

Abstract
Pediatric neuromuscular diseases encompass all disorders with onset in childhood and where the primary area of pathology is in the peripheral nervous system. These conditions are largely genetic in etiology, and only those with a genetic underpinning will be presented in this review. This includes disorders of the anterior horn cell (e.g., spinal muscular atrophy), peripheral nerve (e.g., Charcot–Marie–Tooth disease), the neuromuscular junction (e.g., congenital myasthenic syndrome), and the muscle (myopathies and muscular dystrophies). Historically, pediatric neuromuscular disorders have uniformly been considered to be without treatment possibilities and to have dire prognoses. This perception has gradually changed, starting in part with the discovery and widespread application of corticosteroids for Duchenne muscular dystrophy. At present, several exciting therapeutic avenues are under investigation for a range of conditions, offering the potential for significant improvements in patient morbidities and mortality and, in some cases, curative intervention. In this review, we will present the current state of treatment for the most common pediatric neuromuscular conditions, and detail the treatment strategies with the greatest potential for helping with these devastating diseases.
Keywords: Charcot–Marie–Tooth disease, congenital myopathies, muscular dystrophies, neuromuscular disorders
1. INTRODUCTION
Neuromuscular disorders encompass the spectrum of diseases where the primary abnormality or lesion is in the peripheral nervous system, defined as including the anterior horn cell, the peripheral nerve, the neuromuscular junction, and the muscle (Figure 1). In general, the unifying aspect of neuromuscular disorders is abnormal muscle function and the resulting sequelae from it. This can include chronic signs and symptoms, most typically related to muscle weakness, such as abnormal or impaired ambulation, joint contractures, skeletal deformities (particularly scoliosis), altered sensory perception (in neuropathies) and respiratory failure. It also includes dynamic impairments, such as exercise intolerance, myalgia, rhabdomyolysis, and fatigable weakness, which may exist with normal intervening muscle function, or may instead occur in individuals with persistent neuromuscular manifestations. In total, the genetic neuromuscular disorders are frequently associated with significant lifelong morbidities, which are often severely disabling and associated with premature mortality.
Figure 1.
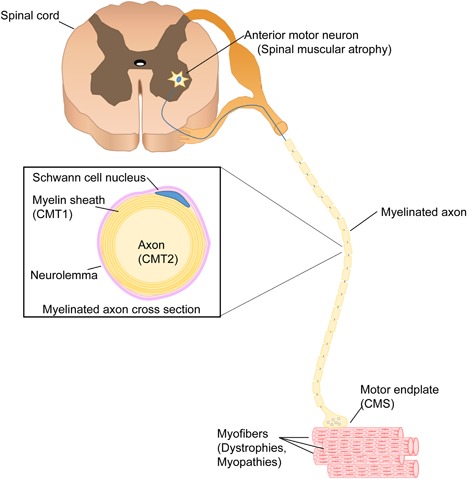
The motor unit. The motor unit is composed by the anterior horn cell (motor neuron) and the skeletal muscle fibers that are innervated by it. All myofibers in one motor unit are of the same type (I, IIA, IIB). The main genetic paediatric disorders of each part of the motor unit are within brackets (CMS, congenital myasthenicsyndromes; CMT, Charcot–Marie–Tooth type 1 or type 2).
In pediatrics, the majority of neuromuscular disorders have a genetic basis, either as a de novo or an inherited pathogenic variant in a single gene (Darras, 2015). The most commonly encountered genetic pediatric neuromuscular condition is Duchenne muscular dystrophy (DMD), a primary muscle disease with an estimated prevalence of approximately 1:5,000 boys (Romitti et al., 2015). Other common disorders include spinal muscular atrophy (a neuronopathy affecting the anterior horn cell), myotonic dystrophy (a multi‐systematic disorder with a significant muscle component), and Charcot–Marie–Tooth disease (a disease of the peripheral nerve). Notable non‐genetic conditions include myasthenia gravis, Guillain–Barré syndrome, and chronic inflammatory polyneuropathy; these conditions, which have helped inform upon aspects of therapeutics for genetic neuromuscular disorders, will not be discussed further in this review. Instead, we will present the major genetic neuromuscular conditions of childhood, discuss presently available therapies, and review key strategies in the therapeutic pipeline.
2. SPINAL MUSCULAR ATROPHY
Spinal muscular atrophies (SMAs) are lower motor neuron diseases that principally affect spinal cord anterior horn cells and brain stem motor nuclei, and they are historically defined by the observation of myofiber atrophy on muscle biopsy (Dubowitz, 1995). While SMAs are a genetically heterogeneous group of conditions with several causative loci, the classic and overwhelmingly most common form of SMA is the autosomal recessive form caused by biallelic pathogenic variants in the SMN1 (survival of motor neuron 1) gene on chromosome 5q13.2. 5q13‐SMA (typically referred to as classic SMA or simply SMA) is the most common cause of lower motor neuron disease (incidence of 1 in 6,000 to 1 in 10,000 live births per year) and one of the most common fatal genetic diseases of childhood (Pearn, 1978). Most of the other SMAs, often termed distal SMAs, are quite rare. The distal SMAs share considerable clinical and genetic overlap with both Charcot–Marie–Tooth disease and hereditary spastic paraplegia. One exception is SMA with respiratory distress (SMARD1), also known as autosomal recessive distal spinal muscular atrophy‐1 (DSMA1), which clinically can resemble classic SMA but with respiratory failure early in the course of disease. The remainder of the discussion will focus on 5q13‐SMA (which will be referred to as SMA) (Table 1).
Table 1.
Novel compounds for SMA in human clinical trials
| Category | Compound | Action | Company | Dosing and delivery | Phase of clinical trial | Results | Safety | Reference |
|---|---|---|---|---|---|---|---|---|
| Genetic based therapies | ||||||||
| Gene Therapy | AVXS‐101 | Gene therapy via AAV‐9 vector systemic and intrathecal routes in development | AveXis | Intravenous single dose | Phase 1 open label | Improved motor milestones and life span compared to natural history data | No significant concerns Liver enzyme elevation | Mendell et al. (2017) |
| RNA splicing manipulation | Nusinersen | Anti‐sense oligonucleotide | Biogen | Intrathecal | Phase 3 | Improved motor function and improved mortality (infants) | No significant concerns identified | Mercuri and Kuntz (2017) |
| RG7916 | Small molecule to alter SMN2 splicing to include exon 7 | Roche | Oral | Phase 2 | Pre‐clinical only | Tolerated in healthy controls | http://Clinicaltrials.gov NCT02908685 | |
| LM1070 | Small molecule to alter SMN2 splicing to include exon 7 | Novartis | Oral | Phase 1/2 | Pre‐clinical only | concerns in animal models | http://Clinicaltrials.gov NCT02268552 | |
| Non genetic based therapies | ||||||||
| Enhancing contraction | CK‐2127107 | Fast skeletal troponin activator | Cytokinetics | Oral | Phase 2 ongoing | Safety only | No significant concerns in healthy controls | http://Clinicaltrials.gov NCT02644668 |
| Neuroprotective | Olesoxime | Binds to mitochondrial membrane channels to mitigate oxidative stress | Roche | Oral | Phase 2 complete | Stabilized motor function over 2 years of trial | No significant concerns | http://Clinicaltrials.gov NCT01302600 |
In SMA, there is dramatic clinical heterogeneity, with four childhood‐onset (types 0, I, II, and III SMA) and one adult‐onset (type IV) form distinguished by age of onset and maximum motor skill achieved (Crawford & Pardo, 1996). Type I SMA, also called Werdnig–Hoffman disease, is the most frequently observed and well‐described subtype. It is a devastating disease with onset in infancy and a progressive, fatal course. Despite their broad spectrum of clinical presentation, all subtypes of classic SMA are caused exclusively by biallelic (usually homozygous) pathogenic variants in SMN1 gene (Lefebvre et al., 1995). Pathogenic variants in SMN1 are most typically exonic deletions in the mid‐region (exon 7) of the gene, with point mutations making up only a small percentage of cases. SMN1 encodes SMN, a ubiquitous protein with a large associated proteome. The normal function(s) of SMN protein, along with the pathomechanisms associated with its loss, are still being unravelled; the protein is known to participate in critical pathways related to RNA processing and transport, and it is believed that motor neurons are particularly vulnerable to impairments in these processes. The end result of the loss of SMN protein is altered motor neuron function and the progressive death of motor neurons.
Importantly, the chromosome 5q13.2 region where SMN1 resides also contains SMN2, a gene centromeric to SMN1 that encodes an essentially identical protein. Compared to SMN1, SMN2 contains an exonic splice enhancer variant that results in preferential skipping of exon 7, leading to a truncated and more unstable protein product that is able to provide approximately 10–20% of total SMN function (Singh, Liew, & Darras, 2013). In healthy controls and in patients, copy number variation at the SMN1 and SMN2 loci is quite variable with nine different genotypes consisting of various combinations of copies of SMN1 and SMN2 alleles. SMN2 gene copy number acts as the main modifier of the SMA clinical phenotype. While there is not a perfect correlation, the higher the SMN2 copy number, the milder the clinical phenotype, with type I patients typically having no more than two copies of SMN2, and type III/IV patients having four or more copies.
2.1. Therapy for SMA: Overview
Current clinical management of SMA relies upon proactive measures in the setting of an interdisciplinary clinic to combat the progressive motor weakness, including aggressive treatment of respiratory infections, pro‐active ventilatory support, adequate nutrition, and interventions for skeletal deformities such as scoliosis as necessary. Consensus care guidelines were completed in 2007 and were recently updated (but are in preparation for publication) at a European Neuromuscular Centre (ENMC) meeting in February 2016 (Finkel, Bertini, Muntoni, Mercuri, & Group ESWS, 2015; Wang et al., 2007). New therapeutic products under development for SMA can be divided broadly into two major categories: genetic based therapies, such as SMN1 gene replacement therapy or SMN2 upregulation or modification; and non‐genetic type therapies, such as neuroprotective strategies or altering downstream motor unit function. Importantly, treatment considerations and care standards are likely to be dramatically altered by the development and clinical implementation of Spinraza (described in the next section), the first disease modifying therapy approved for SMA.
2.2. Genetic based therapies: SMN2 modification as a therapeutic strategy for SMA
The unique genetics of SMA (SMN1 mutations in all patients, with SMN2 copy number as the primary disease modifier) provides a clear and attractive avenue for therapy development, namely increasing protein production from the intact SMN2. Specific strategies include manipulating the promotor region of SMN2 in order to increase the amount of SMN2 and alternating the splicing of SMN2 to include exon 7 and thus generating a fully functional SMN gene transcript. Historical attempts to upregulate SMN2 through the use of histone deacetylase inhibitors that act to increase transcription from the SMN2 locus include the use of valproate (Swoboda et al., 2010), phenylbutyrate (Mercuri et al., 2007), and hydroxyurea (Chen et al., 2010). All of these drugs showed promise in pre‐clinical and open label studies, but failed to demonstrate efficacy in randomized, placebo‐controlled studies of ambulant, and non‐ambulant SMA patients (Chen et al., 2010; Kissel et al., 2014, 2011; Swoboda et al., 2010). While these trials were unsuccessful, they provided a critical roadmap for the current clinical trials in this challenging disease.
New agents aimed at post‐transcriptional mechanisms of modifying splicing of exon 7 appear promising. Nusinersen (Spinraza, Biogen, Cambridge, MA) is an antisense oligonucleotide (AON), originally developed by Ionis Pharmaceuticals (Carlsbad, CA), that targets the splice site of SMN2 exon 7, resulting in inclusion of exon 7 in the final transcript. Due to poor transport across the blood brain barrier it must be given via intrathecal injection. The product has shown much promise in preclinical and early phase human trials (Chiriboga et al., 2016). In an ongoing open label extension study in infants with SMA type 1, the muscle function scores increased on standardized outcome measures and other markers such as electrophysiology and life span showed favorable improvement over what is expected from natural history (Finkel et al., 2016). At present, two phase 3 studies, one in infants and one in later onset SMA, have been completed and on the basis of significant interim results both studies have been stopped, study subjects switched to active treatment in an open label extension trial, and a global exceptional access program developed for type 1 SMA patients. In the study of infants with type I SMA, the risk of progressing to death or permanent ventilation was reduced by approximately half over a 13 month period. There were also improvements in motor milestones, a standardized motor score, and electrophysiology biomarkers (ulnar and peroneal compound muscle action potential) compared to decline in these measures in placebo treated infants (Kuntz et al., 2017).
In the phase 3 study of nusinersen in later onset SMA, or children with SMA type 2 at age 2–7 years, the results were similarly striking at end of study analysis at 15 months. Nusinersen treated children improved by four points on a standardized motor scale for SMA compared to a one point decline for the placebo group. Upper extremity function as measured by the Revised Upper Limb Module, and by the number of new motor milestones achieved, was also significantly better for the treated group (Mercuri et al., 2017). In both cases the safety profile showed very low risk to the actual drug, with no treatment related adverse events, however intrathecal administration comes with a range of typical side effects related to the lumbar puncture and in keeping with the complex medical needs of SMA patients.
Nusinersen (Spinraza) has now received rapid commercial approval in the US, EU, and Canada on the strength of the phase 3 study results. The current regulatory approvals in these jurisdictions do not have any restrictions or criteria for access (e.g., such as start/stop criteria) and minimal post marketing safety mandate beyond standard pharmaco‐vigilence. The arrival of this new therapy into clinical practice will mean a substantial increase in resource requirements, particularly given the drug's high likely cost, as well as given the invasive nature of LPs, the challenge of drug administration to individuals with severe scoliosis, and the frequency of administration. The availability of the drug in clinical care has also created challenges for access and resource allocation and generated new conversations and management strategies as the SMA community is adapting to a therapy that appears to substantially change the trajectory of what was a disease with progressive decline.
There have also been small molecules therapeutics aimed at increasing SMN2 protein levels. One strategy has been to target the SMN2 splicing pattern to include exon 7 (Palacino et al., 2015; Zhao et al., 2016). Both PTC Therapeutics (South Plainfield, NJ)/F. Hoffmann‐La Roche Ltd (Roche, Switzerland) and Novartis AG (Switzerland) have developed such molecules. The PTC Therapeutics/Roche partnership has produced another candidate molecule that started phase II trials in SMA types I–III in 2016 (http://Clinicaltrials.gov identifier: NCT02633709). All these drugs are orally administered, so if safety issues can be resolved and efficacy established they offer an attractive alternative to more invasive drug delivery methods (such as needed for Spinraza). A repurposing drug screen looking for drugs that increased SMN2 identified celecoxib, which appears to work in this context through a p38 mitogen‐activated kinase pathway to stabilize and increase SMN2 transcript levels (Farooq et al., 2013). Celecoxib will be studied further in a forth‐coming clinical trial (NCT02876094). Of note, several previous unsuccessful clinical trials targeting SMN2 have been completed.
2.3. Genetic based therapies: Gene replacement for SMA
Given that SMA results from loss‐of‐function mutations of SMN1 and thus decreased levels of SMN, gene therapy to replace SMN1 is an obvious candidate strategy. In fact, gene therapy with AAV9 virus and full length SMN1 has been attempted via intrathecal delivery in a phase 1 clinical trial in infants with SMA type I at Nationwide Children's Hospital in Ohio (http://Clinicaltrials.gov identifier: NCT02122952. Encouraging results and a favorable safety profile have been presented at recent academic forums; all 15 study participants are still surviving and non require 16 hr or more of ventilation per day (Mendell et al., 2017).
2.4. Other strategies for treating SMA
Beta‐adrenergic medicines (salbutamol/albuterol) have been considered for SMA for a number of years. While their precise mechanism of action is not well understood, results with salbutamol or albuterol have been encouraging. Salbutamol has been examined in an open label trial design in type II SMA patients and found to improve muscle strength over a one‐year period (Pane et al., 2008). Similar results were seen in a mixed cohort of type II/type III patients with albuterol (Kinali et al., 2002). Currently, salbutamol is used off label in many patients with SMA, particularly those with type II, and anecdotal experience suggests it imparts some benefit in terms of strength and motor function.
Two other approaches currently being investigated in human trials include olesoxime, a neuroprotectant, and CK‐2127107, a cytokinetic agent that enhances muscle contractility at the actin myosin interface. While olesoxime is a non‐specific neuro‐protectant (i.e., a drug aimed at preventing neuronal death) that acts at the mitochondrial permeability transition pore to mitigate cell stress reactions with potential application for several neurodegenerative diseases (Kaczmarek, Schneider, Wirth, & Riessland, 2015), it has been shown specifically in SMA to maintain motor function (phase 3 placebo controlled clinical trial in type II and III SMA patients, NCT02628743). CK‐2127107, which is in phase 2 studies (NCT02644668) for SMA, is a small molecule that alters the interaction of calcium and troponin resulting in improved myofibre contractility. This approach of enhancing muscle contraction in SMA clearly targets a downstream pathway (secondary muscle dysfunction from impaired motor neuron signaling), and so its potential value will need to be evaluated not only in short duration studies but also over time, as the progressive loss of motor neurons and their connection with muscle may influence drug efficacy.
Of note, one key challenge to any treatment for SMA is the likely presence of a therapeutic window for the disease (Jablonka & Sendtner, 2017). Elegant experiments from the group of Arthur Burghes (The Ohio State University) and others have shown using a mouse model of SMA that there is a pathophysiologic developmental stage of motor neuron loss that largely sets the disease trajectory of the SMA, and that the disease process cannot be rescued by replacement or upregulation of SMN once this motor neuron loss has progressed beyond a certain critical point (Le et al., 2011). This has implications for the timing of human clinical trials, suggesting that therapeutic interventions need to be given as early as possible, and perhaps even in pre‐symptomatic or fetal stages to treat the disease effectively. In addition, further strategies to treat SMA have been hampered by the difficulty in fully understanding the role of SMN protein, and the ubiquitous nature of SMN suggests a more complex physiologic role.
2.5. Summary
The pivotal clinical trials described above are heralding a new era to SMA clinical treatment with impactful therapies showing improvements in strength and function. Well this is a welcome development for the SMA community it raises new challenges for assuring fair and timely access to treatment for those who could benefit and is a call to ongoing robust data collection of the new natural history of SMA so that the knowledge gaps can be properly filled. One also anticipates an impact on the ability to conduct clinical trials for other emerging therapies, now that a commercial product is available. For multiple reasons, this could potentially negatively affect finding the ideal therapy or combinations of therapies to treat SMA. And so ongoing creative trial design, and thoughtful regulatory consideration for rare diseases such as SMA becomes even more important.
3. CHARCOT–MARIE–TOOTH DISEASE
Charcot–Marie–Tooth disease (CMT), also referred to as hereditary motor and sensory neuropathy, is typically a slowly progressive condition characterized by distal weakness, hand and foot deformities, indolent loss of sensory perceptions, and mild to moderate disabilities (Jani‐Acsadi, Ounpuu, Pierz, & Acsadi, 2015). There is marked clinical and genetic heterogeneity with CMT (Gutmann & Shy, 2015). From a clinical prospective, while the majority of affected individuals have the classical presentation described above, there is a broad range in age of presentation and severity, and there are instances of very severe neuropathy presenting in infancy with profound weakness, as well as cases where symptoms do not occur until adulthood (Jani‐Acsadi et al., 2015). Mutations in more than 80 genes have been associated with CMT (Gutmann & Shy, 2015), though mutations in four genes (PMP22 duplication, which is by far the most common mutation, and variants in MPZ, GJB32, and MFN2) account for the majority of cases (Saporta et al., 2011). The overall prevalence of CMT is thought to be approximately 1:2,500 (Skre, 1974), making it one of the most common neuromuscular conditions of childhood (Table 2).
Table 2.
Therapeutic strategies for Charcot–Marie–Tooth Disease
| Category | Compound | CMT subtype | Action | Company | Dosing and delivery | Phase of clinical trial | Results | Safety | Reference | |
|---|---|---|---|---|---|---|---|---|---|---|
| Genetic based therapies | ||||||||||
| Gene therapy | Neuregulin‐1 | CMT1A | Neuregulin‐1 Enhanced PI3K‐Akt signaling improving differentiation of Schawnn cells | – | Injected intraperitoneally | Rat model | Correction of mRNA splicing | No results in humans yet | Fledrich et al. (2014) | |
| Neurotrophin 3 (NT‐3) | CMT1A | Stimulates neurite outgrowth and myelination | – | Subcutaneous | Pilot human trial of recombinant human NT‐3 | Improved regeneration of myelinated fibers | Not associated with any serious adverse events | Sahenk et al. (2014, 2005) | ||
| GJB1 gene replacement | CMTX | Gene replacement | – | Intrathecal | Mouse model | Improved motor performance, quadriceps muscle contractility, and sciatic nerve conduction velocities | No results in humans yet | Kagiava et al. (2016) | ||
| Non genetic based therapies | ||||||||||
| Ascorbic acid | CMT1A | Lowering PMP22 levels via cAMP‐dependent transcriptional repression | – | Oral | Phase 3 | Failed | Not associated with any serious adverse events | Pareyson et al. (2011) | ||
| PXT3003 (sorbitol, naproxen, baclofen) | CMT1A | Down regulation of PMP22 | Pharnext | Oral | Phase 3 | No results yet. Phase 2 revealed improvement | No significant concerns identified | http://Clinical.trial.gov NCT02579759 NCT03023540 | ||
| Creatine | CMT1A | – | – | Oral | Tested in a small cohort of 20 patients | Failed to showed improvement | No significant concerns identified | Chetlin et al. (2004) | ||
| Vitamin E + fatty acids | CMT1A | Antioxidant | – | Oral | Small randomized trial | Placebo + Vit E increased strength. | No significant concerns identified | Williams et al. (1986) | ||
| Biotin | CMT1A | – | MedDay Pharmaceuticals | – | Phase 1/2 | Pre‐clinical only | No Results yet | http://Clinical.trial.gov NCT02967679 | ||
| Ulipristal Acetate | CMT1A | Decrease transcription of PMP22 | – | Oral | Phase 2 | Pre‐clinical only | Reversible endometrial hyperplasia | http://Clinical.trial.gov NCT02600286 | ||
| Curcumin | CMT1B | Decrease endoplasmic reticulum stress | – | Oral | Rat model | Pre‐clinical only | No results in humans | Patzko et al. (2012) | ||
| Niacin | CMT4B1 | Inhibition of Neuroregulin‐1 increase myelination folds | – | – | Mouse model | Pre‐clinical only | No results in humans | Bolino et al. (2016) | ||
| ACE‐083 | CMTX | Inhibition of members of the TGF‐beta superfamily, specially myostatin, promotes increase in muscle size | Acceleron Pharma | Intramuscular injection | Phase 2 | It generated dose‐dependent increases in muscle volume | Tolerated in healthy controls | http://Clinical.trial.gov NCT03124459 | ||
The primary localization of pathology in CMT is in the peripheral nerve fiber. Classically, CMT is broken down into subcategories based on mode of inheritance (dominant vs. recessive) and whether the primary pathology is in the neuron (e.g., the axon, as in type II CMT) or in the myelinating Schwann cell (e.g., demyelinating CMT, as in CMT Type I and most causes of CMT Type IV). These pathologic subtypes are typically distinguished based on findings from nerve conduction studies and, more rarely, from nerve biopsy results. CMT shares overlap with several conditions that feature prominent peripheral nerve involvement, including the hereditary sensory and autonomic neuropathies, some forms of complicated hereditary spastic paraplegia, some leukodystrophies (such as metachromatic leukodystrophy, which can have a severe peripheral neuropathy), and some motor neuronopathies (such as juvenile amyotrophic lateral sclerosis). This section will focus exclusively on CMT.
While symptoms from CMT are typically not life threatening, they often result in significant disabilities. Despite this, and despite the relative commonality of the condition, presently there are no disease specific therapies for any CMT subtype. Current care is aimed at supportive management and reduction of secondary complications. For example, pes cavus is a common, disabling, and progressive problem in many CMT patients. Both surgical and pharmacologic interventions have been considered; surgery appears to have some benefit, though must be considered with caution and in the context of potential gains in range of motion balanced versus post‐operative loss of motor function and delayed healing (Azmaipairashvili, Riddle, Scavina, & Kumar, 2005). Botulinum toxin injections have been shown in case reports to improve symptoms, but a randomized trial in pediatric patients with CMT1A did not show efficacy (Burns, Scheinberg, Ryan, Rose, & Ouvrier, 2010).
3.1. Therapy development for CMT
The major emphasis in terms of drug development for CMT has focused on CMT1A, the most common form of CMT, though discovery efforts have also yielded interesting candidate therapeutics for other CMT subtypes (Mathis, Magy, & Vallat, 2015). CMT1A is caused by duplication of the PMP22 gene, and is thus a disorder of excessive gene dosage. Most treatment strategies for CMT1A have therefore focused on attempting to lower PMP22 levels. Several small molecules have been tested in CMT1A patients via randomized, placebo controlled clinical trials. Perhaps the most intensely studied has been ascorbic acid, a molecule that has previously shown promise in pre‐clinical models and in pilot patient studies, and it is thought to act by lowering PMP22 levels via cAMP‐dependent transcriptional repression (Passage et al., 2004). However, recent data examining both low‐ and high‐dose treatment have suggested that ascorbic acid does not improve disease status or reduce disease progression (Burns et al., 2009; Micallef et al., 2009; Pareyson et al., 2011). More recently, PXT3003 (a drug that combines naltrexone, baclofen, and sorbitol that act in synergy to lower PMP22 levels and potentially provide pro‐survival signals) has been found in exploratory studies to improve baseline motor function in CMT1A after 1 year versus placebo (Attarian et al., 2014). Additional larger clinical studies are in progress (NCT02579759 and NCT03023540) and needed to fully evaluate this treatment strategy.
Other drugs and supplements have, based on supportive pre‐clinical data, been tested to various extents in patients with CMT. A small randomized trial of essential fatty acids plus vitamin E did not show benefit, though the placebo plus vitamin E group also had improvements in strength, opening the question of whether vitamin E alone may be beneficial (Williams, O'Dougherty, Wright, Bobulski, & Horrocks, 1986). These studies have not been followed up, however, and neither fatty acids nor vitamin E are routinely used in CMT. Creatine has been evaluated in combination with resistance exercise training in a cohort of 20 patients, and did not improve strength (Chetlin, Gutmann, Tarnopolsky, Ullrich, & Yeater, 2004), although exercise alone (as has been examined in several small studies) may improve function in CMT (Sman et al., 2015). High dose biotin has been considered for other types of neuropathy (such as diabetic peripheral neuropathy) (Koutsikos, Agroyannis, & Tzanatos‐Exarchou, 1990), and is now being tested in an open label clinical trial for patients with demyelinating subtypes of CMT (MD 1003, MedDay Pharmaceuticals, NCT02967679).
Several candidate compounds have improved phenotypes in mouse models of CMT, but have yet to be formally tested in patients. One example is curcumin, derivatives of which decrease endoplasmic reticulum (ER) stress and improve neuropathy in a mouse model of CMT1B (a demyelinating CMT subtype due to heterozygous MPZ mutations) (Patzko et al., 2012). Another is progesterone antagonism, a strategy that promotes reduction of PMP22 levels in a rat model of CMT1A and that is currently being examined in a phase II placebo controlled trial (NCT02600286) (Meyer zu Horste et al., 2007). Lastly, a novel strategy has demonstrated efficacy in a mouse model of CMT4B1 (caused by biallelic mutations in Mtmr2): inhibition of neuregulin signaling with niacin (Bolino et al., 2016), the rationale for which is the observation of excessive myelin outfolds in CMT4B1.
Gene based therapies are also under consideration for different subtypes of CMT, though none have entered clinical testing to date. Kagiava et al. (2016), recently demonstrated the ability of intrathecally administered GJB1 to rescue the neuropathy of adult Gjb1 null mice, a model for CMT‐X. Secondary gene therapies with downstream agonist molecules that promote neuro‐protection, such as neurotrophin‐3 or neuregulin‐1 (Fledrich et al., 2014; Sahenk et al., 2014), have also shown efficacy in pre‐clinical studies, specifically improving neuropathy in mouse and rat models of CMT1A.
Lastly, Acceleron has launched a two part (open label dose escalation followed by randomized, placebo controlled) phase II trial of ACE‐083 (NCT03124459), a compound designed to promote/increase muscle mass by inhibiting signaling through members of the TGF‐beta superfamily including particularly myostatin. The rationale of the therapy for CMT is based on the likely potential clinical impact of halting and/or reversing progressive distal muscle wasting. However, preclinical data in CMT animal models to support this strategy is lacking.
3.2. Summary
Currently, there are limited therapeutic options for CMT, and there is a need to expand the pipeline of potential candidate treatment strategies. Treating the disease is complicated by genetic heterogeneity and variability in clinical presentations. However, one major advance has been the recognition of reliable outcome measures for studying the disease, and significant progress has been made recently in clinical trial readiness by establishing the required knowledge baseline of disease natural history (Burns et al., 2012; Fridman et al., 2015; Gutmann & Shy, 2015; Sanmaneechai et al., 2015). These factors should greatly accelerate the translation of promising drugs in the future.
4. CONGENITAL MYASTHENIC SYNDROMES
Congenital myasthenic syndromes (CMSs) are conditions caused by mutations in genes that form and/or regulate the neuromuscular junction (Engel, Shen, Selcen, & Sine, 2015). Patients with CMS commonly have a mixture of static weakness, particularly of the facial and limb girdle musculature, and episodic weakness in a fashion similar to autoimmune myasthenia gravis (Eymard, Hantai, & Estournet, 2013). They most typically present in infancy (Kinali et al., 2008), but the spectrum of disease is quite broad, and individuals come to clinical attention at all stages of life (Garg et al., 2016). The diagnosis of CMS is suspected by the clinical picture (particularly in the setting of fluctuating weakness) and supported by electrodiagnostic abnormalities via electromyography or nerve conduction velocity analysis. CMS is confirmed by the identification of a pathogenic genetic variant, and positive detection of causative mutations are found in the majority (50–70%) of individuals with suspected disease (Abicht, Muller, & Lochmuller, 1993). At present, pathogenic variants in 12 genes are known to cause CMS, with pathogenic variants in the acetylcholine receptor subunit epsilon (CHRNE) being the most common (Figure 2). CMS can be categorized based on the part of the neuromuscular junction where the mutated gene typical functions (i.e., presynaptic, synaptic, or postsynaptic). Patients with postsynaptic CMS and mutations that affect the acetylcholine receptor (CHRNA1, CHRNB1, CHRND, and CHRNE) can be further subdivided into the clinically similar fast channel (the typical result from the majority of mutations) and slow channel syndromes. Slow channel syndrome is caused by prolonged decay of the postsynaptic current and results from prolonged acetylcholine receptor channel opening. It is associated with dominant “gain of function” mutations (Abicht et al., 1993) (Table 3).
Figure 2.
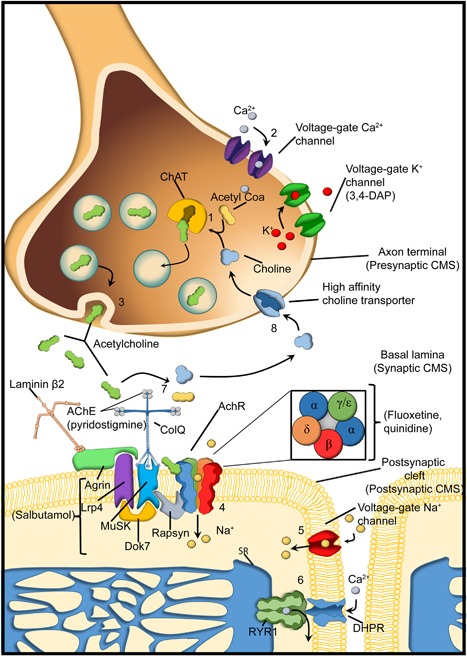
The neuromuscular junction. Components of the neuromuscular junction. In the presynaptic terminal, acetylcholine is synthesized by the enzyme choline acetyltransferase (ChAT) from the compounds choline and acetyl‐CoA (1). When an action potential arrives at the endplate it activates voltage gated Ca2+ channels allowing Ca2+ ions flow into the axon terminal (2) and the release of the acetylcholine into the synaptic cleft (3). Acetylcholine binds to the alpha subunit of the acetylcholine receptor (AchR) to create a Na+ current into the myofiber (4), which then generates an action potential through the activation of the voltage‐gate Na+ channels (5), These leads to activation of the dihydropyridine receptor (DHPR, another Voltage gate Ca2+ channel) and then activation of the ryanodine type 1 receptor (RyR1) that releases Ca2+ from the sarcoplasmic reticulum (SR) into the cytoplasm (6). The acetylcholine will be broken down by the enzyme acetylcholinesterase (7) and choline is then transported into the axon terminal by a high affinity transporter (8). On the postsynaptic membrane AchRs are clustered by a complex of proteins (Rapsyn; docking protein 7, (Dok7); Muscle‐specific kinase (MuSK); Agrin; LDL receptor related protein (Lrp4)). Nerve derived Agrin binds to an LRP4 MuSK complex and induces the Rapsyn mediated clustering of AChR. Twelve catalytic subunits of AchE are attached to Collagen Q (ColQ) to the postsynaptic membrane via binding to MuSK. The congenial myasthenic syndromes (CMS) can be classified by the localization of the protein affected in the neuromuscular junction (presynaptic, synaptic, postsynaptic). The main drugs use as treatment for the CMS are within bracket under the protein/channel where they are acting. Pyridostigmine inhibits the AchE. Fluoxetine and quinidine blocks the AchR. 3–4 diaminopyridine (3–4 DAP) acts in the Voltage gate K+ channel by blocking the repolarization of the terminal axon. It is not know the exact mechanism of action of salbutamol, but it is thought that produce activation of second messenger signaling that partially compensates the instability of the Agrin‐MuSK‐LRP4‐Rapsyn‐DOK7 (Beeson, 2016; Ravenscroft, Laing, & Bönnemann, 2015).
Table 3.
Summary of therapeutic strategies for congenital myasthenic syndromes (CMS)
| Category | Compound | CMS Subtype | Action | Company | Dosing and delivery | Phase of clinical trial | Results | Safety | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Genetic based therapies | |||||||||
| Gene Therapy | AAV‐DOK7 | DOK7 | Gene replacement | – | Intraperitoneal delivery | Mouse model | Successful rescue of the model | No results in humans yet | Arimura et al. (2014) |
| RNA splicing manipulation | Antisense oligonucleotides | CHRNA1 | Induce exon P3A skipping (to produce a functional P3A‐ isoform) | – | – | In vitro | Pre‐clinical only | No results in humans yet | Tei et al. (2015) |
| Tannic acid | CHRNA1 | Induce exon P3A skipping (to produce a functional P3A‐ isoform) | – | – | In vitro | Pre‐clinical only | No results in humans yet | Bian et al. (2009) | |
| Non genetic based therapies | |||||||||
| Cholinesterase inhibitor | Pyridostigmine (mestinon) | ●AChR deficiency (CHRNA1, CHRNB1, CHRND, and CHRNE); fast‐channel mutations ●CHAT ●RAPSN ●GFPT1 ●DAPGT1 ●ALG2 ●ALG14 ●SCN4A ●PREPL | Increase acetylcholine in the NMJ | – | Oral | No formal systematic analyses for CMS | Standard used | To be avoided in ACHE, Slow channel, LAMB2, COLQ, DOK7, and MUSK | Engel et al. (2015) |
| Presynaptic K channel blocker | 3‐4 diaminopyridine (amifampridine) | ●AChR fast syndrome mutations ●RAPSN ●GFPT1 ●DAPGT1 ●ALG2 ●ALG14 | Increases the number of ACh quanta released by nerve impulse | Catalyst (For NCT02562066) | Oral | Phase II clinical trial | Standard use | To be avoid in ACHE, Slow channel, DOK7 | http://Clinical.trial.gov NCT02562066 NCT00872950 NCT03062631 |
| AChR blocker | Fluoxetine, quinine or Quinidine | ●AChR slow channel mutations | Non‐competitive acetylcholine receptor inhibitors with preferential activity against the mutant channels | – | Oral | No formal systematic analyses for CMS | Standard use | No significant concerns identified | Zhu et al. (2015) |
| Unknown | Albuterol or ephedrine | ●AChR mutations ●ACHE ●DOK7 ●LAMB2 ●RAPSN (albuterol) ●MUSK deficiency (albuterol) ●AGRN (partial response to ephedrine) | Not well understood | – | Oral | Open label study | Standard use | No significant concerns identified | Rodriguez Cruz et al. (2015) |
4.1. Pyridostigmine therapy for CMS
The primary therapeutic intervention in CMS is the cholinesterase inhibitor pyridostigmine (Mestinon), typical dosing 1 mg/kg/dose or 7 mg/kg/day total) (Engel et al., 2015; Kinali et al., 2008). The majority of CMS patients derive some positive benefit from this therapy, though response can be quite variable. Some individuals will dramatically improve, though typically response is more modest. In some CMS subtypes, pyridostigmine may not help at all and may potentially worsen symptoms; these include slow channel CMS (due to gain‐of‐function mutations in the acetylcholine receptor) (Chaouch et al., 2012), and patients with pathogenic variants in LAMB2, COLQ, DOK7, and MUSK (Engel et al., 2015). This variability in responsiveness underscores the importance of establishing a specific diagnosis when CMS is suspected. Of note, there have been no formal systematic analyses of pyridostigmine in CMS, and knowledge concerning this therapy is based on retrospective analyses.
4.2. Other treatment approaches for CMS
In patients with CMS who have no improvement or incomplete response to anticholinesterase inhibitors, other therapies have been considered. 3,4‐diaminopyridine (3,4 DAP) (amifampridine), a drug that increases the release of acetylcholine at the neuromuscular junction, has shown efficacy (as reported via case studies), either alone or in combination, in several genetic subtypes of CMS (Banwell, Ohno, Sieb, & Engel, 2004; Engelet al., 2015; Eymard et al., 2013). Based on these promising observations, the drug is now in a phase II clinical trial for CMS (NCT00872950) as well as in an open label expanded access trial (NCT03062631). However, caution should be used with this medication until further clinical data is available, particularly in pediatrics (Beeson, Hantai, Lochmuller, & Engel, 2005), as two children with fast channel CMS died after starting it (cause of death unknown and may have been unrelated to treatment).
The beta adrenergic agonists, ephedrine and salbutamol/albuterol, have also been examined in pyridostigmine‐refractory CMS. A recent open label trial of these medicines in patients with severe recessive (fast channel) CHRNE‐associated CMS showed safety and clinical improvement in terms of strength and fatigue (Rodriguez Cruz et al., 2015). This study echoes previous case reports for both medicines in several CMS subtypes including particularly those caused by mutations in COLQ and DOK7 (often poorly responsive to pyridostigmine) (Burke et al., 2013; Lashley, Palace, Jayawant, Robb, & Beeson, 2010; Liewluck, Selcen, & Engel, 2011; Yeung, Lam, & Ng, 2010). At present, the mechanism(s) by which beta‐agonist medications improve disorders of the neuromuscular junction are not known.
Of note, the combination of fluoxetine and salbutamol has been shown to improve symptoms in one case of slow‐channel CMS (Finlayson et al., 2013), where pyridostigmine and 3,4‐DAP are contraindicated. There is also a single case report showing clinical improvement with quinine in a patient with slow channel syndrome (Peyer, Abicht, Heinimann, Sinnreich, & Fischer, 2013). Of note, both fluoxetine and quinine are thought to improve slow channel CMS by acting as non‐competitive acetylcholine receptor inhibitors with preferential activity against the mutant channels (Zhu et al., 2015).
4.3. Experimental therapies for CMS
Whether it is because of the rarity of these conditions or the relative effectiveness of current therapeutic strategies, there are fewer experimental therapies in the discovery pipeline for CMS than for many of the other neuromuscular diseases. One strategy that has been examined is exon skipping. For example, using a cell culture model, Tei, Ishii, Mitsuhashi, & Ishiura (2015) showed that AONs can be used to target an alternative splice product of CHRNA1, promoting the production of the favorable P3A‐ isoform at the expense of the non‐functional (and typically increased in patients) P3A+ isoform (Tei et al., 2015). Another pre‐clinical study demonstrated that tannic acid can promote similar exon shifting (Bian et al., 2009). An alternative strategy to exon skipping for CMS is gene therapy; one successful example demonstrating pre‐clinical feasibility of gene therapy is the successful rescue of a mouse model of DOK7‐associated CMS with intraperitoneal delivery of AAV9‐DOK7 (Arimura et al., 2014). Interestingly, the authors used the same strategy with positive effect on a mouse model LMNA muscular dystrophy (Arimura et al., 2014), a condition with previously described defects in the NMJ (Mejat et al., 2009).
4.4. Summary of CMS therapy
Given the many potential treatment options for CMS (Figure 2), a high index of suspicion must be maintained for these conditions, particularly in infants with weakness and hypotonia (where therapy may provide significant benefit), and in older individuals with antibody negative myasthenia. In addition, the fact that these therapies have variable benefits depending on genotype makes genetic subtype confirmation paramount. Moving forward, additional delineation of subtype‐specific benefit of therapies will be important, as well the continued development and translation of novel targets that address the underlying pathogenesis of the disease.
5. MUSCULAR DYSTROPHIES
Muscular dystrophies (MDs) (Figure 3) are a clinically and genetically heterogeneous group of diseases that are united by the presence of clinical features of muscle disease (primarily extremity muscle weakness and the disabilities resulting from it) and pathologic presence of dystrophic muscle (as defined by muscle biopsy features consistent with a dystrophy and/or significantly elevated serum creatine kinase levels) (Mercuri & Muntoni, 2013). Duchenne muscular dystrophy (DMD) is the most common pediatric MD, while myotonic dystrophy is the most common adult onset form (though it often presents in infancy and childhood). In general, muscular dystrophies can be separated into categories: dystrophinopathies (DMD and Becker MD), myotonic dystrophy, limb girdle muscular dystrophies, Emery‐Dreifuss muscular dystrophies, and congenital muscular dystrophies. There are some common themes related to treatment and therapy development, and also some very disease and subtype‐specific therapies. We will begin by discussing DMD, where the majority of different therapeutic strategies have been identified and/or tested, and then consider the other less common and well studied forms of muscular dystrophy.
Figure 3.
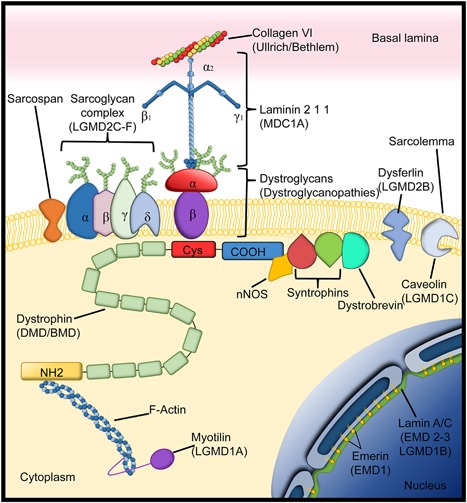
Dystrophin associated glycoprotein complex. Dystrophin associated glycoprotein complex and related proteins that help the anchoring of the sarcolemma to the basal lamina. Within brackets under the different proteins are the different diseases that result from deficiency of the respective proteins. (Limb girdle muscle dystrophies (LGDMD); Duchenne muscular dystrophy DMD; Becker muscular dystrophy (BMD); Congenital muscular dystrophy type 1A (MDC1A); Emery–Dreifuss muscular dystrophy (EMD)) (Adapted from Diseases of Muscle and the Neuromuscular Junction Part 1).
6. DUCHENNE MUSCULAR DYSTROPHY (DMD)
DMD is an X‐linked recessive disorder caused by out‐of‐frame mutations of the dystrophin (DMD) gene. These mutations result in a deficiency of the protein dystrophin. Fifty years ago, Dubowitz (1965) described physiotherapy, splints, and antibiotics to treat infections as the only treatments available for DMD. Since then, the disease trajectory of patients with DMD has steadily improved. Over the last few decades, mainly due to a combination of multidisciplinary care, improved management of cardiac and respiratory complications, and glucocorticoid therapy, DMD is no longer life‐limiting in the pediatric age range, but rather a life‐threatening disorder, with affected boys living into their 30 s. In addition to the current standards of care, a number of therapeutic strategies have been, and continue to be explored, ranging from pharmacological treatments that target disease‐modifying pathways to strategies aimed at correcting the underlying genetic mutation(s) (Table 4).
Table 4.
Summary of therapeutic strategies for Duchenne muscular dystrophy
| Category | Compound | Action | Company | Dosing and delivery | Phase of clinical trial | Results | Safety | Reference |
|---|---|---|---|---|---|---|---|---|
| Genetic based therapies | ||||||||
| Gene Therapy | Micro and mini dystrophin gene delivery | Expression of truncated but functional DMD | – | Intramuscular | Phase 1 | Pre‐clinical only | No data in humans | http://Clinical.trials.gov NCT02376816 |
| CRISPR Cas9 | Gene Editing | – | – | Mouse model | Restoration of DMD open reading frame | No data in humans | Long et al. (2016) | |
| SERCA1 delivery | Over expression of sarcoplasmic/endoplasmic reticulum Ca2+‐ATPase 1 (SERCA1) reduce intracellular Ca2+ | – | – | Mouse model | Ameliorates the model | No data in humans | Goonasekera et al. (2011) | |
| Stop codon read through | Ataluren | Reduces sensitivity of ribsosomes to premature stop codons | PTC Therapeutics | Oral | New phase 3 ongoing Approval in the European Medicines Agency and the European Commission | A second phase 3 study failed to meet its primary endpoint of stabilization of the 6 min walk. A meta‐analysis of the first and second trial identified clinical and statistical benefits in a subgroup of DMD patients | No significant concerns in healthy controls | http://Clinical.trials.gov NCT03179631 |
| Antisense therapy | Drisapersen | Exon skipping in order to reproduce an in frame mutation. Focus on exon 51, including patients with deletions of exons 45–50, 47–50, 48–50, 49–50, 50; 52, or 52–63 | GlaxoSmith‐Kline | Subcutaneous | Phase 3 | It was withdrawn for further development of the molecule (results were not compelling) | At 9 mg/kg dose, pyrexia and transient elevations in inflammatory parameters were seen | Voit et al. (2014) |
| Eteplirsen | Sarepta therapeutics | Once‐weekly IV infusions | Phase 3 | FDA approval although more clinical data is required to prove efficacy | No significant concerns | http://Clinicaltrials.gov NCT02286947 NCT02255552 NCT03218995 | ||
| Non genetic based therapies | ||||||||
| Standard of care drugs Cardio‐protective | Prednisone Predinosolone Deflazacort | It's not fully understood | – | Oral | Ongoing trials to compare daily vs alternative doses | The only drugs that help maintain muscle strength and function in children with DMD | Weight gain and retardation in vertical growth are frequent Elevated blood pressure, glycosuria, pathological fractures, gastrointestinal lesions, and adrenal crises are rare but serious | http://Clinical.trials.gov NCT01603407 |
| ACE inhibitors Beta blockers Angiotensin blockers Aldosterone agonists | Decreased in fibrosis Cardiac afterload reduction. | – | Oral | Randomized, double‐blind trial of lisinopril and losartan | Improves or preserves left ventricular systolic function and delays the progression of cardiomyopathy, No difference in outcome between Lisinopril and losartan | No significant concerns | Allen et al. (2013) | |
| Dissociative steroids | Vamorolone | Inhibits inflammatory NF‐κB signaling but reduces the activation of glucocorticoid response elements | ReveraGen BioPharma | Oral | Phase 2 | Pre‐clinical data | No results yet | http://Clinical.trials.gov NCT02760264 NCT02760277 NCT03038399 |
| Edasalonexent | Catabasis pharmaceuticals | Oral | Phase 1/2 | Pre‐clinical data | No significant concerns | http://Clinical.trials.gov NCT02439216 | ||
| Ezutromid | Up regulation of utrophin, a homologue of dystrophin | Summit | Oral | Phase2 | Pre‐clinical data | No significant concerns 1 Patient on phase one had elevation of liver enzymes | http://Clinical.trials.gov NCT02858362 | |
| Small Molecule | Poloxamer 188 (P188) | Repairing membrane damage and resealing membrane lesions | – | Subcutaneous | Mouse and dog model | Pre‐clinical data | No human data | Markham, Kernodle, Nemzek, Wilkinson, & Sigler, (2015) |
| Laminin‐111 | Repairing membrane damage and resealing membrane lesions | – | Intramuscular | Mouse model | Pre‐clinical data | No human data | Goudenege, Lamarre, Dumont, Rousseau, Frenette, Skuk, & Tremblay, (2010) | |
| Rycals | Improves FKBP binding to RyRs reducing Ca2+ leak and improving excitation and contraction coupling | – | Oral | Mouse model | Pre‐clinical data | No data in patients with DMD | Bellinger et al. (2009) | |
| Myostatin Inhibitors | PF‐06252616 | Inhibition of myostatin promotes muscle differentiation | Pfizer | Intravenous | Phase 2 | Pre‐clinical data | In a previous study myostatin inhibitor ACE‐031 (Acceleron) showed a trend toward improvement but the study was stopped after the second dose due to concerns of epistaxis and telangiectasias | http://Clinical.trials.gov NCT02907619 |
| BMS‐986089 | Bristol‐Myers Squibb | Subcutaneous | Phase 2/3 | Pre‐clinical data | http://Clinical.trials.gov NCT03039686 | |||
| Osteopontin inhibitor | Inhibition of osteopontin disrupts the TGF‐beta signalling, reducing fibrosis and inflammation | – | – | Mouse model | Ameliorates dystrophic signs | No human data | Capote et al. (2016) | |
6.1. Current standard treatments for DMD: glucocorticoids and ACE inhibitors
Bushby et al. (2010a, 2010b) published comprehensive consensus guidelines covering all aspects of managing boys with DMD, which were adopted by the National Institute for Health and Care Excellence (NICE). These care standards cover the many aspects of DMD care, include the need for multi‐disciplinary management including pulmonology, cardiology, bone health, dietary management, orthopaedic issues such as joint contractures and scoliosis, and psychosocial support.
In terms of specific therapeutic interventions, glucocorticoids (GCs) are currently the only drugs that help maintain muscle strength and function in children with DMD. It is important to note that the pharmacological effects of GCs on dystrophic muscle fibres are not fully understood and, in fact, some of the benefits of GCs seen in DMD patients have not been replicated in the dystrophin‐deficient mdx mouse model (Bauer, Straub, Blain, Bushby, & MacGowan, 2009). GCs are tolerated relatively well; however, significant weight gain and retardation in vertical growth are still observed quite frequently, while elevated blood pressure, glycosuria, pathological fractures, gastrointestinal lesions, and adrenal crises are rare but serious side effects. Daily regimes of prednisolone/prednisone or deflazacort are considered superior to intermittent dosing by many with respect to ambulation and long term benefits, and are thus the current recommended standard of care (Bushby et al., 2010a, 2010b). However, long‐term daily treatment is associated in many cases with significant side effects, and the potential efficacy of alternative dosing strategies that minimize side effects is being actively tested in an ongoing clinical trial (FOR‐DMD; NCT01603407). To address GC side effects, patients should have annual bone density scans (dual‐energy x‐ray absorptiometry) to monitor for osteoporosis, and receive supplementation with vitamin D (and potentially oral bisphosphonates) when bone density is diminished or pathologic fractures occur. Of note, there is a continued search for drugs that promote the same benefits as GCs in DMD (i.e., prolonged ambulation, reduced respiratory and cardiac disease, increased survival) but reduce/eliminate the side effects. For example, clinical trials for the “dissociative” steroid Vamorolone (ReveraGen BioPharma, Rockville, MD), studied by Hoffman and colleagues in the mdx mouse model of DMD) NCT02760264, NCT02760277, NCT03038399) (Heier et al., 2013), and edasalonexent (Hammers et al., 2016) (CAT‐1004, NCT02439216; Catabasis Pharmaceuticals, Cambridge, MA), have now commenced and some of the trials started to analyze the clinical data sets.
Most patients with DMD develop dilated cardiomyopathy, the severity and the age of onset of which vary significantly, without apparent DMD genotype‐phenotype correlation (van Westering, Betts, & Wood, 2015). Recently, a systematic review has shown that the use of various cardioprotective agents, such as ACE inhibitors, beta‐blockers, angiotension blockers, and aldosterone agonists, used in DMD patients as either preventative therapy or for treatment of established cardiomyopathy, tended to improve or preserve left ventricular systolic function, and delay the progression of cardiomyopathy (El‐Aloul et al., 2017). This review draws in part from a randomized, double‐blind trial comparing the angiotensin converting enzyme (ACE) inhibitor lisinopril to the angiotensin II receptor blocker losartan that found similar degrees of improvement in DMD cardiomyopathy after 1 year (Allen et al., 2013). These current data are not clearly pointing toward cardiac interventions at the time of diagnosis, though therapy should be considered early in the disease process as part of the standard of care, given that current imaging technologies do not adequately identify very early structural cardiac defects. Overall, while this concurs with care guidelines advocating management of DMD cardiomyopathy, the heterogeneity of the studies do not enable precise guidelines for initiation and dosing of treatments and for monitoring their impact on disease progression.
6.2. Small molecule therapeutics for DMD
Dystrophin is a large cytoskeletal linker protein that interacts with numerous other molecules and participates in the regulation of myriad cellular functions. Perhaps the primary function of dystrophin is to stabilize the myofiber membrane during muscle contraction, and loss of dystrophin leads to small breaks and tears of the sarcolemmal membrane that over time results in membrane instability, alterations in key ion gradients, and disruption of second messenger pathways. Dystrophin also participates in other cellular processes, including regulation of nitric oxide synthesis (Allen, Whitehead, & Froehner, 2016). It also has roles related to the maintenance of the muscle stem cell compartment (Dumont et al., 2015).
Many therapeutic approaches have sought to either improve membrane stability or alter second messenger signaling pathways within the muscle that are disturbed by loss of dystrophin. In fact, over the last decade, numerous re‐purposed drugs and novel compounds have been developed using pre‐clinical models (particularly the mdx mouse) and found to have various degrees of benefit in these model systems. A fraction of these drugs have been tested in clinical trials, and none, unfortunately, have been found to be significantly effective. It is beyond the scope of this review to discuss all of the small molecules that have been assessed, but a few themes of targeting disease modifying pathways are worth emphasizing.
Utrophin up‐regulation was one of the first approaches considered for DMD. Utrophin is the homologue of dystrophin with a molecular weight of 395 kDa and with similar structural organization and protein binding properties. Utrophin is ubiquitously localized at the sarcolemma in utero and is progressively replaced by dystrophin when the muscle matures. In adult muscle, utrophin is localized to the neuromuscular and myotendinous junctions. In repairing muscle as observed in DMD patients and mdx mice, utrophin expression is naturally increased due to the absence of dystrophin in order to re‐establish the continuity of muscle fibers (Janghra et al., 2016). Despite some different functional characteristics between the two proteins, studies have demonstrated that utrophin can act as an effective substitute for dystrophin in mdx muscles. Recently, an orally bioavailable small molecule, SMT C1100 (Ezutromid) (2‐arylbenzoxazole [5‐(ethyl sulfonyl)‐2‐(naphthalen‐2‐yl) benzo[d]oxazole]), has been found to promote utrophin upregulation. Studies of SMT C1100 in healthy volunteers have demonstrated that this drug is safe and well tolerated (Ricotti et al., 2016), and further clinical trials to test efficacy of this compound in DMD are currently under way (NCT02858362). Of note, other approaches aimed at utrophin upregulation are also in various stages of pre‐clinical development.
Some other small molecule drug discovery efforts are also worth highlighting. Houang et al. (2015) have been working to develop a membrane polymer that targets repair/re‐sealing of damaged sarcolemmal membranes in DMD. One candidate poloxamer, P188, has shown promise in mdx mice and in dog models of DMD, particularly in terms of repairing/improving cardiomyopathy. Another molecule based on repairing membrane damage and resealing membrane lesions is laminin‐111, a naturally occurring extracellular matrix (ECM) protein that promotes interaction between the ECM and the sarcolemmal membrane. Intramuscular injection or systemic infusion of laminin‐111 ameliorates aspects of the dystrophic phenotype in the mdx mouse. However, as with P188, laminin‐111 faces challenges related to delivery and distribution, and neither drug has yet to enter clinical trial.
Both laminin‐111 and P188 address the issue of membrane fragility. Other strategies have addressed membrane instability by promoting membrane repair, and include improving lysosome‐mediated repair capacity (e.g., agonists to a lysosomal calcium channel TRPML1) and targeting ubiquitin ligase tripartite motif‐containing (TRIM) proteins which contribute to the repair process (Cheng et al., 2014; Weisleder et al., 2012). Several other strategies are aimed at normalizing dysregulated intracellular pathways. For example, there is evidence in DMD of chronic disturbance of calcium regulation, as well as post‐translational changes in ryanodine receptors (RyR1 and RyR2) that alter excitation contraction coupling and calcium homeostasis. Genetic upregulation of sarcoplasmic/endoplasmic reticulum calcium ATPase 1 (SERCA1) rescues the mdx mouse phenotype (Goonasekera et al., 2011), as do a class of drugs (Rycals) that improve FK506 binding protein (FKBP) binding to RyRs (Bellinger et al., 2009). Another important signaling cascade that has been shown to play a role in muscle regeneration and formation of tissue fibrosis is the family of canonical and non‐canonical TGF‐beta signaling, including myostatin and osteopontin (Amthor & Hoogaars, 2012; Capote et al., 2016; Vetrone et al., 2009), and therapies that modulate TGF‐beta signaling, such as myostatin inhibition, are currently the subject of clinical trials for DMD (NCT 02907619, NCT 03039686). Other critical downstream pathways that have been targeted include NF‐kB and anti‐inflammatory signaling as well as compounds that target mitochondrial function and histone deacetylase (HDAC) inhibitors (Consalvi, Saccone, & Mozzetta, 2014).
6.3. Exon skipping using antisense oligonucleotides (AONs)
Most mutations in the DMD gene disrupt the open reading frame and result in incomplete translation of the dystrophin protein. Exon skipping provides an opportunity for mutation repair that targets the pre‐mRNA transcript, introducing alternative splice sites that result in skipping one or more targeted exons leading to restoration of the dystrophin reading frame. AONs are synthetically modified strands of nucleic acids, typically 20–30 nucleotides in length, composed of complementary sequences to dystrophin pre‐mRNA. The first evidence that exon skipping may indeed be a therapeutic avenue to pursue in DMD was derived from the observation of small clusters of dystrophin positive fibers called “revertant fibers” in muscle biopsies. These fibers express dystrophin due to intrinsic alternative splicing but are not sufficient to improve the clinical phenotype (Wilton, Dye, Blechynden, & Laing, 1997). In the past few years, a number of synthetically designed AONs have been developed to simulate the naturally occurring alternative splicing, with the overall goal of achieving this in enough fibers to demonstrate clinical benefit for the large group (80%) of DMD patients with mutations amenable to exon skipping.
Current clinical trials have been designed to target the hotspot region for deletions mainly focusing on exon 51, which would be applicable to 13% of DMD patients including those with deletions of exons 45–50, 47–50, 48–50, 49–50, 50; 52, or 52–63 (Wilton, Veedu, & Fletcher, 2015). A number of successful pre‐clinical studies have shown efficacy of AONs in mice and dogs using 2′O‐methyl‐ribo‐oligonucleoside‐phosphorothioate (2′OMePS) and phosphorodiamidate morpholino oligomers (PMOs) (Wilton et al., 2015). These two oligomers share a common mechanism of action but differ in their biochemical structure, stability against endonucleases, and toxicity profiles.
Two products, a 2′OMePS oligomer (drisapersen, Prosensa Therapeutics and Biomarin) and a PMO (eteplirsen; Sarepta Therapeutics) targeting exon 51 have been tested systemically in a series of clinical trials. Drisapersen has been tested in three double‐blind, placebo‐controlled trials (DEMAND studies). In one 48‐week study, drisapersen was administered subcutaneously at a dose of 6 mg/kg weekly to 18 patients (continuous regimen); another group of 17 patients received nine doses over 6 weeks followed by a 4‐week break (intermittent regimen); and one group of 18 patients was given placebo. After 24 weeks but not at 48 weeks, the continuous regimen resulted in a statistically significant increase in walking distance (approximately 35 m) compared with placebo. Proteinuria was a common adverse effect (Voit et al., 2014). These data were not sufficiently compelling and currently drisapersen has been withdrawn from further development. Eteplirsen has been extensively studied in 12 boys with DMD on GCs in a double‐blind trial that divided them into three groups. One group received 30 mg/kg intravenous eteplirsen weekly, another 50 mg/kg weekly and a third group placebo. The patients had up to three muscle biopsies throughout the 48‐week trial period. The boys in the eteplirsen groups showed increased dystrophin production of 40–50% on biopsy and were able to walk ∼67 m further than control after 48 weeks of treatment. The drug was well tolerated (Mendell et al., 2016). At the time of writing this manuscript, the FDA announced accelerated approval of eteplirsen for the clinical use in boys with DMD; this approval is contingent on successful completion of additional trials of the drug and other post marketing commitments (Dowling, 2016).
6.4. Stop‐codon read‐through: Mutation suppression
About 15% of DMD patients have a premature stop codon that leads to nonsense mediated decay of DMD mRNA and/or premature cessation of protein translation resulting in a truncated and non‐functional protein. PTC124 (ataluren, now Tranlsarna™, PTC Therapeutics (South Plainfield, NJ)) is a polycyclic organic molecule that reduces the sensitivity of ribsosomes toward premature stop codons, resulting in a so‐called “stop‐codon read‐through” (Peltz, Morsy, Welch, & Jacobson, 2013). However, there has been some controversy about whether this proposed mechanism of action of ataluren is indeed accurate (McElroy et al., 2013). In 2014, the drug was approved under the brand name Translarna by the European Medicines Agency and the European Commission for treatment of DMD patients with nonsense mutations (Ryan, 2014). Bushby et al. (2014) conducted a double‐blind, randomised, placebo‐controlled multicentre trial of oral ataluren involving 173 patients with DMD aged 5–20 years (Bushby et al., 2014). After 48 weeks of treatment, study subjects on 40 mg/kg/day ataluren but not those on 80 mg/kg/day showed a slower decline in their walking distance (decline of only 13 m) compared with the placebo group (decline of 44 m). No serious adverse events were observed. A second phase III trial of ataluren was recently completed and failed to meet its primary endpoint of stabilization of the 6 min walk distance, but it again showed a non‐statistically significant benefit favoring ataluren on all outcome measures. A meta‐analysis of the first and second trial identified both clinical and statistical benefits of the drug and compelling benefits in a group of boys in a specific ambulatory decline phase (McDonald et al., 2017). Based on these suggestive data, the company has initiated regulatory pathways in multiple countries.
6.5. Gene therapy
Gene therapy holds great promise as a potential treatment for most DMD patients by delivering a functional DMD gene to skeletal and cardiac muscle and thus restoring dystrophin protein. Currently, recombinant adeno‐associated virus (rAAV) is the preferred vehicle for delivery due to the persistence of injected AAV in striated muscles and lack of pathogenicity. Given the large size of the dystrophin gene however, reduced‐size dystrophins (mini‐ or micro‐dystrophin, distinguished by total size and inclusion/exclusion of specific DMD subdomains, especially as it relates to nNOS functional binding sites) have been developed stemming from the observations in BMD patients that some truncated dystrophin leads to a mild phenotype (McGreevy, Hakim, McIntosh, & Duan, 2015).
AAV‐mediated, micro‐dystrophin gene therapy is currently at the cutting edge of DMD gene‐replacement therapy. Initially, a number of groups showed that local injection of mini‐dystrophin can protect limb muscles and the heart in mdx mice despite the absence of ∼70% of the coding sequence (Harper et al., 2002; Wang, Li, & Xiao, 2000; Yoshimura et al., 2004; Yue et al., 2003). A number of studies have now shown that newly developed AAV serotypes 6, 8, and 9 vectors (Gao et al., 2002; Rutledge, Halbert, & Russell, 1998) achieve widespread whole‐body muscle gene transfer in rodent and dog models of DMD (Bostick, Ghosh, Yue, Long, & Duan, 2007; Gregorevic et al., 2004; Hakim et al., 2014; Kornegay et al., 2010; Lai, Li, Yue, & Duan, 2008; Pacak et al., 2006; Shin et al., 2013). Most of these studies demonstrate that micro‐dystrophin gene therapy reduces creatine kinase (CK) levels and significantly ameliorates the histological and physiological signs of muscular dystrophy in these models. A number of efforts are currently under way to develop an AAV‐9 based microdystrophin gene product to be used for clinical trials in boys with DMD, and an encouraging phase I/II trial of limited intramuscular injection has been completed (NCT02376816).
An alternative approach to reframe the DMD gene is offered by the discovery of clustered regularly‐interspaced short palindromic repeats (CRISPR) technology, in which an endonuclease called Cas9 can cleave the genome in a precise manner when coupled with a strand of guide RNA (gRNA) (Cong et al., 2013). The cleaved DNA will be rejoined via either a non‐homologous end joining (NHEJ) mechanism facilitated by the cells’ own repair machinery, or homologous‐directed repair (HDR) if a repair template is provided. While the latter is extremely inefficient in post‐mitotic cells such as skeletal muscle, NHEJ appears to be achieved in an efficient manner. A number of in vitro studies have shown that the CRISPR/Cas9 technology can be employed for a number of different mutations including patients with exon or multi‐exon duplications that comprise approximately 12–15% of the DMD mutation spectrum (Ousterout et al., 2015; Wojtal et al., 2016). Given the head‐to‐tail orientation of most of the duplicated region, one gRNA can be sufficient to remove the mutation and lead to restoration of full‐length dystrophin protein, which would be beneficial given the packaging limitation of an AAV vector. More recently, convincing evidence for the therapeutic potential of CRISPR/Cas9‐mediated restoration of DMD open reading frame in vivo has been demonstrated in mdx mice by three independent groups (Long et al., 2016; Nelson et al., 2016; Tabebordbar et al., 2016). Despite these significant findings, a number of hurdles will still have to be overcome to move these therapies into clinic. The versatility of the technology allows editing to be implemented in virtually all DMD‐causing mutations, providing tremendous potential for individualized treatment for DMD patients. One important issue that requires thorough evaluation with regulatory bodies and industry is whether extensive pre‐clinical assessment would be necessary for each guide RNA designed. Creating animal models for each individual mutation to perform pre‐clinical assessment is costly, time consuming, and thus largely impractical. Patient‐derived cells, on the other hand, are easy to obtain and may be sufficient to assess the efficacy and off‐target activities. In addition, development of constructs with tissue‐specific promoters would be advantageous in reducing off‐target effects, as well as minimizing/eliminating concerns of targeting in the germline/embryonic stage. Furthermore, given that a number of these therapies will have to be developed on an individual basis, novel cost‐models will need to be developed in collaboration of industry and academia to provide and develop these treatments.
In all, the therapeutic landscape for DMD has changed dramatically over the last decade and the overall outlook is incredibly exciting. However, DMD is a complex disease, which is often not appreciated enough. Thus, as the field is developing therapies using small molecules and gene correction strategies, it will be critical to begin a process of evaluating combinatorial therapies that work synergistically to improve the clinical symptoms and overall quality of life of DMD.
7. CONGENITAL MUSCULAR DYSTROPHIES
Congenital muscular dystrophies (CMDs) are defined by onset of symptoms in the first 12–18 months of life and the presence of diagnostic features of a muscular dystrophy (dystrophic biopsy and/or elevated CK) (Gilbreath, Castro, & Iannaccone, 2014). There are three major genetic subclasses of CMDs (Figure 3): LAMA2‐related (or merosin‐deficient CMD or MDC1A), collagen‐VI deficient (or Ullrich CMD and Bethlem myopathy), and dystroglycanopathies (due to mutations in dystroglycan or the numerous gene products that glycosylate it). While patients with mutations in these subtypes typically present in infancy and with dystrophic features, they can also present at older ages and with a range of signs and symptoms (Bonnemann et al, 2014). Additional CMD subtypes include LMNA‐related CMD (overlapping with other LMNA disorders such as Emery–Dreifuss muscular dystrophy), Marinesco–Sjogren syndrome (due to SIL1 mutations) and rigid spine muscular dystrophy (overlapping with congenital myopathies due to SEPN1, RYR1, and MYH7 mutations) (Mercuri & Muntoni, 2012). This section will deal primarily with the three major CMD groups, as the other conditions will be discussed in separate sections (Table 5).
Table 5.
Summary of therapeutic strategies for congenital muscular dystrophies (CMD)
| Category | Compound | CMD subtype | Action | Company | Dosing and delivery | Phase of clinical trial | Results | Safety | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Genetic based therapies | |||||||||
| Membrane stabilizer | Mini‐ Agrin and multi linker | MDC1A | Improves cell‐matrix adhesion | – | – | Mouse model | Pre‐clinical data | No human data available | Meimen et al. (2011) |
| Gene modulation | Small interfering RNA | UCMD | allele specific silencing | – | – | In vitro | Pre‐clinical data | No human data available | Bolduc et al. (2014), Noguchi et al. (2014) |
| Splicing | AON | Fukuyama muscular dystrophy | Interferes with splicing to prevent pathogenic exon trapping | – | – | In vitro | Successful rescue of the model | No in vivo data available | Taniguchi‐Ikeda et al. (2011) |
| Antisense theraphy | AON | MDC1A | Exon skipping | – | – | Mouse model | Successful rescue of the model | No human data available | Aoki et al. (2013) |
| Gene overexpression | LARGE | LARGE FKRP POMGNT1 | Improves dystroglycan glycosylation | – | – | Mice models | Successful rescue of models. | Worsening of models of FCMD, and FKRP related CMD | Yu et al. (2013) |
| Gene editing | CRISPR Cas9 | MDC1A | Correction of pathogenic splice site | – | – | Mouse model | Successful rescue of the model | No human data available | Kemaladewi et al. (2017) |
| Non genetic based therapies | |||||||||
| Anti‐fibrotic | Losartan | MDC1A | Inhibition of TGF‐beta pathway which reduces fibrosis | – | Oral | Mouse model | Pre‐clinical data | No results in patients with MDC1A | Elbaz et al. (2012), Meinen et al. (2012) |
| Anti‐apoptotic | Cyclosporin A | UCMD | Corrects mitochondrial dysfunction, increased muscle regeneration, and decreased the number of apoptotic nuclei | – | Oral | Cohort | Increased in strength No changes in motor funcionts and respiratory function kept deteriorating. Decreased number of apoptotic nuclei | Renal dysfunction, hypertension, headache, gastrointes‐tinal disturbances, and hirsutism hypertrichosis | Merlini et al. (2008) |
| Omigapil | UCMD MDC1A | Inhibition of GAPDH acting as anti‐apoptotic agent | Santhera | Oral | Phase 1 | Pre‐clinical Data | No available data yet | http://Clinical.trials.gov NCT01805024 | |
| Membrane stabilizer | Laminin‐111 | MDC1A | Repairing membrane damage and resealing membrane lesions | – | Intramuscular | Mouse model | Pre‐clinical data | No human data | Van Ry, Minogue, Hodges, & Burkin, (2014) |
| Non pharmaco‐logical | Low protein diet | UCMD | Promotes autophagy | – | – | Phase 2 | Pre‐clinical data | No data available | http://Clinical.trials.gov NCT01438788 |
| Hyperinsuffla‐tion therapy | UCMD MDC1A | Slows the loss of breathing function | – | – | Interven‐tional study | Complete no results yet | Complete no results yet | http://Clinical.trials.gov NCT01836627 | |
At present, there are no disease specific therapies for CMDs (Kang et al., 2015). Furthermore, there is only a single active clinical trial examining a potential pharmacologic intervention for any of the CMD subtypes. This is a phase 1 trial examining the safety of omigapil (NCT01805024), a glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) inhibitor that acts primarily as an anti‐apoptotic agent. This is being tested in patients with either Ullrich CMD (i.e., patients with COL6A1, COL6A2, or COL6A3 mutations) or with MDC1A, reflecting the fact that increased apoptosis has been observed in muscle from patients and mouse models for both of these CMD subtypes (Irwin et al., 2003; Miller & Girgenrath, 2006). In addition, omigapil has been shown to improve strength and histopathologic changes in mouse models of MDC1A (Erb et al., 2009; Yu, Sali, et al., 2013).
Several other potential therapeutics are in pre‐clinical development. These can be roughly broken down into treatments aimed at improving cell‐matrix adhesion, treatments targeted at cellular salvage pathways including apopotosis, autophagy, and ubiquitin‐proteosome system, and gene‐based therapies (Collins & Bonnemann, 2010). For LAMA2‐related CMD (i.e., MDC1A), the loss of muscle‐matrix interaction is at the crux of the disease, and thus molecules that re‐establish this interaction have high therapeutic potential. Rooney, Knapp, Hodges, Wuebbles, and Burkin (2012) have been working to develop therapy with laminin 111, a protein similar in function to laminin 211 (a subunit of which is encoded by LAMA2). Intramuscular administration of laminin 111 to Lama2 deficient mice reduces apoptosis, increases fiber size, and improves muscle strength. Another “pro‐adhesion” approach has been developed by Ruegg and colleagues using various “mini‐agrin” formulations (Meinen et al., 2011; Moll et al., 2001). Agrin is an extracellular matrix protein that can serve as a bridge between the ECM and muscle membrane receptors, and mini‐agrin has been shown to ameliorate symptomatology in MDC1A mouse models. Most recently, Reinhard et al. (2017) have successfully tested a novel combinatorial linker molecule approach that results in dramatic improvements in the phenotype and survival of an MDC1A mouse model, reinforcing the potential promise of the linker strategy. In terms of clinical translatability, however, key questions for these strategies center on systemic delivery, scalability, and the potential for adverse immune response.
Several studies have been done in both MDC1A and UCMD mouse models looking at the potential efficacy of modulating various degradation pathways, including inhibiting apoptosis (as with omigapil) or altering autophagy. The effect size is modest in the mouse with these interventions, though it is possible that such changes may result in meaningful benefits to patients. CyclosporinA, which likely has both anti‐apopototic and pro‐autophagic flux properties, has shown efficacy in UCMD mouse and zebrafish models (Irwin et al., 2003; Telfer, Busta, Bonnemann, Feldman, & Dowling, 2010), and has been shown to reduce apoptosis in a pilot trial in patients with UCMD (Merlini et al., 2008). Interestingly, an open label phase II trial of low protein diet (which promotes autophagy and improved aspects of the UCMD mouse phenotype (Grumati et al., 2010)) in patients with UCMD was recently completed. The results of this trial have yet to be disclosed, though it will be interesting to see if (a) autophagy was altered by this approach and (b) if that correlated with any clinical improvement.
One additional therapeutic strategy to mention is anti‐fibrotic therapy. Fibrosis is a significant component of most dystrophic biopsies, and thus reducing or eliminating it has been considered not just for CMDs but also for DMD and the LGMDs. The TGF‐beta pathway appears critical for the development of the fibrotic process; thus pharmacologic strategies that block or inhibit it have been considered for various muscular dystrophies. For the CMDs, losartan, an angiotensin II inhibitor, has been shown to reduce fibrosis and ameliorate aspects of disease in the MDC1A mouse model (Elbaz et al., 2012; Meinen, Lin, & Ruegg, 2012). It has not been examined in models of UCMD or dystroglycanopathies. The consideration of clinical translation of this drug is still under active discussion, and the question of whether a placebo controlled trial versus an open label/case controlled analysis for a repurposed drug such as losartan is being debated.
There are also gene‐based therapies in pre‐clinical development for several CMDs. Two studies have demonstrated restoration of normal collagen VI fibril production using oligonucleotide based allele specific silencing in patient derived cell lines with dominant mutations associated with UCMD (Bolduc, Zou, Ko, & Bonnemann, 2014; Noguchi, Ogawa, Kawahara, Malicdan, & Nishino, 2014). Antisense oligonucleotide based exon skipping has also been considered for MDC1A, in which removing a stop codon and producing a truncated protein product promotes merosin re‐expression and mild improvement in a mouse model of MDC1A (Aoki et al., 2013). The use of antisense oligonucleotides to alter splicing has also been examined for Fukuyama muscular dystrophy, a dystroglycanopathy subtype caused by a transposon insertion mutation in the 3′UTR of fukutin, and shown to ameliorate phenotypic features of the mouse model of the disease and restore protein expression in patient cells (Taniguchi‐Ikeda et al., 2011).
Importantly, AAV based gene replacement therapy is not feasible for MDC1A or UCMD because of the large size of these genes. However, it is being examined for some of the dystroglycanopathy subtypes, and after showing success in a mouse model (Xu et al., 2013)) clinical trials are planned for FKRP‐related muscular dystrophies. In addition, overexpression of the glycosyltransferase LARGE, mutations in which are a rare cause of congenital muscular dystrophy, has been tested in mouse models of dystroglycanopathy. LARGE overexpression (delivered either transgenically or through viral vector) can restore glycosylation and improve pathology in several different mouse models, including those due to Large, Fkrp, and Pomgnt1 mutations (Yu, He et al., 2013). However, it has also been shown that LARGE overexpression can aggravate the dystrophic phenotype, specifically in mouse models of MDC1A, FCMD, and Fkrp related CMD (Saito et al., 2014; Whitmore et al., 2014). Resolution of this discrepancy (which may relate to a requirement for Fukutin expression (Ohtsuka et al., 2015)) is key to considering whether (or for which subtypes) such a therapy could be advanced to patients with dystroglycanopathies.
More recently, CRISPR/Cas9 has been employed as a therapeutic strategy in MDC1A with a specific emphasis on splice site mutations (which account for up to 40% of mutations in MDC1A patients). Previous studies from other diseases have shown correction of splice site mutations utilizing the homology‐directed repair (HDR) pathway, but HDR is extremely inefficient in post‐mitotic tissues such as skeletal muscle (Hsu, Lander, & Zhang, 2014). In a new study by Kemaladewi et al. (2017), CRISPR/Cas9 was used to correct a pathogenic splice site mutation using the non‐homologous end‐joining (NHEJ) repair pathway. Specifically, a splice site mutation that causes exclusion of exon two in the Lama2 gene and truncation of Lama2 protein in the dy2J/dy2J mouse model of MDC1A has been corrected using AAV9 delivery of Staphylococcus aureus Cas9 and two guide RNAs. These guides simultaneously excised the intronic region containing the mutation, thus creating a functional splice donor site through NHEJ. This strategy led to successful inclusion of exon 2 in the Lama2 transcript, restoration of full‐length Laminin‐α2 protein and significant improvement in muscle histopathology, strength, and function.
It should be noted that non‐pharmacologic interventions have an important place as disease‐specific therapeutic modalities for CMDs (Kang et al., 2015). Most or all patients receive physiotherapy, though its impact on the natural history has not been rigorously studied. There is an ongoing clinical trial for UCMD and MDC1A for a pulmonary intervention called hyperinsufflation (NCT01836627). The concept behind this intervention is that pulmonary function in CMDs becomes progressively worse due in part to increasing chest wall stiffness and diminished compliance, and that daily use of cough assist can counteract these changes. The results of this trial should be available in the next year or two.
One key challenge with CMDs is that the primary disease mechanism (i.e., altered cell‐matrix adhesion due either to mutation of components of the ECM or to ECM receptors on the sarcolemmal membrane) is difficult to overcome. This is particularly the case for dystroglycanopathies, where the ideal therapy would restore the normal glycosylation pattern to the dystroglycan protein, though perhaps strategies like LARGE overexpression will surmount this barrier. Another important challenge facing not only CMDs but also other rare congenital muscle conditions (CMS and congenital myopathies) is the paucity of natural history data and clinical trial outcome measures. These conditions have lagged greatly behind in this arena as compared to other rare diseases (including DM1, DMD, and SMA), though there are concerted efforts underway to remedy this (Meilleur et al., 2015). The importance of comprehensive, longitudinal natural history data cannot be underestimated, as it informs on current clinical care, disease specific prognostication, and clinical trial development. A recent example of the potential power of this type of information is a study from Foley et al. (2013), which examined pulmonary function longitudinally in patients with Collagen VI muscular dystrophies. They found a reproducible, age related decline in pulmonary status, data that has helped advance clinical care and that identifies an important outcome measure for future clinical trials.
8. LIMB GIRDLE MUSCULAR DYSTROPHIES
LGMDs are defined as conditions with presentation after the first year of life, limb girdle pattern of weakness, elevated CK levels, and dystrophic biopsies, and usually progressive weakness (Narayanaswami et al., 2014). LGMDs are quite clinically heterogeneous and genetically diverse (Magri et al., 2016). In pediatrics, the major subtypes are the sarcoglycanopathies (which present quite similar to DMD and are due to mutations in the genes that encode the sarcoglycan membrane protein complex), the dystroglycanopathies (which were presented in the CMD section), and the calpainopathies (due to Calpain3 mutations) (Straub & Bushby, 2006) (Figure 3). Other genetic causes are rarer, though important to mention as they are common in adult onset LGMDs. In particular, these include dysferlinopathies and ANO5 related LGMD (Table 6).
Table 6.
Summary of therapeutic strategies for LGMD
| Category | Compound | LGMD Subtype | Action | Company | Dosing and delivery | Phase of clinical trial | Results | Safety | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Genetic based therapies | |||||||||
| Gene therapy | rAAV1.tMCK.hSGCA | LGMD2D | Gene delivery | – | Intramuscular | Pilot clinical trial | Restoration of sarcoclycan complex | No adverse events reported | Mendell et al. (2009) |
| MG53 delivery | LGMD2F | Increase membrane repair | – | Hamster model | Ameliorates the model | No data in humans | He et al. (2012) | ||
| Mono‐clonal antibodies | PF‐06252616 | LGMD2I | Inhibition of myostatin promotes muscle differentiation | Pfizer | Intravenous | Phase 1/2 | Pre‐clinical data | No data in humans yet | http://Clinical.trials.gov NCT02841267 |
| Antisense therapy | AOS | Sarcoglycano‐pathies | Exon skipping of exons 4 and 7 of the gamma sarcoglycan produce a functional truncated protein | – | Subcutaneous | In vitro Drosophila model Mouse model | Pre‐clinical data | No results in humans | Gao et al. (2015) |
| Non genetic based therapies | |||||||||
| Steroids | Prednisone Deflazacort | Sarcoglycanopathy Dystroglycanopathy | It's not fully understood | – | Oral | Mouse model Non‐systematic cohort studies | Contradictory results with the mouse model No current recommendation to use in LGMDs | Weight gain and retardation in vertical growth are frequent Elevated blood pressure, glycosuria, pathological fractures, gastrointestinal lesions, and adrenal crises are rare but serious | |
| Anti‐fibrotic | ACE inhibitors | Sarcoglycanopathies LGMD2I LGMD2I LGM | Decreased in fibrosis Cardiac afterload reduction. | – | Oral | Phase 2/3 | Pre‐clinical data available | No significant concerns | http://Clinical.trials.gov NCT01126697 |
Perhaps the biggest therapeutic conundrum in pediatric muscle disease is whether or not patients with LGMD would respond like DMD patients to chronic glucocorticoid therapy. Prednisone has been tried on a non‐systematic basis in sarcoglycanopathies and dystroglycanopathies, with some reports of positive response (Connolly, Pestronk, Mehta, & Al‐Lozi, 1998; Godfrey et al., 2006). On the other hand, studies in mouse models of sarcoglycanopathy suggest steroids could be deleterious, particularly to cardiac function (Bauer, Macgowan, Blain, Bushby, & Straub, 2008). A randomized trial is planned for LGMD2I (due to FKRP mutations), and it will be of great interest to see if prednisone impacts disease in this context. At present, though, there is no recommendation to consider steroids in LGMD patients.
Other therapeutic strategies mirror those considered for DMD and for the congenital muscular dystrophies, reflecting the overlap in pathogenic mechanisms with DMD and the shared spectrum of genetic causes with CMDs. For example, aberrant TGF‐beta signaling and fibrosis have been described in models of both DMD and sarcoglycanopathy, and modification of the TGF pathway (either genetically or pharmacologically with ACE inhibitors) has been demonstrated to ameliorate the dystrophic phenotype (Accornero et al., 2014; Goldstein et al., 2014). Facilitating membrane repair, as done for example with AAV delivered MG53, has also shown promise in pre‐clinical models of LGMD (e.g., testing in a hamster model with mutation of delta sarcoglycan) (He et al., 2012). However, few of these strategies have been brought into the clinical arena for testing. There is an ongoing trial focused on cardiac outcomes of lisinopril + co‐enzyme Q10 for recessive LGMDs (sarcoglycanopathies and LGMD2I) that is part of a large study that includes Duchenne/Becker muscular dystrophy (NCT01126697). There is also a phase I/II study of a humanized anti‐myostatin in ambulant patients with LGMD2I (Pfizer, NCT02841267).
As with many neuromuscular diseases, gene therapy holds great promise for certain LGMD subtypes (Bengtsson, Seto, Hall, Chamberlain, & Odom, 2016). A pilot clinical trial for LGMD2D has been performed, with local intramuscular injection of the gene vector showing restoration of not only β‐sarcoglycan but also the whole sarcoglycan complex (Mendell et al., 2009). A larger clinical trial is planned, and development is underway for AAV based gene therapy for other LGMD subtypes as well, including (as mentioned in the CMD section) FKRP/LGMD2I and Calpainopathy (LGMD2A) (Bartoli et al., 2006). There is also consideration of non‐replacement genetic strategies. Gao et al. (2015) recently demonstrated that exon exclusion of 4 of the 7 exons of gamma sarcoglycan produces a truncated protein with retained functionality, thus opening the possibility of antisense mediated exon skipping for patients with mutations in this subtype of LGMD.
9. EMERY‐DREIFUSS MUSCULAR DYSTROPHIES
Emery–Dreifuss muscular dystrophy (EDMD) is associated with scapulo‐peroneal weakness that ranges in severity and progression and can be quite severe and life limiting in many cases (Bonne, Leturcq, & Ben Yaou, 1993). The most common causes of EDMD are mutations in either LMNA or emerin (EMD) (Bonne, 2014), though there are several other rare causes as well as significant clinical overlap with certain genes more commonly associated with either congenital muscular dystrophies (such as COL6) or congenital myopathies (ACTA1, MYH7, and RYR1). In addition to EDMD, there is a very broad spectrum of disease associated with LMNA mutations, including a congenital presentation, classic EDMD, limb girdle muscular dystrophy, and non muscle diseases such as CMT, lipodystrophy and (with one specific mutation) progeria (Worman & Bonne, 2007). LMNA‐associated CMD is a particularly severe disease, with relatively rapid progression, severe contractures, and early death (Quijano‐Roy et al., 2008) (Table 7).
Table 7.
Summary of therapeutic strategies for Emery Dreifuss muscular dystrophy (EDMD)
| Category | Compound | Action | Company | Dosing and delivery | Phase of clinical trial | Results | Safety | Reference |
|---|---|---|---|---|---|---|---|---|
| Genetic based therapies | ||||||||
| Antisense therapy | AON | Exon skipping (skipping of exon 3 or 5 of LMNA to remove dominant mutation | – | – | In vitro | Preclinical data | No data in humans | Scharner et al. (2015) |
| Non genetic based therapies | ||||||||
| Steroids | Prednisone Deflazacort | It's not fully understood | – | Oral | Open label observational study | No Data yet | Weight gain and retardation in vertical growth are frequent Elevated blood pressure, glycosuria, pathological fractures, gastrointestinal lesions, and adrenal crises are rare but serious | ‐ |
| Cardio‐protective | ACE inhibitors | Decreased in fibrosis | – | Oral | Open label observational study | Improve left ventricular function | No significant concerns | Muchir et al. (2014) |
| Early defibrillator placement | Preventive | – | – | Prospective study | Prophylactic implantable cardioverter‐defibrillator is an effective treatment | – | Anselme et al. (2013) | |
| Small molecule | Temsirolimus | Inhibition of mTOR signaling | – | Intraperitoneal injections | Mouse model | Ameliorates cardiomyopathy | No data in human patients | Choi et al. (2012) |
| Rapamycin | Increase autophagy | – | Intraperitoneal injections | Mouse model | Rescue cardiac and muscle function | No data in human patients | Ramos et al. (2012) | |
Most of the effort related to therapy development for EDMD has centered on LMNA. Several of the strategies that have been considered for DMD have also been examined in pre‐clinical models of LMNA mutations. For example, ACE inhibition (which targets fibrosis), either alone or in combination with MEK1/2 inhibition, has been shown to improve left ventricular function in Lmna knock in mice (Muchir, Wu, Sera, Homma, & Worman, 2014). Activation of autophagy can also improve cardiomyopathy in this same mouse model (Choi et al., 2012), and inhibition of mTOR signaling with rapamycin improves both skeletal and cardiac myopathy in Lmna knockout mice (Ramos et al., 2012). However, none of these therapies have yet been tested in patients with LMNA mutations. One treatment strategy used in DMD that is now being examined via an open label observational study in LMNA patients is glucocorticoid therapy (i.e., prednisone or deflazacort); this is based in part on the high degree of inflammation seen in biopsies in many patients with LMNA mutations (Komaki et al., 2011).
One area where a significant therapeutic advance has been made is cardiac management in EDMD. Patients are at risk of developing fatal arrhythmias, and thus early monitoring and intervention are key means of prevention in this disease. There has been a prospective trial of early defibrillator placement in patients with EDMD, and it showed that defibrillators can reduce pathologic arrhythmias and prevent sudden death (Anselme et al., 2013). As cardiomyopathy can also occur (either along with arrhythmias or in isolation), close assessment (using by echocardiography) and intervention using standard cardiac management strategies for heart failure are indicated.
As with many of the myopathies and muscular dystrophies, gene based therapeutic strategies are being considered for Emery‐Dreifuss muscular dystrophy as well. Recently, it was shown in cell culture that exon skipping of either exon 3 or 5 of LMNA can produce a functional truncated protein, and thus could be consider for patients with dominant mutations in those exons (Scharner, Figeac, Ellis, & Zammit, 2015). Other untested strategies that could potentially be affective for EDMD are AAV based gene replacement of emerin and allele specific silencing of dominant LMNA mutations.
10. MYOTONIC DYSTROPHY
Myotonic dystrophy is a dominantly inherited multiple system disease caused by triplet repeat expansion mutations that is separated into two subtypes based on the location of the mutation. Type 2 myotonic dystrophy (or DM2) manifests purely in adulthood and will not be covered in this review, nor will the non‐dystrophic myotonic conditions such as myotonia congenita, paramyotonia congenita and issacs disease. Type 1 myotonic dystrophy (or DM1) is caused by a CTG triplet repeat expansion mutation in the 3′ UTR of the DMPK gene locus. It is commonly characterized by early adult onset of distal extremity and facial muscle weakness plus myotonia, but can present at all ages with variable severity and multi systemic signs and symptoms. Some of the non‐neuromuscular symptoms include: cognitive impairment, cataract formation, premature balding, insulin resistance, infertility, cardiac arrhythmia, dysphagia, and hypersomnolence. Congenital myotonic dystrophy (cDM), resulting from the extreme expansion of the triplet repeat when inherited from an affected mother (i.e., genetic anticipation) is the more dramatic presentation of myotonic dystrophy. In this scenario the affected neonate has hypotonia (floppy infant syndrome), weakness, feeding difficulties, and mechanical respiratory failure, often requiring intubation and ventilation immediately after birth (Table 8).
Table 8.
Summary of therapeutic strategies for myotonic dystrophy type I (DM1)
| Category | Compound | Action | Company | Dosing and delivery | Phase of clinical trial | Results | Safety | Reference |
|---|---|---|---|---|---|---|---|---|
| Genetic based therapies | ||||||||
| Gene Therapy | VAL‐0411 | Correct RNA splicing by using a decoy protein that will bind to the toxic RNA | Valerion | – | In vitro Mouse model | Correction of mRNA splicing | No results in humans yet | |
| Artificial site specific RNA endonucleases | Disrupt toxic RNA | – | – | In vitro studies | Reverse the missplicing of many genes affected in DM1 patients | No results in humans yet | Zhang et al. (2014) | |
| RNA splicing manipulation | IONIS‐DMPKRx | Anti‐sense oligonucleotide | IONIS | Subcutaneous | Phase 1/2 | Drug measured in biopsy tissue does not achieved the necessary therapeutic levels | The program was discontinued | http://Clinicaltrials.gov NCT02312011 |
| Non genetic based therapies | ||||||||
| Antimyotonic | Mexiletine | Antagonist of the Sodium channel reducing membrane depolarization | – | Oral | Randomized, cross over designed clinical trial | Reduce frequency and severity of myotonia. It is routinely used | Not associated with any serious adverse events | Logigian et al. (2010) |
| Daytime sleepiness treatment | Methyl‐phenidate | Interferes with the metabolism of dopamine + noradrenaline | – | Oral | Small, randomized cross‐over trial | Improves daytime fatigue | Loss of appetite, nausea, and palpitations were the most common adverse events | Puymirat et al. (2012) |
The pathogenesis of DM1 is related to the effects of accumulated mutant mRNA in the nucleus of cells. In accordance with this theory, animal models replicate human disease when either long CTG repeat mutations or high levels of expressed CTG expansions accumulate in the nucleus (Mankodi et al., 2000). This toxic mRNA has an impact on a class of nuclear proteins that regulate splicing of multiple proteins. The Muscleblind (MBNL) family and CELF CUG‐binding protein 1 (CELF/CUG‐BP1), are two such proteins implicated in the pathophysiology of DM1. The splicing differences induced by a reduction in MNBL are directed toward an arrest of the normal transition to adult isoforms of a set of MBNL sensitive proteins (Lin et al., 2006), whereas elevated levels of CELF/CUG‐BP1 causes RNA splicing changes that appear to be equally responsible for the DM1 phenotype (Faustino & Cooper, 2003). A fine balance between the roles of MNBL and CELF/CUG‐BP1 appears to exist. Under expression of MNBL or over expression of CELF/CUG‐BP1 in mouse models, or a combination of both situations as exists in human DM1, results in RNA mis‐splicing and the multisystemic manifestations of DM1.
At present there is no clinical care consensus or evidence based guidelines, but an ongoing effort by the myotonic dystrophy foundation (http://www.mdf.org) is bringing together experts in the field to delineate such a document. However, similar to other pediatric NM diseases, there are no treatments that alter or reverse disease progression, and current clinical care paradigms are focused on symptomatic therapy for the consequences of DM1. DM1 is a multisystem disease and the monitoring and management are beyond the scope of this article but there are several recent reviews outlining care recommendations in adults (Turner & Hilton‐Jones, 2014) and children (Campbell, Levin, Siu, Venance, & Jacob, 2013). Comprehensive models of health care have been developed by Gagnon et al. (2010) that take into account the extent of disease impact on the family across generations, and the unique and challenging cognitive, personality and motivation differences experienced by those with DM1. One aspect of care that needs to be emphasized is the need for genetic education and counselling for the whole family, which may need to be repeated and revisited during youth and adult lifespan, especially around risks of having children with congenital myotonic dystrophy. A multicenter study examining exercise and lifestyle modifications is currently underway (NCT02118779) (van Engelen & Consortium, 2015).
Emerging therapeutic paradigms have been largely aimed at one of several mechanisms to modify the toxic properties of the CTG repeat expansion. An important and exciting observation in relation to therapy development is the recognition from pre‐clinical models that the cardiac and muscle histopathology of DM1 may be able to be completely reversed when toxic mRNA is reduced or eliminated (Mahadevan et al., 2006), a phenomenon that would be ideal, if true of human disease as well.
10.1. Genetic‐based approaches
Presently only one product that aims to directly disrupt the toxic mRNA is at the stage of human clinical trial. This drug is an AON against the CUG repeat domain of the DMPK RNA (Pandey et al., 2015). Although it principally binds to CUG repeats and is intended to disrupt MBNL sequestration, a reduction in nuclear accumulation of toxic RNA foci has been demonstrated, suggesting that in the absence of MBNL sequestration the toxic RNA is degraded more easily or transported from the nucleus more efficiently (Gao & Cooper, 2013). Although there is potential for reduction of the normal DMPK protein, it does not appear to target the wide type transcript enough to cause concerns, nor is there noted off‐target effects. There is an ongoing phase 2 clinical trial of this AON (Biogen/Ionis, NCT02312011), which involves a dose finding component, with AON delivered systemically through sub‐ cutaneous route on a weekly basis.
Other pre‐clinical toxic RNA disruptive therapeutic avenues are being developed, and include interference RNA and small molecule strategies (Gao & Cooper, 2013). In addition, other strategies in preclinical development have used AON to block the binding sites of the CUG binding protein muscleblind (MBNL) on the mutant mRNA thus preventing sequestration of MBNL and ultimately restoring splicing (Wojtkowiak‐Szlachcic et al., 2015). A separate approach is to create a MBNL decoy effectively releasing MBNL from sequestration (Valerion Therapeutics). Endonuclease targeting of the CUG repeat in the toxic mRNA has been demonstrated to be helpful in DM1 animal models (Zhang et al., 2014).
10.2. Non‐gene based approaches
Given that DM1 is a multi‐system disease there are many target signs and symptoms that can be addressed to improve the lives of patients. For the muscle pathology, three separate clinical trials in adults with DM1 have been performed, evaluating (1) creatine (Tarnopolsky, 2007); (2) insulin like growth factor and a binding protein combination IGF‐1/IGFBP3 (Heatwole et al., 2011); and (3) Dehydroepiandrosterone (Penisson‐Besnier et al., 2008). Unfortunately, none have had positive results, and these agents are not widely used clinically. Another particularly disabling symptom of DM1 is myotonia, which is thought to result from altered expression of the chloride channel CLCN1. A randomized, cross over designed clinical trial of the sodium channel antagonist mexiletine has been performed in DM1 and shown to significantly reduce the frequency and severity of myotonia (Logigian et al., 2010). This drug is now in routine clinical use, both for DM1 and for other non‐dystrophic conditions such as myotonia congenita. Importantly, while no safety concerns were reported during the clinical trial or after, and no changes on ECG parameters were identified, mexiletine is potentially pro‐arrhythmic, a concerning possible side effect (see below) that should be appropriately monitored and disclosed to patients. Lastly, excessive daytime sleepiness is a tremendous burden on DM1 patients, and a small cross‐over RCT comparing a single dose of methylphenidate to placebo showed improvements in standardized measures of daytime fatigue (Puymirat, Bouchard, & Mathieu, 2012). Modafinil, which is used to combat fatigue in many neurological conditions, has been evaluated in DM1 in a RCT, but was not found to be effective (Orlikowski et al., 2009).
Perhaps the most concern complication of DM1 is sudden cardiac death or life threatening arrhythmia, and it is clear that determining the risk of such events from standard EEG and other cardiac monitoring is not straightforward. A body of literature has emerged, including a recent systematic review (Lau, Sy, Corbett, & Kritharides, 2015), assessing different interventions for cardiac manifestations that is beyond the scope of this review, however it is imperative that all patients have a comprehensive cardiac review of systems and regular ECGs. Any abnormality should prompt a referral to a cardiologist, ideally with experience in DM1.
11. CONGENITAL MYOPATHIES
Like congenital muscular dystrophies, congenital myopathies are a genetically heterogeneous group of muscle diseases that primarily present in infancy with hypotonia, muscle weakness, and often respiratory compromise (Dowling, North, Goebel, & Beggs, 2015). They may come to clinical attention at ages other than infancy, and newly diagnosed adult cases are described (North et al., 2014). Patients with congenital myopathies may have a range of disabilities, from mild weakness and motor impairment to severe symptomatology that includes wheelchair and ventilator dependence (Wang et al., 2012). Most often the weakness in congenital myopathies is slowly or non‐progressive, though exceptions do exist, and most individuals experience some degree of worsening of disease in the teen years and in adulthood (Table 9).
Table 9.
Summary of therapeutic strategies for congenital myopathies (CM)
| Category | Compound | CM Subtype | Action | Company | Dosing and delivery | Phase of clinical trial | Results | Safety | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Genetic based therapies | |||||||||
| Gene therapy | AT132 | MTM1 | Gene replacement | Audentes | Intravenous | Phase 1 | Pre‐clinical data | No results in humans yet | http://Clinical.trials.gov NCT03199469 |
| Gene modulation | DNM2 PIK3C2b | MTM1 | Gene modulation | – | – | Mice models | Successful models rescue | No results in humans yet | Cowling et al. (2014), Sabha et al. (2016) |
| Enzyme replacement | VAL‐0620 | MTM1 | Enzyme replacement | Valerion | Intramuscular | Mouse model | Successful model rescue | No results in humans yet | Lawlor et al. (2013) |
| Non genetic based therapies | |||||||||
| Molecules | Pyridostigmine (mestinon) | RYR1 MTM1 DNM2 TPM2 TPM3 KLHL40 | Increase acetylcholine in the NMJ | – | Oral | No formal systematic analyses | Evidence based on case reports and animal models | No significant concerns | Dowling et al. (2012b), Gibbs et al. (2013), Illingworth et al. (2014), Robb et al. (2011) |
| Dantrolene | RYR1 mutations associated with MHS | Antagonizes the intracellular release of calcium by RyR1 | – | Oral (Intravenous) | No formal systematic analyses | Systematic analysis have been done for malignant hyperthermia (MH) | Standard use in MH. Case reports of use in patients with myalgias | Dowling et al. (2014) | |
| L‐tyrosine | Nemaline myopathy | ? | – | Oral | No formal systematic analyses | Subjective improvement in sialorrhoea | No significant concerns | Ryan et al. (2008) | |
| Salbutamol | RYR1 (central core disease) | Not well understood | – | Oral | Control study | Off label indication | No significant concerns identified | Messina et al. (2004) | |
| N‐acetylcysteine | SEPN1 RYR1 | Improvement of aberrant oxidative stress | – | Oral | Phase 1/2 Phase 2/3 | Pre‐clinical data | No available data yet | http://Clinical.trials.gov NCT02362425 NCT02505087 | |
| Wortmannin | MTM1 | Inhibition of PIK3C2B enzyme activity restoring PI3P levels | – | Oral | Zebrafish and mouse model | Pre‐clinical data | No available data yet | Sabha et al. (2016) | |
| Small molecule | CK‐2066260 | NEB | Improves actin‐myosin cross bridging dynamics at the sarcomere | – | – | In vitro studies | Increase of contraction | No in vivo studies. | Marieke de Winter et al. (2013) |
Congenital myopathies are distinguished from CMDs clinically in that they often quite prominently involve the facial musculature, and separated diagnostically by the observation of normal or only mildly elevated CK levels and by the appearance of the muscle biopsy (North et al., 2014). This group of diseases is classically defined by the features of the muscle biopsy, and separated into histopathologic subtypes including core myopathies (central core disease, minicore myopathy), nemaline rod myopathies, centronuclear myopathies, and congenital fiber‐type disproportion (Dowling et al., 2015). Increasingly, congenital myopathies are being defined on a genetic basis, and mutations in more than 20 genes have been identified. The most common genetic cause is mutation of the skeletal muscle ryanodine receptor (RYR1), with other frequently encountered gene mutations in MTM1, NEB, ACTA1, MYH7, DNM2, and SEPN1 (Colombo et al., 2015). The two major areas of pathology for congenital myopathies are the triad (which governs excitation contraction coupling) and the thin filament (a portion of the sarcomere that helps govern muscle contraction) (Figure 4).
Figure 4.
The myofiber. Muscle fiber and it different components. Within brackets are the congenital myopathies associated with defects in the different muscle substructures. (Adapted from Ravenscroft et al., 2015).
There are currently no drugs approved for therapy specifically for congenital myopathies. Patients with centronuclear myopathy, and particularly those with mutations in RYR1, MTM1, and DNM2, have shown positive response to pyridostigmine (Dowling JJ, Joubert et al., 2012b; Gibbs et al., 2013; Illingworth et al., 2014; Robb et al., 2011), with the commonest benefit described with fatigue and energy level, though with a few individuals showing more dramatic improvements. The rationale for consideration of pyridostigmine comes from two things. One is the clinical overlap between CNMs and congenital myasthenic syndromes, as CNM patients also have prominent ptosis and ophthalmoparesis, and can additionally have fluctuating symptoms (Illingworth et al., 2014; Robb et al., 2011). The other is the demonstration in animal models of MTM1 and DNM2 related CNM of abnormalities in the structure of the neuromuscular junction (Dowling, Joubert et al., 2012b; Gibbs et al., 2013). Of note, positive response to pyridostigmine has been additionally described in several single case studies including: (1) a patient with TPM2 mutation and cap myopathy (Rodriguez Cruz et al., 2014); (2) a patient with TPM3 mutation and congenital fiber‐type disproportion (Munot et al., 2010); and (3) in a patient with KLHL40 mutation and nemaline myopathy (Natera‐de Benito et al., 2016). Thus pyridostigmine (typically at 7 mg/kg/day though reported at significantly higher doses as well) may represent a therapeutic with potential benefit across the spectrum of congenital myopathies.
The other medication often utilized in an “off label” manner for congenital myopathies is oral salbutamol. Salbutamol is a beta agonist that improves muscle strength through mechanisms that are not completely understood. A limited case control trial of salbutamol (1–2 mg PO TID) was performed on a small number of patients with central core disease and RYR1 mutation (Messina et al., 2004). There was an overall beneficial effect of this treatment, with improvements in muscle strength and motor function. As of yet, no formal clinical trial has followed these promising initial results. The drug continues to be used in many cases of RYR1 related myopathy. The experience outside of RYR1 mutations is much more limited, and its benefit in such settings is unclear.
Dantrolene has also been used in a select group of individuals with RYR1 mutations (Dowling, Lawlor, & Dirksen, 2014). These patients have mutations associated with a hypersensitive RyR1 channel, and often primarily have myalgias and exercise intolerance as their main complaints. In this setting, dantrolene (which antagonizes the intracellular release of calcium by RyR1) has been reported to improve endurance and diminish pain. The more common application of dantrolene is as an agent to abort malignant hyperthermia, which is associated in the majority of cases with (often overlapping) mutations in RYR1 (Rosenberg, Davis, James, Pollock, & Stowell, 2007). Of note, there is reason to believe that dantrolene may not only not work but may be harmful in most myopathies, particularly those associated with reduced RyR1 function and/or abnormalities in the structure of the excitation contraction coupling apparatus (such as centronuclear myopathy). On the other hand, a recent report using a mouse model of DMD suggests that dantrolene may improve muscle disease in other settings (Kendall et al., 2012). Future studies are obviously required to sort out the potentially applicability of this drug.
L‐tyrosine has been considered in patients with nemaline myopathy. A pilot study of five NM patients demonstrated reduced oral secretions in all subjects, and improved muscle strength and function in one of the five (Ryan et al., 2008). This is in keeping with a pre‐clinical study of tyrosine showing improved histopathology and motor function in a severe mouse model of Acta1 related NM (Nguyen et al., 2011). Anecdotally, and in a non‐systematic fashion, tyrosine is used or has been tried by many patients with NM. It is well tolerated (though with some potential difficulties in administration via G tube), and subjective improvement in sialorrhoea has been reported. However, an indepth appraisal of this experience has not been undertaken, and the precise impact (if any) of L‐tyrosine on NM patients uncertain.
Finally, there are two ongoing interventional clinical trials for congenital myopathies. Both trials are testing the anti‐oxidant N‐acetylcysteine; one is being conducted in France in patients with SEPN1 related myopathies, and the other is in the US for RYR1 related myopathies. Both are examining whether NAC improves muscle function, endurance, and quality of life. The genesis of these trials are pre‐clinical studies in SEPN1 (patient cells and a mouse model) and RYR1 myopathy (patient cells and zebrafish) model systems that uncovered aberrant oxidative stress and phenotypic improvement with anti‐oxidants (Arbogast et al., 2009; Dowling, Arbogast et al., 2012a; Rederstorff et al., 2011).
There are several molecules and therapeutic strategies in pre‐clinical development for congenital myopathies. Perhaps the most exciting is gene therapy for myotubular myopathy (XLMTM, a X‐linked centronuclear myopathy caused by mutations in MTM1). Childers et al. (2014) have shown that AAV based gene therapy can both prevent and reverse disease in mouse and canine models of myotubular myopathy. A clinical trial for this therapy is in active planning (Audentes, NCT03199469). Also under consideration for XLMTM is enzyme replacement therapy (Lawlor et al., 2013), using concepts based on the successful treatment of Pompe Disease with myozyme. Lastly, there is evidence from XLMTM mouse models that reducing levels and/or activity of enzymes (e.g., PIK3C2B or DNM2) that act to balance the cellular function(s) of MTM1 (a phosphoinositide phosphatase that regulates endosomal sorting) provides rescue in the mouse model of the disease (Ketel et al., 2016; Sabha et al., 2016; Tasfaout et al., 2017).
In terms of small molecule approaches, troponin activators seem to hold promise for several congenital myopathies (de Winter et al., 2013). These drugs improve actin‐myosin cross bridging dynamics at the sarcomere and thus result in increased force generation. They are particularly attractive to consider for nemaline myopathies, which as a group are caused by abnormalities in the structure and/or function of the actin thin filament part of the sarcomere.
11.1. Summary
Childhood neuromuscular diseases have long been considered untreatable, life limiting diseases with little or no hope for meaningful therapeutic interventions. Starting with glucocorticoid use in DMD, this picture has evolved over time, with improved outcomes in many neuromuscular disorders. Much of the improvement can be attributed to better general care and attention to respiratory health. However, until recently there were few specific disease‐modulating therapies. This is set to change, as many exciting treatment approaches are currently in clinical trial, and several have achieved conditional or full market approval in various regions of the world. Many other treatments are in the pipeline, and we predict that over the next decade meaningful therapies will become widespread across the neuromuscular disease spectrum. With the excitement of new therapies comes also the recognition that (a) comprehensive multi‐disciplinary care is a must for neuromuscular patients and (b) in‐depth knowledge of disease natural history is critical for present day care and for clinical trial readiness.
Dowling JJ, D. Gonorazky H, Cohn RD, Campbell C. Treating pediatric neuromuscular disorders: The future is now. Am J Med Genet Part A. 2018;176: 804–841. https://doi.org/10.1002/ajmg.a.38418
REFERENCES
- Abicht A., Muller J. S., Lochmuller H. (1993). Congenital Myasthenic syndromes. In R. A. Pagon, M. P. Adam, H. H. Ardinger, S. E.Wallace, A. Amemiya, L. J. H. Bean, T. D. Bird, C. T. Fong, H. C. Mefford, R. J. H. Smith, & K. Stephens (Eds). Seattle WA: GeneReviews(R).
- Accornero, F. , Kanisicak, O. , Tjondrokoesoemo, A. , Attia, A. C. , McNally, E. M. , & Molkentin, J. D. (2014). Myofiber‐specific inhibition of TGFbeta signaling protects skeletal muscle from injury and dystrophic disease in mice. Human Molecular Genetics, 23(25), 6903–6915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen, D. G. , Whitehead, N. P. , & Froehner, S. C. (2016). Absence of dystrophin disrupts skeletal muscle signaling: Roles of Ca2+, reactive oxygen species, and nitric oxide in the development of muscular dystrophy. Physiological Reviews, 96(1), 253–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen, H. D. , Flanigan, K. M. , Thrush, P. T. , Dvorchik, I. , Yin, H. , Canter, C. , … Mendell, J. R. (2013). A randomized, double‐blind trial of lisinopril and losartan for the treatment of cardiomyopathy in duchenne muscular dystrophy. PLoS Currents, 5, pii: ecurrents.md.2cc69a1dae4be7dfe2bcb420024ea865 https://doi.org/10.1371/currents.md.2cc69a1dae4be7dfe2bcb420024ea865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amthor, H. , & Hoogaars, W. M. (2012). Interference with myostatin/ActRIIB signaling as a therapeutic strategy for Duchenne muscular dystrophy. Current Gene Therapy, 12(3), 245–259. [DOI] [PubMed] [Google Scholar]
- Anselme, F. , Moubarak, G. , Savoure, A. , Godin, B. , Borz, B. , Drouin‐Garraud, V. , & Gay, A. (2013). Implantable cardioverter‐defibrillators in lamin A/C mutation carriers with cardiac conduction disorders. Heart Rhythm, 10(10), 1492–1498. [DOI] [PubMed] [Google Scholar]
- Aoki, Y. , Nagata, T. , Yokota, T. , Nakamura, A. , Wood, M. J. , Partridge, T. , & Takeda, S. (2013). Highly efficient in vivo delivery of PMO into regenerating myotubes and rescue in laminin‐alpha2 chain‐null congenital muscular dystrophy mice. Human Molecular Genetics, 22(24), 4914–4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbogast, S. , Beuvin, M. , Fraysse, B. , Zhou, H. , Muntoni, F. , & Ferreiro, A. (2009). Oxidative stress in SEPN1‐related myopathy: From pathophysiology to treatment. Annals of Neurology, 65(6), 677–686. [DOI] [PubMed] [Google Scholar]
- Arimura, S. , Okada, T. , Tezuka, T. , Chiyo, T. , Kasahara, Y. , Yoshimura, T. , … Yamanashi, Y. (2014). Neuromuscular disease. DOK7 gene therapy benefits mouse models of diseases characterized by defects in the neuromuscular junction. Science, 345(6203), 1505–1508. [DOI] [PubMed] [Google Scholar]
- Attarian, S. , Vallat, J. M. , Magy, L. , Funalot, B. , Gonnaud, P. M. , Lacour, A. , … Cohen, D. (2014). An exploratory randomised double‐blind and placebo‐controlled phase 2 study of a combination of baclofen, naltrexone and sorbitol (PXT3003) in patients with Charcot‐Marie‐Tooth disease type 1A. Orphanet Journal of Rare Diseases, 9, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azmaipairashvili, Z. , Riddle, E. C. , Scavina, M. , & Kumar, S. J. (2005). Correction of cavovarus foot deformity in Charcot‐Marie‐Tooth disease. Journal of Pediatric Orthopedics, 25(3), 360–365. [DOI] [PubMed] [Google Scholar]
- Banwell, B. L. , Ohno, K. , Sieb, J. P. , & Engel, A. G. (2004). Novel truncating RAPSN mutations causing congenital myasthenic syndrome responsive to 3,4‐diaminopyridine. Neuromuscular Disorders, 14(3), 202–207. [DOI] [PubMed] [Google Scholar]
- Bartoli, M. , Roudaut, C. , Martin, S. , Fougerousse, F. , Suel, L. , Poupiot, J. , … Richard, I. (2006). Safety and efficacy of AAV‐mediated calpain 3 gene transfer in a mouse model of limb‐girdle muscular dystrophy type 2A. Molecular Therapy, 13(2), 250–259. [DOI] [PubMed] [Google Scholar]
- Bauer, R. , Macgowan, G. A. , Blain, A. , Bushby, K. , & Straub, V. (2008). Steroid treatment causes deterioration of myocardial function in the {delta}‐sarcoglycan‐deficient mouse model for dilated cardiomyopathy. Cardiovascular Research, 79(4), 652–661. [DOI] [PubMed] [Google Scholar]
- Bauer, R. , Straub, V. , Blain, A. , Bushby, K. , & MacGowan, G. A. (2009). Contrasting effects of steroids and angiotensin‐converting‐enzyme inhibitors in a mouse model of dystrophin‐deficient cardiomyopathy. European Journal of Heart Failure, 11(5), 463–471. [DOI] [PubMed] [Google Scholar]
- Beeson, D. , Hantai, D. , Lochmuller, H. , & Engel, A. G. (2005). 126th international workshop: Congenital myasthenic syndromes, 24–26 september 2004, naarden, the Netherlands. Neuromuscular Disorders, 15(7), 498–512. [DOI] [PubMed] [Google Scholar]
- Beeson, D. (2016). Congenital myasthenic syndromes: Recent advances. Current Opinion in Neurology, 29(5), 565 –571. [DOI] [PubMed] [Google Scholar]
- Bellinger, A. M. , Reiken, S. , Carlson, C. , Mongillo, M. , Liu, X. , Rothman, L. , … Marks, A. R. (2009). Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nature Medicine, 15(3), 325–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtsson, N. E. , Seto, J. T. , Hall, J. K. , Chamberlain, J. S. , & Odom, G. L. (2016). Progress and prospects of gene therapy clinical trials for the muscular dystrophies. Human Molecular Genetics, 25(R1), R9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian, Y. , Masuda, A. , Matsuura, T. , Ito, M. , Okushin, K. , Engel, A. G. , & Ohno, K. (2009). Tannic acid facilitates expression of the polypyrimidine tract binding protein and alleviates deleterious inclusion of CHRNA1 exon P3A due to an hnRNP H‐disrupting mutation in congenital myasthenic syndrome. Human Molecular Genetics, 18(7), 1229–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolduc, V. , Zou, Y. , Ko, D. , & Bonnemann, C. G. (2014). SiRNA‐mediated allele‐specific silencing of a COL6A3 mutation in a cellular model of dominant ullrich muscular dystrophy. Molecular Therapy. Nucleic Acids, 3, e147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolino, A. , Piguet, F. , Alberizzi, V. , Pellegatta, M. , Rivellini, C. , Guerrero‐Valero, M. , … Previtali, S. C. (2016). Niacin‐mediated tace activation ameliorates CMT neuropathies with focal hypermyelination. EMBO Molecular Medicine, 8(12), 1438–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonne, G. (2014). Nuclear envelope proteins in health and diseases. Seminars in Cell and Developmental Biology, 29, 93–94. [DOI] [PubMed] [Google Scholar]
- Bonne G., Leturcq F., Ben Yaou R. (1993). Emery‐Dreifuss Muscular Dystrophy. In R. A. Pagon, M. P. Adam, H. H. Ardinger, S. E. Wallace, A. Amemiya, L. J. H. Bean, T. D. Bird, C. T. Fong, H. C. Mefford, R. J. H. Smith, & K. Stephens, (Eds.). Seattle WA: GeneReviews(R).
- Bonnemann, C. G. , Wang, C. H. , Quijano‐Roy, S. , Deconinck, N. , Bertini, E. , … Ferreiro, A. (2014). Diagnostic approach to the congenital muscular dystrophies. Neuromuscular Disorders, 24(4), 289–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostick, B. , Ghosh, A. , Yue, Y. , Long, C. , & Duan, D. (2007). Systemic AAV‐9 transduction in mice is influenced by animal age but not by the route of administration. Gene Therapy, 14(22), 1605–1609. [DOI] [PubMed] [Google Scholar]
- Burke, G. , Hiscock, A. , Klein, A. , Niks, E. H. , Main, M. , Manzur, A. Y. , … Robb, S. (2013). Salbutamol benefits children with congenital myasthenic syndrome due to DOK7 mutations. Neuromuscular Disorders, 23(2), 170–175. [DOI] [PubMed] [Google Scholar]
- Burns, J. , Ouvrier, R. , Estilow, T. , Shy, R. , Laura, M. , Pallant, J. F. , … Finkel, R. S. (2012). Validation of the Charcot‐Marie‐Tooth disease pediatric scale as an outcome measure of disability. Annals of Neurology, 71(5), 642–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns, J. , Ouvrier, R. A. , Yiu, E. M. , Joseph, P. D. , Kornberg, A. J. , Fahey, M. C. , & Ryan, M. M. (2009). Ascorbic acid for Charcot‐Marie‐Tooth disease type 1A in children: A randomised, double‐blind, placebo‐controlled, safety and efficacy trial. Lancet Neurology, 8(6), 537–544. [DOI] [PubMed] [Google Scholar]
- Burns, J. , Scheinberg, A. , Ryan, M. M. , Rose, K. J. , & Ouvrier, R. A. (2010). Randomized trial of botulinum toxin to prevent pes cavus progression in pediatric Charcot‐Marie‐Tooth disease type 1A. Muscle and Nerve, 42(2), 262–267. [DOI] [PubMed] [Google Scholar]
- Bushby, K. , Finkel, R. , Birnkrant, D. J. , Case, L. E. , Clemens, P. R. , Cripe, L. , … Group DMDCCW. (2010a). Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and pharmacological and psychosocial management. Lancet Neurology, 9(1), 77–93. [DOI] [PubMed] [Google Scholar]
- Bushby, K. , Finkel, R. , Birnkrant, D. J. , Case, L. E. , Clemens, P. R. , Cripe, L. , … Group DMDCCW. (2010b). Diagnosis and management of Duchenne muscular dystrophy, part 2: Implementation of multidisciplinary care. Lancet Neurology, 9(2), 177–189. [DOI] [PubMed] [Google Scholar]
- Bushby, K. , Finkel, R. , Wong, B. , Barohn, R. , Campbell, C. , … Comi, G. P. (2014). Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle and Nerve, 50(4), 477–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, C. , Levin, S. , Siu, V. M. , Venance, S. , & Jacob, P. (2013). Congenital myotonic dystrophy: Canadian population‐based surveillance study. Jornal De Pediatria, 163(1), 120–125. [DOI] [PubMed] [Google Scholar]
- Capote, J. , Kramerova, I. , Martinez, L. , Vetrone, S. , Barton, E. R. , Sweeney, H. L. , … Spencer, M. J. (2016). Osteopontin ablation ameliorates muscular dystrophy by shifting macrophages to a pro‐regenerative phenotype. The Journal of Cell Biology, 213(2), 275–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaouch, A. , Muller, J. S. , Guergueltcheva, V. , Dusl, M. , Schara, U. , Rakocevic‐Stojanovic, V. , … Lochmuller, H. (2012). A retrospective clinical study of the treatment of slow‐channel congenital myasthenic syndrome. Journal of Neurology, 259(3), 474–481. [DOI] [PubMed] [Google Scholar]
- Chen, T. H. , Chang, J. G. , Yang, Y. H. , Mai, H. H. , Liang, W. C. , Wu, Y. C. , … Jong, Y. J. (2010). Randomized, double‐blind, placebo‐controlled trial of hydroxyurea in spinal muscular atrophy. Neurology, 75(24), 2190–2197. [DOI] [PubMed] [Google Scholar]
- Cheng, X. , Zhang, X. , Gao, Q. , Ali Samie, M. , Azar, M. , Tsang, W. L. , … Xu, H. (2014). The intracellular Ca(2)(+) channel MCOLN1 is required for sarcolemma repair to prevent muscular dystrophy. Nature Medicine, 20(10), 1187–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chetlin, R. D. , Gutmann, L. , Tarnopolsky, M. A. , Ullrich, I. H. , & Yeater, R. A. (2004). Resistance training exercise and creatine in patients with Charcot‐Marie‐Tooth disease. Muscle and Nerve, 30(1), 69–76. [DOI] [PubMed] [Google Scholar]
- Childers, M. K. , Joubert, R. , Poulard, K. , Moal, C. , Grange, R. W. , Doering, J. A. , … Buj‐Bello, A. (2014). Gene therapy prolongs survival and restores function in murine and canine models of myotubular myopathy. Science Translational Medicine, 6(220), 220ra210.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiriboga, C. A. , Swoboda, K. J. , Darras, B. T. , Iannaccone, S. T. , Montes, J. , De Vivo, D. C. , … Bishop, K. M. (2016). Results from a phase 1 study of nusinersen (ISIS‐SMN(Rx)) in children with spinal muscular atrophy. Neurology, 86(10), 890–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, J. C. , Muchir, A. , Wu, W. , Iwata, S. , Homma, S. , Morrow, J. P. , & Worman, H. J. (2012). Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Science Translational Medicine, 4(144), 144ra102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins, J. , & Bonnemann, C. G. (2010). Congenital muscular dystrophies: Toward molecular therapeutic interventions. Current Neurology and Neuroscience Reports, 10(2), 83–91. [DOI] [PubMed] [Google Scholar]
- Colombo, I. , Scoto, M. , Manzur, A. Y. , Robb, S. A. , Maggi, L. , Gowda, V. , … Muntoni, F. (2015). Congenital myopathies: Natural history of a large pediatric cohort. Neurology, 84(1), 28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong, L. , Ran, F. A. , Cox, D. , Lin, S. , Barretto, R. , Habib, N. , … Zhang, F. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science, 339(6121), 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly, A. M. , Pestronk, A. , Mehta, S. , & Al‐Lozi, M. (1998). Primary alpha‐sarcoglycan deficiency responsive to immunosuppression over three years. Muscle and Nerve, 21(11), 1549–1553. [DOI] [PubMed] [Google Scholar]
- Consalvi, S. , Saccone, V. , & Mozzetta, C. (2014). Histone deacetylase inhibitors: A potential epigenetic treatment for Duchenne muscular dystrophy. Epigenomics, 6(5), 547–560. [DOI] [PubMed] [Google Scholar]
- Crawford, T. O. , & Pardo, C. A. (1996). The neurobiology of childhood spinal muscular atrophy. Neurobiology of Disease, 3(2), 97–110. [DOI] [PubMed] [Google Scholar]
- Darras, B. T. (2015). Neuromuscular disorders of infancy, childhood, and adolescence a clinician's approach. 2nd ed Amsterdam: Elsevier/Academic Press, p xxxii (p. 1123). [Google Scholar]
- de Winter, J. M. , Buck, D. , Hidalgo, C. , Jasper, J. R. , Malik, F. I. , Clarke, N. F. , … Granzier, H. (2013). Troponin activator augments muscle force in nemaline myopathy patients with nebulin mutations. Journal of Medical Genetics, 50(6), 383–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling, J. J. (2016). Eteplirsen therapy for Duchenne muscular dystrophy: Skipping to the front of the line. Nature Reviews. Neurology, 12(12), 675–676. [DOI] [PubMed] [Google Scholar]
- Dowling, J. J. , Arbogast, S. , Hur, J. , Nelson, D. D. , McEvoy, A. , Waugh, T. , … Ferreiro, A. (2012a). Oxidative stress and successful antioxidant treatment in models of RYR1‐related myopathy. Brain: A Journal of Neurology, 135(Pt 4), 1115–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling, J. J. , Joubert, R. , Low, S. E. , Durban, A. N. , Messaddeq, N. , Li, X. , … Pierson, C. R. (2012b). Myotubular myopathy and the neuromuscular junction: A novel therapeutic approach from mouse models. Disease Models & Mechanisms, 5(6), 852–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling, J. J. , Lawlor, M. W. , & Dirksen, R. T. (2014). Triadopathies: An emerging class of skeletal muscle diseases. Neurotherapeutics, 11(4), 773–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling, J. J. , North, K. N. , Goebel, H. H. , & Beggs, A. H. , (2015). Congenital and other structural myopathies In Darras B. T., Jones H. R., Ryan M. M. & De Vivo D. C. (Eds.), Neuromuscular disorders of infancy, childhood, and adolescence: A clinician's approach. Boston: Elsevier. [Google Scholar]
- Dubowitz, V. (1965). Muscular dystrophy and related disorders. Postgraduate Medical Journal, 41(476), 332–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubowitz, V. (1995). Muscle disorders in childhood. London; Philadelphia: Saunders, x (p. 540). [Google Scholar]
- Dumont, N. A. , Wang, Y. X. , von Maltzahn, J. , Pasut, A. , Bentzinger, C. F. , Brun, C. E. , & Rudnicki, M. A. (2015). Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nature Medicine, 21(12), 1455–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Aloul, B. , Altamirano‐Diaz, L. , Zapata‐Aldana, E. , Rodrigues, R. , Malvankar‐Mehta, M. S. , Nguyen, C. T. , & Campbell, C. (2017). Pharmacological therapy for the prevention and management of cardiomyopathy in Duchenne muscular dystrophy: A systematic review. Neuromuscular Disorders, 27(1), 4–14. [DOI] [PubMed] [Google Scholar]
- Elbaz, M. , Yanay, N. , Aga‐Mizrachi, S. , Brunschwig, Z. , Kassis, I. , Ettinger, K. , … Nevo, Y. (2012). Losartan, a therapeutic candidate in congenital muscular dystrophy: Studies in the dy(2J) /dy(2J) mouse. Annals of Neurology, 71(5), 699–708. [DOI] [PubMed] [Google Scholar]
- Engel, A. G. , Shen, X. M. , Selcen, D. , & Sine, S. M. (2015). Congenital myasthenic syndromes: Pathogenesis, diagnosis, and treatment. Lancet Neurology, 14(4), 420–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erb, M. , Meinen, S. , Barzaghi, P. , Sumanovski, L. T. , Courdier‐Fruh, I. , Ruegg, M. A. , & Meier, T. (2009). Omigapil ameliorates the pathology of muscle dystrophy caused by laminin‐alpha2 deficiency. Journal of Pharmacology and Experimental Therapeutics, 331(3), 787–795. [DOI] [PubMed] [Google Scholar]
- Eymard, B. , Hantai, D. , & Estournet, B. (2013). Congenital myasthenic syndromes. Handbook of Clinical Neurology, 113, 1469–1480. [DOI] [PubMed] [Google Scholar]
- Farooq, F. , Abadia‐Molina, F. , MacKenzie, D. , Hadwen, J. , Shamim, F. , O'Reilly, S. , … MacKenzie, A. (2013). Celecoxib increases SMN and survival in a severe spinal muscular atrophy mouse model via p38 pathway activation. Human Molecular Genetics, 22(17), 3415–3424. [DOI] [PubMed] [Google Scholar]
- Faustino, N. A. , & Cooper, T. A. (2003). Pre‐mRNA splicing and human disease. Genes & Development, 17(4), 419–437. [DOI] [PubMed] [Google Scholar]
- Finkel, R. , Bertini, E. , Muntoni, F. , Mercuri, E. , & Group ESWS. (2015). 209th ENMC international workshop: Outcome measures and clinical trial readiness in spinal muscular atrophy 7–9 november 2014, heemskerk, The Netherlands. Neuromuscular Disorders, 25(7), 593–602. [DOI] [PubMed] [Google Scholar]
- Finkel, R. S. , Chiriboga, C. A. , Vajsar, J. , Day, J. W. , Montes, J. , De Vivo, D. C. , … Bishop, K. M. (2016). Treatment of infantile‐onset spinal muscular atrophy with nusinersen: A phase 2, open‐label, dose‐escalation study. The Lancet, 388(10063), 3017–3026. [DOI] [PubMed] [Google Scholar]
- Finlayson, S. , Spillane, J. , Kullmann, D. M. , Howard, R. , Webster, R. , Palace, J. , & Beeson, D. (2013). Slow channel congenital myasthenic syndrome responsive to a combination of fluoxetine and salbutamol. Muscle and Nerve, 47(2), 279–282. [DOI] [PubMed] [Google Scholar]
- Fledrich, R. , Stassart, R. M. , Klink, A. , Rasch, L. M. , Prukop, T. , Haag, L. , … Sereda, M. W. (2014). Soluble neuregulin‐1 modulates disease pathogenesis in rodent models of Charcot‐Marie‐Tooth disease 1A. Nature Medicine, 20(9), 1055–1061. [DOI] [PubMed] [Google Scholar]
- Foley, A. R. , Quijano‐Roy, S. , Collins, J. , Straub, V. , McCallum, M. , Deconinck, N. , … Bonnemann, C. G. (2013). Natural history of pulmonary function in collagen VI‐related myopathies. Brain: A Journal of Neurology, 136(Pt 12), 3625–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridman, V. , Bundy, B. , Reilly, M. M. , Pareyson, D. , Bacon, C. , … Burns, J. (2015). CMT subtypes and disease burden in patients enrolled in the inherited neuropathies consortium natural history study: A cross‐sectional analysis. Journal of Neurology, Neurosurgery, and Psychiatry, 86(8), 873–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon, C. , Chouinard, M. C. , Laberge, L. , Veillette, S. , Begin, P. , Breton, R. , … Panel, D. M. I. E. (2010). Health supervision and anticipatory guidance in adult myotonic dystrophy type 1. Neuromuscular Disorders, 20(12), 847–851. [DOI] [PubMed] [Google Scholar]
- Gao, G. P. , Lu, F. , Sanmiguel, J. C. , Tran, P. T. , Abbas, Z. , Lynd, K. S. , … Wilson, J. M. (2002). Rep/Cap gene amplification and high‐yield production of AAV in an A549 cell line expressing Rep/Cap. Molecular Therapy, 5(5 Pt 1), 644–649. [DOI] [PubMed] [Google Scholar]
- Gao, Q. Q. , Wyatt, E. , Goldstein, J. A. , LoPresti, P. , Castillo, L. M. , Gazda, A. , … McNally, E. M. (2015). Reengineering a transmembrane protein to treat muscular dystrophy using exon skipping. The Journal of Clinical Investigation, 125(11), 4186–4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, Z. , & Cooper, T. A. (2013). Antisense oligonucleotides: Rising stars in eliminating RNA toxicity in myotonic dystrophy. Human Gene Therapy, 24(5), 499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg, N. , Yiannikas, C. , Hardy, T. A. , Belaya, K. , Cheung, J. , Beeson, D. , & Reddel, S. W. (2016). Late presentations of congenital myasthenic syndromes—how many do we miss? Muscle and Nerve, 54(4), 721–727. [DOI] [PubMed] [Google Scholar]
- Gibbs, E. M. , Clarke, N. F. , Rose, K. , Oates, E. C. , Webster, R. , Feldman, E. L. , & Dowling, J. J. (2013). Neuromuscular junction abnormalities in DNM2‐related centronuclear myopathy. Journal of Molecular Medicine (Berlin, Germany), 91(6), 727–737. [DOI] [PubMed] [Google Scholar]
- Gilbreath, H. R. , Castro, D. , & Iannaccone, S. T. (2014). Congenital myopathies and muscular dystrophies. Neurologic Clinics, 32(3), 689 –703. [DOI] [PubMed] [Google Scholar]
- Godfrey, C. , Escolar, D. , Brockington, M. , Clement, E. M. , Mein, R. , Jimenez‐Mallebrera, C. , … Muntoni, F. (2006). Fukutin gene mutations in steroid‐responsive limb girdle muscular dystrophy. Annals of Neurology, 60(5), 603–610. [DOI] [PubMed] [Google Scholar]
- Goldstein, J. A. , Bogdanovich, S. , Beiriger, A. , Wren, L. M. , Rossi, A. E. , Gao, Q. Q. , … McNally, E. M. (2014). Excess SMAD signaling contributes to heart and muscle dysfunction in muscular dystrophy. Human Molecular Genetics, 23(25), 6722–6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goonasekera, S. A. , Lam, C. K. , Millay, D. P. , Sargent, M. A. , Hajjar, R. J. , Kranias, E. G. , & Molkentin, J. D. (2011). Mitigation of muscular dystrophy in mice by SERCA overexpression in skeletal muscle. The Journal of Clinical Investigation, 121(3), 1044–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudenege, S. , Lamarre, Y. , Dumont, N. , Rousseau, J. , Frenette, J. , Skuk, D. , & Tremblay, J. P. (2010). Laminin‐111: A potential therapeutic agent for Duchenne muscular dystrophy. Molecular Therapy, 18(12), 2155–2163. https://doi.org/10.1038/mt.2010.165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorevic, P. , Blankinship, M. J. , Allen, J. M. , Crawford, R. W. , Meuse, L. , Miller, D. G. , … Chamberlain, J. S. (2004). Systemic delivery of genes to striated muscles using adeno‐associated viral vectors. Nature Medicine, 10(8), 828–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumati, P. , Coletto, L. , Sabatelli, P. , Cescon, M. , Angelin, A. , Bertaggia, E. , … Bonaldo, P. (2010). Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nature Medicine, 16(11), 1313–1320. [DOI] [PubMed] [Google Scholar]
- Gutmann, L. , & Shy, M. (2015). Update on Charcot‐Marie‐Tooth disease. Current Opinion in Neurology, 28(5), 462–467. [DOI] [PubMed] [Google Scholar]
- Hakim, C. H. , Yue, Y. , Shin, J. H. , Williams, R. R. , Zhang, K. , Smith, B. F. , & Duan, D. (2014). Systemic gene transfer reveals distinctive muscle transduction profile of tyrosine mutant AAV‐1, ‐6, and ‐9 in neonatal dogs. Molecular Therapy. Methods & Clinical Development, 1, 14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammers, D. W. , Sleeper, M. M. , Forbes, S. C. , Coker, C. C. , Jirousek, M. R. , Zimmer, M. , … Sweeney, H. L. (2016). Disease‐modifying effects of orally bioavailable NF‐kappaB inhibitors in dystrophin‐deficient muscle. Journal of Clinical Investigation Insight, 1(21), e90341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper, S. Q. , Hauser, M. A. , DelloRusso, C. , Duan, D. , Crawford, R. W. , Phelps, S. F. , … Chamberlain, J. S. (2002). Modular flexibility of dystrophin: Implications for gene therapy of Duchenne muscular dystrophy. Nature Medicine, 8(3), 253–261. [DOI] [PubMed] [Google Scholar]
- He, B. , Tang, R. H. , Weisleder, N. , Xiao, B. , Yuan, Z. , Cai, C. , … Xiao, X. (2012). Enhancing muscle membrane repair by gene delivery of MG53 ameliorates muscular dystrophy and heart failure in delta‐sarcoglycan‐deficient hamsters. Molecular Therapy, 20(4), 727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heatwole, C. R. , Eichinger, K. J. , Friedman, D. I. , Hilbert, J. E. , Jackson, C. E. , Logigian, E. L. , … Moxley, R. T., 3rd (2011). Open‐label trial of recombinant human insulin‐like growth factor 1/recombinant human insulin‐like growth factor binding protein 3 in myotonic dystrophy type 1. Archives of Neurology, 68(1), 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heier, C. R. , Damsker, J. M. , Yu, Q. , Dillingham, B. C. , Huynh, T. , Van der Meulen, J. H. , … Nagaraju, K. (2013). VBP15, a novel anti‐inflammatory and membrane‐stabilizer, improves muscular dystrophy without side effects. EMBO Molecular Medicine, 5(10), 1569–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houang, E. M. , Haman, K. J. , Filareto, A. , Perlingeiro, R. C. , Bates, F. S. , Lowe, D. A. , & Metzger, J. M. (2015). Membrane‐stabilizing copolymers confer marked protection to dystrophic skeletal muscle in vivo. Molecular Therapy. Methods & Clinical Development, 2, 15042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu, P. D. , Lander, E. S. , & Zhang, F. (2014). Development and applications of CRISPR‐Cas9 for genome engineering. Cell, 157(6), 1262–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illingworth, M. A. , Main, M. , Pitt, M. , Feng, L. , Sewry, C. A. , Gunny, R. , … Robb, S. A. (2014). RYR1‐related congenital myopathy with fatigable weakness, responding to pyridostigimine. Neuromuscular Disorders, 24(8), 707–712. [DOI] [PubMed] [Google Scholar]
- Irwin, W. A. , Bergamin, N. , Sabatelli, P. , Reggiani, C. , Megighian, A. , Merlini, L. , … Bonaldo, P. (2003). Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nature Genetics, 35(4), 367–371. [DOI] [PubMed] [Google Scholar]
- Jablonka, S. , & Sendtner, M. (2017). Developmental regulation of SMN expression: Pathophysiological implications and perspectives for therapy development in spinal muscular atrophy. Gene Therapy, https://doi.org/10.1038/gt.2017.46 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- Janghra, N. , Morgan, J. E. , Sewry, C. A. , Wilson, F. X. , Davies, K. E. , Muntoni, F. , & Tinsley, J. (2016). Correlation of utrophin levels with the dystrophin protein complex and muscle fibre regeneration in duchenne and becker muscular dystrophy muscle biopsies. PLoS ONE, 11(3), e0150818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jani‐Acsadi, A. , Ounpuu, S. , Pierz, K. , & Acsadi, G. (2015). Pediatric Charcot‐Marie‐Tooth disease. Pediatric Clinics of North America, 62(3), 767–786. [DOI] [PubMed] [Google Scholar]
- Kaczmarek, A. , Schneider, S. , Wirth, B. , & Riessland, M. (2015). Investigational therapies for the treatment of spinal muscular atrophy. Expert Opin Investigational Drugs, 24(7), 867–881. [DOI] [PubMed] [Google Scholar]
- Kagiava, A. , Sargiannidou, I. , Theophilidis, G. , Karaiskos, C. , Richter, J. , Bashiardes, S. , … Kleopa, K. A. (2016). Intrathecal gene therapy rescues a model of demyelinating peripheral neuropathy. Proceedings of the National Academy of Sciences of the United States of America, 113(17), E2421–E2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, P. B. , Morrison, L. , Iannaccone, S. T. , Graham, R. J. , Bonnemann, C. G. , Rutkowski, A. , … Electrodiagnostic M. (2015). Evidence‐based guideline summary: Evaluation, diagnosis, and management of congenital muscular dystrophy: Report of the Guideline Development Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Neurology, 84(13), 1369–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemaladewi, D. U. , Maino, E. , Hyatt, E. , Hou, H. , Ding, M. , Place, K. M. , … Cohn, R. D. (2017). Correction of a splicing defect in a mouse model of congenital muscular dystrophy type 1A using a homology‐directed‐repair‐independent mechanism. Nature Medicine, 23(8), 984–989. [DOI] [PubMed] [Google Scholar]
- Kendall, G. C. , Mokhonova, E. I. , Moran, M. , Sejbuk, N. E. , Wang, D. W. , Silva, O. , … Miceli, M. C. (2012). Dantrolene enhances antisense‐mediated exon skipping in human and mouse models of Duchenne muscular dystrophy. Science Translational Medicine, 4(164), 164ra160. [DOI] [PubMed] [Google Scholar]
- Ketel, K. , Krauss, M. , Nicot, A. S. , Puchkov, D. , Wieffer, M. , Muller, R. , & Haucke, V. (2016). A phosphoinositide conversion mechanism for exit from endosomes. Nature, 529(7586), 408–412. [DOI] [PubMed] [Google Scholar]
- Kinali, M. , Beeson, D. , Pitt, M. C. , Jungbluth, H. , Simonds, A. K. , Aloysius, A. , … Robb, S. A. (2008). Congenital myasthenic syndromes in childhood: Diagnostic and management challenges. Journal of Neuroimmunology, 201–202, 6 –12. [DOI] [PubMed] [Google Scholar]
- Kinali, M. , Mercuri, E. , Main, M. , De Biasia, F. , Karatza, A. , Higgins, R. , … Muntoni, F. (2002). Pilot trial of albuterol in spinal muscular atrophy. Neurology, 59(4), 609–610. [DOI] [PubMed] [Google Scholar]
- Kissel, J. T. , Elsheikh, B. , King, W. M. , Freimer, M. , Scott, C. B. , Kolb, S. J. , … Project Cure Spinal Muscular Atrophy Investigators N. (2014). SMA valiant trial: A prospective, double‐blind, placebo‐controlled trial of valproic acid in ambulatory adults with spinal muscular atrophy. Muscle and Nerve, 49(2), 187––192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissel, J. T. , Scott, C. B. , Reyna, S. P. , Crawford, T. O. , Simard, L. R. , Krosschell, K. J. , … Project Cure Spinal Muscular Atrophy Investigators N. (2011). SMA CARNIVAL TRIAL PART II: a prospective, single‐armed trial of L‐carnitine and valproic acid in ambulatory children with spinal muscular atrophy. PLoS ONE, 6(7), e21296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komaki, H. , Hayashi, Y. K. , Tsuburaya, R. , Sugie, K. , Kato, M. , Nagai, T. , … Nishino, I. (2011). Inflammatory changes in infantile‐onset LMNA‐associated myopathy. Neuromuscular Disorders, 21(8), 563–568. [DOI] [PubMed] [Google Scholar]
- Kornegay, J. N. , Li, J. , Bogan, J. R. , Bogan, D. J. , Chen, C. , Zheng, H. , … Xiao, X. (2010). Widespread muscle expression of an AAV9 human mini‐dystrophin vector after intravenous injection in neonatal dystrophin‐deficient dogs. Molecular Therapy, 18(8), 1501 –1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutsikos, D. , Agroyannis, B. , & Tzanatos‐Exarchou, H. (1990). Biotin for diabetic peripheral neuropathy. Biomedicine and Pharmacotherapy, 44(10), 511–514. [DOI] [PubMed] [Google Scholar]
- Kuntz N., Finkel R. S., Mercuri E, et al. (2017). Final results of the phase 3 endear study assessing the efficacy and safety of nusinersen in infants with Spinal Muscular Atrophy (SMA). Presentation at: 69th annual meeting of the American Academy of Neurology; April 22–28, 2017; Boston, MA. PIPE‐15727.
- Lai, Y. , Li, D. , Yue, Y. , & Duan, D. (2008). Design of trans‐splicing adeno‐associated viral vectors for Duchenne muscular dystrophy gene therapy. Methods in Molecular Biology, 433, 259–275. [DOI] [PubMed] [Google Scholar]
- Lashley, D. , Palace, J. , Jayawant, S. , Robb, S. , & Beeson, D. (2010). Ephedrine treatment in congenital myasthenic syndrome due to mutations in DOK7. Neurology, 74(19), 1517–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau, J. K. , Sy, R. W. , Corbett, A. , & Kritharides, L. (2015). Myotonic dystrophy and the heart: A systematic review of evaluation and management. International Journal of Cardiology, 184, 600–608. [DOI] [PubMed] [Google Scholar]
- Lawlor, M. W. , Armstrong, D. , Viola, M. G. , Widrick, J. J. , Meng, H. , Grange, R. W. , … Beggs, A. H. (2013). Enzyme replacement therapy rescues weakness and improves muscle pathology in mice with X‐linked myotubular myopathy. Human Molecular Genetics, 22(8), 1525–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le, T. T. , McGovern, V. L. , Alwine, I. E. , Wang, X. , Massoni‐Laporte, A. , Rich, M. M. , & Burghes, A. H. (2011). Temporal requirement for high SMN expression in SMA mice. Human Molecular Genetics, 20(18), 3578–3591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre, S. , Burglen, L. , Reboullet, S. , Clermont, O. , Burlet, P. , Viollet, L. , Benichou, B. , Cruaud, C. , Millasseau, P. , Zeviani, M. , et al. (1995). Identification and characterization of a spinal muscular atrophy‐determining gene. Cell, 80(1), 155–165. [DOI] [PubMed] [Google Scholar]
- Liewluck, T. , Selcen, D. , & Engel, A. G. (2011). Beneficial effects of albuterol in congenital endplate acetylcholinesterase deficiency and Dok‐7 myasthenia. Muscle and Nerve, 44(5), 789–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, X. , Miller, J. W. , Mankodi, A. , Kanadia, R. N. , Yuan, Y. , Moxley, R. T. , … Thornton, C. A. (2006). Failure of MBNL1‐dependent post‐natal splicing transitions in myotonic dystrophy. Human Molecular Genetics, 15(13), 2087–2097. [DOI] [PubMed] [Google Scholar]
- Logigian, E. L. , Martens, W. B. , Moxley, R. T. , McDermott, M. P. , Dilek, N. , Wiegner, A. W. , … Moxley, R. T., 3rd (2010). Mexiletine is an effective antimyotonia treatment in myotonic dystrophy type 1. Neurology, 74(18), 1441–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long, C. , Amoasii, L. , Mireault, A. A. , McAnally, J. R. , Li, H. , Sanchez‐Ortiz, E. , … Olson, E. N. (2016). Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science, 351(6271), 400–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magri, F. , Nigro, V. , Angelini, C. , Mongini, T. , Mora, M. , Moroni, I. , … Comi, G. P. (2016). The Italian LGMD registry: Relative frequency, clinical features, and differential diagnosis. Muscle and Nerve, 55(1), 55–68. [DOI] [PubMed] [Google Scholar]
- Mahadevan, M. S. , Yadava, R. S. , Yu, Q. , Balijepalli, S. , Frenzel‐McCardell, C. D. , Bourne, T. D. , & Phillips, L. H. (2006). Reversible model of RNA toxicity and cardiac conduction defects in myotonic dystrophy. Nature Genetics, 38(9), 1066–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankodi, A. , Logigian, E. , Callahan, L. , McClain, C. , White, R. , Henderson, D. , … Thornton, C. A. (2000). Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science, 289(5485), 1769–1773. [DOI] [PubMed] [Google Scholar]
- Markham, B. E. , Kernodle, S. , Nemzek, J. , Wilkinson, J. E. , & Sigler, R. (2015). Chronic dosing with membrane Sealant Poloxamer 188 NF improves respiratory dysfunction in Dystrophic Mdx and Mdx/Utrophin‐/‐ mice. PLoS ONE, 10(8), e0134832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathis, S. , Magy, L. , & Vallat, J. M. (2015). Therapeutic options in Charcot‐Marie‐Tooth diseases. Expert Review of Neurotherapeutics, 15(4), 355–366. [DOI] [PubMed] [Google Scholar]
- McDonald, C. M. , Campbell, C. , Torricelli, R. E. , Finkel, R. S. , Flanigan, K. M. , Goemans, N. , … Group ADS. (2017). Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): a multicentre, randomised, double‐blind, placebo‐controlled, phase 3 trial. The Lancet, pii: S0140–6736(17), 31611–31612. [DOI] [PubMed] [Google Scholar]
- McElroy, S. P. , Nomura, T. , Torrie, L. S. , Warbrick, E. , Gartner, U. , Wood, G. , & McLean, W. H. (2013). A lack of premature termination codon read‐through efficacy of PTC124 (Ataluren) in a diverse array of reporter assays. PLoS Biology, 11(6), e1001593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGreevy, J. W. , Hakim, C. H. , McIntosh, M. A. , & Duan, D. (2015). Animal models of Duchenne muscular dystrophy: From basic mechanisms to gene therapy. Disease Models & Mechanisms, 8(3), 195–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meilleur, K. G. , Jain, M. S. , Hynan, L. S. , Shieh, C. Y. , Kim, E. , Waite, M. , … Bonnemann, C. G. (2015). Results of a two‐year pilot study of clinical outcome measures in collagen VI‐ and laminin alpha2‐related congenital muscular dystrophies. Neuromuscular Disorders, 25(1), 43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinen, S. , Lin, S. , & Ruegg, M. A. (2012). Angiotensin II type 1 receptor antagonists alleviate muscle pathology in the mouse model for laminin‐alpha2‐deficient congenital muscular dystrophy (MDC1A). Skelet Muscle, 2(1), 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinen, S. , Lin, S. , Thurnherr, R. , Erb, M. , Meier, T. , & Ruegg, M. A. (2011). Apoptosis inhibitors and mini‐agrin have additive benefits in congenital muscular dystrophy mice. EMBO Molecular Medicine, 3(8), 465–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejat, A. , Decostre, V. , Li, J. , Renou, L. , Kesari, A. , Hantai, D. , … Misteli, T. (2009). Lamin A/C‐mediated neuromuscular junction defects in Emery‐Dreifuss muscular dystrophy. The Journal of Cell Biology, 184(1), 31–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell, J. R. , Goemans, N. , Lowes, L. P. , Alfano, L. N. , Berry, K. , Shao, J. , … Telethon Foundation DMDIN. (2016). Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Annals of Neurology, 79(2), 257–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell, J. R. , Rodino‐Klapac, L. R. , Rosales‐Quintero, X. , Kota, J. , Coley, B. D. , Galloway, G. , … Clark, K. R. (2009). Limb‐girdle muscular dystrophy type 2D gene therapy restores alpha‐sarcoglycan and associated proteins. Annals of Neurology, 66(3), 290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell, et al. (2017). AVXS‐101 Clinical Update Presented at: 69th Annual Meeting of the American Academy of Neurology; April 22–28, 2017; Boston, MA.
- Mercuri, E. , Bertini, E. , Messina, S. , Solari, A. , D'Amico, A. , Angelozzi, C. , … Brahe, C. (2007). Randomized, double‐blind, placebo‐controlled trial of phenylbutyrate in spinal muscular atrophy. Neurology, 68(1), 51–55. [DOI] [PubMed] [Google Scholar]
- Mercuri, E. , & Muntoni, F. (2012). The ever‐expanding spectrum of congenital muscular dystrophies. Annals of Neurology, 72(1), 9–17. [DOI] [PubMed] [Google Scholar]
- Mercuri, E. , & Muntoni, F. (2013). Muscular dystrophies. The Lancet, 381(9869), 845–860. [DOI] [PubMed] [Google Scholar]
- Mercuri E., Finkel R. S., & Kirschner J., et al. (2017). Efficacy and Safety of nusinersen in children with later‐onset spinal muscular atrophy (SMA): end of study results from the phase 3 CHERISH study. Presented at: 69th Annual Meeting of the American Academy of Neurology; April 22–28, 2017; Boston, MA. ES1‐009.
- Merlini, L. , Angelin, A. , Tiepolo, T. , Braghetta, P. , Sabatelli, P. , Zamparelli, A. , … Bernardi, P. (2008). Cyclosporin A corrects mitochondrial dysfunction and muscle apoptosis in patients with collagen VI myopathies. Proceedings of the National Academy of Sciences of the United States of America, 105(13), 5225–5229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messina, S. , Hartley, L. , Main, M. , Kinali, M. , Jungbluth, H. , Muntoni, F. , & Mercuri, E. (2004). Pilot trial of salbutamol in central core and multi‐minicore diseases. Neuropediatrics, 35(5), 262–266. [DOI] [PubMed] [Google Scholar]
- Meyer zu Horste, G. , Prukop, T. , Liebetanz, D. , Mobius, W. , Nave, K. A. , & Sereda, M. W. (2007). Antiprogesterone therapy uncouples axonal loss from demyelination in a transgenic rat model of CMT1A neuropathy. Annals of Neurology, 61(1), 61–72. [DOI] [PubMed] [Google Scholar]
- Micallef, J. , Attarian, S. , Dubourg, O. , Gonnaud, P. M. , Hogrel, J. Y. , Stojkovic, T. , … Blin, O. (2009). Effect of ascorbic acid in patients with Charcot‐Marie‐Tooth disease type 1A: a multicentre, randomised, double‐blind, placebo‐controlled trial. Lancet Neurology, 8(12), 1103–1110. [DOI] [PubMed] [Google Scholar]
- Miller, J. B. , & Girgenrath, M. (2006). The role of apoptosis in neuromuscular diseases and prospects for anti‐apoptosis therapy. Trends in Molecular Medicine, 12(6), 279–286. [DOI] [PubMed] [Google Scholar]
- Moll, J. , Barzaghi, P. , Lin, S. , Bezakova, G. , Lochmuller, H. , Engvall, E. , … Ruegg, M. A. (2001). An agrin minigene rescues dystrophic symptoms in a mouse model for congenital muscular dystrophy. Nature, 413(6853), 302–307. [DOI] [PubMed] [Google Scholar]
- Muchir, A. , Wu, W. , Sera, F. , Homma, S. , & Worman, H. J. (2014). Mitogen‐activated protein kinase kinase 1/2 inhibition and angiotensin II converting inhibition in mice with cardiomyopathy caused by lamin A/C gene mutation. Biochemical and Biophysical Research Communications, 452(4), 958–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munot, P. , Lashley, D. , Jungbluth, H. , Feng, L. , Pitt, M. , Robb, S. A. , … Muntoni, F. (2010). Congenital fibre type disproportion associated with mutations in the tropomyosin 3 (TPM3) gene mimicking congenital myasthenia. Neuromuscular Disorders, 20(12), 796–800. [DOI] [PubMed] [Google Scholar]
- Narayanaswami, P. , Weiss, M. , Selcen, D. , David, W. , Raynor, E. , … Carter, G. (2014). Evidence‐based guideline summary: Diagnosis and treatment of limb‐girdle and distal dystrophies: Report of the guideline development subcommittee of the American Academy of Neurology and the practice issues review panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Neurology, 83(16), 1453–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natera‐de Benito, D. , Nascimento, A. , Abicht, A. , Ortez, C. , Jou, C. , Muller, J. S. , … Lochmuller, H. (2016). KLHL40‐related nemaline myopathy with a sustained, positive response to treatment with acetylcholinesterase inhibitors. Journal of Neurology, 263(3), 517–523. [DOI] [PubMed] [Google Scholar]
- Nelson, C. E. , Hakim, C. H. , Ousterout, D. G. , Thakore, P. I. , Moreb, E. A. , Castellanos Rivera, R. M. , … Gersbach, C. A. (2016). In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science, 351(6271), 403–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, M. A. , Joya, J. E. , Kee, A. J. , Domazetovska, A. , Yang, N. , Hook, J. W. , … Hardeman, E. C. (2011). Hypertrophy and dietary tyrosine ameliorate the phenotypes of a mouse model of severe nemaline myopathy. Brain: A Journal of Neurology, 134(Pt 12), 3516–3529. [DOI] [PubMed] [Google Scholar]
- Noguchi, S. , Ogawa, M. , Kawahara, G. , Malicdan, M. C. , & Nishino, I. (2014). Allele‐specific gene silencing of mutant mRNA restores cellular function in ullrich congenital muscular dystrophy fibroblasts. Molecular Therapy. Nucleic Acids, 3, e171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North, K. N. , Wang, C. H. , Clarke, N. , Jungbluth, H. , Vainzof, M. , Dowling, J. J. , … Bonnemann, C. G. (2014). Approach to the diagnosis of congenital myopathies. Neuromuscular Disorders, 24(2), 97–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsuka, Y. , Kanagawa, M. , Yu, C. C. , Ito, C. , Chiyo, T. , Kobayashi, K. , … Toda, T. (2015). Fukutin is prerequisite to ameliorate muscular dystrophic phenotype by myofiber‐selective LARGE expression. Scientific Reports, 5, 8316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlikowski, D. , Chevret, S. , Quera‐Salva, M. A. , Laforet, P. , Lofaso, F. , Verschueren, A. , … Annane, D. (2009). Modafinil for the treatment of hypersomnia associated with myotonic muscular dystrophy in adults: A multicenter, prospective, randomized, double‐blind, placebo‐controlled, 4‐week trial. Clinical Therapeutics, 31(8), 1765–1773. [DOI] [PubMed] [Google Scholar]
- Ousterout, D. G. , Kabadi, A. M. , Thakore, P. I. , Majoros, W. H. , Reddy, T. E. , & Gersbach, C. A. (2015). Multiplex CRISPR/Cas9‐based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nature Communications, 6, 6244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacak, C. A. , Mah, C. S. , Thattaliyath, B. D. , Conlon, T. J. , Lewis, M. A. , Cloutier, D. E. , … Byrne, B. J. (2006). Recombinant adeno‐associated virus serotype 9 leads to preferential cardiac transduction in vivo. Circulation Research, 99(4), e3–e9. [DOI] [PubMed] [Google Scholar]
- Palacino, J. , Swalley, S. E. , Song, C. , Cheung, A. K. , Shu, L. , Zhang, X. , … Sivasankaran, R. (2015). SMN2 splice modulators enhance U1‐pre‐mRNA association and rescue SMA mice. Nature Chemical Biology, 11(7), 511–517. [DOI] [PubMed] [Google Scholar]
- Pandey, S. K. , Wheeler, T. M. , Justice, S. L. , Kim, A. , Younis, H. S. , Gattis, D. , … MacLeod, A. R. (2015). Identification and characterization of modified antisense oligonucleotides targeting DMPK in mice and nonhuman primates for the treatment of myotonic dystrophy type 1. Journal of Pharmacology and Experimental Therapeutics, 355(2), 329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pane, M. , Staccioli, S. , Messina, S. , D'Amico, A. , Pelliccioni, M. , Mazzone, E. S. , … Mercuri, E. (2008). Daily salbutamol in young patients with SMA type II. Neuromuscular Disorders, 18(7), 536–540. [DOI] [PubMed] [Google Scholar]
- Pareyson, D. , Reilly, M. M. , Schenone, A. , Fabrizi, G. M. , Cavallaro, T. , Santoro, L. , … groups, C.‐T. (2011). Ascorbic acid in Charcot‐Marie‐Tooth disease type 1A (CMT‐TRIAAL and CMT‐TRAUK): a double‐blind randomised trial. Lancet Neurology, 10(4), 320–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passage, E. , Norreel, J. C. , Noack‐Fraissignes, P. , Sanguedolce, V. , Pizant, J. , Thirion, X. , … Fontes, M. (2004). Ascorbic acid treatment corrects the phenotype of a mouse model of Charcot‐Marie‐Tooth disease. Nature Medicine, 10(4), 396–401. [DOI] [PubMed] [Google Scholar]
- Patzko, A. , Bai, Y. , Saporta, M. A. , Katona, I. , Wu, X. , Vizzuso, D. , … Shy, M. E. (2012). Curcumin derivatives promote Schwann cell differentiation and improve neuropathy in R98C CMT1B mice. Brain: A Journal of Neurology, 135(Pt 12), 3551–3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearn, J. (1978). Incidence, prevalence, and gene frequency studies of chronic childhood spinal muscular atrophy. Journal of Medical Genetics, 15(6), 409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltz, S. W. , Morsy, M. , Welch, E. M. , & Jacobson, A. (2013). Ataluren as an agent for therapeutic nonsense suppression. Annual Review of Medicine, 64, 407–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penisson‐Besnier, I. , Devillers, M. , Porcher, R. , Orlikowski, D. , Doppler, V. , Desnuelle, C. , … Annane, D. (2008). Dehydroepiandrosterone for myotonic dystrophy type 1. Neurology, 71(6), 407–412. [DOI] [PubMed] [Google Scholar]
- Peyer, A. K. , Abicht, A. , Heinimann, K. , Sinnreich, M. , & Fischer, D. (2013). Quinine sulfate as a therapeutic option in a patient with slow channel congenital myasthenic syndrome. Neuromuscular Disorders, 23(7), 571–574. [DOI] [PubMed] [Google Scholar]
- Puymirat, J. , Bouchard, J. P. , & Mathieu, J. (2012). Efficacy and tolerability of a 20‐mg dose of methylphenidate for the treatment of daytime sleepiness in adult patients with myotonic dystrophy type 1: a 2‐center, randomized, double‐blind, placebo‐controlled, 3‐week crossover trial. Clinical Therapeutics, 34(5), 1103–1111. [DOI] [PubMed] [Google Scholar]
- Quijano‐Roy, S. , Mbieleu, B. , Bonnemann, C. G. , Jeannet, P. Y. , Colomer, J. , Clarke, N. F. , … Estournet, B. (2008). De novo LMNA mutations cause a new form of congenital muscular dystrophy. Annals of Neurology, 64(2), 177–186. [DOI] [PubMed] [Google Scholar]
- Ramos, F. J. , Chen, S. C. , Garelick, M. G. , Dai, D. F. , Liao, C. Y. , Schreiber, K. H. , … Kennedy, B. K. (2012). Rapamycin reverses elevated mTORC1 signaling in lamin A/C‐deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci Transl Med, 4(144), 144ra103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravenscroft, G. , Laing, N. G. , & Bönnemann, C. G. (2015). Pathophysiological concepts in the congenital myopathies: Blurring the boundaries, sharpening the focus. Brain, 138(2), 246–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rederstorff, M. , Castets, P. , Arbogast, S. , Laine, J. , Vassilopoulos, S. , Beuvin, M. , … Lescure, A. (2011). Increased muscle stress‐sensitivity induced by selenoprotein N inactivation in mouse: A mammalian model for SEPN1‐related myopathy. PLoS ONE, 6(8), e23094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhard, J. R. , Lin, S. , McKee, K. K. , Meinen, S. , Crosson, S. C. , Sury, M. , … Ruegg, M. A. (2017). Linker proteins restore basement membrane and correct LAMA2‐related muscular dystrophy in mice. Science Translational Medicine, 9(396). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricotti, V. , Spinty, S. , Roper, H. , Hughes, I. , Tejura, B. , Robinson, N. , … Tinsley, J. (2016). Safety, tolerability, and pharmacokinetics of SMT C1100, a 2‐Arylbenzoxazole utrophin modulator, following single‐ and multiple‐Dose administration to pediatric patients with duchenne muscular dystrophy. PLoS ONE, 11(4), e0152840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robb, S. A. , Sewry, C. A. , Dowling, J. J. , Feng, L. , Cullup, T. , Lillis, S. , … Muntoni, F. (2011). Impaired neuromuscular transmission and response to acetylcholinesterase inhibitors in centronuclear myopathies. Neuromuscular Disorders, 21(6), 379–386. [DOI] [PubMed] [Google Scholar]
- Rodriguez Cruz, P. M. , Palace, J. , Ramjattan, H. , Jayawant, S. , Robb, S. A. , & Beeson, D. (2015). Salbutamol and ephedrine in the treatment of severe AChR deficiency syndromes. Neurology, 85(12), 1043–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez Cruz, P. M. , Sewry, C. , Beeson, D. , Jayawant, S. , Squier, W. , McWilliam, R. , & Palace, J. (2014). Congenital myopathies with secondary neuromuscular transmission defects; a case report and review of the literature. Neuromuscular Disorders, 24(12), 1103–1110. [DOI] [PubMed] [Google Scholar]
- Romitti, P. A. , Zhu, Y. , Puzhankara, S. , James, K. A. , Nabukera, S. K. , Zamba, G. K. , … STARnet, M. D. (2015). Prevalence of Duchenne and Becker muscular dystrophies in the United States. Pediatrics, 135(3), 513–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney, J. E. , Knapp, J. R. , Hodges, B. L. , Wuebbles, R. D. , & Burkin, D. J. (2012). Laminin‐111 protein therapy reduces muscle pathology and improves viability of a mouse model of merosin‐deficient congenital muscular dystrophy. The American Journal of Pathology, 180(4), 1593–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg, H. , Davis, M. , James, D. , Pollock, N. , & Stowell, K. (2007). Malignant hyperthermia. Orphanet Journal of Rare Diseases, 2, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutledge, E. A. , Halbert, C. L. , & Russell, D. W. (1998). Infectious clones and vectors derived from adeno‐associated virus (AAV) serotypes other than AAV type 2. Journal of Virology, 72(1), 309–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan, M. M. , Sy, C. , Rudge, S. , Ellaway, C. , Ketteridge, D. , Roddick, L. G. , … North, K. N. (2008). Dietary L‐tyrosine supplementation in nemaline myopathy. Journal of Child Neurology, 23(6), 609–613. [DOI] [PubMed] [Google Scholar]
- Ryan, N. J. (2014). Ataluren: First global approval. Drugs, 74(14), 1709–1714. [DOI] [PubMed] [Google Scholar]
- Sabha, N. , Volpatti, J. R. , Gonorazky, H. , Reifler, A. , Davidson, A. E. , Li, X. , … Dowling, J. J. (2016). PIK3C2B inhibition improves function and prolongs survival in myotubular myopathy animal models. The Journal of Clinical Investigation, 126(9), 3613–3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahenk, Z. , Galloway, G. , Clark, K. R. , Malik, V. , Rodino‐Klapac, L. R. , Kaspar, B. K. , … Mendell, J. R. (2014). AAV1.NT‐3 gene therapy for charcot‐marie‐tooth neuropathy. Molecular Therapy, 22(3), 511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahenk, Z. , Nagaraja, H. N. , McCracken, B. S. , King, W. M. , Freimer, M. L. , Cedarbaum, J. M. , & Mendell, J. R. (2005). NT‐3 promotes nerve regeneration and sensory improvement in CMT1A mouse models and in patients. Neurology, 65(5), 681–689. [DOI] [PubMed] [Google Scholar]
- Saito, F. , Kanagawa, M. , Ikeda, M. , Hagiwara, H. , Masaki, T. , Ohkuma, H. , … Matsumura, K. (2014). Overexpression of LARGE suppresses muscle regeneration via down‐regulation of insulin‐like growth factor 1 and aggravates muscular dystrophy in mice. Human Molecular Genetics, 23(17), 4543–4558. [DOI] [PubMed] [Google Scholar]
- Sanmaneechai, O. , Feely, S. , Scherer, S. S. , Herrmann, D. N. , Burns, J. , … Muntoni, F. (2015). Genotype‐phenotype characteristics and baseline natural history of heritable neuropathies caused by mutations in the MPZ gene. Brain: A Journal of Neurology, 138(Pt 11), 3180–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saporta, A. S. , Sottile, S. L. , Miller, L. J. , Feely, S. M. , Siskind, C. E. , & Shy, M. E. (2011). Charcot–Marie–Tooth disease subtypes and genetic testing strategies. Annals of Neurology, 69(1), 22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharner, J. , Figeac, N. , Ellis, J. A. , & Zammit, P. S. (2015). Ameliorating pathogenesis by removing an exon containing a missense mutation: A potential exon‐skipping therapy for laminopathies. Gene Therapy, 22(6), 503–515. [DOI] [PubMed] [Google Scholar]
- Shin, J. H. , Pan, X. , Hakim, C. H. , Yang, H. T. , Yue, Y. , Zhang, K. , … Duan, D. (2013). Microdystrophin ameliorates muscular dystrophy in the canine model of duchenne muscular dystrophy. Molecular Therapy, 21(4), 750–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, P. , Liew, W. K. , & Darras, B. T. (2013). Current advances in drug development in spinal muscular atrophy. Current Opinion in Pediatrics, 25(6), 682–688. [DOI] [PubMed] [Google Scholar]
- Skre, H. (1974). Genetic and clinical aspects of Charcot‐Marie‐Tooth's disease. Clinical Genetics, 6(2), 98–118. [DOI] [PubMed] [Google Scholar]
- Sman, A. D. , Hackett, D. , Fiatarone Singh, M. , Fornusek, C. , Menezes, M. P. , & Burns, J. (2015). Systematic review of exercise for Charcot‐Marie‐Tooth disease. Journal of the Peripheral Nervous System, 20(4), 347–362. [DOI] [PubMed] [Google Scholar]
- Straub, V. , & Bushby, K. (2006). The childhood limb‐girdle muscular dystrophies. Seminars in Pediatric Neurology, 13(2), 104–114. [DOI] [PubMed] [Google Scholar]
- Swoboda, K. J. , Scott, C. B. , Crawford, T. O. , Simard, L. R. , Reyna, S. P. , Krosschell, K. J. , … Project Cure Spinal Muscular Atrophy Investigators N. (2010). SMA CARNI‐VAL trial part I: Double‐blind, randomized, placebo‐controlled trial of L‐carnitine and valproic acid in spinal muscular atrophy. PLoS ONE, 5(8), e12140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabebordbar, M. , Zhu, K. , Cheng, J. K. , Chew, W. L. , Widrick, J. J. , Yan, W. X. , … Wagers, A. J. (2016). In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science, 351(6271), 407–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi‐Ikeda, M. , Kobayashi, K. , Kanagawa, M. , Yu, C. C. , Mori, K. , Oda, T. , … Toda, T. (2011). Pathogenic exon‐trapping by SVA retrotransposon and rescue in Fukuyama muscular dystrophy. Nature, 478(7367), 127–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarnopolsky, M. A. (2007). Clinical use of creatine in neuromuscular and neurometabolic disorders. Sub‐Cellular Biochemistry, 46, 183–204. [DOI] [PubMed] [Google Scholar]
- Tasfaout, H. , Buono, S. , Guo, S. , Kretz, C. , Messaddeq, N. , Booten, S. , … Laporte, J. (2017). Antisense oligonucleotide‐mediated Dnm2 knockdown prevents and reverts myotubular myopathy in mice. Nature Communications, 8, 15661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tei, S. , Ishii, H. T. , Mitsuhashi, H. , & Ishiura, S. (2015). Antisense oligonucleotide‐mediated exon skipping of CHRNA1 pre‐mRNA as potential therapy for Congenital Myasthenic Syndromes. Biochemical and Biophysical Research Communications, 461(3), 481–486. [DOI] [PubMed] [Google Scholar]
- Telfer, W. R. , Busta, A. S. , Bonnemann, C. G. , Feldman, E. L. , & Dowling, J. J. (2010). Zebrafish models of collagen VI‐related myopathies. Human Molecular Genetics, 19(12), 2433–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner, C. , & Hilton‐Jones, D. (2014). Myotonic dystrophy: Diagnosis, management and new therapies. Current Opinion in Neurology, 27(5), 599–606. [DOI] [PubMed] [Google Scholar]
- van Engelen, B. , & Consortium, O. (2015). Cognitive behaviour therapy plus aerobic exercise training to increase activity in patients with myotonic dystrophy type 1 (DM1) compared to usual care (OPTIMISTIC): study protocol for randomised controlled trial. Trials, 16, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Ry P. M., Minogue, P. , Hodges, B. L. , & Burkin, D. J. (2014). Laminin‐111 improves muscle repair in a mouse model of merosin‐deficient congenital muscular dystrophy. Human Molecular Genetics, 23(2), 383–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Westering, T. L. , Betts, C. A. , & Wood, M. J. (2015). Current understanding of molecular pathology and treatment of cardiomyopathy in duchenne muscular dystrophy. Molecules, 20(5), 8823–8855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetrone, S. A. , Montecino‐Rodriguez, E. , Kudryashova, E. , Kramerova, I. , Hoffman, E. P. , … Spencer, M. J. (2009). Osteopontin promotes fibrosis in dystrophic mouse muscle by modulating immune cell subsets and intramuscular TGF‐beta. The Journal of Clinical Investigation, 119(6), 1583–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voit, T. , Topaloglu, H. , Straub, V. , Muntoni, F. , Deconinck, N. , Campion, G. , … Kraus, J. E. (2014). Safety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): an exploratory, randomised, placebo‐controlled phase 2 study. Lancet Neurology, 13(10), 987–996. [DOI] [PubMed] [Google Scholar]
- Wang, B. , Li, J. , & Xiao, X. (2000). Adeno‐associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model. Proceedings of the National Academy of Sciences of the United States of America, 97(25), 13714–13719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, C. H. , Dowling, J. J. , North, K. , Schroth, M. K. , Sejersen, T. , Shapiro, F. , … Yuan, N. (2012). Consensus statement on standard of care for congenital myopathies. Journal of Child Neurology, 27(3), 363–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, C. H. , Finkel, R. S. , Bertini, E. S. , Schroth, M. , Simonds, A. , … Wong, B. (2007). Consensus statement for standard of care in spinal muscular atrophy. Journal of Child Neurology, 22(8), 1027–1049. [DOI] [PubMed] [Google Scholar]
- Weisleder, N. , Takizawa, N. , Lin, P. , Wang, X. , Cao, C. , Zhang, Y. , … Ma, J. (2012). Recombinant MG53 protein modulates therapeutic cell membrane repair in treatment of muscular dystrophy. Science Translational Medicine, 4(139), 139ra185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmore, C. , Fernandez‐Fuente, M. , Booler, H. , Parr, C. , Kavishwar, M. , Ashraf, A. , … Brown, S. C. (2014). The transgenic expression of LARGE exacerbates the muscle phenotype of dystroglycanopathy mice. Human Molecular Genetics, 23(7), 1842–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, L. L. , O'Dougherty, M. M. , Wright, F. S. , Bobulski, R. J. , & Horrocks, L. A. (1986). Dietary essential fatty acids, vitamin E, and Charcot‐Marie‐Tooth disease. Neurology, 36(9), 1200–1205. [DOI] [PubMed] [Google Scholar]
- Wilton, S. D. , Dye, D. E. , Blechynden, L. M. , & Laing, N. G. (1997). Revertant fibres: A possible genetic therapy for Duchenne muscular dystrophy? Neuromuscular Disorders, 7(5), 329–335. [DOI] [PubMed] [Google Scholar]
- Wilton, S. D. , Veedu, R. N. , & Fletcher, S. (2015). The emperor's new dystrophin: Finding sense in the noise. Trends in Molecular Medicine, 21(7), 417–426. [DOI] [PubMed] [Google Scholar]
- Wojtal, D. , Kemaladewi, D. U. , Malam, Z. , Abdullah, S. , Wong, T. W. , Hyatt, E. , … Cohn, R. D. (2016). Spell checking nature: Versatility of CRISPR/Cas9 for developing treatments for inherited disorders. American Journal of Human Genetics, 98(1), 90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtkowiak‐Szlachcic, A. , Taylor, K. , Stepniak‐Konieczna, E. , Sznajder, L. J. , Mykowska, A. , Sroka, J. , … Sobczak, K. (2015). Short antisense‐locked nucleic acids (all‐LNAs) correct alternative splicing abnormalities in myotonic dystrophy. Nucleic Acids Research, 43(6), 3318–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worman, H. J. , & Bonne, G. (2007). Laminopathies”: a wide spectrum of human diseases. Experimental Cell Research, 313(10), 2121–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, L. , Lu, P. J. , Wang, C. H. , Keramaris, E. , Qiao, C. , Xiao, B. , … Lu, Q. L. (2013). Adeno‐associated virus 9 mediated FKRP gene therapy restores functional glycosylation of alpha‐dystroglycan and improves muscle functions. Molecular Therapy, 21(10), 1832–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung, W. L. , Lam, C. W. , & Ng, P. C. (2010). Intra‐familial variation in clinical manifestations and response to ephedrine in siblings with congenital myasthenic syndrome caused by novel COLQ mutations. Developmental Medicine and Child Neurology, 52(10), e243–e244. [DOI] [PubMed] [Google Scholar]
- Yoshimura, M. , Sakamoto, M. , Ikemoto, M. , Mochizuki, Y. , Yuasa, K. , Miyagoe‐Suzuki, Y. , & Takeda, S. (2004). AAV vector‐mediated microdystrophin expression in a relatively small percentage of mdx myofibers improved the mdx phenotype. Molecular Therapy, 10(5), 821–828. [DOI] [PubMed] [Google Scholar]
- Yu, M. , He, Y. , Wang, K. , Zhang, P. , Zhang, S. , & Hu, H. (2013). Adeno‐associated viral‐mediated LARGE gene therapy rescues the muscular dystrophic phenotype in mouse models of dystroglycanopathy. Human Gene Therapy, 24(3), 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, Q. , Sali, A. , Van der Meulen, J. , Creeden, B. K. , Gordish‐Dressman, H. , Rutkowski, A. , … Spurney, C. F. (2013). Omigapil treatment decreases fibrosis and improves respiratory rate in dy(2J) mouse model of congenital muscular dystrophy. PLoS ONE, 8(6), e65468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue, Y. , Li, Z. , Harper, S. Q. , Davisson, R. L. , Chamberlain, J. S. , & Duan, D. (2003). Microdystrophin gene therapy of cardiomyopathy restores dystrophin‐glycoprotein complex and improves sarcolemma integrity in the mdx mouse heart. Circulation, 108(13), 1626–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, W. , Wang, Y. , Dong, S. , Choudhury, R. , Jin, Y. , & Wang, Z. (2014). Treatment of type 1 myotonic dystrophy by engineering site‐specific RNA endonucleases that target (CUG)(n) repeats. Molecular Therapy, 22(2), 312–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, X. , Feng, Z. , Ling, K. K. , Mollin, A. , Sheedy, J. , Yeh, S. , … Weetall, M. (2016). Pharmacokinetics, pharmacodynamics, and efficacy of a small‐molecule SMN2 splicing modifier in mouse models of spinal muscular atrophy. Human Molecular Genetics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, H. , Grajales‐Reyes, G. E. , Alicea‐Vazquez, V. , Grajales‐Reyes, J. G. , Robinson, K. , Pytel, P. , … Gomez, C. M. (2015). Fluoxetine is neuroprotective in slow‐channel congenital myasthenic syndrome. Experimental Neurology, 270, 88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]