

ABSTRACT
Introduction: 3,4‐diaminopyridine has been used to treat Lambert‐Eaton myasthenia (LEM) for 30 years despite the lack of conclusive evidence of efficacy. Methods: We conducted a randomized double‐blind placebo‐controlled withdrawal study in patients with LEM who had been on stable regimens of 3,4‐diaminopyridine base (3,4‐DAP) for ≥ 3 months. The primary efficacy endpoint was >30% deterioration in triple timed up‐and‐go (3TUG) times during tapered drug withdrawal. The secondary endpoint was self‐assessment of LEM–related weakness (W‐SAS). Results: Thirty‐two participants were randomized to continuous 3,4‐DAP or placebo groups. None of the 14 participants who received continuous 3,4‐DAP had > 30% deterioration in 3TUG time versus 72% of the 18 who tapered to placebo (P < 0.0001). W‐SAS similarly demonstrated an advantage for continuous treatment over placebo (P < 0.0001). Requirement for rescue and adverse events were more common in the placebo group. Discussion: This trial provides significant evidence of efficacy of 3,4‐DAP in the maintenance of strength in LEM. Muscle Nerve 57: 561–568, 2018
Keywords: 3,4‐diaminopyridine; amifampridine; clinical trial; Eaton‐Lambert syndrome; efficacy; ELS; Lambert‐Eaton myasthenia; Lambert‐Eaton myasthenic syndrome; Lambert‐Eaton syndrome; LEMS; LES; timed up‐and‐go
Abbreviations
- 3,4‐DAP
3,4‐diaminopyridine base
- 3TUG
triple timed up‐and‐go
- ACh
acetylcholine
- AE
adverse event
- CMAP
compound muscle action potential
- DAPPER
DAP Prospective Efficacy Research
- ECG
electrocardiogram
- IM
immunomodulators/immunosuppressants
- IND
investigational new drug
- LEFS
lower extremity functional scale
- LEM
Lambert‐Eaton myasthenia
- QMG
Quantitative Myasthenia Gravis
- QTcF
electrocardiographic QT interval with Fridericia's correction
- TEAE
treatment‐emergent adverse event
- VGCC
voltage‐gated calcium channel
- W‐SAS
LEM‐related weakness self‐assessment scale
Lambert‐Eaton myasthenia (LEM) is a rare autoimmune disorder often associated with a malignancy, usually small cell lung cancer.1 Epidemiological studies suggest that there may now be approximately 800 patients with LEM in the USA and up to 170 new cases annually.2, 3
The symptoms that characterize LEM result from reduced release of acetylcholine (ACh) from the presynaptic terminals of the neuromuscular junctions.1 Autoantibodies that target presynaptic voltage‐gated calcium channels (VGCCs) impair entry of calcium into nerve terminals, thereby decreasing ACh release.4 By blocking presynaptic potassium channels, 3,4‐diaminopyridine, also known as amifampridine, prolongs depolarization from impulses arriving at the nerve ending, allowing VGCCs to remain open longer, thus increasing entry of calcium. Because the quantal release of ACh depends on the intracellular concentration of calcium, 3,4‐diaminopyridine increases the release of ACh.
The first use of 3,4‐diaminopyridine formulated as the 3,4‐diaminopyridine free base (3,4‐DAP) in the treatment of LEM was described in 1983,5 and subsequent uncontrolled studies reported benefits in small numbers of patients.6 In a double‐blind, placebo‐controlled crossover study of 3,4‐DAP in 12 patients with LEM in 1989,7 oral doses up to 100 mg/day were effective in treating both the motor and the autonomic deficits, and the amplitude of compound muscle action potentials (CMAPs) nearly doubled. A randomized, placebo‐controlled study of 26 patients with LEM in 2000 demonstrated that those who received oral 3,4‐DAP had a greater improvement in Quantitative Myasthenia Gravis (QMG) score and in the summated amplitude of CMAPs in 3 sentinel muscles,8 although the magnitude of change in QMG score in this study was not clinically significant. A randomized, placebo‐controlled crossover study demonstrated efficacy of intravenous 3,4‐DAP with dynamometry and CMAP in 9 patients with LEM.9 None of these studies were used to file for regulatory approval in the USA or in Europe. Other clinical studies failed to provide definitive objective evidence of efficacy of 3,4‐diaminopyridine.10, 11
In 1990, the US Food and Drug Administration granted orphan designation to 3,4‐diaminopyridine, and compassionate use of the free base formulation 3,4‐DAP became available through physician‐held investigational new drugs (IND) for named patients in the United States and through special governmental approvals in other countries. To provide definitive evidence of efficacy, we designed the 3,4‐DAP Product Efficacy Research (DAPPER) trial, a prospective, placebo‐controlled withdrawal study of 3,4‐DAP in LEM.
MATERIALS AND METHODS
Outcome Measures
Because physiological endpoints of dynamometry and CMAP do not capture function and QMG performance in LEM had proven questionable in previous clinical trials, we used the triple timed up‐and‐go (3TUG) walking test, a modification of the validated Timed Up‐and‐Go Test,12 as the quantitative measure of proximal muscle function.13 Based on minimal detectable change data reported in Parkinson disease, the primary efficacy outcome was defined as a >30% deterioration in the 3TUG time.12, 14
Study Participants
Given the paucity of eligible patients, we targeted patients with LEM who were being actively treated with 3,4‐DAP and for whom responsiveness could be objectively documented before randomization.15 To be eligible, participants had to indicate a need to wait briefly after the first morning dose of 3,4‐DAP for improvement in LEM‐related dysfunction, and they also needed to display quantifiable functional benefit after the first morning dose.15 This approach avoided entering participants whose LEM may have improved or remitted over time.16
Fifty‐two patients with LEM, all participants in the Jacobus Pharmaceutical Company (Sponsor) 3,4‐DAP compassionate use program, were screened for eligibility. Fifty of the 52 patients were receiving treatment under 21 different physician‐held INDs in the United States. One participant each from Canada and Argentina had special access through appropriate governmental agencies and local neuromuscular specialists. The diagnosis of LEM was confirmed by independent neurologist (D.B.S.) review of clinical records, including VGCC antibody and electromyography test results. The dose of 3,4‐DAP was at least 10 mg 3 times per day up to a total daily dose of 100 mg and permitted participants to walk, with or without an assistive device. All LEM‐related treatments were stable for at least 3 months, and other concomitant medications were stable for at least 1 month before study entry. Patients were excluded if they had previous respiratory failure while on 3,4‐DAP or an insufficient 3TUG response to 3,4‐DAP during the baseline observation period (see Supp. Info. Tables 1, 2 for detailed inclusion and exclusion criteria).
Study Oversight
Written informed consent was obtained from all study participants. The study was conducted in accordance with International Conference on Harmonization guidelines and principles of the Declaration of Helsinki. The protocol was approved by the institutional review board at each study site and registered with https://clinicaltrials.gov/ (NCT01511978).
Criteria for rescue requiring reinstitution of baseline dosing were new dysphagia, a drop in pulse oximetry of 5% from baseline, or a decrease in oxygen saturation to <90% with accompanying shortness of breath. Inability to get out of bed or inability to rise from a chair, even with assistance, after 2 efforts about 1 h apart, also merited reinstitution of baseline 3,4‐DAP. Continuous safety monitoring was built into the study design, precluding the requirement for a formal safety monitoring committee.
The definition for eligibility was concealed during the study. Individual 3TUG results were shared with the Sponsor, who determined if the eligibility criterion had been met. The initial eligibility criterion for randomization required ≥30% improvement in 3TUG after the first 3,4‐DAP dose on 2 consecutive mornings. This criterion threshold was lowered to ≥27% after approximately 20% of participants had been randomized because of the constrained number of potential participants and concern that participants who were responsive to 3,4‐DAP were being excluded. The eligibility criterion was further modified after approximately 30%–40% of participants had been randomized to accommodate a potential stacking effect of 3,4‐DAP (i.e., the additive effect of multiple doses of 3,4‐DAP throughout the day) and allowing consideration of 3TUGs performed in the afternoon as well as in the morning to determine whether subsequent participants displayed a sufficiently large response to 3,4‐DAP during the baseline observation period. A later modification also permitted consideration of the evening postdose 3TUG. The 4 thresholds used for each participant are detailed in Supporting Information Tables 3 and 4. Each modification made it easier for patients to qualify for participation and less likely that they would weaken enough during the drug taper to reach the study endpoint.
Vital signs, including pulse oximetry, were measured at least 6 times daily in conjunction with 3TUG testing, with continuous pulse oximetry monitoring overnight. Postdose LEM‐related weakness self‐assessment scale (W‐SAS) score was obtained 3 times daily as was an inventory of LEM‐related signs or symptoms. Electrocardiograms (ECG) and CMAPs were obtained before and after the first doses of the morning and afternoon throughout the study. A Lower Extremity Functional Scale (LEFS) score was obtained at baseline and at the end of the study.17 Blood for plasma drug and metabolite levels was drawn at regular intervals surrounding doses during the baseline observation period and after randomization.
Participants were classified into 1 of 4 baseline treatment categories according to their baseline LEM treatment regimen: 3,4‐DAP plus pyridostigmine; 3,4‐DAP alone; 3,4‐DAP plus pyridostigmine plus immunomodulators/immunosuppressants (IM); and 3,4‐DAP plus IMs. To assure that the 2 study arms were balanced for possible disease severity, participants within each category were randomized centrally in a 1:1 ratio.
Study Design
See Figure 1 for a diagram of the study design. A screening visit was scheduled up to 4 weeks before admission to the inpatient clinical research unit. After determination of initial eligibility, participants were hospitalized and observed for 2.5 days on their usual dose and schedule of 3,4‐DAP. Responsiveness to 3,4‐DAP, the final determinant of eligibility, was assessed by 3TUG testing before and after the first doses of the morning, afternoon, and evening, regardless of a participant's total number of daily doses. The 2‐h postdose period was selected as the estimated time for peak pharmacodynamic effect.9 Because patients with LEM usually do not take 3,4‐DAP during the night and the half‐life following oral doses is approximately 3.5–4 h, patients effectively experience a withdrawal from drug every night and frequently have mobility issues before their first dose of the morning. This suggested that morning would be the ideal time to detect drug responsiveness. Patients with a sufficient 3TUG response were randomly assigned to the taper‐to‐placebo group or the continuous 3,4‐DAP group on the afternoon of the second day.
Figure 1.
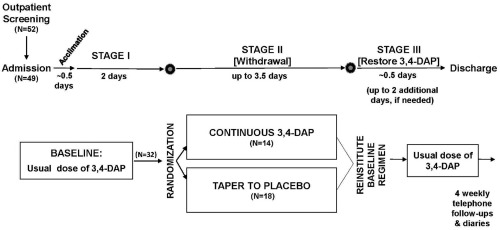
Study design schematic. 3,4‐DAP, 3,4‐diaminopyridine base.
Tapered withdrawal began with the last dose of the second full hospital day and was decreased to 90% of the participant's usual dose. Each consecutive dose was then decreased so that it was 50% of the usual dose by the end of the third full day, 20% at the end of the fourth day, and 0% for the third dose of the fifth full day (Supp. Info. Fig. 1). Thereafter, there was an additional up to 16 h with no 3,4‐DAP before the participant's usual dose was restored. Participants were then observed for 0.5 days or until deemed clinically stable and then discharged. Standardized weekly follow‐ups continued for 1 month after discharge. Enrollment was planned to cease when approximately 15 participants were enrolled in each of the 2 study groups.
Treatments
The Sponsor prepared tablets of 0.5, 2, 3, 4, 5, and 10 mg 3,4‐DAP and placebo, all identical in appearance. Combinations of 4 tablets were prepackaged into a series of blisters to permit a smooth taper (Supp. Info. Fig. 2) according to each participant's individualized prerandomization dosing regimen.
Efficacy Endpoints
Primary Endpoint
The primary efficacy measure was the percentage change in the last completed postdose 3TUG during the treatment period compared with the average of the 2 time‐matched 3TUGs obtained during the baseline. The 3TUG involves 3 repetitions (without rest between laps) of rising from an 18‐in. straight‐backed armchair, walking to a line 10 feet from the chair, and returning to sit in the chair.15 Participants are instructed to walk at their normal pace and only prompted with the word “Go” for the first lap and “Go again” after they come to a full stop in the chair for the subsequent laps. Each lap is timed and the score is the average of the 3 lap times. In addition to timing the 3TUGs “live” with a traceable stop watch, the tests were videotaped, and the lap times were later measured in triplicate by a remote assessor blinded to the treatment and to the date, time, and sequence of the 3TUGs. The blinded assessor's times were used to determine the primary endpoint. The calculation of percentage change in 3TUG for the primary endpoint used the last completed postdose 3TUG during the withdrawal period: Percentage change = 100 × [1 − (final postdose 3TUG/time‐matched baseline 3TUGs)]. Rescue was an anticipated event, and rescued participants were considered to have completed the trial.
Secondary Endpoint
The secondary efficacy measure was the W‐SAS, a 7‐level categorical scale developed by the Sponsor to demonstrate participant‐perceived change in overall strength from study entry; responses ranged from “much much weaker (−3)” to “much much stronger (+3)”. The W‐SAS score was obtained 3 times daily, corresponding to the postdose 3TUGs. The last completed postdose W‐SAS during the randomized treatment period was the endpoint.
Additional Efficacy Endpoints
Change in CMAP was measured in the nerve–muscle pair determined to be most responsive to 3,4‐DAP during acclimation (ulnar nerve–abductor digiti minimi, median nerve–abductor pollicis brevis, or peroneal nerve–extensor digitorum brevis). A reviewer blinded to the treatment and to the date, time, and sequence of the CMAPs used predetermined criteria to assess CMAP test quality. Tests approved by this reviewer were used to determine change in CMAP amplitude at the end of the blinded treatment period compared with baseline.
The LEFS at the end of the blinded treatment period was compared with baseline. This questionnaire consists of 20 items, each scored from 0 (extreme difficulty) to 4 (no difficulty); a change of 9 points has been determined to represent a clinically meaningful functional change in patients with a lower extremity musculoskeletal condition.17 Requirement for rescue was considered as both an efficacy and a safety endpoint. Blinded physician assessment of treatment effect was performed once at the end of the study.
Safety Endpoints
Safety assessments included requirement for rescue during blinded treatment and/or the emergence of LEM‐related signs and symptoms or predosage LEM‐associated weakness, 3TUG, W‐SAS, and treatment‐emergent adverse events. Changes in ECG parameters, vital signs, and pulse oximetry were also examined.
Plasma Drug Levels
Pharmacokinetic analyses of plasma drug and metabolite levels were used to confirm the taper.
Statistical Analysis
With α at 0.05 and the power at 80%, 10 participants were required in each study group. To allow for departures in the assumptions, 30 participants were planned, 15 for each treatment arm. Thirty‐two participants were actually enrolled. No interim analysis was performed. Statistical analyses followed the intention‐to‐treat principle. Fisher's exact test was used to compare primary efficacy outcomes between treatment groups (i.e., > 30% prolongation in 3TUG times), and a t test was used to compare W‐SAS assessments. ECGs were analyzed for changes in cardiac intervals, and a concentration response analysis exploring changes in QT interval with Fridericia's correction (QTcF) and plasma drug concentrations was performed. Statistical analysis of the other outcome measures was not performed.
RESULTS
Participants
Fifty‐two patients were screened at 7 study sites and agreed to participate (Fig. 2). Eighteen were ineligible because of insufficient improvement in 3TUG (Supp. Info. Table 4). Two patients were excluded because of positive toxicology testing. Thirty‐two participants were eligible and were randomized, 14 to the continuous 3,4‐DAP group and 18 to the taper‐to‐placebo group. There were no important differences between the 2 treatment groups in demographics (Table 1), LEM‐related history, or LEM treatments (Supp. Info. Tables 5, 6). All randomized participants completed the study.
Figure 2.
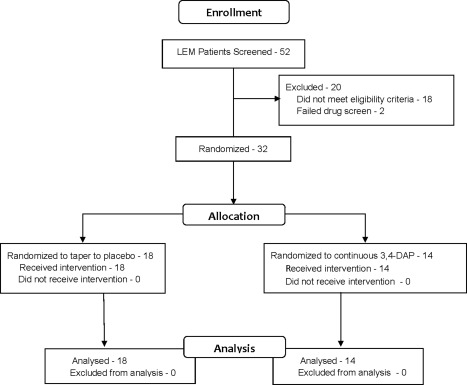
CONSORT (Consolidated Standards of Reporting Trials) diagram. 3,4‐DAP, 3,4‐diaminopyridine base.
Table 1.
Demographics and baseline characteristics
| Variable | Taper‐to‐placebo | Continuous 3,4‐DAP |
|---|---|---|
| Patients, n | 18 | 14 |
| Age, y, mean/SD (range) | 59.3/14.99 (28–78) | 50.7/15.97 (23–83) |
| Sex | ||
| Men, n (%) | 7 (38.9) | 4 (30.8) |
| Women, n (%) | 11 (61.1) | 10 (71.4) |
| Race | ||
| White, n (%) | 18 (100) | 11 (78.6) |
| Black, n (%) | 0 (0.0) | 3 (21.4) |
| Ethnicity | ||
| Hispanic or Latino, n (%) | 1 (5.6) | 0 (0.0) |
| Not Hispanic or Latino, n (%) | 17 (94.4) | 14 (100) |
| BMI, kg/m2, Mean/SD (range) | 27.7/5.14 (18.9–35.4) | 27.3/5.92 (20.3–39.0) |
| Positive P/Q VGCC‐Ab at screening, n (%) | 17 (94.4) | 12 (85.7) |
| CMAP facilitation > 100% at screening, n (%) | 10 (55.6) | 7 (50.0) |
| Paraneoplastic LEM, n (%) | 1 (5.6) | 0 (0.0) |
| Duration of LEM prior to randomization, y, mean/SD (range) | 6.7/6.08 (0.3–22.3) | 6.7/5.70 (1.1–19.8) |
| Duration of 3,4‐DAP treatment at entry, y, mean/SD (range) | 5.5/4.92 (0.3–18.3) | 6.2/5.30 (0.7–18.9) |
| TDD of 3,4‐DAP at entry, mg, mean/SD (range) | 74.7/22.26 (30–100) | 76.4/19.46 (35–100) |
| LEM treatment before and during study | ||
| 3,4‐DAP + PB, n (%) | 11 (61.1) | 9 (64.3) |
| 3,4‐DAP, n (%) | 0 (0.0) | 1 (7.1) |
| 3,4‐DAP + PB + IM, n (%) | 4 (22.2) | 2 (14.2) |
| 3,4‐DAP + IM, n (%) | 3 (16.7) | 2 (14.2) |
3,4‐DAP, 3,4‐diaminopyridine base; BMI, body mass index; CMAP, compound muscle action potential; IM, immunomodulators/immunosuppressants; LEM, Lambert‐Eaton myasthenia; PB, pyridostigmine bromide; TDD, total daily dose; VGCC‐Ab, voltage‐gated calcium channel antibodies.
Efficacy
Primary Outcome Measure
The primary outcome measure was >30% deterioration in final 3TUG time during tapered withdrawal of study drug. The 3TUG primary efficacy endpoint demonstrated a highly significant difference between treatment groups in favor of continuous 3,4‐DAP (Table 2, Fig. 3). There was very strong agreement as well as a high correlation between the blinded 3TUGs and the on‐site 3TUGs (correlation coefficient = 0.9192; Supp. Info. Fig. 2).
Table 2.
Primary and secondary efficacy endpoints: change in the final 3TUG times and W‐SAS score upon withdrawal of study drug
| Study group | Score | Taper‐to‐placebo, n = 18, n (%) | Continuous 3,4‐DAP, n = 14, n (%) |
|---|---|---|---|
| 3TUG change | No change or faster | 5 (27.8) | 14 (100) |
| >30% Slower | 13 (72.2) | 0 (0) | |
| P < 0.0001a | |||
| Final W‐SAS | Much much weaker (−3) | 10 (55.6) | 1 (7.1) |
| Much weaker (−2) | 6 (33.3) | 1 (7.1) | |
| Somewhat weaker (−1) | 1 (5.6) | 1 (7.1) | |
| About the same (0) | 1 (5.6) | 9 (64.3) | |
| Somewhat stronger (+1) | 0 (0.0) | 1 (7.1) | |
| Much stronger (+2) | 0 (0.0) | 1 (7.1) | |
| Much much stronger (+3) | 0 (0.0) | 0 (0.0) | |
| P < 0.0001b | |||
3,4‐DAP, 3,4‐diaminopyridine base; 3TUG, triple timed up‐and‐go; CMH, Cochrane‐Mantel‐Haenszel; W‐SAS, LEM‐related weakness self‐assessment scale.
Fisher's exact test.
CMH test for categorical data.
Figure 3.
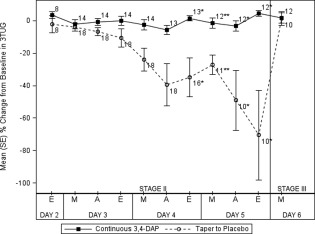
Percentage change from baseline in 3TUG test at 2 h after dosing versus time, by treatment group. 3TUG, triple timed up‐and‐go. *P < 0.05, **P < 0.01, one‐way ANCOVA, with the baseline 3TUG as the covariate. 3,4‐DAP, 3,4‐diaminopyridine base; A, afternoon; ANCOVA, analysis of covariance; 3TUG, triple timed up ‐and ‐go; E, evening; M, morning.
A significantly greater proportion of patients tapered to placebo had >30% deterioration in the final postdose 3TUG test compared with patients in the continuous 3,4‐DAP group. Results were consistent for efficacy, intent‐to‐treat, and per‐protocol populations.
Secondary Outcome Measure
The W‐SAS secondary efficacy endpoint (Table 2) also demonstrated a highly significant difference in favor of the continuous 3,4‐DAP group, with most participants who continued 3,4‐DAP being “about the same” and most of those who tapered to placebo being “much weaker” or “much much weaker” at the last W‐SAS assessment. Results were consistent for the efficacy, intent‐to‐treat, and per‐protocol populations.
Additional Outcome Measures
The final postdose CMAP measurements approved by the blinded reviewer favored the continuous 3,4‐DAP group, which had a median CMAP of 3.4 mV (all nerve–muscle pairs) and a median percentage change of −9.5%, whereas the taper‐to‐placebo group, for whom the median final CMAP was 2.3 mV, had a median percentage change from baseline of −42.1%.
The continuous 3,4‐DAP group had a median change in LEFS score of −1.5 points compared with −27 points for the taper‐to‐placebo group. In a repeat LEFS assessment 1 week postdischarge in the taper‐to‐placebo group, the median score increased from 10 to 42, returning to baseline.
Two participants in the continuous treatment group (14.3%) and 5 in the taper‐to‐placebo group (44.4%) were rescued for new dysphagia during the withdrawal phase without having reached the primary study endpoint (Supp. Info. Table 3). In addition, 1 participant in the tapered group was rescued because of a 5% drop in oxygen saturation, and another required rescue because of inability to rise from a chair. Two of the 7 rescued participants in the tapered group did not meet the primary endpoint prior to rescue (Supp. Info. Table 3a). Another participant in the tapered group had baseline medication reinstituted upon request because of anxiety and a sense of impending doom, preceded by intermittent nocturnal hypoxemia.
Blinded physician assessment of treatment effect at the end of the study favored the continuous 3,4‐DAP group, with physicians indicating that 12 of 14 (86%) patients had no change, whereas 1 was “somewhat worse” and 1 was “much worse” than during baseline. In contrast, 14 of 18 (78%) participants in the taper‐to‐placebo group were considered “much worse,” 3 (17%) were “somewhat worse,” and only 1 had “no change” from baseline.
Recovery of 3TUG After Reinstitution of 3,4‐DAP
The morning postdose 3TUG returned to baseline after reinstitution of baseline 3,4‐DAP doses in all participants who tapered to placebo and in those who were rescued or advanced early (Fig. 3).
Recovery of W‐SAS After Reinstitution of 3,4‐DAP
Participants in the taper‐to‐placebo group reported return to their baseline strength after the first dose of their baseline 3,4‐DAP. A similar recovery in the W‐SAS assessment was observed in rescued participants.
Unblinding
There were no identified instances of unblinding during the study. At the end of the study, participants and investigators were independently asked to which treatment arm they believed they had been randomized. Participants and physicians agreed in all cases; both guessed treatment assignment incorrectly in 3 cases.
Pharmacokinetic Results
Pharmacokinetic sampling was assayed post hoc and confirmed the randomization. First morning predose plasma drug levels confirmed that most participants, regardless of their treatment arm, had effectively withdrawn from the drug overnight.
Safety
Treatment‐Emergent Adverse Events
Treatment‐emergent adverse events (TEAE) were reported with a higher frequency in the taper‐to‐placebo group, with 23 adverse events (AE) in 12 of 18 participants (67%) versus 9 AEs in 5 of 14 participants (36%) in the continuous 3,4‐DAP group. The most common non–LEM‐related signs and symptoms of TEAEs were abdominal discomfort and respiratory tract infection (2 in the taper‐to‐placebo group). One patient in each treatment group had back pain, headache, nasopharyngitis, or oropharyngeal pain. One serious AE of pneumonia occurred in a participant in the taper‐to‐placebo group more than 3 weeks after completing the inpatient trial. There were no deaths in this study.
Re‐emergence of LEM‐Related Signs and Symptoms
The most common LEM‐related signs and symptoms to emerge during drug withdrawal were decreased oxygen saturation in 3 participants in the taper‐to‐placebo group, muscle spasms and nausea in 2 participants in the taper‐to‐placebo group, and arthralgia in 1 participant in each group. One participant in the taper‐to‐placebo group had a severe episode of anxiety with a sense of impending doom. Results of the morning predose 3TUGs indicated that participants who were not rescued were no weaker at the end of the 3.5‐day taper than they were every morning on their usual steady regimen of 3,4‐DAP.
Clinical Laboratory Tests
With the exception of minor blood glucose increases, there were no clinically meaningful laboratory value changes. End of study measurements of blood glucose were increased compared with screening in both treatment groups, with mean blood glucose increased by 21.3 and 15.9 mg/dl in the taper‐to‐placebo group and continuous 3,4‐DAP group, respectively.
Vital Signs
Small increases in average pulse rate of 4–6 bpm were observed at some time points postdose in both treatment groups. Low oxygen saturation occurred in 5 participants in the taper‐to‐placebo group and was reported as an LEM‐related sign/symptom AE in 3 participants who had preexisting pulmonary disease and/or sleep apnea and who received supplemental oxygen. Low oxygen saturation resolved spontaneously or with reinstitution of 3,4‐DAP treatment. Oxygen saturation did not worsen significantly in any participant who received continuous 3,4‐DAP.
Electrocardiograms
None of the participants had clinically significant abnormal ECG findings. All ECG parameters (RR, QRS, PR, QT, and QTcF) as well as the interpretative statements by the investigators showed little or no difference between baseline and study days or between the treatment groups.
An increased heart rate up to 5 bpm was observed in the taper‐to‐placebo group. Short‐term withdrawal of 3,4‐DAP had no apparent effect on standard ECG parameters. Linear regression analysis indicated that there was no relationship between changes in QTcF interval and the respective plasma drug concentrations, regardless of treatment group.
Tolerability
3,4‐DAP in doses from 30 to 100 mg daily was well tolerated by participants in the continuous 3,4‐DAP group.
DISCUSSION
This trial provides highly significant evidence of efficacy and demonstrates that 3,4‐DAP is essential for the maintenance of strength in patients with LEM. Lowering 3,4‐DAP resulted in significant deterioration of strength when doses reached approximately 50% of the usual individual dosage (Fig. 3). Rescue was required in 44% of participants who tapered 3,4‐DAP and resulted in prompt resolution of weakness.
DAPPER contained 2 embedded efficacy studies in addition to the overall randomized withdrawal trial: (1) a series of N‐of‐1 trials of each participant's daily responses to 3,4‐DAP and (2) the responses to reinstitution of 3,4‐DAP at the end of the study. The recovery of strength and function with reinstitution of 3,4‐DAP supports the lack of deconditioning, rebound weakness, or sustained negative effects from short‐term withdrawal of 3,4‐DAP. Key to the potential success of the withdrawal design was the identification of 3,4‐DAP‐responsive patients with LEM, which ensured an enriched population for the study.
Although the TUG has been validated and used to study a variety of conditions, the use of 3 repetitions of the TUG (3TUG) for the DAPPER study was developed to accommodate the neuromuscular fatigue or facilitation that may affect patients with LEM to different degrees. Initial pilot testing indicated the ability of the 3TUG to detect drug effect 1–2 h postdose. A subsequent study demonstrated the reliability of the 3TUG in controls, in non‐LEM patients with neuromuscular disease, and in patients with LEM.13 Pharmacokinetic/pharmacodynamic analysis of plasma levels of 3,4‐DAP from the DAPPER study demonstrated a concentration‐response relationship with 3TUG times, supporting the use of the 3TUG as a measure of disease‐related weakness in patients with LEM.18
The 3TUG data used in the analysis of the DAPPER study were timed from videos made on site. To eliminate a potential source of bias in outcome assessment, the video reader was remote from any of the study sites and blinded to the sequence of the tests and treatment assignments There was very strong agreement as well as a high correlation between the blinded 3TUGs and the on‐site 3TUGs.
This study can be criticized for changing the eligibility criterion after the study had started. However, the study endpoint was not changed, and decreasing the required level of responsiveness for eligibility to <30% should have made it less likely that participants would have >30% deterioration when tapered to placebo. To avoid potential biasing of the 3TUG results used to determine eligibility, site personnel were blinded to the eligibility threshold. The Sponsor determined eligibility from data reported by the sites using preset criteria that were not subject to bias.
Although only 1 study participant had paraneoplastic LEM (Supp. Info. Table 3a, Screen No. 50.0), previous reports have demonstrated clinical responsiveness to 3,4‐DAP in paraneoplastic LEM and no difference in responsiveness in paraneoplastic and nonparaneoplastic LEM.7, 19
That 2 participants in the continuous 3,4‐DAP treatment group were rescued for dysphagia is evidence for the effectiveness of the blind. Both participants and study personnel may have been overly attuned to swallowing issues, knowing that participants would be rescued under the protocol to avoid aspiration. The onset of dysphagia in these 2 participants demonstrates that, even with usual treatment, patients with LEM may experience unexpected weakness that can be addressed by an extra dose of 3,4‐DAP.
The highly significant findings for both the primary and secondary outcome measures are strengthened by the rapid recovery of function after resumption of the participants' usual regimens and by the finding that the majority of patients in the continuous 3,4‐DAP group had daily demonstrable benefit with >30% improvement in 3TUG times compared with the morning predose 3TUG.
In conclusion, 3,4‐DAP is a safe and effective treatment for LEM‐related weakness.
The content of this study was presented in part at the annual meetings of the American Academy of Neurology, April 2015, Washington, DC; the American Neurological Association, September 2015, Chicago, Illinois; and the American Association of Neuromuscular & Electrodiagnostic Medicine, October 2015, Honolulu, Hawaii.
The authors thank study coordinators Katherine Beck, RN, Claire MacAdam, PT, Charles Latner, Meg Lessard, Kay Artibee, RN, Med, Diana Dimitrova, PhD, Angela Micheels PT, Sandra Guingrich, Janelle Butters, RN; Bradstreet Clinical Research Associates, Inc for study monitoring; Ready Clinical, LLC for 3TUG blinded readings; Brightech International for statistical consultation; EDETEK, Inc for data management and statistical services; Erik Stålberg, MD, PhD (Uppsala University) for CMAP blinded reviews; nabios GMBH for ECG analysis; and DAPPER Study Team coinvestigators Jeffrey T. Guptill, MD, Janice M. Massey, MD, Lisa D. Hobson‐Webb, MD, Ali Arvantaj, MD, Cecile Phan, MD, Shima Mousavi, MD, J. Robinson Singleton, MD, Peter D. Donofrio, MD, Christopher Lee, MD, MPH, Alexandra Dimitrova, MD, Brent Burroughs, MD, Ghazaleh Jafari, MD, Julie Khoury, MD, Mananya Satayaprasert, MD, Cynthia Bodkin, MD, John Kincaid, MD, Riley Snook, MD, Mark A. Agius, MD.
Ethical Publication Statement: We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
Additional supporting information may be found in the online version of this article
Supporting Information
Supporting Information
Supporting Information
Funding: Jacobus Pharmaceutical Co., Inc. provided funding for this work.
Conflicts of Interest: Jacobus Pharmaceutical Company, Inc, was the sponsor for this research. Drs Sanders, Demmel, and Xie are consultants for Jacobus Pharmaceutical Company; Laura Jacobus is Vice President, Jacobus Pharmaceutical Company; Dr Aleš is Medical Director, Jacobus Pharmaceutical Company; Dr David Jacobus is President, Jacobus Pharmaceutical Company; the remaining authors have no conflicts of interest.
REFERENCES
- 1. Juel VC, Sanders DB. Lambert‐Eaton myasthenic syndrome In: Engel AG, editor. Myasthenia gravis and myasthenic disorders, 2nd ed. Oxford, United Kingdom: Oxford University Press; 2015. p 156–172. [Google Scholar]
- 2. Deenen JCW, Horlings CGC, Verschuuren JJGM, Verbeek ALM. The edipemiology of neuromuscular disorders: a comprehensive overview of the literature. J Neuromuscular Dis 2015;2:73–85. [PubMed] [Google Scholar]
- 3. Abenroth DC, Smith AG, Greenlee JE, Austin SD, Clardy SL. Lambert–Eaton myasthenic syndrome: epidemiology and therapeutic response in the national veterans affairs population. Muscle Nerve 2017;56(3):421–426. [DOI] [PubMed] [Google Scholar]
- 4. Lang B, Waterman S, Pinto A, Jones D, Moss F, Boot J, et al The role of autoantibodies in Lambert‐Eaton myasthenic syndrome. Ann N Y Acad Sci 1998;841:596–605. [DOI] [PubMed] [Google Scholar]
- 5. Lundh H, Nilsson O, Rosén I. Novel drug of choice in Eaton‐Lambert syndrome. J Neurol Neurosurg Psychiatry 1983;46:684–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lundh H, Nilsson O, Rosén I. Treatment of Lambert‐Eaton syndrome: 3,4‐diaminopyridine and pyridostigmine. Neurology 1984;34:1324–1330. [DOI] [PubMed] [Google Scholar]
- 7. McEvoy KM, Windebank AJ, Daube JR, Low PA. 3,4‐Diaminopyridine in the treatment of Lambert‐Eaton myasthenic syndrome. New Engl J Med 1989;321:1567–1571. [DOI] [PubMed] [Google Scholar]
- 8. Sanders DB, Massey JM, Sanders LL, Edwards LJ. A randomized trial of 3,4‐diaminopyridine in Lambert‐Eaton myasthenic syndrome. Neurology 2000;54:603–607. [DOI] [PubMed] [Google Scholar]
- 9. Wirtz PW, Verschuuren J, van Kijk JG, de Kam ML, Schoenmaker RC, van Hasselt JG, et al Efficacy of 3,4–diaminopyridine and pyridostigmine in the treatment of Lambert‐Eaton myasthenic syndrome: a randomized, double‐blind, placebo‐controlled, crossover study. Clin Pharmacol Ther 2009;86:44–48. [DOI] [PubMed] [Google Scholar]
- 10. Oh SJ, Claussen GG, Hatanaka Y, Morgan MB. 3,4‐Diaminopyridine is more effective than placebo in a randomized, double‐blind, cross‐over drug study in LEMS. Muscle Nerve 2009;40(5):795–800. [DOI] [PubMed] [Google Scholar]
- 11. Oh SJ, Shcherbakova N, Kostera‐Pruszczyk A, Alsharabati M, Dimachkie M, Blanco JM, et al Amifampridine phosphate (Firdapse®) is effective and safe in a phase 3 clinical trial in LEMS. Muscle Nerve 2016;53(5):717–725. [DOI] [PubMed] [Google Scholar]
- 12. Morris S, Morris ME, Iansek R. Reliability of measurements obtained with the timed “up & go” test in people with Parkinson disease. Phys Ther 2001;81:810–818. [DOI] [PubMed] [Google Scholar]
- 13. Sanders DB, Guptill JT, Ales KL, Hobson‐Webb LD, Jacobus DP, Mahmood R, et al Reliability of the triple timed‐up‐and ‐go (3TUG) test. Muscle Nerve 2018;57(1):136‐139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huang SL, Hsieh C‐L, Wu R‐M, Tai C‐H, Lin C‐H, Lu W‐S. Minimal detectable change of the timed “up & go” test and the dynamic gait index in people with Parkinson disease. Phys Ther 2011;91:114–121. [DOI] [PubMed] [Google Scholar]
- 15. Sanders DB, Jacobus LR, Aleš KL, Jacobus DP, DAPPER Study Team . Predicting responsiveness to study drug before randomization in the DAPPER trial of 3,4‐diaminopyridine in Lambert‐Eaton myasthenic syndrome (Abstract). Neurology 2015;84(14 Suppl):P7.066. [Google Scholar]
- 16. Punter MNM, Zermansky A, Ramdass R, MacDonagh RJ, Marshall AG, Sussman JD, et al Successful withdrawal of 3,4‐diaminopyridine: a report on six patients with Lambert‐Eaton myasthenic syndrome. J Neurol Neurosurg Psychiatry 2011;82:e1. [Google Scholar]
- 17. Binkley JM, Stratford PW, Lott SA, Riddle DL, North American Orthopaedic Rehabilitation Network . The Lower Extremity Functional Scale (LEFS): scale development, measurement properties and clinical application. Phys Ther 1999;79:371–383. [PubMed] [Google Scholar]
- 18. Thakkar N, Guptill JT, Aleš K, Jacobus D, Jacobus L, Peloquin C, et al Population pharmacokinetics/pharmacodynamics of 3,4‐diaminopyridine free base in patients with Lambert‐Eaton myasthenia. CPT Pharmacometrics Syst Pharmacol 2017;6(9):625–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lundh H, Nilsson O, Rosén I, Johansson S. Practical aspects of 3,4‐diaminopyridine treatment of the Lambert‐Eaton myasthenic syndrome. Acta Neurol Scand 1993;88:136–140. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article
Supporting Information
Supporting Information
Supporting Information