year 1 HBsAg seroconversion | 2% @ 24 wk post Rx | 0.5% | 0/0% | <1%/0% | |
| Long‐term HBsAg loss / sero‐conversion | N/A |
2.9% in genotype B 0.9% in genotype C |
2.28% | |||
| Histological Improvement | Year 1 histological improvement | 38% | 71% | 77% | ||
| Long‐term histological improvement | N/A | 100% | N/A | |||
7.1.1. Pegylated interferon Alfa‐2a
Pegylated interferon alfa‐2a (Pegasys) was licensed by the FDA for the treatment of HBV infection in 2005. Two phase 3 clinical trials provided the evidence base supporting approval. In the first, Lau et al50 reported the findings in 814 HBeAg positive patients which showed that after 48 weeks of treatment, pegylated interferon alfa‐2a alone and pegylated interferon alfa‐2b plus LAM were superior to LAM alone in achieving HBeAg seroconversion (32% vs 27% vs 19%, P = 0.02) and suppression of HBV DNA to <100 000 copies/mL (32% vs 34% vs. 22%, P = 0.003). Eight patients in each of the pegylated interferon groups achieved HBsAg seroconversion, whereas no patients treated with LAM alone achieved this endpoint (3% vs. 3% vs. 0%, P = 0.004). In the second, Marcellin et al76 reported the findings in 537 HBeAg negative patients which showed that after 48 weeks of treatment, pegylated interferon alfa‐2b alone and pegylated interferon alfa‐2b plus LAM were superior to LAM alone in achieving normalisation of serum ALT (59% vs 60% vs 44%, P = 0.004), and suppression of HBV DNA to <400 copies/mL (19% vs 20% vs 7%, P < 0.001). Seven patients in the pegylated interferon alone group and five patients in the pegylated interferon plus LAM group achieved HBsAg seroconversion, whereas no patients treated with LAM achieved this endpoint (4% vs 2.8% vs 0%, P = 0.029). Additional studies combined pegylated interferon alfa‐2a with NAs such as LAM,77 ADV,78 telbivudine,79 entecavir,80 and TDF,81 but due to limited evidence, these regimens have not been incorporated into practice guidelines.
7.1.2. Entecavir
Entecavir is an oral NA which was licensed by the FDA for the treatment of HBV infection in 2005. Two phase 3 clinical trials provided the evidence base supporting FDA approval. In the first, Chang et al82 reported the findings in 715 treatment‐naïve HBeAg positive patients which showed that after 52 weeks of treatment, entecavir was superior to LAM in achieving the primary efficacy endpoint of histological improvement (72% vs 62%, P = 0.009), as well as in achieving secondary endpoints including undetectable HBV DNA (67% vs 36%, P < 0.001), normalisation of serum ALT (68% vs 60%, P = 0.02), and mean log reduction in serum HBV DNA between baseline and week 48 (6.9 vs 5.4 log copies/mL, P < 0.001).82 No statistical difference was observed in HBeAg seroconversion rates (21% vs 18%, P = 0.33). In the second, Lai et al83 reported the findings in 648 HBeAg negative patients which showed that after 52 weeks of treatment, entecavir was superior to LAM in achieving the primary efficacy endpoint of histological improvement (70% vs 61%, P = 0.01), as well as in achieving secondary endpoints including undetectable HBV DNA (90% vs 72%, P < 0.001), normalisation of serum ALT (78% vs 71%, P = 0.045), and mean log reduction in serum HBV DNA (5.0 vs 4.5 log copies/mL, P < 0.001).83 Multiple studies have confirmed the long‐term safety and efficacy of entecavir in USA cohorts with sustained virological suppression to undetectable levels and histological improvement.84, 85
7.1.3. Tenofovir disoproxil fumarate (TDF)
Tenofovir disoproxil fumarate is an oral nucleotide analogue, which was licensed by the FDA for the treatment of HBV infection in 2008. Two phase 3 clinical trials provided the evidence base supporting FDA approval. Marcellin et al86 reported the findings of both phase 3 clinical trials in which 276 treatment‐naïve HBeAg positive patients and 375 treatment‐naïve HBeAg negative patients were randomised to receive 48 weeks of treatment with either TDF 300 mg daily or ADV 10 mg daily. TDF was superior to ADV in achieving the primary efficacy endpoint of suppression of HBV DNA to <400 copies/mL (69 IU/mL) and histological improvement in both HBeAg positive (66% vs 12%, P < 0.001) and HBeAg negative patients (71% vs 49%, P < 0.001). TDF was also superior to ADV for other secondary endpoints including undetectable HBV DNA (76% vs 13%, P < 0.001 in HBeAg positive patients and 93% vs 63%, P < 0.001 in HBeAg negative patients). TDF was associated with higher rates of normalisation of serum ALT (68% vs 54%, P = 0.03) in HBeAg positive patients, but not in HBeAg negative patients (76% vs 77%, P = 0.86). Rates of HBeAg seroconversion were not statistically different between TDF and ADV (21% vs 18%, P = 0.36), although TDF was associated with a higher rate of HBsAg loss (3.2% vs 0%, P = 0.02). Long‐term studies have further confirmed the sustained safety and efficacy of TDF including virological suppression and histological regression at 5 years of therapy.87, 88, 89
7.1.4. Tenofovir alafenamide (TAF)
Tenofovir alafenamide represents a prodrug which enhances delivery of active tenofovir to hepatocytes and reduces circulating levels of tenofovir relative to TDF, and thereby decreases the risk of renal dysfunction and bone mineral density (BMD) decline. Two phase 3 clinical trials provided the evidence base supporting FDA approval. Chan et al90 reported the results in 873 HBeAg positive patients randomised 2:1 to TAF 25 mg daily (n = 581) or TDF 300 mg daily (n = 292) for a period of 96 weeks. Based on a noninferiority study design, no difference was observed between groups in the primary therapeutic endpoint of suppression of HBV DNA to <29 IU/mL (64% TAF vs 67% TDF, P = 0.25), or secondary endpoints such as normalisation of serum ALT (72% TAF vs 67% TDF, P = 0.18), HBeAg loss (14% TAF vs TDF 12%, P = 0.47), HBeAg seroconversion (10% TAF vs 8% TDF, P = 0.32) or HBsAg loss (<1% TAF vs <1% TDF, P = 0.52). Although total AEs, SAEs, and discontinuations due to AEs were similar between groups, fewer patients on TAF experienced a > 3% decrease in bone mineral density at week 48 in the spine (18% TAF vs 38% TDF, P < 0.001) and hip (8% TAF vs 24% TDF, P < 0.001), and fewer changes were observed in bone turnover markers. In addition, TAF was associated with less change in serum creatinine (0.009 vs 0.026, P = 0.02) and fewer patients with decline in the estimated glomerular filtration rate (eGFR) to <50 mL/min (0 vs 2%, P = 0.004). Buti et al91 reported the results in 425 HBeAg negative patients randomised 2:1 to TAF 25 mg daily (n = 285) or TDF 300 mg daily (n = 140) for a period of 96 weeks. Based on a noninferiority study design, no difference was observed between groups in the primary therapeutic endpoint of suppression of HBV DNA to <29 IU/mL (94% TAF vs 93% TDF, P = 0.47), or secondary endpoints such as normalisation of serum ALT (83% TAF vs 75% TDF, P = 0.076). Although total AEs, SAEs, and discontinuations due to AEs were similar between groups, fewer patients on TAF experienced a > 3% decrease in bone mineral density at week 48 in the spine (22% TAF vs 39% TDF, P < 0.001) and hip (10% TAF vs 33% TDF, P < 0.001), and fewer changes were observed in bone turnover markers. The mean change in serum creatinine at week 48 was minimal in both groups (TAF 0.01 mg/dL vs TDF 0.02 mg/dL, P = 0.32), but patients receiving TAF, had a smaller reduction in creatinine clearance (median change in eGFR −1.8 mL/min vs −4.8 mL/min, P = 0.04), with minimal changes in markers of proximal renal tubular function. Based on a superior safety profile and non‐inferiority of efficacy endpoints, TAF represents an attractive alternative to TDF in the treatment of CHB.
7.1.5. Treatment for decompensated cirrhosis
The efficacy and safety of oral NAs has been studied in patients with decompensated cirrhosis, although the evidence base is limited with historical anti‐viral agents.92 Interferon alfa‐2b and pegylated interferon alfa‐2a are contraindicated in this population. Due to superior potency and low rates of virological resistance, and more robust evidence in prospective trials,71, 74, 93 either entecavir or TDF are preferred agents for use in patients with decompensated cirrhosis. Long‐term studies documenting the impact of anti‐viral therapy in this population on both pre‐transplant and post‐transplant clinical outcomes (eg, hepatic decompensation, hospitalisations, HCC, liver‐related and all‐cause mortality) remain limited and would be helpful in clarifying the role and timing of treatment in this population.
8. MONITORING HBV TREATMENT
Prior to initiating anti‐viral treatment, patients should be informed that therapy is long‐term and adherence and regular follow‐up visits are essential. Anti‐viral medications for hepatitis B are usually well tolerated with minimal side effects.11, 93, 94, 95, 96, 97 Timely prescription refills and appropriate patient support will improve patient compliance and treatment outcomes.
8.1. Laboratory monitoring
After initiating anti‐viral therapy, liver tests should be performed every 3 months until serum ALT levels are within normal limits and thereafter at 3‐6 month intervals. ALT flares may occur after initiation of treatment, but usually do not require discontinuation of medication. Renal impairment including cases of acute renal failure and Fanconi syndrome (renal tubular injury) with hypophosphatemia have been reported in a few cases treated with TDF.93 If clinically indicated, serum creatinine, estimated creatinine clearance, phosphorus and urine glucose and protein should be measured.11, 93, 94 Similar assessment of renal function also is recommended for patients treated with TAF.98 In patients receiving pegylated interferon therapy, complete blood cell counts including absolute neutrophil and platelet counts, and thyroid function testes are required. Serum HBV DNA should be measured using a sensitive PCR test with preferred detection limit of 20‐50 IU/mL every 3 months until undetectable, and then every 3‐6 months thereafter. HBeAg positive patients on anti‐viral treatment should have tests for HBeAg every 6 months until seronegative before initiating anti‐HBe testing. HBsAg should be tested every 12 months after seroconversion from HBeAg to anti‐HBe positive.11, 93 Although on‐treatment HBsAg clearance is rare in Asian patients, it is recommended that testing for HBsAg be performed every 12 months after sustained suppression of HBV DNA in HBeAg negative patients.94 Recent reports have indicated HBsAg quantification may be useful in predicting HBV treatment responses, but such tests are not widely available at present.99
8.2. Treatment duration and endpoints
Hepatitis B virus treatment duration with anti‐viral regimens is dependent on the clinical stage of disease and HBeAg status.100 The decision to discontinue anti‐viral therapy should be made only after careful assessment of patients’ compliance for continued monitoring since only 38% of patients will maintain their clinical endpoints within 36 months of follow‐up, as described in a recent meta‐analysis of 25 studies for NA cessation.101
The optimal duration of anti‐viral therpy for HBeAg positive and HBeAg negative chronic hepatitis patients without cirrhosis is unknown. Also, risk factors associated with relapse after stopping anti‐viral therapy have not yet been identified. However, based on current information, our recommendations for duration of treatment and the importance of close post‐treatment monitoring are described below.
8.2.1. HBeAg positive CHB Infection
For HBeAg positive CHB infection without histological or imaging evidence of advanced CHB infection, it had been recommended that anti‐viral treatment may be discontinued with at least 12‐24 months of consolidation treatment after HBeAg seroconversion.102 However, studies have indicated that HBV progression may continue after HBeAg seroconversion, and a substantial number of patients who discontinue therapy after completing consolidation therapy can experience recurrent viraemia, biochemical flares, and clinical relapse.103 In a retrospective study of 124 Chinese patients treated with LAM who achieved HBeAg serconversion with HBV DNA <200 copies/mL, the relapse rates (HBeAg servconversion) at 1 and 2 years post‐treatment were 54% and 68% respectively.104 In a recent report in HBeAg positive Asian American patients who discontinued anti‐viral therapy after loss of HBeAg and development of anti‐HBe, 90% developed recurrent viraemia, 38% had elevated ALT levels and 8% had seroreversion to HBeAg positivity.105 Another study of HBeAg positive Chinese patients who discontinued LAM after HBeAg seroconversion showed that all patients experienced reappearance of HBV DNA and 44% of patients had ALT flares.106 Due to the risk of recurrent viraemia and biochemical flares after discontinuing therapy, a long‐term anti‐viral treatment is now recommended until 6‐12 months after HBsAg clearance or HBsAg seroconversion.103, 104, 105 However, if treatment discontinuation is being considered after HBeAg seroconversion and a period of consolidated treatment but before HBsAg clearance, we recommend that the patient meet the following criteria:
Histological or elastography evidence of hepatic fibrosis stage <2
Close post‐treatment monitoring at monthly intervals for the first 3 months, and then 3‐6 months thereafter. Close attention for recurrent viraemia, with or without HBeAg seroreversion, ALT elevations and clinical relapse will determine if anti‐viral therapy needs to be reinstituted. Recently, in patients treated with entecavir, HBsAg titres of ≤2 to ≤2.5 log10 IU/mL at the time of HBeAg seroconversion were predictive of a low virological relapse rate in Asian patients.107, 108 Further studies quantifying HBsAg levels will be useful in confirming these findings.
8.2.2. HBeAg negative CHB infection
Although NA treatment is highly effective in suppressing HBV replication, it does not eliminate HBV infection. The recurrent viraemia rate is high even after a long‐term treatment‐induced suppression of HBV replication, and may result in biochemical flare or even hepatic decompensation. A recent systematic review study on 1732 HBeAg negative CHB‐infected patients who received anti‐viral treatment reported that 1‐year off‐therapy virological relapse and clinical relapse occurred in <70% and <50% of the patients, respectively, and <40% of the patients received re‐treatment.100 In patients who received 5 years of treatment with ADV, up to 60% had virological relapse with elevated ALT levels after stopping treatment.109 Recently, two Asian studies also showed high relapse rates after ETV therapy. One reported a virological relapse rate of 57.8%, a clinical relapse rate of 45.2%, and a re‐treatment rate of 35.8% after 2 years of ETV therapy.110 Another study reported a virological relapse rate of 91.4%, and a retreatment rate of 88.5% after 3 years of ETV therapy.111 Therefore, for HBeAg negative CHB patients with histological or imaging evidence of advanced CHB infection, indefinite NA treatment is recommended to maintain HBV DNA suppression. However, if stopping anti‐viral therapy is being considered, then close follow‐up with frequent laboratory testing to detect relapse and reinstitution of anti‐viral treatment is strongly recommended. Although HBsAg loss or HBsAg seroconversion is desired, it is rarely achievable in Asian patients. If HBsAg clearance does occur, NA treatment discontinuation with close follow‐up may be considered.
8.2.3. Cirrhosis
Discontinuation of anti‐viral treatment in patients with cirrhosis carries a risk of hepatic decompensation or even liver failure.11, 93 Furthermore, a large retrospective study indicated that even among patients already on anti‐viral therapy, a low‐level viraemia, defined as persistent or intermittent episodes of HBV DNA > than the lower detection limit of 12 IU/mL but, <2000 IU/mL was associated with a higher risk of developing HCC, particularly in patients with cirrhosis, when compared to those who maintained virological response with persistently undetectable HBV DNA levels.112 It is recommended that all patients with baseline clinical evidence of compensated or decompensated HBV cirrhosis, regardless of HBeAg and HBV DNA status, remain on a life‐long course of anti‐viral therapy.
8.2.4. Monitoring and managing anti‐viral treatment nonresponse or suboptimal response
As summarised in Table S5, primary virological non‐response to anti‐viral treatment is defined as a <1 log10 drop from baseline after 3 months of therapy, whereas a suboptimal response is defined as a >1 log10 decline, but detectable HBV DNA after 3‐6 months of treatment. Primary nonresponse or suboptimal response may occur if:
The patient is noncompliant to the treatment regimen, or is having difficulty with prescription refills.
The anti‐viral agent had limited potency, or
There is pre‐existing anti‐viral resistance.
These potential scenarios must be determined during clinic visits.
8.2.5. Virological breakthrough or rebound and anti‐viral resistance
Virological breakthrough is defined as a > 1 log10 increase from the nadir HBV DNA (IU/mL) level while on therapy in compliant patients. Virological rebound is defined as an increase in serum HBV DNA to >20 000 IU/mL or above the pre‐treatment baseline level during continued therapy. Both virological breakthrough and rebound may occur after achieving an initial virological response. Once noncompliance is excluded, virological breakthrough and rebound usually denotes anti‐viral resistance.
Hepatitis B virus anti‐viral resistance can be divided into two categories; (1) genotypic resistance which refers to substitutions at conserved sites within the polymerase gene targeted by the drug, and (2), phenotypic resistance which refers to a reduction in the in vitro susceptibility to anti‐viral agents associated with genotypic resistance.113 Anti‐viral resistance can be single, multiple, or cross‐resistant. Cross‐resistance is defined as decreased susceptibility to more than one HBV anti‐viral drug conferred by the same amino acid substitution or combination of amino acid substitutions. The clinical consequences of drug resistance include a reduced rate of HBeAg seroconversion, reversal of virological and histological improvement, an increased rate of disease progression and severe exacerbations especially in patients with cirrhosis.114, 115 The potential exists for transmission of drug resistant HBV, and for the appearance of HBsAg mutations that may lead to vaccine failure.116, 117
It is recommended that any patient with a virological breakthrough or rebound undergo drug resistance analysis. Currently, there are two types of genotypic HBV resistance tests; direct sequencing allowing the identification of mutations that comprise >20% of the total HBV population, and a line probe assay that identifies resistance mutations using real‐time PCR analysis with specific probes and polymorphism analysis that allows for the identification of mutations that comprise 5% of the total HBV population. In a recent report, up to 40% of virological breakthroughs in CHB‐infected patients receiving NA were related to medication non‐adherence and not anti‐viral genotypic resistance.118 Thus, close counselling of patients on adherence to their anti‐viral medications as well as confirmation of virological breakthrough and drug resistance testing are recommended to avoid unnecessary changes in anti‐viral mediations.
Table 4 summarises recommended therapy for anti‐viral resistance and cross‐resistance. When a single‐drug genotypic resistance is confirmed, the preferred approach in patients with either LAM, telbivudine, ADV or ETV resistance is to switch to TDF therapy.119 Patients with ADV resistance may also be switched to ETV.11, 93 However, switching to ADV is not recommended in patients who developed renal dysfunction during ADV treatment. If multidrug resistance is identified, the combination of ETV and TDF is preferred.120 Recently, TDF genetic resistance was reported in a few treatment experienced Korean patients.121 However, identification of a triple mutation that confers tenofovir resistance in HBV patients has not yet been established.
Table 4.
Anti‐viral resistance, cross resistance and recommended therapy
| NA‐based resistance | Common mutation sites | Resistance rates | Recommended therapy |
|---|---|---|---|
| LAM | M204V/I/S, L180M/I, M204V/I/S, A181T/V | 74% at 5 years | Switch to TDF |
| ADV | A181T/V, N236T, I233V | 29% at 3 years | Switch to TDF |
| LdT | M204I/V, rtA81T/V, rtL229W/V | 21.6% at 2 years | Switch to TDF |
| ETV | M204V/I, L180M, M250V, I169T | 1.2% at 6 years | Switch to TDF |
| TDF | No known mutation | 0% at 8 years | Continue TDF |
| LAM+ADV | A181V and N236T | Variable | Switch to TDF |
| Multiple resistance | Variable combination | Variable | Switch to TDF + ETV |
LAM, lamivudine; ADV, adefovir, LdT, telbivudine; ETV, entecavir; TDF, tenofovir.
Patients with a suboptimal response to ADV may be switched to ETV or TDF,11, 93, 103, 122 those with suboptimal response to telbivudine or LAM may be switched to TDF;11, 93, 119 and patients with suboptimal response to ETV may be switched to TDF.123
9. SPECIAL POPULATIONS
9.1. Chronic hepatitis B infection during childhood and adolescence
Chronic HBV infection in children is a major health problem not only in high endemic regions, but also in Western countries with large proportions of immigrants from endemic areas, as well as in cases of international adoption from regions where HBV is prevalent.124 HBV exposure leading to chronicity occurs either via perinatal transmission from infected mothers or during early childhood. If immunoprophylaxis is not administered at birth, 90% of infants born to HBeAg positive highly viremic mothers, and 5% born to HBeAg negative mothers will become chronic carriers.125 In addition, 25%‐50% of children exposed to HBV before 5 years of age via horizontal transmission from non‐maternal sources, ie, HBsAg positive family members, also will become chronic carriers. When infected beyond 5 years of age or during adolescence, 2.5%‐10% become HBV carriers. HBV infection during infancy and childhood is usually clinically asymptomatic with minimal or normal liver histological changes. However, although rare, acute and fulminant hepatitis have been reported in Asian infants born to HBeAg negative carrier mothers.126, 127, 128
Similar to adults, chronic HBV infection in children is classified into four phases: the HBeAg positive immune tolerant and immune active phases, and the HBeAg negative low replication and reactivation phases.129 HBsAg positive children who acquired HBV by vertical transmission or during early childhood begin in the immune tolerant phase. In Asian HBsAg positive children, HBeAg seroconversion is uncommon and only occurs at an annual rate of less than 2% per year in infants less than 3 years of age, and at 4%‐5% per year in children over the age of 3 years.129 Progression to the HBeAg positive immune active phase may occur at any age in children and is accompanied by ALT elevation and inflammatory activity in the liver.130 In a recent report, up to 40% of children in the HBeAg positive immune active phase experienced seroconversion to HBeAg negative and entered into the low replication phase.131 In the study, approximately 50% of the children remained in the HBeAg positive immune tolerant phase during the course of follow‐up. Also, other reports showed that by 17 years of age, 25% of Asian American children and 40% of Japanese children had experienced spontaneous HBeAg seroconversion.131, 132
Progression to cirrhosis and development of HCC in HBsAg positive Asian children is rare. In a report from Taiwan, only three of 426 (0.7%) of chronic carrier children progressed to cirrhosis.133 Two reports from Asia showed low rates of 0.5% to 1.5% for HCC development.129, 133 In one report, early HBeAg seroconversion and presence of cirrhosis were risk factors for HCC in HBsAg positive children. The youngest reported Asian American child to develop HCC was a 6‐year‐old male born to an HBeAg positive carrier mother.127 By the age of 3 years, a liver biopsy showed cirrhosis and seroconversion to anti‐HBe positivity had already occurred.
Anti‐viral treatment is recommended for children and adolescents who are in the HBeAg positive immune active phase of CHB, with elevated serum ALT values and HBV DNA levels >2000 IU/mL.124, 129 The primary endpoint of therapy in HBeAg positive children is HBeAg seroconversion. Children with HBeAg negative hepatitis (reactivation phase) with elevated ALT levels and HBV DNA levels >2000 IU/mL to >20 000 IU/mL also are recommended for anti‐viral treatment.124 In HBeAg negative children, the primary endpoints are reduction in HBV DNA and normalisation of ALT levels. Children with compensated cirrhosis with any detectable HBV DNA value should receive immediate anti‐viral treatment regardless of ALT levels or HBeAg status. The primary endpoint of treatment in children with compensated cirrhosis is undetectable HBV DNA. Children with decompensated cirrhosis should be immediately referred to liver transplant centres.
At present, children in the HBeAg positive immunotolerant phase are not recommended for treatment since anti‐viral therapies have not been proven to be effective.124 Also, anti‐viral therapy is not recommended in children with HBeAg negative low replication (HBV DNA <2000 IU/mL) “inactive carrier” phase.124, 134 More detailed information for the treatment of HBV in children and adolescents are summarised in recent reports.124, 129
Currently, there are five drugs that are approved for treatment of children with CHB.124 Standard interferon is approved for use in children ages 1‐17 years. Entecavir is approved for use in children 2 to <18 years and tenofovir is recommended for use in children ≥12 to 18 years of age. Entecavir and tenofovir have low cumulative rates of resistant variants of 2.6% and 0% respectively, and both drugs are well tolerated with minimal side effects. Lamivudine and adefovir are seldom used in the USA. Only nucleos(t)ide analogs should be used in children with cirrhosis.
9.2. HBV infection and pregnancy
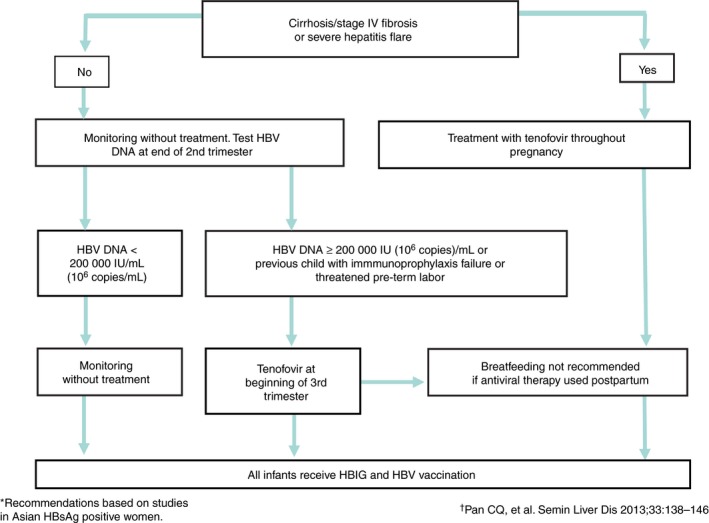
Pregnant mothers with CHB infection can vertically transmit HBV to their infants,125 and if untreated, CHB infection will develop in up to 90% of infants born to HBeAg positive mothers. The combination of postnatal passive and active immunisation reduces the rate of MTCT from 90% to 10%.135, 136, 137 However, even with passive‐active immunoprophylaxis, 7%‐32% of infants born to carrier mothers with HBV DNA levels ≥200 000 IU/mL (≥106 copies/mL) will still become HBsAg positive.138, 139, 140 To reduce immunoprophylaxis failure rates among infants born to mothers with high viral loads, anti‐viral therapy with TDF, LAM or telbivudine is indicated during the third trimester of pregnancy.141, 142, 143, 144 Another indication for anti‐viral treatment during pregnancy is maternal liver disease progression or reactivation, especially if clinically severe.145 Candidates for anti‐viral therapy during pregnancy are as follows:
Women with HBV DNA levels ≥200 000 IU/mL (≥106 copies/mL) at the third trimester of pregnancy.
Mothers with exacerbation of ALT (>5 × ULN of laboratory reference value), or severe fibrosis (ie, stage IV at Metavir scale; I).146, 147 Anti‐viral treatment can be initiated at any time point during pregnancy as indicated.
The use of LAM and telbivudine to reduce the transmission of HBV from mothers with high viral loads during the third trimester appears to be effective and well tolerated.139, 141, 142, 143 However, high‐quality data are lacking. Recently, a prospective randomised control study with TDF demonstrated that the rate of MTCT was significantly reduced from 7% to 0% when TDF was given during the third trimester to HBeAg positive mothers with high viral loads. The safety profiles were comparable between the TDF‐treated group and the control group at 28 weeks of postpartum follow‐up.144
Although few studies investigated the safety of using LAM or telbivudine during the first or second trimester to control active CHB disease, the treatment appears to be effective at normalising ALT or reducing viraemia with a comparable safety profile146, 147 Anti‐viral resistance has been reported in mothers who received LAM therapy, although this could be managed by switching to telbivudine or TDF during pregnancy. Therefore, the authors recommend the following treatment selection for mothers, and an algorithm for anti‐viral therapy during pregnancy is presented in Figure 6. These recommendations are based on studies in HBsAg positive pregnant Asian women.
Figure 6.
Anti‐viral therapy for mothers during pregnancy*,†
TDF should be considered as the first line treatment option for mothers with high viraemia and started at gestational week 30, or earlier if maternal liver disease required treatment.
As the second line treatment, telbivudine may be used during pregnancy. LAM should be limited to late second trimester or third trimester treatment for patients with HBV DNA between 6 and 9 log10 copies/mL due to the potential for resistance and lower potency.139, 148
In a retrospective study by Pan et al, a significant reduction in the MTCT rate of HBV was observed in HBeAg positive mothers with high viral loads when elective caesarean section was performed.149 However, further studies are warranted to confirm these findings. Mothers with high viral loads who missed the opportunity for third trimester anti‐viral treatment may consider this option and careful assessment of the risk‐benefit ratio with the participation of patient's obstetrician is recommended.
For consultation on pre‐conception, antepartum, or postpartum care, and infant outcomes, the discussion may include the following:
Treatment naïve patients with a treatment indication may consider interferon therapy to seroconvert HBeAg prior to becoming pregnant if the pregnancy is planned more than 1 year in advance.
For patients who are receiving oral anti‐viral treatment and found to be pregnant, therapy should either be stopped or switched to TDF if the patient was receiving non‐TDF‐based treatment.146, 147, 150
Amniocentesis may increase the risk of HBV transmission in mothers with viral loads >107 copies/mL. The decision to perform this procedure should be made with careful assessment on the risk‐benefit ratio for these mothers.151
As gestational diabetes, antepartum haemorrhage, and threatened preterm labor frequently occur among HBV‐infected mothers, close monitoring with at least one HBV DNA quantitative testing at gestational week 28 is highly recommend for assessing the risk of HBV transmission.152
In mothers who prefer breast‐feeding, anti‐viral therapy should be discontinued since these agents may be secreted in breast milk, and long‐term data on the safety of exposing infants to anti‐viral agents are not currently available. Stopping anti‐viral treatment in mothers after delivery for lactation appears to be safe, as significant hepatitis flares have not been observed.153, 154, 155 Postpartum monitoring on ALT should be performed on weeks 4 and 12.144, 156, 157
9.3. HBV coinfection with HCV or HDV
Since HBV and HCV share similar routes of transmission, coinfection with either virus is detected in 7%‐22% of CHB‐infected patients. Coinfection with HCV is found predominantly among individuals at high risk for parenteral infection, including injection drug use.158 Coinfection with HDV is often seen in those residing in areas where HBV infection is highly endemic.159 HBV patients co‐infected with HCV or HDV are at higher risk for rapid progression to decompensated liver disease and HCC.160, 161 In dual HBV/HCV infection, the treatment strategy should be directed towards the dominant replicating virus, though HCV eradication should be considered if appropriate treatment is accessible.158, 162, 163 When receiving direct acting anti‐viral therapy for HCV infection, dual HBV/HCV‐infected patients may experience an acute exacerbation of HBV unless anti‐viral treatment for HBV is also provided.164 The response to HBV treatment with oral anti‐viral agents does not appear to be effected by concurrent HCV infection. In addition, it has been reported that there is a risk for HBV reactivation in patients with HCV/HBV co‐infection whose HCV has been cleared by direct acting anti‐viral therapy (DAA).165 The FDA recently issued a warning on the risk of HBV reactivation and recommend that HCV patients should begin treatment for HBV prior to initiating DAA therapies (https://www.fda.gov/downloads/Drugs/DrugSafety/UCM523499.pdf).
Among CHB‐infected Asian patients with HDV super‐infection, lower levels of HBV DNA are common. HDV antigen or antibody tests are often used for screening. Weekly injection of pegylated interferon is currently used for 12‐18 months to treat co‐infected patients.166 The HDV RNA PCR qualitative test is available for monitoring the treatment response. Sustained viral response at 6 months post‐treatment, which correlates better with long‐term cure in the absence of advanced fibrosis, is only seen in 17% of patients. However, late HDV RNA relapse may occur.166
9.4. HBV and HIV coinfection
CHB infection affects nearly 15% of HIV‐infected individuals. HIV accelerates the course of HBV infection to end‐stage liver disease with more rapid progression of fibrosis.167, 168 The treatment of HBV in HBV/HIV co‐infected patients should take into account both viruses. Initiation of treatment for HBV infection is based on the presence of significant fibrosis or meeting the HBV treatment criteria.169
Treatment for HBV has been shown to improve clinical outcomes.170 In patients not requiring highly active antiretroviral therapy (HAART), drugs with dual activity against HBV and HIV such as LAM, entecavir, emtricitabine, or TDF should not be used as monotherapy since these drugs may lead to mutation selection and development of antiretroviral resistance.171, 172 Pegylated interferon monotherapy or ADV in combination with telbivudine may have a role in treating these patients.168, 173 In contrast, TDF and emtricitabine or other TDF based regimens should be initiated in co‐infected patients with low CD4 counts who require HAART.169, 172 Reactivation of HBV has been observed in patients on HAART therapy after immune reconstitution.174
9.5. HBV reactivation during chemotherapy or immunosuppressive therapy
Treatment with cancer chemotherapy or immunomodulatory agents may induce potentially fatal hepatic flares in HBsAg positive patients or those who are HBsAg negative but maintain a detectable level of HBV DNA. The acute flare or “reactivation” results from immune reconstitution directed towards the rise in HBV DNA following the use of immunosuppressive or immunomodulating agents.175 A recent meta‐analysis of clinical trials suggested that treatment with LAM or entecavir may prevent HBV reactivation during chemotherapy.176, 177 It is recommended that Asian patients be screened for HBsAg prior to initiation of chemotherapy or immunosuppressive therapy. Patients who are HBsAg negative but anti‐HBc positive should be tested for HBV DNA. When the level of HBV DNA level is undetectable in these patients, they may have the option of being monitored closely without anti‐viral therapy if a moderate or low level of immunosuppressant therapy is used. In addition, CHB patients who will require long‐term corticosteroid therapy alone or in combination with other immunosuppressive or anti‐neoplastic medications, should receive concomitant oral anti‐viral HBV drugs.178, 179 An algorithm for selecting patients for anti‐viral therapy is shown in Figure S3.
It is recommended that all HBsAg positive patients who require chemotherapy or immunomodulatory agents should receive anti‐viral therapy.177
Among HBsAg negative but anti‐HBc positive patients, anti‐viral prophylaxis is recommended when the HBV DNA level is detectable and the patient is to receive immunosuppressant agents such as rituximab, or will be receiving bone marrow transplantation.180
For better efficacy of viral suppression and preventing ALT flares, entecavir is preferred over LAM in patients with B cell lymphoma,178 or other solid tumours.181
10. RECOMMENDATIONS
Based on the evidence above, the authors have agreed to the following recommendations regarding the management of CHB infection in Asian Americans:
For HBeAg positive or negative chronic hepatitis patients with HBV DNA >2000 IU/mL (>104 copies/mL) and ALT > ULN, treat with a first‐line agent (entecavir, TDF, TAF) (I,A).
For compensated cirrhosis patients with detectable HBV DNA, treat with entecavir, TDF or TAF. For decompensated cirrhotic patients, entecavir or TDF are currently the agents of choice (I,A).
In HBeAg negative patients with HBV DNA >2000 IU/mL (>104 copies/mL) and normal ALT, a liver biopsy, transient elastography or serum marker for fibrosis is recommended. If not available, further stratification for risk factors including measurement of serum albumin values and platelet counts should be conducted prior to treatment (IIa, C‐LD).
To monitor treatment, test for serum ALT every 3 months. Measure HBV DNA every 3 months until negative, then every 3‐6 months. Measure HBeAg every 6 months until negative, then test for anti‐HBe (I, C‐LD).
After seroconversion from HBeAg positive to anti‐HBe, test for HBsAg every 12 months. In HBeAg negative patients, test for HBsAg every 12 months after sustained suppression of HBV DNA (I, C‐EO).
Regarding monitoring for resistance, viral breakthrough with confirmation of single drug resistance requires switching to another first‐line oral anti‐viral agent (IIa, B‐NR).
Surveillance for HCC with alpha‐fetoprotein and abdominal ultrasound should be performed every 6 months in HBsAg positive patients with chronic hepatitis, cirrhosis, and for patients with a family history of HCC (I, B‐R).
HBsAg positive pregnant women should be tested for HBV DNA. If the HBV DNA level is ≥ 200 000 IU/mL (≥106 copies/mL at gestational week 28, TDF treatment is preferred and should be initiated at gestational week 30 (I, B‐R).
HBsAg positive patients or patients with any detectable levels of HBV DNA requiring chemotherapy or treatment with immunomodulatory agents should receive anti‐viral treatment (I, B‐NR).
AUTHORSHIP
Guarantor of the article: Myron J. Tong.
Author contributions: The participation of the authors in the preparation of this manuscript were as follows. All of the authors were assigned a section of the paper so the data acquisition, analysis and interpretation had the participation of Drs. Myron Tong, Calvin Pan, Steven Han, David Lu, Steven Raman, Ke‐Qin Hu, Joseph Lim, Hie‐Won Hann and Albert Min. The impetus and research design for this paper included Drs. Myron Tong and Calvin Pan.
All authors approved the final version of the manuscript.
Supporting information
ACKNOWLEDGEMENTS
Editorial services were provided by William Perlman, PhD, and Thomas Saenz.
Declaration of personal interests: Myron J. Tong has served on advisory boards for Gilead, and is a speaker for Gilead and Merck. Calvin Q. Pan has served on advisory boards for AbbVie and Gilead. He has served as a consultant for Gilead and has received research grants from Merck and Gilead. He has served as a speaker for AbbVie and Gilead. Steven‐Huy B. Han has served as a speaker for Gilead and has received research grants from Gilead. David S‐K Lu has served as a speaker, a consultant and an advisory board member for Neuwave, Amgen, Perseon, and has received research funding from Neuwave. Steven Raman has served as a consultant for Bayer, Johnson and Johnson and Amgen. Ke‐Qin Hu has served as a speaker, a consultant and an advisory board member for Gilead, Merck, and Intercept. Joseph K. Lim has received research contracts (to institution) from Allergan, Conatus, Genfit, Gilead, Hologic, Intercept, and Prometheus, and has served as a consultant to AbbVie, Bristol‐Myers Squibb, and Gilead. Hie‐Won Hann has served on the advisory board for Gilead and has received clinical research grant support from Gilead. Albert D. Min has served as a speaker for AbbVie, Gilead and Merck and an advisory board member for AbbVie, Gilead and Merck.
Tong MJ, Pan CQ, Han S‐HB, et al. An expert consensus for the management of chronic hepatitis B in Asian Americans. Aliment Pharmacol Ther. 2018;47:1181–1200. https://doi.org/10.1111/apt.14577
Funding information
Independent grants to the Gi Health Foundation from Bristol Myers Squibb and Gilead Sciences, Inc. were used to help support the development of these recommendations
The Handling Editor for this article was Professor Grace Wong, and it was accepted for publication after full peer‐review.
REFERENCES
- 1. MacLachlan JH, Cowie BC. Hepatitis B virus epidemiology. Cold Spring Harb Perspect Med. 2015;5:a021410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. World Health Organization . Guidelines for the Prevention, Care and Treatment of Persons with Chronic Hepatitis B Infection. http://apps.who.int/iris/bitstream/10665/155081/1/WHO_HIV_2015.5_eng.pdf?ua=1&ua=1. Accessed August 18, 2017. [PubMed]
- 3. Kowdley KV, Wang CC, Welch S, Roberts H, Brosgart CL. Prevalence of chronic hepatitis B among foreign‐born persons living in the United States by country of origin. Hepatology. 2012;56:422‐433. [DOI] [PubMed] [Google Scholar]
- 4. Mitchell T, Armstrong GL, Hu DJ, Wasley A, Painter JA. The increasing burden of imported chronic hepatitis B–United States, 1974‐2008. PLoS ONE. 2011;6:e27717 https://doi.org/10.1371/journal.pone.0027717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tong MJ, Blatt LM, Tyson KB, Kao VW. Death from liver disease and development of hepatocellular carcinoma in patients with chronic hepatitis B virus infection: a prospective study. Gastroenterol Hepatol. 2006;2:41‐47. [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang S, Ristau JT, Trinh HN, Garcia RT, Nguyen HA, Nguyen MH. Undertreatment of Asian chronic hepatitis B patients on the basis of standard guidelines: a community‐based study. Dig Dis Sci. 2012;57:1373‐1383. [DOI] [PubMed] [Google Scholar]
- 7. Hu DJ, Xing J, Tohme RA, et al. Hepatitis B testing and access to care among racial and ethnic minorities in selected communities across the United States, 2009‐2010. Hepatology. 2013;58:856‐862. [DOI] [PubMed] [Google Scholar]
- 8. Kwong SL, Stewart SL, Aoki CA, Chen MS Jr. Disparities in hepatocellular carcinoma survival among Californians of Asian ancestry, 1988 to 2007. Cancer Epidemiol Biomarkers Prev. 2010;19:2747‐2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Halperin JL, Levine GN, Al‐Khatib SM, et al. Further Evolution of the ACC/AHA Clinical Practice Guideline Recommendation Classification System: a Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2016;67:1572‐1574. [DOI] [PubMed] [Google Scholar]
- 10. Pollack HJ, Kwon SC, Wang SH, Wyatt LC, Trinh‐Shevrin C; AAHBP Coalition . Chronic hepatitis B and liver cancer risks among Asian immigrants in New York City: results from a large community‐based screening, evaluation, and treatment program. Cancer Epidemiol Biomarkers Prev. 2014;23:2229‐2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tong MJ, Pan CQ, Hann HW, et al. The management of chronic hepatitis B in Asian Americans. Dig Dis Sci. 2011;56:3143‐3162. [DOI] [PubMed] [Google Scholar]
- 12. Liang X, Bi S, Yang W, et al. Epidemiological serosurvey of hepatitis B in China–declining HBV prevalence due to hepatitis B vaccination. Vaccine. 2009;27:6550‐6557. [DOI] [PubMed] [Google Scholar]
- 13. Chang MH, Chen CJ, Lai MS, et al. Universal hepatitis B vaccination in Taiwan and the incidence of hepatocellular carcinoma in children. Taiwan Childhood Hepatoma Study Group. N Engl J Med. 1997;336:1855‐1859. [DOI] [PubMed] [Google Scholar]
- 14. Chae HB, Kim JH, Kim JK, Yim HJ. Current status of liver diseases in Korea: hepatitis B. Korean J Hepatol. 2009;15:S13‐S24. [DOI] [PubMed] [Google Scholar]
- 15. Lau GK, Lai CL, Wu PC. The natural history of chronic hepatitis B infection. Hong Kong Med J. 1997;3:283‐288. [PubMed] [Google Scholar]
- 16. Lin CL, Kao JH. Hepatitis B viral factors and clinical outcomes of chronic hepatitis B. J Biomed Sci. 2008;15:137‐145. [DOI] [PubMed] [Google Scholar]
- 17. Iino S. Natural history of hepatitis B and C virus infections. Oncology. 2002;62:18‐23. [DOI] [PubMed] [Google Scholar]
- 18. Hann HW, Kim CY, London WT, Whitford P, Blumberg BS. Hepatitis B virus and primary hepatocellular carcinoma: family studies in Korea. Int J Cancer. 1982;30:47‐51. [DOI] [PubMed] [Google Scholar]
- 19. Yuen MF, Hou JL, Chutaputti A. Hepatocellular carcinoma in the Asia‐Pacific region. J Gastroenterol Hepatol. 2009;24:346‐353. [DOI] [PubMed] [Google Scholar]
- 20. Tong MJ, Hwang SJ. Hepatitis B virus infection in Asian Americans. Gastroenterol Clin North Am. 1994;23:523‐536. [PubMed] [Google Scholar]
- 21. World Health Organization . Hepatitis B. Department of Communicable Diseases Surveillance and Response. http://www.who.int/csr/disease/hepatitis/HepatitisB/_whocdscsrlyo2002_2.pdf. Accessed August 18, 2017.
- 22. Tong MJ, Hsu L, Hsien C, et al. A comparison of hepatitis B viral markers of patients in different clinical stages of chronic infection. Hepatol Int. 2010;4:516‐522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Takkenberg RB, Jansen L, de Niet A, et al. Baseline hepatitis B surface antigen (HBsAg) as predictor of sustained HBsAg loss in chronic hepatitis B patients treated with peginterferon alfa‐2a and adefovir. Antivir Ther. 2013;18:895‐904. [DOI] [PubMed] [Google Scholar]
- 24. Liew Y, Tai D, Chen T, et al. Alpha‐fetoprotein changes in the course of chronic hepatitis: relation to bridging hepatic necrosis and hepatocellular carcinoma. Liver. 1986;6:133‐137. [DOI] [PubMed] [Google Scholar]
- 25. Tong MJ, Huynh TT, Siripongsakun S, et al. Predicting clinical outcomes in patients with HBsAg‐positive chronic hepatitis. Hepatol Int. 2015;9:567‐577. [DOI] [PubMed] [Google Scholar]
- 26. Loomba R, Liu J, Yang HI, et al. Synergistic effects of family history of hepatocellular carcinoma and hepatitis B virus infection on risk for incident hepatocellular carcinoma. Clin Gastroenterol Hepatol 2013;11:1636‐1645. e1‐3. https://doi.org/10.1016/j.cgh.2013.04.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen CJ, Yang HI, Su J, et al. Risk of hepatocellular carcinoma across a biological gradient of serum hepatitis B virus DNA level. JAMA. 2006;295:65‐73. [DOI] [PubMed] [Google Scholar]
- 28. Beasley RP, Hwang LY, Lin CC, Chien CS. Hepatocellular carcinoma and hepatitis B virus. A prospective study of 22 707 men in Taiwan. Lancet. 1981;2:1129‐1133. [DOI] [PubMed] [Google Scholar]
- 29. Liaw YF, Leung N, Kao JH, et al. Asian‐Pacific consensus statement on the management of chronic hepatitis B: a 2008 update. Hepatol Int. 2008;2:263‐283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen CJ, Yang HI. Natural history of chronic hepatitis B REVEALed. J Gastroenterol Hepatol. 2011;26:628‐638. [DOI] [PubMed] [Google Scholar]
- 31. Yuen MF, Tanaka Y, Fong DY, et al. Independent risk factors and predictive score for the development of hepatocellular carcinoma in chronic hepatitis B. J Hepatol. 2009;50:80‐88. [DOI] [PubMed] [Google Scholar]
- 32. Park BK, Park YN, Ahn SH, et al. Long‐term outcome of chronic hepatitis B based on histological grade and stage. J Gastroenterol Hepatol. 2007;22:383‐388. [DOI] [PubMed] [Google Scholar]
- 33. Tong MJ, Blatt LM, Kao JH, Cheng JT, Corey WG. Basal core promoter T1762/A1764 and precore A1896 gene mutations in hepatitis B surface antigen‐positive hepatocellular carcinoma: a comparison with chronic carriers. Liver Int. 2007;27:1356‐1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tong MJ, Blatt LM, Kao JH, Cheng JT, Corey WG. Precore/basal core promoter mutants and hepatitis B viral DNA levels as predictors for liver deaths and hepatocellular carcinoma. World J Gastroenterol. 2006;12:6620‐6626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang BH, Yang BH, Tang ZY. Randomized controlled trial of screening for hepatocellular carcinoma. J Cancer Res Clin Oncol. 2004;130:417‐422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tong MJ, Sun HE, Hsien C, Lu DS. Surveillance for hepatocellular carcinoma improves survival in Asian‐American patients with hepatitis B: results from a community‐based clinic. Dig Dis Sci. 2010;55:826‐835. [DOI] [PubMed] [Google Scholar]
- 37. Yu NC, Chaudhari V, Raman SS, et al. CT and MRI improve detection of hepatocellular carcinoma, compared with ultrasound alone, in patients with cirrhosis. Clin Gastroenterol Hepatol. 2011;9:161‐167. [DOI] [PubMed] [Google Scholar]
- 38. McClune AC, Tong MJ. Chronic hepatitis B and hepatocellular carcinoma. Clin Liver Dis. 2010;14:461‐476. [DOI] [PubMed] [Google Scholar]
- 39. Tong MJ, Hsien C, Song JJ, et al. Factors associated with progression to hepatocellular carcinoma and to death from liver complications in patients with HBsAg‐positive cirrhosis. Dig Dis Sci. 2009;54:1337‐1346. [DOI] [PubMed] [Google Scholar]
- 40. Tong MJ, Rosinski AA, Huynh CT, et al. Long‐term survival after surveillance and treatment in patients with chronic viral hepatitis and hepatocellular carcinoma. Hepatol Commun 2017;1:595‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen MF, Tsai HP, Jeng LB, et al. Prognostic factors after resection for hepatocellular carcinoma in noncirrhotic livers: univariate and multivariate analysis. World J Surg. 2003;27:443‐447. [DOI] [PubMed] [Google Scholar]
- 42. Chang CH, Chau GY, Lui WY, Tsay SH, King KL, Wu CW. Long‐term results of hepatic resection for hepatocellular carcinoma originating from the noncirrhotic liver. Arch Surg 2004;139:320‐325; discussion 326. [DOI] [PubMed] [Google Scholar]
- 43. Tong MJ, Chavalitdhamrong D, Lu DS, et al. Survival in Asian Americans after treatments for hepatocellular carcinoma: a seven‐year experience at UCLA. J Clin Gastroenterol. 2010;44:e63‐e70. [DOI] [PubMed] [Google Scholar]
- 44. European Association for the Study of the Liver . EASL 2017 clinical practice guidelines on the management of hepatitis B virus infection. J Hepatol. 2017;67:370‐398. [DOI] [PubMed] [Google Scholar]
- 45. Sarin SK, Kumar M, Lau GK, et al. Asian‐Pacific clinical practice guidelines on the management of hepatitis B: a 2015 update. Hepatol Int. 2016;10:1‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Terrault NA, Bzowej NH, Chang KM, et al. AASLD guidelines for treatment of chronic hepatitis B. Hepatology. 2016;63:261‐283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Martin P, Lau DT, Nguyen MH, et al. A treatment algorithm for the management of chronic hepatitis B virus infection in the United States: 2015 update. Clin Gastroenterol Hepatol. 2015;13:2071‐2087. [DOI] [PubMed] [Google Scholar]
- 48. Lok AS, Wu PC, Lai CL, et al. A controlled trial of interferon with or without prednisone priming for chronic hepatitis B. Gastroenterology. 1992;102:2091‐2097. [DOI] [PubMed] [Google Scholar]
- 49. Lok AS, Lai CL, Wu PC, Leung EK. Long‐term follow‐up in a randomised controlled trial of recombinant alpha 2‐interferon in Chinese patients with chronic hepatitis B infection. Lancet. 1988;2:298‐302. [DOI] [PubMed] [Google Scholar]
- 50. Lau GK, Piratvisuth T, Luo KX, et al. Peginterferon alfa‐2a, lamivudine, and the combination for HBeAg positive chronic hepatitis B. N Engl J Med. 2005;352:2682‐2695. [DOI] [PubMed] [Google Scholar]
- 51. Chien RN, Liaw YF, Atkins M. Pretherapy alanine transaminase level as a determinant for hepatitis B e antigen seroconversion during lamivudine therapy in patients with chronic hepatitis B. Asian Hepatitis Lamivudine Trial Group. Hepatology. 1999;30:770‐774. [DOI] [PubMed] [Google Scholar]
- 52. Perrillo RP, Lai CL, Liaw YF, et al. Predictors of HBeAg loss after lamivudine treatment for chronic hepatitis B. Hepatology. 2002;36:186‐194. [DOI] [PubMed] [Google Scholar]
- 53. Marcellin P, Chang TT, Lim SG, et al. Baseline ALT predicts histologic and serological response in patients with HBeAg+ chronic hepatitis B treated with adefovir dipivoxil (ADV). J Hepatol. 2002;36(Suppl 1):122‐123. [Google Scholar]
- 54. Lai CL, Chien RN, Leung NW, et al. A one‐year trial of lamivudine for chronic hepatitis B. Asia Hepatitis Lamivudine Study Group. N Engl J Med. 1998;339:61‐68. [DOI] [PubMed] [Google Scholar]
- 55. Wang C, Deubner H, Shuhart MC, Manansala JN, Corey L, Kowdley KV. High prevalence of significant fibrosis in patients with immunotolerance to chronic hepatitis B infection. Hepatology. 2005;42:573A. [Google Scholar]
- 56. Nguyen MH, Trinh H, Ahmed A. Significant histologic disease in HBV‐infected patients with normal to minimally elevated ALT levels at initial evaluation. Hepatology. 2005;42:593A. [Google Scholar]
- 57. Lai CL, Hyatt BJ, Afdhal NH. Role of liver biopsy in patients with normal ALT and high HBV DNA. Hepatology. 2005;42:720A. [Google Scholar]
- 58. Chao DT, Lim JK, Ayoub WS, Nguyen LH, Nguyen MH. Systematic review with meta‐analysis: the proportion of chronic hepatitis B patients with normal alanine transaminase ≤ 40 IU/L and significant hepatic fibrosis. Aliment Pharmacol Ther. 2014;39:349‐358. [DOI] [PubMed] [Google Scholar]
- 59. Yuen MF, Yuan HJ, Wong DK, et al. Prognostic determinants for chronic hepatitis B in Asians: therapeutic implications. Gut. 2005;54:1610‐1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Fattovich G, Rugge M, Brollo L, et al. Clinical, virologic and histologic outcome following seroconversion from HBeAg to anti‐HBe in chronic hepatitis type B. Hepatology. 1986;6:167‐172. [DOI] [PubMed] [Google Scholar]
- 61. Park YM, Kim BS, Tabor E. Precore codon 28 stop mutation in hepatitis B virus from patients with hepatocellular carcinoma. Korean J Intern Med. 1997;12:201‐207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fang ZL, Yang J, Ge X, et al. Core promoter mutations (A(1762)T and G(1764)A) and viral genotype in chronic hepatitis B and hepatocellular carcinoma in Guangxi, China. J Med Virol. 2002;68:33‐40. [DOI] [PubMed] [Google Scholar]
- 63. Lin CL, Liao LY, Wang CS, et al. Basal core‐promoter mutant of hepatitis B virus and progression of liver disease in hepatitis B e antigen‐negative chronic hepatitis B. Liver Int. 2005;25:564‐570. [DOI] [PubMed] [Google Scholar]
- 64. Iloeje UH, Yang HI, Su J, Jen CL, You SL, Chen CJ. Predicting cirrhosis risk based on the level of circulating hepatitis B viral load. Gastroenterology. 2006;130:678‐686. [DOI] [PubMed] [Google Scholar]
- 65. Yuen MF, Tanaka Y, Shinkai N, et al. Risk for hepatocellular carcinoma with respect to hepatitis B virus genotypes B/C, specific mutations of enhancer II/core promoter/precore regions and HBV DNA levels. Gut. 2008;57:98‐102. [DOI] [PubMed] [Google Scholar]
- 66. Murata K, Sugimoto K, Shiraki K, Nakano T. Relative predictive factors for hepatocellular carcinoma after HBeAg seroconversion in HBV infection. World J Gastroenterol. 2005;11:6848‐6852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tong MJ, Hsien C, Hsu L, Sun HE, Blatt LM. Treatment recommendations for chronic hepatitis B: an evaluation of current guidelines based on a natural history study in the United States. Hepatology. 2008;48:1070‐1078. [DOI] [PubMed] [Google Scholar]
- 68. Prati D, Taioli E, Zanella A, et al. Updated definitions of healthy ranges for serum alanine aminotransferase levels. Ann Intern Med. 2002;137:1‐10. [DOI] [PubMed] [Google Scholar]
- 69. Lai CL, Gane E, Liaw YF, et al. Telbivudine versus lamivudine in patients with chronic hepatitis B. N Engl J Med. 2007;357:2576‐2588. [DOI] [PubMed] [Google Scholar]
- 70. Tong MJ, Siripongsakun S, Stanford‐Moore G, Hsu L, Chang PW, Blatt LM. Tumor factors associated with clinical outcomes in patients with hepatitis B virus infection and hepatocellular carcinoma. Gastroenterol Hepatol (N Y). 2012;8:808‐819. [PMC free article] [PubMed] [Google Scholar]
- 71. Liaw YF, Sheen IS, Lee CM, et al. Tenofovir disoproxil fumarate (TDF), emtricitabine/TDF, and entecavir in patients decompensated chronic hepatitis B liver disease. Hepatology. 2011;53:62‐72. [DOI] [PubMed] [Google Scholar]
- 72. Gish R, Tsai N, Pan C, et al. Efficacy and safety of entecavir in nucleos(t)ide naive Asians and HBeAg positive and ‐negative chronic hepatitis B: Results from studies ETV‐022/027. Hepatology. 2010;52:561A. [Google Scholar]
- 73. Liaw YF, Sung JJ, Chow WC, et al. Lamivudine for patients with chronic hepatitis B and advanced liver disease. N Engl J Med. 2004;351:1521‐1531. [DOI] [PubMed] [Google Scholar]
- 74. Shim JH, Lee HC, Kim KM, et al. Efficacy of entecavir in treatment‐naïve patients with hepatitis B virus‐related decompensated cirrhosis. J Hepatol. 2010;52:176‐182. [DOI] [PubMed] [Google Scholar]
- 75. Sinn DH, Lee J, Goo J, et al. Hepatocellular carcinoma risk in chronic hepatitis B virus‐infected compensated cirrhosis patients with low viral load. Hepatology. 2015;62:694‐701. [DOI] [PubMed] [Google Scholar]
- 76. Marcellin P, Lau GK, Bonino F, et al. Peginterferon alfa‐2a alone, lamivudine alone, and the two in combination in patients with HBeAg negative chronic hepatitis B. N Engl J Med. 2004;351:1206‐1217. [DOI] [PubMed] [Google Scholar]
- 77. Wei W, Wu Q, Zhou J, Kong Y, You H. A better antiviral efficacy found in nucleos(t)ide analog combinations with interferon therapy than NA monotherapy for HBeAg positive chronic hepatitis B: a meta‐analysis. Int J Environ Res Public Health. 2015;12:10039‐10055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wursthorn K, Lutgehetmann M, Dandri M, et al. Peginteferon alpha‐2b plus adefovir induce strong cccDNA decline and HBsAg reduction in patients with chronic hepatitis B. Hepatology. 2006;44:675‐684. [DOI] [PubMed] [Google Scholar]
- 79. Marcellin P, Wursthorn K, Wedemeyer H, et al. Telbivudine plus pegylated interferon alfa‐2a in a randomized study in chronic hepatitis B is associated with an unexpected high rate of peripheral neuropathy. J Hepatol. 2015;62:41‐47. [DOI] [PubMed] [Google Scholar]
- 80. Xie QL, Zhu Y, Wu LH, Fu LL, Xiang Y. The efficacy and safety of entecavir and interferon combination therapy for chronic hepatitis B virus infection a meta‐analysis. PLoS ONE. 2015;10:e0132219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Marcellin P, Ahn SH, Ma X, et al. Combination of tenofovir disoproxil fumarate and peginterferon alpha‐2a increases loss of hepatitis B surface antigen in patients with chronic hepatitis B. Gastroenterology. 2016;150: 134‐144. e10. [DOI] [PubMed] [Google Scholar]
- 82. Chang TT, Gish RG, de Man R, et al. A comparison of entecavir and lamivudine for HBeAg positive chronic hepatitis B. N Engl J Med. 2006;354:1001‐1010. [DOI] [PubMed] [Google Scholar]
- 83. Lai CL, Shouval D, Lok AS, et al. Entecavir versus lamivudine for patients with HBeAg negative chronic hepatitis B. N Engl J Med. 2006;354:1011‐1020. [DOI] [PubMed] [Google Scholar]
- 84. Pan CQ, Tong M, Kowdley KV, et al. High rates of viral suppression after long‐term entecavir treatment of Asian patients with hepatitis B e antigen‐positive chronic hepatitis B. Clin Gastroenterol Hepatol. 2012;10:1047‐1050. e1. [DOI] [PubMed] [Google Scholar]
- 85. Ahn J, Lee HM, Lim JK, et al. Entecavir safety and effectiveness in a national cohort of treatment‐naïve chronic hepatitis B patients in the US – the ENUMERATE study. Aliment Pharmacol Ther. 2016;43:134‐144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Marcellin P, Heathcote EJ, Buti M, et al. Tenofovir disoproxil fumarate versus adefovir dipivoxil for chronic hepatitis B. N Engl J Med. 2008;359:2442‐2455. [DOI] [PubMed] [Google Scholar]
- 87. Heathcote EJ, Marcellin P, Buti M, et al. Three‐year efficacy and safety of tenofovir disoproxil treatment for chronic hepatitis B. Gastroenterology. 2011;140:132‐143. [DOI] [PubMed] [Google Scholar]
- 88. Marcellin P, Gane E, Buti M, et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: a 5‐year open‐label follow‐up study. Lancet. 2013;381:468‐475. [DOI] [PubMed] [Google Scholar]
- 89. Pan CQ, Chan S, Trinh H, Yao A, Bae H, Lou L. Similar efficacy and safety of tenofovir in Asians and non‐Asians with chronic hepatitis B. World J Gastroenterol. 2015;21:5524‐5531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Chan HLY, Fung S, Seto WK, et al. Tenofovir alafenamide versus tenofovir disoproxil fumarate for the treatment of HBeAg positive chronic hepatitis B virus infection: a randomized, double‐blind, phase 3, non‐inferiority trial. Lancet Gastroenterol Hepatol. 2016;1:185‐195. [DOI] [PubMed] [Google Scholar]
- 91. Buti M, Gane E, Seto WK, et al. Tenofovir alafenamide versus tenofovir disoproxil fumarate for the treatment of patients with HBeAg negative chronic hepatitis B virus infection: a randomized, double‐blind, phase 3, non‐inferiority trial. Lancet Gastroenterol Hepatol. 2016;1:196‐206. [DOI] [PubMed] [Google Scholar]
- 92. Hyun JJ, Seo YS, Yoon E, et al. Comparison of the efficacies of lamivudine versus entecavir in patients with hepatitis B virus‐related decompensated cirrhosis. Liver Int. 2012;32:656‐664. [DOI] [PubMed] [Google Scholar]
- 93. Keeffe EB, Dieterich DT, Han SH, et al. A treatment algorithm for the management of chronic hepatitis B virus infection in the United States: 2008 update. Clin Gastroenterol Hepatol. 2008;6:1315‐1341. [DOI] [PubMed] [Google Scholar]
- 94. HEPSERA® (adefovir dipivoxil) Tablets, [package insert]. Foster City, CA: Gilead Sciences Inc.; 2012. [Google Scholar]
- 95. Viread® (Tenofovir Disoproxil Fumarate) Tablets, US Package Insert, Gilead Sciences, Inc., Foster City, CA, 2017.
- 96. Fontana RJ. Side effects of long‐term oral antiviral therapy for hepatitis B. Hepatology. 2009;49(5 Suppl):S185‐S195. [DOI] [PubMed] [Google Scholar]
- 97. Lampertico P, Chan HLY, Janssen HLA, Strasser SI, Schindler R, Berg T. Review article: long‐term safety of nucleoside and nucleotide analogues in HBV‐monoinfected patients. Aliment Pharmacol Ther. 2016;44:16‐34. [DOI] [PubMed] [Google Scholar]
- 98. Vemlidy® (tenofovir alafenamide) tablets [prescribing information]. Foster City, CA: Gilead Sciences, Inc.; 11/2016.
- 99. Seto W‐K, Wong DK‐H, Fung J, Huang F‐Y, Lai C‐L, Yuen M‐F. Reduction of hepatitis B surface antigen levels and hepatitis B surface antigen seroclearance in chronic hepatitis B patients receiving 10 years of nucleoside analogue therapy. Hepatology. 2013;58:923‐931. [DOI] [PubMed] [Google Scholar]
- 100. Chang M‐L, Liaw Y‐F, Hadziyannis SJ. Systematic review: cessation of long‐term nucleos(t)ide analogue therapy in patients with hepatitis B e antigen‐ negative chronic hepatitis B. Aliment Pharmacol Ther. 2015;42:243‐257. [DOI] [PubMed] [Google Scholar]
- 101. Papatheodoridis G, Vlachogiannakos I, Cholongitas E, et al. Discontinuation of oral antivirals in chronic hepatitis B: a systematic review. Hepatology. 2016;63:1481‐1492. [DOI] [PubMed] [Google Scholar]
- 102. Chi H, Hansen BE, Yim C, et al. Reduced risk of relapse after long‐term nucleos(t)ide analogue consolidation therapy for chronic hepatitis B. Aliment Pharmacol Ther. 2015;41:867‐876. [DOI] [PubMed] [Google Scholar]
- 103. Reijnders JG, Deterding K, Petersen J, et al. Antiviral effect of entecavir in chronic hepatitis B: influence of prior exposure to nucleos(t)ide analogues. J Hepatol. 2010;52:493‐500. [DOI] [PubMed] [Google Scholar]
- 104. Kuo YH, Chen CH, Wang JH, et al. Extended lamivudine consolidation therapy in hepatitis B e antigen‐positive chronic hepatitis B patients improves sustained hepatitis B e antigen seroconversion. Scand J Gastroenterol. 2010;45:75‐81. [DOI] [PubMed] [Google Scholar]
- 105. Chaung KT, Ha NB, Trinh HN, et al. High frequency of recurrent viremia after hepatitis B e antigen seroconversion and consolidation therapy. J Clin Gastroenterol. 2012;46:865‐870. [DOI] [PubMed] [Google Scholar]
- 106. Fung J, Lai CL, Tanaka Y, et al. The duration of lamivudine therapy for chronic hepatitis B: cessation vs. continuation of treatment after HBeAg seroconversion. Am J Gastroenterol. 2009;104:1940‐1946. [DOI] [PubMed] [Google Scholar]
- 107. Qiu YW, Huang LH, Yang WL, et al. Hepatitis B surface antigen quantification at hepatitis B e antigen seroconversion predicts virological relapse after the cessation of entecavir treatment in hepatitis B e antigen‐positive patients. Int J Infect Dis. 2016;43:43‐48. [DOI] [PubMed] [Google Scholar]
- 108. Lee HA, Seo YS, Park SW, et al. Hepatitis B surface antigen titer is a good indicator of durable viral response after entecavir off‐treatment for chronic hepatitis B. Clin Mol Hepatol. 2016;22:382‐389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Hadziyannis SJ, Sevastianos V, Rapti I, Vassilopoulos D, Hadziyannis E. Sustained responses and loss of HBsAg in HBeAg negative patients with chronic hepatitis B who stop long‐term treatment with adefovir. Gastroenterology. 2012;143:629‐636. [DOI] [PubMed] [Google Scholar]
- 110. Jeng WJ, Sheen IS, Chen YC, et al. Off‐therapy durability of response to entecavir therapy in hepatitis B e antigen‐negative chronic hepatitis B patients. Hepatology. 2013;58:1888‐1896. [DOI] [PubMed] [Google Scholar]
- 111. Seto WK, Hui AJ, Wong VW, et al. Treatment cessation of entecavir in Asian patients with hepatitis B e antigen negative chronic hepatitis B: a multicentre prospective study. Gut. 2015;64:667‐672. [DOI] [PubMed] [Google Scholar]
- 112. Kim JH, Sinn DH, Kang W, et al. Low‐level viremia and the increased risk of hepatocellular carcinoma in patients receiving entecavir treatment. Hepatology. 2017;66:335‐343. [DOI] [PubMed] [Google Scholar]
- 113. Lok AS. Antiviral drug‐resistant HBV: standardization of nomenclature and assays and recommendations for management. Hepatology. 2007;46:254‐265. [DOI] [PubMed] [Google Scholar]
- 114. Yuen MF, Kato T, Mizokami M, et al. Clinical outcome and virologic profiles of severe hepatitis B exacerbation due to YMDD mutations. J Hepatol. 2003;39:850‐855. [DOI] [PubMed] [Google Scholar]
- 115. Nafa S, Ahmed S, Tavan D, et al. Early detection of viral resistance by determination of hepatitis B virus polymerase mutations in patients treated by lamivudine for chronic hepatitis B. Hepatology. 2000;32:1078‐1088. [DOI] [PubMed] [Google Scholar]
- 116. Mutimer D, Pillay D, Shields P, et al. Outcome of lamivudine resistant hepatitis B virus infection in the liver transplant recipient. Gut. 2000;46:107‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Thibault V, Aubron‐Olivier C, Agut H, Katlama C. Primary infection with a lamivudine‐resistant hepatitis B virus. AIDS. 2002;16:131‐133. [DOI] [PubMed] [Google Scholar]
- 118. Hongthanakorn C, Chotiyaputta W, Oberhelman K, et al. Virological breakthrough and resistance in patients with chronic hepatitis B receiving nucleos(t)ide analogues in clinical practice. Hepatology. 2011;53:1854‐1863. [DOI] [PubMed] [Google Scholar]
- 119. Patterson SJ, George J, Strasser SI, et al. Tenofovir disoproxil fumarate rescue therapy following failure of both lamivudine and adefovir dipivoxil in chronic hepatitis B. Gut. 2011;60:247‐254. [DOI] [PubMed] [Google Scholar]
- 120. Choe WH, Hong SP, Kim BK, et al. Evolution of hepatitis B virus mutation during entecavir rescue therapy in patients with antiviral resistance to lamivudine and adefovir. Antivir Ther. 2009;14:985‐993. [DOI] [PubMed] [Google Scholar]
- 121. First patient resistant to Viread found in Korea. Korean Biomedical Review Web cite. http://www.koreabiomed.com/news/articleView.html?idxno=758. Published June 26, 2017. Updated June 26, 2017. Accessed Januanry 16, 2018.
- 122. Nguyen MH, Trinh HN, Do ST, et al. Response to entecavir (ETV) in patients with chronic hepatitis B (CHB) and prior suboptimal response to adefovir (ADV): interim report of a multicenter study. Gastroenterology 2009;136(5 Suppl 1):A‐798. [Google Scholar]
- 123. Pan CQ, Hu K‐Q, Yu AS, Chen W, Bunchorntavakul C, Reddy KR. Response to tenofovir monotherapy in chronic hepatitis B patients with prior suboptimal response to entecavir. J Viral Hepat. 2012;19:213‐219. [DOI] [PubMed] [Google Scholar]
- 124. Clemente MG, Vajro P. An update on the strategies used for the treatment of chronic hepatitis B in children. Expert Rev Gastroenterol Hepatol. 2016;10:649‐658. [DOI] [PubMed] [Google Scholar]
- 125. Stevens CE, Beasley RP, Tsui J, Lee WC. Vertical transmission of hepatitis B antigen in Taiwan. N Engl J Med. 1975;292:771‐774. [DOI] [PubMed] [Google Scholar]
- 126. Shiraki K, Yoshihara N, Kawana T, Yasui H, Sakurai M. Hepatitis B surface antigen and chronic hepatitis in infants born to asymptomatic carrier mothers. Am J Dis Child. 1977;131:644‐647. [DOI] [PubMed] [Google Scholar]
- 127. Tong MJ, Sinatra FR, Thomas DW, Nair PV, Merritt RJ, Wang DW. Need for immunoprophylaxis in infants born to HBsAg‐positive carrier mothers who are HBeAg negative. J Pediatr. 1984;105:945‐947. [DOI] [PubMed] [Google Scholar]
- 128. Sinatra FR, Shah P, Weissman JY, Thomas DW, Merritt RJ, Tong MJ. Perinatal transmitted acute icteric hepatitis B in infants born to hepatitis B surface antigen‐positive and anti‐hepatitis Be‐positive carrier mothers. Pediatrics. 1982;70:557‐559. [PubMed] [Google Scholar]
- 129. Komatsu H, Inui A, Fujisawa T. Pediatric hepatitis B treatment. Ann Transl Med. 2017;5:37 https://doi.org/10.21037/atm.2016.11.52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Chang MH. Natural history and clinical management of chronic hepatitis B virus infection in children. Hepatol Int. 2008;2(Suppl 1):28‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Takano T, Tajiri H, Hosono S, et al. Natural history of chronic hepatitis B virus infection in children in Japan: a comparison of mother‐to‐child transmission with horizontal transmission. J Gastroenterol. 2017;52:1041‐1050. https://doi.org/10.1007/s00535-017-1315-4 [DOI] [PubMed] [Google Scholar]
- 132. Evans AA, Fine M, London WT. Spontaneous seroconversion in hepatitis B e antigen‐positive chronic hepatitis B: implications for interferon therapy. J Infect Dis. 1997;176:845‐850. [DOI] [PubMed] [Google Scholar]
- 133. Wen WH, Chang MH, Hsu HY, Ni YH, Chen HL. The development of hepatocellular carcinoma among prospectively followed children with chronic hepatitis B virus infection. J Pediatr. 2004;144:397‐399. [DOI] [PubMed] [Google Scholar]
- 134. Della Corte C, Comparcola D, Nobili V. Hepatitis B virus infection in children. Clin Res Hepatol Gastroenterol. 2012;36:291‐293. [DOI] [PubMed] [Google Scholar]
- 135. Beasley RP, Trepo C, Stevens CE, Szmuness W. The e antigen and vertical transmission of hepatitis B surface antigen. Am J Epidemiol. 1977;105:94‐98. [DOI] [PubMed] [Google Scholar]
- 136. Lee C, Gong Y, Brok J, Boxall EH, Gluud C. Hepatitis B immunisation for newborn infants of hepatitis B surface antigen‐positive mothers. Cochrane Database Syst Rev. 2006;(2):CD004790. [DOI] [PubMed] [Google Scholar]
- 137. Mast EE, Weinbaum CM, Fiore AE, et al. A comprehensive immunization strategy to eliminate transmission of hepatitis B virus infection in the United States: recommendations of the Advisory Committee on Immunization Practices (ACIP) Part II: immunization of adults. MMWR Recomm Rep 2006;16:1‐33; quiz CE31‐CE34. [PubMed] [Google Scholar]
- 138. Zou H, Chen Y, Duan Z, Zhang H, Pan CQ. Virologic factors associated with failure to passive active immunoprophylaxis in infants born to HBsAg‐positive mothers. J Viral Hepat. 2012;19:e18‐e25. [DOI] [PubMed] [Google Scholar]
- 139. Xu WM, Cui YT, Wang L, et al. Lamivudine in late pregnancy to prevent perinatal transmission of hepatitis B virus infection: a multicentre, randomized, double‐blind, placebo‐controlled study. J Viral Hepat. 2009;16:94‐103. [DOI] [PubMed] [Google Scholar]
- 140. Wiseman E, Fraser MA, Holden S, et al. Perinatal transmission of hepatitis B virus: an Australian experience. Med J Aust. 2009;190:489‐492. [DOI] [PubMed] [Google Scholar]
- 141. Pan CQ, Han GR, Jiang HX, et al. Telbivudine prevents vertical transmission from HBeAg positive women with chronic hepatitis B. Clin Gastroenterol Hepatol. 2012;10:520‐526. [DOI] [PubMed] [Google Scholar]
- 142. Pan CQ, Mi LJ, Bunchorntavakul C, et al. Tenofovir disoproxil fumarate for prevention of vertical transmission of hepatitis B virus infection by highly viremic pregnant women: a case series. Dig Dis Sci. 2012;57:2423‐2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Zhang H, Pan CQ, Pang Q, Tian R, Yan M, Liu X. Telbivudine or lamivudine use in late pregnancy safely reduces perinatal transmission of hepatitis B virus in real‐life practice. Hepatology. 2014;69:468‐476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Pan CQ, Duan Z, Dai E, et al. Tenofovir to prevent hepatitis B transmission in mothers with high viral load. N Engl J Med. 2016;374:2324‐2334. [DOI] [PubMed] [Google Scholar]
- 145. Hung JH, Chu CJ, Sung PL, et al. Lamivudine therapy in the treatment of chronic hepatitis B with acute exacerbation during pregnancy. J Chin Med Assoc. 2008;71:155‐158. [DOI] [PubMed] [Google Scholar]
- 146. Liu M, Cai H, Yi W. Safety of telbivudine treatment for chronic hepatitis B for the entire pregnancy. J Viral Hepat. 2013;20(Suppl 1):65‐70. [DOI] [PubMed] [Google Scholar]
- 147. Yi W, Liu M, Cai HD. Safety of lamivudine treatment for chronic hepatitis B in early pregnancy. World J Gastroenterol. 2012;18:6645‐6650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Pan CQ, Yi W, Liu M, et al. Lamivudine therapy during the second vs the third trimester for preventing transmission of chronic hepatitis B. J Viral Hepat. 2017;24:246‐252. [DOI] [PubMed] [Google Scholar]
- 149. Pan CQ, Zou H‐B, Chen Y, et al. Cesarean section reduces perinatal transmission of hepatitis b virus infection from hepatitis B surface antigen–positive women to their infants. Clin Gastroenterol Hepatol. 2013;11:1349‐1355. [DOI] [PubMed] [Google Scholar]
- 150. Brown RS Jr, Verna EC, Pereira MR, et al. Hepatitis B virus and human immunodeficiency virus drugs in pregnancy: findings from the Antiretroviral Pregnancy Registry. J Hepatol. 2012;57:953‐959. [DOI] [PubMed] [Google Scholar]
- 151. Yi W, Pan CQ, Hao J, et al. Risk of vertical transmission of hepatitis B after amniocentesis in HBs antigen‐positive mothers. J Hepatol. 2014;60:523‐529. [DOI] [PubMed] [Google Scholar]
- 152. Tse KY, Ho LF, Lao T. The impact of maternal HBsAg carrier status on pregnancy outcomes: a case‐control study. J Hepatol. 2005;43:771‐775. [DOI] [PubMed] [Google Scholar]
- 153. Park JS, Pan C. Current recommendations of managing HBV infection in preconception or pregnancy. Front Med. 2014;8:158‐165. [DOI] [PubMed] [Google Scholar]
- 154. Pan CQ, Lee HM. Antiviral therapy for chronic hepatitis B in pregnancy. Semin Liver Dis. 2013;33:138‐146. [DOI] [PubMed] [Google Scholar]
- 155. Pan CQ, Duan ZP, Bhamidimarri KR, et al. An algorithm for risk assessment and intervention of mother to child transmission of hepatitis B virus. Clin Gastroenterol Hepatol. 2012;10:452‐459. [DOI] [PubMed] [Google Scholar]
- 156. Lin HH, Wu WY, Kao JH, et al. Hepatitis B post‐partum e antigen clearance in hepatitis B carrier mothers: correlation with viral characteristics. J Gastroenterol Hepatol. 2006;21:605‐609. [DOI] [PubMed] [Google Scholar]
- 157. Ter Bogr MJ, Leemans WF, de Man RA, et al. Exacerbation of chronic hepatitis B infection after delivery. J Viral Hepat. 2008;15:37‐41. [DOI] [PubMed] [Google Scholar]
- 158. Konstantinou D, Deutsch M. The spectrum of HBV/HCV coinfection: epidemiology, clinical characteristics, viral interactions and management. Ann Gastroenterol. 2015;28:221‐228. [PMC free article] [PubMed] [Google Scholar]
- 159. Abbas Z, Jafri W, Raza S. Hepatitis D: scenario in the Asia‐Pacific region. World J Gastroenterol. 2010;16:554‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Liu CJ, Liou JM, Chen DS, Chen PJ. Natural course and treatment of dual hepatitis B virus and hepatitis C virus infections. J Formos Med Assoc. 2005;104:783‐791. [PubMed] [Google Scholar]
- 161. Wedemeyer H. Hepatitis D revival. Liver Int. 2011;31(Suppl 1):140‐144. [DOI] [PubMed] [Google Scholar]
- 162. Chu CJ, Lee SD. Hepatitis B virus/hepatitis C virus coinfection: epidemiology, clinical features, viral interactions and treatment. J Gastroenterol Hepatol. 2008;23:512‐520. [DOI] [PubMed] [Google Scholar]
- 163. Yu JW, Sun LJ, Zhao YH, Kang P, Gao J, Li SC. Analysis of the efficacy of treatment with peginterferon alpha‐2a and ribavirin in patients coinfected with hepatitis B virus and hepatitis C virus. Liver Int. 2009;29:1485‐1493. [DOI] [PubMed] [Google Scholar]
- 164. Takayama H, Sato T, Ikeda F, Fujiki S. Reactivation of hepatitis B virus during interferon‐free therapy with daclatasvir and asunaprevir in patient with hepatitis B virus/hepatitis C virus co‐infection. Hepatol Res. 2016;46:489‐491. [DOI] [PubMed] [Google Scholar]
- 165. De Monte A, Courjon J, Anty R, et al. Direct‐acting antiviral treatment in adults infected with hepatitis C virus: reactivation of hepatitis B virus coinfection as a further challenge. J Clin Virol. 2016;78:27‐30. [DOI] [PubMed] [Google Scholar]
- 166. Abbas Z, Memon MS, Mithani H, Jafri W, Hamid S. Treatment of chronic hepatitis D patients with pegylated interferon: a real‐world experience. Antivir Ther. 2014;19:463‐468. [DOI] [PubMed] [Google Scholar]
- 167. Nikolopoulos GK, Paraskevis D, Hatzitheodorou E, et al. Impact of hepatitis B virus infection on the progression of AIDS and mortality in HIV‐infected individuals: a cohort study and meta‐analysis. Clin Infect Dis. 2009;48:1763‐1771. [DOI] [PubMed] [Google Scholar]
- 168. Thio CL. Hepatitis B and human immunodeficiency virus coinfection. Hepatology. 2009;49(5 Suppl):S138‐S145. [DOI] [PubMed] [Google Scholar]
- 169. Sherman KE. Management of the Hepatitis B Virus/HIV‐Coinfected Patient. Top Antivir Med. 2015;23:111‐114. [PMC free article] [PubMed] [Google Scholar]
- 170. Martin‐Carbonero L, Teixeira T, Poveda E, et al. Clinical and virological outcomes in HIV‐infected patients with chronic hepatitis B on long‐term nucleos(t)ide analogues. AIDS. 2011;25:73‐79. [DOI] [PubMed] [Google Scholar]
- 171. McMahon MA, Jilek BL, Brennan TP, et al. The HBV drug entecavir ‐ effects on HIV‐1 replication and resistance. N Engl J Med. 2007;356:2614‐2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV‐1‐infected adults and adolescents. Department of Health and Human Services. January 10, 2011;1‐166. Available at http://www.aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf. Accessed August 18, 2017.
- 173. Soriano V, Puoti M, Sulkowski M, et al. Care of patients coinfected with HIV and hepatitis C virus: 2007 updated recommendations from the HCV‐HIV International Panel. AIDS. 2007;21:1073‐1089. [DOI] [PubMed] [Google Scholar]
- 174. Manegold C, Hannoun C, Wywiol A, et al. Reactivation of hepatitis B virus replication accompanied by acute hepatitis in patients receiving highly active antiretroviral therapy. Clin Infect Dis. 2001;32:144‐148. [DOI] [PubMed] [Google Scholar]
- 175. Hoofnagle JH, Dusheiko GM, Schafer DF, et al. Reactivation of chronic hepatitis B virus infection by cancer chemotherapy. Ann Intern Med. 1982;96:447‐449. [DOI] [PubMed] [Google Scholar]
- 176. Liu JY, Sheng YJ, Ding XC, et al. The efficacy of lamivudine prophylaxis against hepatitis B reactivation in breast cancer patients undergoing chemotherapy: a meta‐analysis. J Formos Med Assoc. 2015;114:164‐173. [DOI] [PubMed] [Google Scholar]
- 177. Di Bisceglie AM, Lok AS, Martin P, Terrault N, Perrillo RP, Hoofnagle JH. Recent US Food and Drug Administration warnings on hepatitis B reactivation with immune‐suppressing and anticancer drugs: just the tip of the iceberg? Hepatology. 2015;61:703‐711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Huang H, Li X, Zhu J, et al. Entecavir vs lamivudine for prevention of hepatitis B virus reactivation among patients with untreated diffuse large B‐cell lymphoma receiving R‐CHOP chemotherapy: a randomized clinical trial. JAMA. 2014;312:2521‐2530. [DOI] [PubMed] [Google Scholar]
- 179. Perrillo RP, Gish R, Falck‐Ytter YT. American Gastroenterological Association Institute technical review on prevention and treatment of hepatitis B virus reactivation during immunosuppressive drug therapy. Gastroenterology. 2015;148(221–244):e3. [DOI] [PubMed] [Google Scholar]
- 180. Centers for Disease Control . Recommendations for identification and public health management of persons with chronic hepatitis B virus infection. MMWR. 2008;57:1‐20. [PubMed] [Google Scholar]
- 181. Chen WC, Cheng JS, Chiang PH, et al. A comparison of entecavir and lamivudine for the prophylaxis of hepatitis B virus reactivation in solid tumor patients undergoing systemic cytotoxic chemotherapy. PLoS ONE. 2015;10:e0131545. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials