

Abstract
Background and Purpose
Chemokines and their receptors form an intricate interaction and signalling network that plays critical roles in various physiological and pathological cellular processes. The high promiscuity and apparent redundancy of this network makes probing individual chemokine/receptor interactions and functional effects, as well as targeting individual receptor axes for therapeutic applications, challenging. Despite poor sequence identity, the N‐terminal regions of chemokines, which play a key role in their activity and selectivity, contain several conserved features. Thus far little is known regarding the molecular basis of their interactions with typical and atypical chemokine receptors or the conservation of their contributions across chemokine‐receptor pairs.
Experimental Approach
We used a broad panel of chemokine variants and modified peptides derived from the N‐terminal region of chemokines CXCL12, CXCL11 and vCCL2, to compare the contributions of various features to binding and activation of their shared receptors, the two typical, canonical G protein‐signalling receptors, CXCR4 and CXCR3, as well as the atypical scavenger receptor CXCR7/ACKR3, which shows exclusively arrestin‐dependent activity.
Key Results
We provide molecular insights into the plasticity of the ligand‐binding pockets of these receptors, their chemokine binding modes and their activation mechanisms. Although the chemokine N‐terminal region is a critical determinant, neither the most proximal residues nor the N‐loop are essential for binding and activation of ACKR3, as distinct from binding and activation of CXCR4 and CXCR3.
Conclusion and Implications
These results suggest a different interaction mechanism between this atypical receptor and its ligands and illustrate its strong propensity to activation.
Abbreviations
- ACKR
atypical chemokine receptor
- CRS
chemokine recognition site
- ECL
extracellular loop
- HIV
human immunodeficiency virus
- Rluc
Renilla luciferase
- TM
transmembrane segment
- vCCL2
viral CCL2
- YFP
yellow fluorescent protein
Introduction
http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=950 are a superfamily of small (7–12 kDa), secreted, chemo‐attractant cytokines that regulate vital cellular mechanisms including migration, adhesion and growth and survival (Rossi and Zlotnik, 2000). They play critical roles in many physiological and pathological processes including immune responses and surveillance, development, atherosclerosis, human immunodeficiency virus (HIV) infection and cancer (Balkwill, 2004; Romagnani et al., 2004). Despite their low sequence similarity, all chemokines display a common fold consisting of a flexible N terminus followed by a conserved cysteine motif, an N‐loop, three anti‐parallel β‐strands and a C‐terminal α‐helix (Fernandez and Lolis, 2002; Allen et al., 2007) (Figure 1D). The biological effects of chemokines are mediated through specific interactions with http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=14, which belong to the superfamily of seven transmembrane http://www.guidetopharmacology.org/GRAC/ReceptorFamiliesForward?type=GPCR. To date, 47 chemokines and 19 chemokine receptors have been identified in humans (Zlotnik and Yoshie, 2012; Bachelerie et al., 2014a). The chemokine‐receptor network is highly intricate, and a given chemokine may bind to several receptors while a single chemokine receptor usually has several ligands. Based on the conserved cysteine motifs present in their N termini, chemokines are divided into four subfamilies (XC, CC, CXC and CX3C) and the receptors are named according to the subfamily of chemokines they bind (XCR, CCR, CXCR and CX3CR) (Zlotnik and Yoshie, 2012). In addition to canonical receptors, four receptors referred to as atypical chemokine receptors (ACKR1–4) can act as scavengers, regulating chemokine availability, or signal through alternative G protein‐independent pathways, further contributing to the complexity of the chemokine network (Bachelerie et al., 2014a,b).
Figure 1.
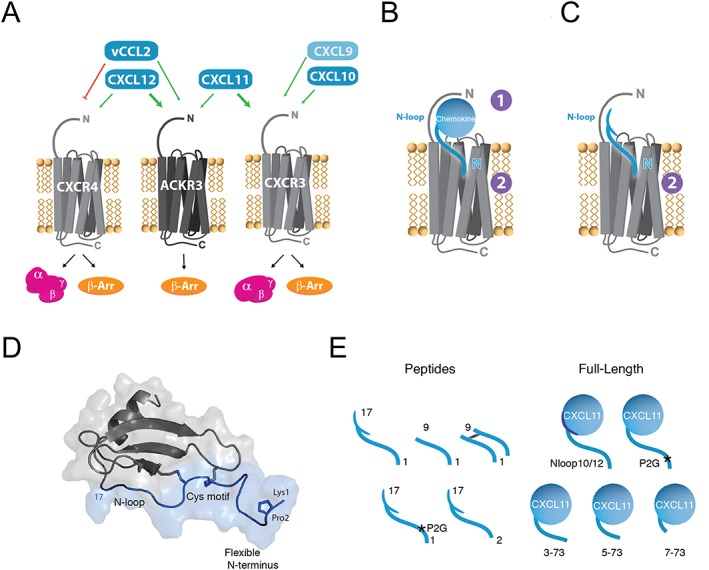
CXCR4‐ACKR3‐CXCR3 receptor‐ligand interaction network and chemokine N‐terminal features. (A) Selectivity and crosstalk between the two canonical G protein‐signalling chemokine receptors, CXCR4 and CXCR3, the atypical β‐arrestin‐biased receptor ACKR3 and their shared ligands. CXCL12 is the only endogenous chemokine ligand for CXCR4. It is also the highest affinity chemokine for ACKR3 but does not bind CXCR3. The viral chemokine vCCL2 is a CXCR4 antagonist but acts as an agonist of ACKR3. CXCL11 is the dominant ligand for CXCR3 and binds also to ACKR3. CXCL10 and CXCL9 bind and activate CXCR3, but not ACKR3. (B) Two‐site/two‐step model for the interactions of full‐length chemokines with their cognate receptors. In the first step, the body of the chemokine and the N‐loop are specifically recognized by the N terminus of the receptor (CRS1). During the second step, the insertion of the chemokine N terminus into the receptor‐binding pocket (CRS2) stabilizes its active form triggering downstream signalling. (C) Peptides derived from the N‐terminal region of chemokines, which represent useful probes to investigate the interaction between chemokines and receptors. (D) Schematic representation of CXCL12 and location of the N‐terminal features investigated in this study (blue). The N‐terminal region encompasses the flexible N terminus (1–8), the CXC cysteine motif (9–11) and the N‐loop (13–17). (E) Chemokine‐derived peptides and CXCL11 variants investigated in this study.
Because of its implication in HIV infection and in many cancers, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=71&familyId=14&familyType=GPCR is one of the most studied chemokine receptors and is often considered the model for CXC chemokine receptors as a whole (Ganju et al., 1998; Zou et al., 1998; Veldkamp et al., 2008; Wu et al., 2010; Chevigne et al., 2014). It binds a unique endogenous agonist chemokine, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4358, as well as the human herpesvirus 8‐encoded broad‐spectrum antagonist chemokine, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?tab=summary&ligandId=768 (vMIP‐II) (Kledal et al., 1997; Ganju et al., 1998; Szpakowska and Chevigne, 2015; Szpakowska et al., 2016). CXCL12 and vCCL2 are both agonists of http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=80&familyId=14&familyType=GPCR, formerly designated as CXCR7, one of the most recently deorphanized chemokine receptors (Balabanian et al., 2005; Burns et al., 2006; Szpakowska et al., 2016). In addition, ACKR3 shares one ligand, the chemokine http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=836, with http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=70&familyId=14&familyType=GPCR, to which http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=835 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=837 also bind, albeit with lower affinities than CXCL11 (Loetscher et al., 1996; Cole et al., 1998; Burns et al., 2006) (Figure 1). Unlike CXCR4 and CXCR3, which signal via both the canonical G protein pathways that modulate cAMP production and induce intracellular calcium release and the arrestin pathways, ACKR3 has been proposed to exclusively trigger arrestin‐dependent signalling (Ganju et al., 1998; Kalatskaya et al., 2009; Rajagopal et al., 2010; Sun et al., 2002). ACKR3 has also been shown to act as a scavenger receptor for CXCL12, CXCL11 and vCCL2, thus regulating their availability for other chemokine receptors (Boldajipour et al., 2008; Luker et al., 2010; Naumann et al., 2010; Berahovich et al., 2014; Szpakowska et al., 2016). Thus far however, the molecular basis accounting for its atypical functions and signalling remains unclear.
Based in part on the large amount of data regarding CXCL12 and CXCR4, the interaction between chemokines and their receptors is generally described as a multi‐step process with extensive contacts between the two partners and 1:1 stoichiometry (Crump et al., 2001; Veldkamp et al., 2008; Szpakowska et al., 2012; Kufareva et al., 2014; Qin et al., 2015). During the initial step of the interaction, the N terminus of the receptor (chemokine recognition site 1, CRS1) binds the core of the chemokine including the N‐loop region, allowing for optimal orientation with respect to the top of the ligand‐binding pocket (CRS1.5) (Qin et al., 2015). This enables the insertion of the flexible chemokine N terminus into the receptor transmembrane cavity (CRS2), stabilizing an active state of the receptor, that in turn triggers intracellular signalling (Crump et al., 1997; Veldkamp et al., 2008; Chevigne et al., 2011, 2014; Qin et al., 2015). Although chemokine receptors can function as monomeric signalling units, direct evidence suggests that they are able to form both homo‐ and heterodimers in a ligand‐independent manner (Levoye et al., 2009; Wu et al., 2010; Watts et al., 2013). Such oligomeric states were also observed for chemokines, possibly adding another level of fine tuning to the already highly intricate interplay between chemokines and receptors (Stephens and Handel, 2013; Dyer et al., 2016; Kleist et al., 2016).
The N‐terminal portion of chemokines is a key determinant of their activity and selectivity that harbors several features involved in chemokine‐receptor interactions, including the flexible N terminus, the cysteine motif and the N‐loop (Fernandez and Lolis, 2002; Allen et al., 2007; Chevigne et al., 2011). In addition, approximately one‐third of CC and CXC chemokines possess a proline in their proximal N terminus, usually at position 2, proposed to play an essential role not only in receptor activation but also in the regulation of chemokine availability through their degradation by extracellular proteases (Crump et al., 2001; Ludwig et al., 2002). Peptides derived from the N‐terminal region of CXCL12 and vCCL2 have been shown to be sufficient to specifically bind to CXCR4, while also conserving the agonist or antagonist activity of the parental chemokine (Heveker et al., 1998; Loetscher et al., 1998; Zhou et al., 2000; Chevigne et al., 2011). Notably, the introduction of further modifications to these peptides, such as mutations, truncations, dimerization or D‐amino acid replacement, emerged as a means by which to assess the importance of specific residues for receptor binding and activation, the propensity of the receptor to dimerize or the plasticity of CRS2 (Heveker et al., 2001; Zhou et al., 2002; Choi et al., 2012). Moreover, N‐terminal deletions or the P2G mutation convert peptides derived from the N terminus of CXCL12 to CXCR4 antagonists, illustrating their therapeutic potential (Crump et al., 1997; Heveker et al., 1998; Luo et al., 2000; Chevigne et al., 2011). However, with the exception of CXCR4, little is known regarding the importance of these proximal features of chemokines in receptor recognition and activation or the conservation of their contributions across chemokine‐receptor pairs. Therefore, CXCR4, ACKR3 and CXCR3 in conjunction with their ligands CXCL12, CXCL11, CXCL10, CXCL9 and vCCL2, of which some are shared or display opposite activities, offer the opportunity to investigate these questions.
In this study, using peptides derived from the N‐terminal regions of chemokines and modified full‐length chemokines, we compared the role of various N‐terminal features in their interactions with canonical receptors, CXCR3 and CXCR4, and the atypical receptor ACKR3. Our data provide insights into the plasticity of receptor ligand‐binding pockets and their activation mechanisms. Unlike CXCR4, CXCR3 and other classical chemokine receptors, ACKR3 was relatively insensitive to chemokine N‐terminal modifications maintaining a high propensity for activation. These results show that ligand recognition and activation of ACKR3 differs significantly from that of the classical chemokine receptors CXCR4 and CXCR3, suggesting that the ACKRs may employ an interaction mechanism that departs from the well‐established two‐step, two‐site model.
Methods
Cells and antibodies
HEK293E cells were obtained from Invitrogen and U87 cells from Dr. Deng and Dr. Littman through the NIH AIDS programme (Bjorndal et al., 1997). U87.CXCR3, U87.CXCR4 and U87.ACKR3 cell lines were established by Lipofectamine transfection (Life Technologies, Carlsbad, CA) of U87 cells with pBABE‐puro (Addgene, Cambridge, MA) or pcDNA3.1‐hygro (Invitrogen, Carlsbad, CA) vectors encoding the different receptors, subsequent puromycin (1 or 0.5 μg·mL−1) or hygromycin selection (250 μg·mL−1) and single‐cell sorting. For each cell line, receptor surface expression was verified by flow cytometry using monoclonal antibodies specific for ACKR3 [clones 11G8 (R&D Systems, Minneapolis, MN) and 8F11 (BioLegend, San Diego, CA)], CXCR4 [clones 4G10 (Santa Cruz Biotechnology, Dallas, TX) and 12G5 (BD Biosciences, Franklin Lakes, NJ)] and CXCR3 [clone 1C6 (BD Biosciences)].
Cloning and purification of recombinant modified CXCL11 chemokines
The N‐loop‐swapped CXCL11 chimeras, N‐terminally truncated and P2G‐mutated CXCL11 chemokines were cloned into previously described pQE30 vectors that incorporate an N‐terminal His6 and Saccharomyces cervisiae SUMO protein (Smt3) fusion tag for use in purification (Veldkamp et al., 2008; Takekoshi et al., 2012).
Recombinant wild‐type (CXCL111–73) and modified CXCL11 (CXCL11Nloop12, CXCL11Nloop10, CXCL113–73, CXCL115–73 and CXCL117–73) were purified as N‐terminal His6SUMO fusion proteins in Escherichia coli as previously described (Veldkamp et al., 2008; Takekoshi et al., 2012; Bachelerie et al., 2014a). Cells were grown in Terrific Broth and induced with 1 mM isopropyl β‐D‐1‐thiogalactopyranoside before being harvested and stored at −80°C. Cell pellets were lysed, and lysates were clarified by centrifugation (12 000× g for 20 min). The supernatant and solubilized inclusion body pellets were loaded onto Ni‐nitrilotriacetic acid resin, and after 1 h, proteins were eluted with 6 M guanidinium chloride, 50 mM Na2PO4 (pH 7.4), 300 mM NaCl, 500 mM imidazole, 0.2% sodium azide and 0.1% β‐mercaptoethanol. The eluate was pooled and refolded via dilution overnight before cleavage of the His6SUMO fusion tag by Ulp1 protease for 4 h. The His6SUMO fusion tag and chemokine were separated using cation‐exchange chromatography (SP Sepharose Fast Flow resin; GE Healthcare UK Ltd. Little Chalfont, UK) and the eluate subjected to reverse‐phase high‐performance liquid chromatography as a final purification. Proteins were frozen, lyophilized and stored at −20°C. Purification, folding and homogeneity of recombinant proteins were verified by SDS‐PAGE, matrix‐assisted laser desorption/ionization time of flight spectroscopy and 1H‐15N heteronuclear single quantum correlation NMR spectroscopy.
Binding competition assays with labelled CXCL12 and CXCL11
Binding of full‐length chemokines and peptides derived from chemokine N termini to CXCR4 and ACKR3 expressed at the surface of U87 cells was evaluated by competition with Alexa Fluor 647 (AF647)‐labelled CXCL12. For ACKR3 binding, U87.ACKR3 cells were incubated with CXCL12‐AF647 (40 ng·mL−1) and chemokines or peptides for 90 min at 4°C. CXCR4 binding was evaluated by incubation of U87.CXCR4 cells with CXCL12‐AF647 (100 ng·mL−1) and chemokines or peptides for 45 min at 37°C. All binding experiments were performed in PBS containing 1% BSA and 0.1% NaN3 (FACS buffer). Non‐specific chemokine binding was evaluated by the addition of 250‐fold excess unlabelled CXCL12 or CXCL11. Simultaneous staining with Zombie NIR™ Fixable Viability dye (BioLegend) was allowed to determine peptide cytotoxicity. Chemokine binding was quantified as the mean fluorescence intensity on a BD FACS Canto or Fortessa cytometer (BD Biosciences).
Binding of wild‐type and modified recombinant CXCL11 chemokines to CXCR3 and ACKR3 was assessed in competition studies using radiolabeled CXCL11 (Perkin Elmer, Waltham, MA). Membranes from CXCR3‐ or ACKR3‐expressing HEK293 cells were prepared as previously described (Kaya et al., 2012; Montpas et al., 2015). Binding was performed using 5 μg of membrane protein per point with either 50 pM 125I–CXCL11 or 50 pM 125I–CXCL12 (PerkinElmer). Samples were equilibrated for 2 h at 4°C prior to the separation of unbound radioligand from bound radioligand using filtration and counting.
cAMP modulation assay
U87 cells stably transfected with cAMP GloSensor 22F vector (Promega, Madison, WI) (U87.Glo) were selected using hygromycin resistance (10 μg·mL−1), and the forskolin‐induced luminescence response was assessed. U87.Glo cells were then stably transfected with pBABE or pIRES vectors encoding CXCR3, CXCR4 or ACKR3 and selected using puromycin (0.5 or 1 μg·mL−1) or hygromycin (250 μg·mL−1) respectively. Single clones were isolated by cell sorting, using the corresponding monoclonal antibodies, and further validated by flow cytometry. For cAMP modulation measurements, cells were incubated for 90 min in the dark at 37°C in phenol red‐free DMEM containing IBMX (500 μM) and 2% luciferin (GloSensor reagent; Promega); 15 × 104 cells per well were distributed onto white 96‐well LumitracTM plates (Greiner, Kremsmünster, Austria) already containing chemokines or peptides at different concentrations in phenol red‐free DMEM containing IBMX (500 μM) and 2% luciferin. Luminescence was recorded at different time points under forskolin‐free conditions (Gilissen et al., 2015) using a POLARstar Omega (BMG LABTECH, Ortenberg, Germany).
cAMP modulation induced by CXCL11 variants was assessed as previously described (Leduc et al., 2009). HEK293E cells transiently expressing the BRET reporter fusion construct protein GFP10‐Epac‐Rluc3 and either CXCR3 or ACKR3 were seeded onto poly‐D‐lysine‐coated 96‐well plates. At 48 h post‐transfection, culture medium was replaced with PBS supplemented with 0.1% BSA, 20 μM forskolin (Sigma‐Aldrich, St Louis, MO), and cells stimulated with chemokines. cAMP modulation was measured after 10 min by the addition of Coelenterazine 400A at a final concentration of 5 μM (NanoLight Technology, Pinetop, AZ). Fluorescence and luminescence readings were collected using a Mithras LB940 plate reader (Berthold Technologies, Bad Wildbad, Germany).
Ligand‐induced calcium mobilization
Intracellular calcium mobilization, induced by chemokines or chemokine‐derived peptides, was evaluated using a calcium‐responsive fluorescent probe and an FLIPR Tetra device. U87.CXCR3, U87.CXCR4 and U87.ACKR3 cells were seeded in black‐wall, gelatin‐coated 96‐well plates at 2 × 104 cells per well and incubated for 12 h. Cells were then loaded with either the fluorescent calcium probe Fluo‐2 acetoxymethyl (AM; TefLabs, Austin, TX) at a final concentration of 4 μM in assay buffer (HBSS containing 20 mM HEPES buffer and 0.2% bovine serum albumin, pH 7.4) for 45 min at 37°C. The intracellular calcium mobilization induced by the chemokines (2 nM to 1 μM) or chemokine‐derived peptides (200 nM to 100 μM) was then measured at room temperature by monitoring the fluorescence as a function of time in all wells simultaneously using a fluorescence imaging plate reader (FLIPR Tetra, Molecular Devices Sunnyvale, CA) as previously described (Princen et al., 2003).
Signalling of chimeric CXCL11 chemokines through CXCR3 was assessed using Ready‐to‐Assay™ CXCR3‐expressing Chem‐1 cells (Eurofins Pharma Bioanalytics, Abingdon, UK). Cells were seeded onto 0.3 mL v‐bottom 96‐well plates (Costar, Corning, NY) according to the manufacturer's protocol. The medium was removed 24 h later, and cells were washed with 200 μL HBSS. Assay buffer (HBSS, 20 mM HEPES, 0.1% BSA pH 7.4) and FLIPR 4 Calcium Flux kit dye (Molecular Devices) were added to each well in a 1:1 ratio. Plates were centrifuged for 15 s at 250× g and subsequently incubated for 1 h at 37°C. Fluorescence was measured at 37°C using a FlexStation2 microplate reader with excitation and emission of 485 nm and 515 nm respectively.
Arrestin recruitment assays
Chemokine‐ and peptide‐induced β‐arrestin‐2 recruitment to CXCR3, CXCR4 and ACKR3 was monitored by NanoLuc complementation assay (NanoBiT, Promega) (Dixon et al., 2016); 1.2 × 106 U87 cells were plated in 10 cm culture dishes and, 48 h later, transfected with pNBe vectors containing human β‐arrestin‐2 N‐terminally fused to LgBiT and receptors C‐terminally fused to SmBiT. Forty‐eight hours post‐transfection cells were harvested, incubated 40 min at 37°C with 200‐fold diluted Nano‐Glo Live Cell substrate and distributed into white 96‐well plates (5 × 104 cells per well). β‐arrestin‐2 recruitment in response to chemokines (0.1 nM to 1000 nM) or peptides (2 nM to 100 μM) was evaluated with a Mithras LB9 40 luminometer (Berthold Technologies).
Modified recombinant CXCL11‐induced β‐arrestin2 recruitment to CXCR3 and ACKR3 was monitored by BRET measurements as previously described (Berchiche et al., 2011). Briefly, HEK293E cells were transiently transfected with receptor yellow fluorescent protein (YFP) fusion constructs and β‐arrestin‐2‐Rluc. Transfected cells were seeded onto poly‐D‐lysine‐treated 96‐well plates. At 48 h post‐transfection, the culture medium was replaced with PBS supplemented with 0.1% BSA. Cells expressing receptor‐YFP and β‐arrestin‐2‐Rluc at a ratio resulting in BRETMAX were stimulated with chemokine ligands for 5 min at 37°C followed by the addition of Coelenterazine H to a final concentration of 5 μM (NanoLight technology). Fluorescence and luminescence were measured using a Mithras LB940 plate reader (Berthold Technologies). The Net‐BRET signal was calculated by subtracting the background BRET signal from the signal obtained with the expression of β‐arrestin‐2‐Rluc alone. The maximum signal of the mutants is reported as a percentage of WT, with CXCL11WT being set to 100%.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Concentration–response curves were fitted to the four‐parameter Hill equation using an iterative, least‐squares method (GraphPad Prism version 7.02) to provide mean (with SEM) values of pEC50, pIC50, EC50 or IC50. Unpaired t‐tests were used to analyse the differences in pEC50/ pIC50 obtained from at least five experiments (n = 5). Peptides 1–17 (for CXCL12 and CXCL11) and 1–21 (for vCCL2) were considered as reference. Dimeric peptides were compared with their monomeric counterparts. CXCL11 variants were compared with the wild‐type chemokine. P value of <0.05 was considered as statistically significant.
Materials
All peptides were purchased from JPT (Berlin, Germany) and contain a free amine at the N terminus and an amide group at the C terminus to avoid additional negative charge. Chemokines CXCL12, CXCL11, vCCL2 (vMIP‐II), CXCL10 and CXCL9 were purchased from PeproTech (Rocky Hill, NJ). Alexa Fluor 647‐labelled CXCL12 (CXCL12‐AF647) and CXCL11 (CXCL11‐AF647) were purchased from Almac (Craigavon, UK) and radiolabeled CXCL12 (125I–CXCL12) and CXCL11 (125I–CXCL11) from PerkinElmer. Peptide and chemokine cytotoxicity was monitored using an amino‐reactive cell viability dye (Life Technologies) and an ATP quantification‐based cell viability assay (Promega).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org/, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b).
Results
The ability of synthetic peptides derived from CXCL12, CXCL11, CXCL10, CXCL9 and vCCL2 to interact with CXCR4, CXCR3 and ACKR3 was evaluated in competition studies with fluorescently labelled chemokines and in various G protein signalling or β‐arrestin‐2 recruitment assays and compared with that of the parental chemokines (Figure 1).
Binding and activity of chemokine N‐terminal peptides towards CXCR4, CXCR3 and ACKR3
First, peptides comprising the flexible N terminus, the cysteine motif and the N‐loop of CXCL12, CXCL11, CXCL10, CXCL9 and vCCL2 chemokines (CXCL121–17, CXCL111–17, CXCL101–17, CXCL91–17 and vCCL21–21) were examined (Figure 1C–E).
In the CXCR4 binding assay, peptides CXCL121–17 and vCCL21–21 showed an 8000‐ and 100‐fold weaker interaction, respectively, compared with full‐length chemokines (Table 1, Figure 2A, B). vCCL21–21 was approximately 50 times more potent (IC50 = 2.0 μM, pIC50 = 5.71 ± 0.24) in displacing the labelled CXCL12 from CXCR4 than CXCL121–17 (IC50 ~100 μM, pIC50 ~4), whereas the parental chemokines of the two peptides interacted with the receptor with similar IC50 values (IC50 = 13 nM, pIC50 = 7.89 ± 0.04 vs. IC50 = 20 nM, pIC50 = 7.70 ± 0.06 for CXCL12 and vCCL2, respectively). In functional assays, CXCL12‐ and vCCL2‐derived peptides retained the activity of the parental chemokine, with CXCL121–17 inducing a decrease in basal cAMP levels (EC50 = 2.1 μM, pEC50 = 5.67 ± 0.06) and intracellular calcium release (EC50 = 2.6 μM, pEC50 = 5.58 ± 0.04) (Figure 2C, D) and vCCL21–21 acting as antagonist of CXCL12‐induced calcium release (IC50 = 7.0 μM, pIC50 = 5.15 ± 0.05) (Table 2). Although its capacity to trigger G protein signalling was maintained, the ability of CXCL121–17 to induce β‐arrestin‐2 recruitment to CXCR4 was markedly reduced and, at the highest concentration tested (100 μM), reached only 18% of the maximal signal observed with CXCL12 (Table 3, Figure 2E).
Table 1.
Binding properties of full‐length chemokines and peptides derived from their N‐terminal regions towards CXCR4 and ACKR3
| Name | Sequence | Binding competition | |
|---|---|---|---|
| CXCR4 | ACKR3 | ||
| pIC50 ± SEM | pIC50 ± SEM | ||
| CXCL12 | 7.89 ± 0.04 | 8.74 ± 0.07 | |
| CXCL121–17 | KPVSLSYRCPCRFFESH | ~4.00 | 5.39 ± 0.10 |
| CXCL121–9 | KPVSLSYRS | <3.00 | 5.05 ± 0.13ns |
| (CXCL121–9)2 | (KPVSLSYRC)2 | 5.31 ± 0.16 | 5.65 ± 0.09* |
| CXCL121–17D | KPVSLSYRCPCRFFESH | ~4.00 | <4.00 |
| CXCL122–17 | ‐ PVSLSYRCPCRFFESH | ~4.00 | 5.23 ± 0.25ns |
| CXCL121–17/P2G | KGVSLSYRCPCRFFESH | <4.00 | 4.22 ± 1.19ns |
| vCCL2 | 7.70 ± 0.06 | 7.47 ± 0.07 | |
| vCCL21–21 | LGASWHRPDKCCLGYQKRPLP | 5.71 ± 0.24 | 5.77 ± 0.13 |
| vCCL21–11 | LGASWHRPDKS | ~4.00 | 5.27 ± 0.24ns |
| (vCCL21–11)2 | (LGASWHRPDKC)2 | 6.67 ± 0.06 | 5.59 ± 0.13ns |
| vCCL21–21 D | LGASWHRPDKCCLGYQKRPLP | 6.21 ± 0.21ns | ~4.00 |
| CXCL11 | <6.00 | 8.45 ± 0.03 | |
| CXCL111–17 | FPMFKRGRCLCIGPGVK | <4.52a | 5.46 ± 0.12a |
| CXCL111–9 | FPMFKRGRS | ND | 5.09 ± 0.13ns |
| (CXCL111–9)2 | (FPMFKRGRC)2 | ND | 6.03 ± 0.14* |
| CXCL111–17D | FPMFKRGRCLCIGPGVK | ND | 5.06 ± 0.12*, a |
| CXCL112–17 | ‐ PMFKRGRCLCIGPGVK | ND | <4.52a |
| CXCL111–17/P2G | FGMFKRGRCLCIGPGVK | ND | 6.72 ± 0.09*, a |
| CXCL10 | <6.00 | <6.00 | |
| CXCL101–17 | VPLSRTVRCTCISISNQ | <4.52a | <4.52*, a |
Binding competition studies with fluorescently labelled CXCL12 were performed in U87 cells (n = 5). Peptides 1‐17 (for CXCL12 and CXCL11) and 1‐21 (for vCCL2) were considered as the standard, reference peptides and monomeric mutant peptides were compared with these. Where dimeric peptides were assessed, these were compared with their corresponding monomeric peptides.
P < 0.05, significantly different from standard peptides or from monomeric peptides; ns, not significant, P > 0.05.
Highest concentration tested 30 μM. ND: not determined.
Figure 2.
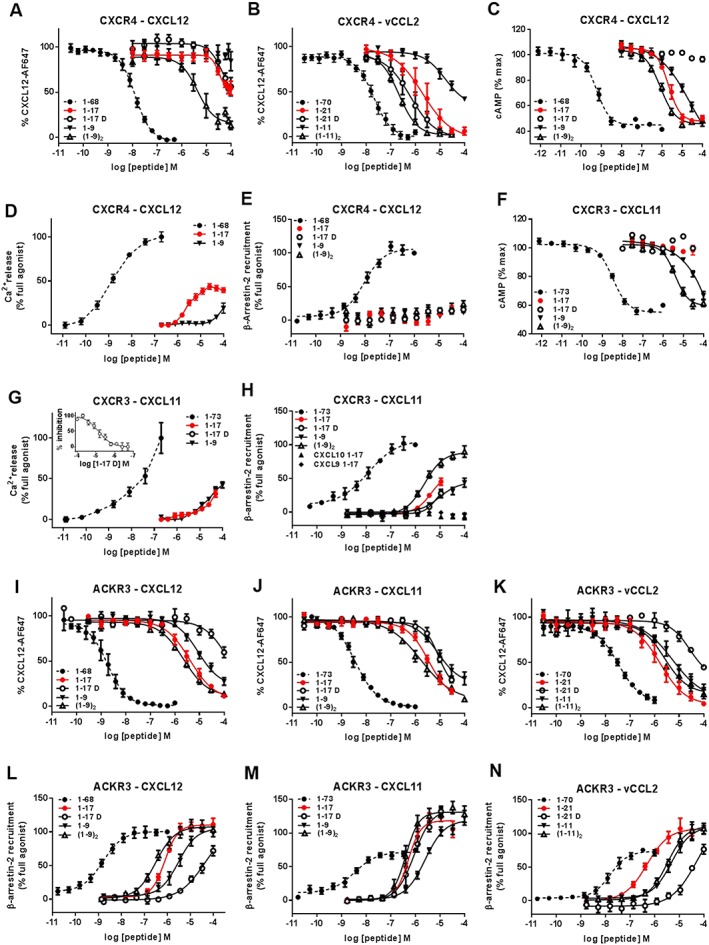
Binding, G protein signalling and β‐arrestin‐2 recruitment to CXCR4, CXCR3 and ACKR3 induced by full‐length chemokines and chemokine N‐terminal peptides. Binding and modulation of CXCR4 (A–E), CXCR3 (F–H) and ACKR3 (I–N) by full‐length CXCL12, CXCL11, vCCL2 and peptides derived from their N‐terminal regions. Binding to CXCR4 (A and B) and ACKR3 (I–K) was assessed by binding competition studies with Alexa Fluor 647‐coupled CXCL12 in U87 cells stably expressing the receptors and analysed by flow cytometry. G protein signalling induced by full‐length chemokines and peptides derived from their N‐terminal regions towards CXCR4 (C and D) and CXCR3 (F and G) was evaluated by measuring the modulation of the basal intracellular cAMP concentration using Glo‐cAMP sensor (C and F) or the release of intracellular calcium using Fluo‐2 dye and FLIPR platform (D and G). (G‐inset) Antagonist properties of peptide CXCL111–17D monitored in calcium assay. β‐arrestin‐2 recruitment to CXCR4 (E), CXCR3 (H) and ACKR3 (L–N) induced by full‐length chemokines and N‐terminal peptides was monitored using a Nanoluciferase‐based complementation assay (NanoBIT). Each experiment was performed five times, and the data shown are means ± SEM.
Table 2.
G protein signalling properties of full‐length chemokines and peptides derived from their N‐terminal regions towards CXCR4 and CXCR3
| Name | Sequence | cAMP | Calcium | ||||||
|---|---|---|---|---|---|---|---|---|---|
| CXCR4 | CXCR3 | CXCR4 | CXCR3 | ||||||
| pEC50 ± SEM | Max (%) | pEC50 ± SEM | Max (%) | pEC50 (pIC50) ± SEM | Max (%) | pEC50 (pIC50) ± SEM | Max (%) | ||
| CXCL12 | – | 8.97 ± 0.09 | 100 | <6.00 | 2 | 8.84 ± 0.05 | 100 | <6.00 | 0 |
| CXCL121–17 | KPVSLSYRCPCRFFESH | 5.67 ± 0.06 | 103 | <4.00 | 1 | 5.58 ± 0.04 | 44 | <4.00 | 0 |
| CXCL121–9 | KPVSLSYRS | 5.11 ± 0.09* | 101 | ND | <4.00 | 16 | ND | ||
| (CXCL121–9)2 | (KPVSLSYRC)2 | 6.07 ± 0.06* | 105 | ND | ND | ND | |||
| CXCL121–17D | KPVSLSYRCPCRFFESH | <4.00 | 6 | ND | (<4.00) | 0 | ND | ||
| CXCL122–17 | ‐PVSLSYRCPCRFFESH | <4.00 | 1 | ND | (4.50 ± 0.12) | 0 | ND | ||
| CXCL121–17/P2G | KGVSLSYRCPCRFFESH | <4.00 | 1 | ND | (4.45 ± 0.06) | 0 | ND | ||
| vCCL2 | – | <6 | 0 | <6.00 | (7.67 ± 0.08) | 0 | <6.00 | 0 | |
| vCCL21–21 | LGASWHRPDKCCLGYQKRPLP | <4.00 | 0 | <4.00 | 0 | (5.15 ± 0.05) | 0 | <4.00 | 0 |
| vCCL21–11 | LGASWHRPDKS | <4.00 | 0 | ND | (<4) | 0 | ND | ||
| (vCCL21–11)2 | (LGASWHRPDKC)2 | <4.00 | 0 | ND | ND | ND | |||
| vCCL21–21D | LGASWHRPDKCCLGYQKRPLP | <4.00 | 0 | ND | (5.16 ± 0.04) | 0 | ND | ||
| CXCL11 | – | <6.00 | 1 | 8.46 ± 0.06 | 100 | <6.00 | 0 | 7.72 ± 0.13 | 100 |
| CXCL111–17 | FPMFKRGRCLCIGPGVK | <4.52a | 0 | <4.52a | 4 | <4.30b | 0 | 4.32 ± 0.04b | 78 |
| CXCL111–9 | FPMFKRGRS | ND | 0 | 4.45 ± 0.08 | 89 | ND | 4.79 ± 0.05* | 37 | |
| (CXCL111–9)2 | (FPMFKRGRC)2 | ND | 0 | 5.35 ± 0.07* | 95 | ND | ND | ||
| CXCL111–17D | FPMFKRGRCLCIGPGVK | ND | 0 | <4.52a | 4 | ND | (5.13 ± 0.06)b | 0 | |
| CXCL112–17 | ‐PMFKRGRCLCIGPGVK | ND | 0 | <4.52a | 2 | ND | (5.32 ± 0.15)b | 0 | |
| CXCL111–17/P2G | FGMFKRGRCLCIGPGVK | ND | 0 | <4.52a | 0 | ND | (5.73 ± 0.19)b | 0 | |
| CXCL10 | – | <6.00 | 0 | 8.36 ± 0.12 | 97 | <6.00 | 0 | 7.76 ± 0.19 | 100 |
| CXCL101–17 | VPLSRTVRCTCISISNQ | <4.00 | 0 | <4.52a | 0 | <4.30b | 0 | 4.58 ± 0.03 | 112 |
| CXCL9 | – | <6.00 | 0 | 6.47 ± 0.10 | 85 | <6.00 | 0 | 6.73 ± 0.15 | 100 |
| CXCL91–17 | TPVVRKGRCSCISTNQG | <4.00 | 0 | <4.00 | 0 | <4.00 | 0 | <4.00 | 0 |
cAMP modulation measurements were performed in U87 cells using GloSensor technology (n = 5). Peptides 1‐17 (for CXCL12 and CXCL11) and 1‐21 (for vCCL2) were considered as the standard, reference peptides and monomeric mutant peptides were compared with these. Where dimeric peptides were assessed, these were compared with their corresponding monomeric peptides.
P < 0.05, significantly different from standard peptides or from monomeric peptides; ns, not significant, P > 0.05.
Highest concentration tested 30 μM. Intracellular calcium release was evaluated in U87 cells using a calcium‐responsive fluorescent probe and an FLIPR system (n = 5).
Highest concentration tested 50 μM. ND: not determined. (Data antagonist mode).
Table 3.
Recruitment of β‐arrestin‐2 induced by full‐length chemokines and peptides derived from their N‐terminal regions to CXCR4, CXCR3 and ACKR3
| Arrestin recruitment | |||||||
|---|---|---|---|---|---|---|---|
| Name | Sequence | CXCR4 | CXCR3 | ACKR3 | |||
| pEC50 ± SEM | Max (%) | pEC50 ± SEM | Max (%) | pEC50 ± SEM | Max (%) | ||
| CXCL12 | −8.03 ± 0.09 | 100 | <6.00 | 0 ± 1 | 8.81 ± 0.10 | 100 | |
| CXCL121–17 | KPVSLSYRCPCRFFESH | <4.00 | 18 ± 1 | <4.00 | 1 ± 1 | 6.11 ± 0.05 | 111 ± 9 |
| CXCL121–9 | KPVSLSYRS | <4.00 | 12 ± 2 | ND | 5.52 ± 0.09* | 97 ± 5 | |
| (CXCL121–9)2 | (KPVSLSYRC)2 | <4.00 | 25 ± 5 | ND | 6.50 ± 0.09* | 103 ± 4 | |
| CXCL121–17D | KPVSLSYRCPCRFFESH | <4.00 | 16 ± 4 | ND | 4.35 ± 0.43* | 72 ± 8 | |
| CXCL122–17 | ‐ PVSLSYRCPCRFFESH | <4.00 | 3 ± 1 | ND | 4.89 ± 0.22* | 100 ± 3 | |
| CXCL121–17/P2G | KGVSLSYRCPCRFFESH | <4.00 | 2 ± 7 | ND | 4.55 ± 0.47* | 63 ± 7 | |
| vCCL2 | <6.00 | 10 ± 1 | <6.00 | 0 ± 5 | 7.80 ± 0.09 | 71 ± 5 | |
| vCCL21–21 | LGASWHRPDKCCLGYQKRPLP | <4.00 | 18 ± 2 | <4.00 | 0 ± 2 | 6.31 ± 0.10 | 107 ± 8 |
| vCCL21–11 | LGASWHRPDKS | <4.00 | 0 ± 2 | ND | 5.31 ± 0.09* | 105 ± 6 | |
| (vCCL21–11)2 | (LGASWHRPDKC)2 | <4.00 | 0 ± 2 | ND | 5.48 ± 0.08ns | 107 ± 8 | |
| vCCL21–21D | LGASWHRPDKCCLGYQKRPLP | <4.00 | 0 ± 6 | ND | 4.27 ± 0.66* | 77 ± 7 | |
| CXCL11 | <6.00 | 8 ± 4 | 7.91 ± 0.16 | 100 | 8.47 ± 0.12 | 75 ± 3 | |
| CXCL111–17 | FPMFKRGRCLCIGPGVK | <4.52a | 13 ± 7 | <4.52a | 45 ± 8 | 6.22 ± 0.08a | 134 ± 6 |
| CXCL111–9 | FPMFKRGRS | ND | <4.00 | 35 ± 9 | 5.57 ± 0.12* | 102 ± 8 | |
| (CXCL111–9)2 | (FPMFKRGRC)2 | ND | 5.64 ± 0.08 | 89 ± 9 | 6.40 ± 0.07* | 132 ± 12 | |
| CXCL111–17D | FPMFKRGRCLCIGPGVK | ND | <4.52a | 28 ± 6 | 6.05 ± 0.10a ,ns | 136 ± 8 | |
| CXCL112–17 | ‐ PMFKRGRCLCIGPGVK | ND | <4.52a | 2 ± 3 | 5.81 ± 0.17a ,ns | 59 ± 15 | |
| CXCL111–17/P2G | FGMFKRGRCLCIGPGVK | ND | <4.52a | 5 ± 5 | 6.78 ± 0.09a , * | 126 ± 10 | |
| CXCL10 | ND | 7.59 ± 1.17 | 45 ± 6 | <6.00 | 21 ± 1 | ||
| CXCL101–17 | VPLSRTVRCTCISISNQ | ND | <4.00 | 0 ± 3 | <4.00 | 2 ± 2 | |
| CXCL9 | ND | 7.03 ± 0.21 | 46 ± 7 | ND | |||
| CXCL91–17 | TPVVRKGRCSCISTNQG | ND | <4.00 | 1 ± 2 | ND | ||
β‐arrestin‐2 recruitment was monitored in U87 cells using split Nanoluciferase complementation assay (n = 5). Peptides 1‐17 (for CXCL12 and CXCL11) and 1‐21 (for vCCL2) were considered as the standard, reference peptides and monomeric mutant peptides were compared with these. Where dimeric peptides were assessed, these were compared with their corresponding monomeric peptides.
P < 0.05, significantly different from standard peptides or from monomeric peptides.
Highest concentration tested 30 μM. ND: not determined. P < 0.05, significantly different from ; ns. not significant, P > 0.05.
Similar results were obtained with peptides derived from CXCL11, CXCL10 and CXCL9 towards CXCR3 (Tables 2 and 3, Figure 2F–H). However, strong non‐specific binding of fluorescently labelled CXCL11, resistant to competition by unlabelled chemokines, made it impossible to evaluate their binding to CXCR3 in competition studies on U87 cells. Moreover, due to peptide cytotoxicity, it was not possible to characterize the activity of CXCL111–17 and its variants as well as CXCL101–17 at concentrations above 30 to 50 μM, depending on the assay. Among the three peptides, CXCL111–17 and CXCL101–17 retained the ability to induce G protein‐mediated signalling as shown in calcium release assay, albeit with an over 1500‐fold reduction in potency (EC50 ≈ 50 μM, pEC50 ≈ 4.30 and EC50 = 26.0 μM, pEC50 = 4.58 ± 0.03) compared with the parental chemokines (EC50 = 19 nM, pEC50 = 7.72 ± 0.13 for CXCL11 and EC50 = 18 nM, pEC50 = 7.76 ± 0.19 for CXCL10) (Figure 2G). This decrease of potency was reminiscent of the 1500‐fold difference observed between CXCL121–17 and the full‐length CXCL12 in G protein signalling through CXCR4 (Table 2). Although it showed no cytotoxicity, CXCL91–17 was unable to induce G protein signalling even at a concentration as high as 100 μM, likely originating from the already low potency of its parental chemokine (EC50 = 190 nM, pEC50 = 6.73 ± 0.15) (Table 2). CXCL111–17 induced 45% of the maximum β‐arrestin‐2 recruitment at a concentration of 10 μM, while CXCL101–17 was surprisingly unable to induce β‐arrestin‐2 recruitment even at a concentration of 30 μM (Table 3, Figure 2H).
In ACKR3 binding experiments, the relative rank order of IC50 values of the three peptides derived from CXCL12, CXCL11 and vCCL2 was different from that of their parental chemokines (Figure 2I–K).
CXCL121–17 and CXCL111–17 showed similar IC50 values of 4.1 μM (pIC50 = 5.39 ± 0.10) and 3.4 μM (pIC50 = 5.46 ± 0.12), corresponding to a 2000‐ and a 1000‐fold loss of binding when compared with their parental chemokines (1.8 nM, pIC50 = 8.74 ± 0.07 and 3.5 nM, pIC50 = 8.45 ± 0.03) (Table 1) (Figure 2I, J). The reduced potency of these peptides was similar to that seen on CXCR4 and CXCR3 respectively. vCCL21–21 was stronger in binding ACKR3 (IC50 = 1.7 μM, pIC50 = 5.77 ± 0.13) than peptides derived from the endogenous chemokines, with only a 50‐fold reduction compared to the full‐length vCCL2 (IC50 = 33.6 nM, pIC50 = 7.47 ± 0.07) (Figure 2K). Unlike their weak activity towards CXCR4 and CXCR3, CXCL121–17, CXCL111–17 and vCCL21–21 all strongly induced β‐arrestin‐2 recruitment to ACKR3, with EC50 values of 0.8 μM (pEC50 = 6.11 ± 0.05), 0.6 μM (pEC50 = 6.22 ± 0.08) and 0.5 μM (pEC50 = 6.31 ± 0.10) respectively (Figure 2L–N). Interestingly, peptides CXCL111–17 and vCCL21–21, in contrast to their parental chemokines showing partial agonist profiles (Emax = 75 and 71% of CXCL12, respectively), were able to induce levels of β‐arrestin‐2 recruitment respectively higher than (134%) or comparable to (107%) the full agonist CXCL12 (Figure 2M, N, Table 3). The maximum efficacy of CXCL121–17 was also comparable to that of the full‐length CXCL12.
Effect of D‐stereoisomer replacement on binding and activity of chemokine N‐terminal peptides
The properties of peptides CXCL121–17, CXCL111–17 and vCCL21–21 in which each amino acid has been replaced by the corresponding D‐stereoisomer (CXCL121–17D, CXCL111–17D and vCCL21–21D) were then evaluated for their action towards the three receptors in order to assess the plasticity of their ligand‐binding pockets and their tolerance to such ligand modifications at the functional level.
D‐amino acid replacement had no effect on the binding of the CXCL12‐derived peptides to CXCR4 and improved the binding of vCCL21–21 three‐fold (IC50 = 0.6 μM, pIC50 = 6.21 ± 0.21) (Figure 2A, B, Table 1). Nevertheless, D‐isomer replacement turned CXCL121–17 into a CXCR4 antagonist (IC50 > 100 μM), while the initial antagonist activity of vCCL21–21 was conserved (IC50 = 0.7 μM, pIC50 = 5.2 ± 0.04) (Figure 2C, Table 2). A similar agonist‐to‐antagonist conversion was observed for CXCL111–17D in G protein signalling through CXCR3 (IC50 = 7.4 μM, pIC50 = 5.13 ± 0.06) (Figure 2G‐inset) (Table 2), although the peptide did retain some of the agonist effect in β‐arrestin recruitment to the receptor (Figure 2G, H, Table 3).
In contrast, D‐isomer replacement significantly impaired the ability of peptides derived from vCCL2 and CXCL12 to bind ACKR3, with a 50‐fold increase in IC50 for vCCL21–21D and CXCL121–17D compared with the L‐stereoisomers (Figure 2I–K, Table 1). For the CXCL11‐derived peptide, this difference was much less marked, with the D‐isomer showing only a twofold decrease in its ability to displace the labelled CXCL12 from ACKR3. Remarkably however, although reduced, all three D‐stereoisomer peptides conserved the parental agonist activity towards ACKR3, inducing β‐arrestin‐2 recruitment with potencies reflecting the effect of the substitution observed in binding competition studies (Figure 2L–N, Table 3).
Binding and activity of N‐loop‐ and cysteine motif‐truncated chemokine N‐terminal peptides
Next, truncated peptides devoid of the N‐loop and the cysteine motif and comprising only the flexible N termini of chemokines CXCL12, CXCL11 and vCCL2 (CXCL121–9, CXCL111–9 and vCCL21–11) were investigated to assess the importance of these different N terminal regions in the binding and activity towards the receptors (Figure 1E).
Truncation of CXCL121–17 after the first cysteine (CXCL121–9) resulted in a loss of binding to CXCR4 (IC50 > 1000 μM, pIC50 < 3.00), a marked decrease of calcium mobilization and a reduced ability to modulate cAMP production (EC50 = 7.4 μM, pEC50 = 5.11 ± 0.09) (Figure 2A, C, D, Tables 1 and 2). The similar truncation of vCCL21–21 to vCCL21–11 affected the peptide's ability to compete with labelled CXCL12 and resulted in an approximately 50‐fold reduction in CXCR4 binding (IC50 ~100 μM, pIC50 ~4.00) and antagonist properties (Figure 2B, Tables 1 and 2). CXCL111–9 retained its ability to induce cAMP modulation and showed a reduced potency in calcium mobilization through CXCR3 as well as β‐arrestin‐2 recruitment to the receptor (Figure 2F–H, Tables 2 and 3). The effect of the truncation, however, could not be precisely evaluated and compared due to the cytotoxicity of the parental peptide CXCL111–17 at concentrations above 30 μM.
CXCL121–9 and CXCL111–9 showed only a slightly reduced binding to ACKR3 (IC50 = 8.9 μM, pIC50 = 5.05 ± 0.13 and 8.1 μM pIC50 = 5.09 ± 0.13) as compared with CXCL121–17 and CXCL111–17, suggesting that the first nine residues of the chemokines support a large part of the binding of CXCL12 and CXCL11 N terminal region to the receptor (Figure 2I, J, Table 1). In accordance with this assumption, a peptide in which the residues following the first cysteine where permutated randomly showed an IC50 comparable with that of CXCL121–17 and CXCL121–9 (data not shown). Similarly, for vCCL2, truncation to vCCL21–11 resulted in a three‐fold weaker binding to ACKR3 (IC50 = 1.7 μM, pIC50 = 5.27 ± 0.24) than vCCL21–21 (IC50 = 5.4 μM, pIC50 = 5.77 ± 0.13) (Figure 2K, Table 1). In β‐arrestin‐2 recruitment experiments, truncation resulted only in a four‐fold reduction of the potency of CXCL121–9 and CXCL111–9 compared with CXCL121–17 and CXCL111–17, whereas it decreased the potency of vCCL21–11 by ten‐fold (Figure 2L–N, Table 3).
In conclusion, N‐loop truncation drastically affected the interactions of peptides derived from CXCL12 and vCCL2 with CXCR4, whereas it only moderately reduced peptide ability to bind and activate ACKR3, suggesting that the two receptors have different chemokine recognition modes.
Effect of dimerization of the flexible chemokine N‐terminal peptides on their binding and activity
Dimerization of the C‐terminally truncated peptides [(CXCL121–9)2, (CXCL111–9)2 and (vCCL21–11)2] via a disulfide bridge between the terminal cysteine residues had a range of effects on the different peptide‐receptor pairs (Figure E).
For CXCR4, dimerization of CXCL121–9 and vCCL21–11 substantially enhanced their binding and signalling potencies, as shown by an improvement of their IC50 of over 100‐fold observed in binding competition and their EC50 of over three‐ to ten‐fold in G protein signalling assays (Figure 2A–C, Tables 1 and 2). Similar ten‐fold higher potency in cAMP modulation and markedly enhanced β‐arrestin‐2 recruitment were observed for CXCR3 interaction with the dimeric CXCL111–9 (Figure 2F, H, Tables 2 and 3).
For ACKR3, dimerization had different effects on CXCL12 and CXCL11 peptides compared to the vCCL2 peptide. Both (CXCL121–9)2 and (CXCL111–9)2 showed an overall 5‐ to 10‐fold improvement in their binding and β‐arrestin‐2 recruitment properties compared with their monomeric counterparts (Figure 2I, J, L, M), whereas dimerization had no effect on the vCCL2‐derived peptide (Figure 2K, N), suggesting that its binding mode differed slightly from that of the CXCL12‐ and CXCL11‐derived peptides.
Altogether, these data showed that dimerization strongly enhanced the binding and activity of N‐terminal peptides not only to CXCR4 but also CXCR3 and ACKR3. Moreover, the N‐terminal domain of vCCL2 seems to interact with CXCR4 and ACKR3 according to different binding modes.
Effect of N‐loop replacement on binding and activity of CXCL11 towards CXCR3 and ACKR3
To circumvent the cytotoxicity problems of CXCL11 peptides described above and to further assess the importance of the N‐loop for CXCR3 and ACKR3 binding and activation, we generated variants of full‐length CXCL11, in which the N‐loop residues were replaced with those of CXCL10 (CXCL11Nloop10) or CXCL12 (CXCL11Nloop12) and evaluated their binding in competition studies with radiolabeled CXCL11 as well as receptor activation (Figure 1E).
CXCL11Nloop10 and CXCL11Nloop12 bound to CXCR3 with IC50 values of 38.2 nM (pEC50 = 7.41 ± 0.11) and 120.5 nM (pEC50 = 6.91 ± 0.08), respectively, compared with 3.2 nM (pEC50 = 8.48 ± 0.09) for CXCL11WT (Table 4). Compared with CXCL11WT, CXCL11Nloop12 also showed reduced potency and efficacy in the induction of β‐arrestin‐2 recruitment to CXCR3, whereas the activity of CXCL11Nloop10 (EC50 = 37.3 nM, pEC50 = 7.42 ± 0.08, Emax = 41%) was equivalent to that of CXCL11WT (26.8 nM, pEC50 = 7.57 ± 0.10) (Figure 3A–C, Table 4). CXCL11Nloop12 showed a ~20‐fold loss of potency in cAMP modulation relative to CXCL11WT, while CXCL11Nloop10 was almost unaffected by the substitution of N‐loop residues from CXCL10 (Figure 3B, Table 4). These data demonstrate that exchanging the N‐loops of CXCL10 and CXCL11, which both bind to CXCR3, did not significantly impair chemokine activity whereas replacement of the CXCL11 N‐loop with residues from a non‐cognate chemokine ligand (i.e. CXCL12) significantly diminished its CXCR3 binding and activation.
Table 4.
Binding and signalling properties of CXCL11 variants towards CXCR3 and ACKR3
| Name | Sequence | Binding competition | cAMP | Calcium | Arrestin recruitment | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CXCR3 | ACKR3 | CXCR3 | CXCR3 | CXCR3 | ACKR3 | ||||||
| pIC50 ± SEM | pIC50 ± SEM | pEC50 ± SEM | Max (%) | pEC50 ± SEM | Max (%) | pEC50 ± SEM | Max (%) | pEC50 ± SEM | Max (%) | ||
| CXCL11 WT | 1‐FPMFKRGRCLCIGPGVKAVK‐73 | 8.48 ± 0.09 | 6.52 ± 0.05 | 8.21 ± 0.11 | 100 | 7.80 ± 0.07 | 100 | 7.57 ± 0.07 | 100 | 7.46 ± 0.09 | 100 |
| CXCL11Nloop10 | 1‐FPMFKRGRCLCISISNQAVK‐73 | 7.41 ± 0.11* | 6.11 ± 0.07* | 7.93 ± 0.13 ns | 81 | 7.98 ± 0.11ns | 92 | 7.42 ± 0.08ns | 89 | 7.68 ± 0.10 ns | 85 |
| CXCL11Nloop12 | 1‐FPMFKRGRCLCRFFESHAVK‐73 | 6.91 ± 0.08* | 6.29 ± 0.04* | 6.85 ± 0.14* | 102 | 7.02 ± 0.06* | 79 | <6.00 | 41 | 7.32 ± 0.09ns | 84 |
| CXCL113–73 | 3‐MFKRGRCLCIGPGVKAVK‐73 | 6.90 ± 0.09* | 5.78 ± 0.07* | 8.39 ± 0.21ns | 54 | <6.00 | 37 | <6.00 | 22 | 7.15 ± 0.08* | 81 |
| CXCL115–73 | 5‐KRGRCLCIGPGVKAVK‐73 | 6.18 ± 0.05* | 5.29 ± 0.01* | <6.00 | <6.00 | <6.00 | 16 | <6.00 | 30 | ||
| CXCL117–73 | 7‐RCLCIGPGVKAVK‐73 | <5.00 | <5.00 | <6.00 | <6.00 | <6.00 | 15 | <6.00 | 22 | ||
| CXCL11P2G | 1‐FGMFKRGRCLCIGPGVKAVK‐73 | 9.63 ± 0.09* | 8.76 ± 0.05* | 7.86 ± 0.15ns | 100 | 7.40 ± 0.04* | 83 | 7.22 ± 0.14ns | 61 | 7.99 ± 0.07* | 136 |
Binding competition studies with radiolabeled CXCL11 (CXCR3) or CXCL12 (ACKR3) were performed in HEK293T cells (n = 5). β‐arrestin‐2 recruitment was monitored by BRET in HEK293T cells expressing YFP‐fused receptors and Rluc‐fused β‐arrestin‐2.
P < 0.05, significantly different from WT peptide; ns. not significant.
Figure 3.
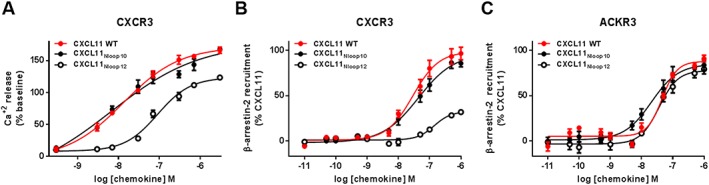
ACKR3 and CXCR3 activation by CXCL11 N‐loop variants. (A) CXCR3 activation by CXCL11 WT and N‐loop variants monitored in HEK293 cells through intracellular calcium release using FLIPR 4 Calcium Flux kit dye. (B and C) Comparison of β‐arrestin‐2 recruitment to CXCR3 (B) and ACKR3 (C) induced by CXCL11WT and N‐loop variants monitored in HEK293 cells by BRET using receptor‐YFP fusion constructs and β‐arrestin‐2‐Rluc. Each experiment was performed five times, and the data shown are means ± SEM.
In contrast to CXCR3, only modest differences were observed for the binding of the N‐loop variants to ACKR3 as well as for their ability to induce the recruitment of β‐arrestin‐2 (Figure 3, Table 4). Indeed, ACKR3 activation by CXCL11WT and CXCL11Nloop12 were indistinguishable (Figure 6A). Surprisingly, N‐loop substitution from the non‐cognate chemokine (i.e. CXCL10) also had no significant effects on ACKR3 β‐arrestin‐2 recruitment (Figure 3C, Table 4), which is consistent with data generated using the N‐loop‐truncated peptides. This suggests that specific N‐loop contacts are not required for ACKR3 binding or activation by CXCL11 and CXCL12. Therefore, unlike CXCR3 and CXCR4, ACKR3 appears relatively insensitive to the N‐loop composition of its CXC chemokine ligands.
Figure 6.
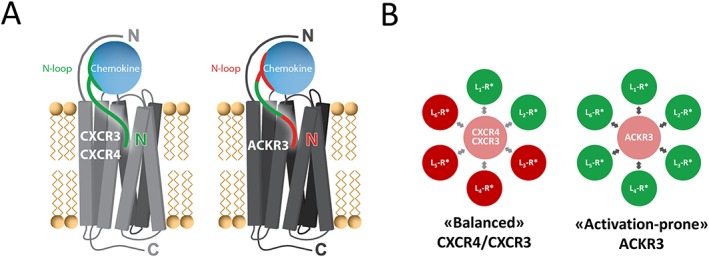
Differential contribution of chemokine N‐terminal features to interactions with CXCR3/CXCR4 and ACKR3 and distribution between receptor active/inactive states. (A) The entire chemokine N‐terminal region (green) is critical for the binding and activation of the canonical receptors CXCR3 and CXCR4, whereas the most N‐terminal residues (red) as well as the N‐loop (red) do not appear important for activation of the atypical receptor ACKR3. (B) Comparison of the distribution of active (R* green) and inactive (R* red) conformations of CXCR4/CXCR3 and ACKR3 stabilized by ligands (L) targeting the receptor transmembrane ligand‐binding pocket (CRS2). CXCR4 and CXCR3 have low basal activity (light red) and are ‘balanced’ receptors as ligand binding to CRS2 can stabilize either the active state (green) or inactive state (red). ACKR3 is an ‘activation‐prone’ receptor as ligand binding preferentially stabilizes the active state (green) of the receptor leading to β‐arrestin‐2 recruitment. Indeed, so far, no ligand targeting the transmembrane pocket of ACKR3 without displaying agonist activity has been reported.
Effects of P2G mutation on binding and activity of chemokine‐derived peptides and modified CXCL11
The proline at position 2 in CXCL12 is critical for CXCR4 activation (Crump et al., 1997). As this proline is also conserved in CXCL11, P2G mutation was introduced in peptides derived from both CXCL12 and CXCL11, and the binding and activity towards their cognate receptors was evaluated (Figure 1D, E).
For CXCR4, P2G mutation (CXCL121–17/P2G) considerably affected the agonist properties of the peptide and turned it into an antagonist (IC50 = 35.5 μM, pIC50 = 4.45 ± 0.06), while it slightly reduced its binding compared with CXCL121–17 (Table 2). A similar shift from CXCR3 agonist to antagonist was observed in calcium assay with CXCL111–17 bearing the P2G mutation (CXCL111–17/P2G) (IC50 = 1.9 μM, pIC50 = 5.73 ± 0.19) (Figure 4F‐inset, Tables 2).
Figure 4.
Binding and activation of ACKR3 and CXCR3 by N‐terminal peptides and full‐length CXCL11 bearing the P2G mutation. (A and B) Comparison of the effects of the P2G mutation in CXCL12‐ and CXCL11‐derived peptides on (A) binding to ACKR3 assessed by competition studies with Alexa Fluor 647‐coupled CXCL12 and (B) β‐arrestin‐2 recruitment to ACKR3 monitored by NanoLuc complementation in U87 cells. (C and D) Comparison of β‐arrestin‐2 recruitment to ACKR3 (C) and CXCR3 (D) induced by CXCL11WT and P2G mutant in HEK293 cells monitored by BRET using receptor‐YFP fusion constructs and β‐arrestin‐2‐Rluc. (E and F) CXCR3 activation by CXCL11WT and P2G mutant monitored in HEK293 cells through (E) adenylate cyclase inhibition using BRET reporter GFP10‐Epac‐ Rluc3 and (F) intracellular calcium release using FLIPR 4 Calcium Flux kit dye. (F‐inset) Antagonist properties of peptide CXCL111–17/P2G monitored in calcium assay. Each experiment was performed five times, and the data shown are means ± SEM.
The P2G mutation had opposite effect on the interaction of the two peptides with ACKR3. CXCL121–17/P2G exhibited a 150‐fold reduction of binding (IC50 = 60.9 μM, pIC50 = 4.22 ± 1.19), while the same substitution in the CXCL11 peptide (CXCL111–17/P2G) resulted in a surprising 20‐fold increase in its binding with an IC50 in the nanomolar range (IC50 = 190 nM, pIC50 = 6.72 ± 0.09) (Figure 4A, Table 1). Moreover, in contrast to what was observed for CXCR4 and CXCR3, P2G mutation in CXCL12 and CXCL11 peptides did not convert them into ACKR3 antagonists but rather improved the potency of CXCL111–17/P2G while it reduced that of CXCL121–17/P2G (Figure 4B, Table 3).
In agreement with the data obtained with the N‐terminal peptides, the replacement of the proline at position 2 in full‐length CXCL11 substantially enhanced its IC50 in ACKR3 binding competition (170‐fold improvement), as well as its potency and efficacy (136%) in recruiting β‐arrestin‐2 to the receptor (Figure 4C, Table 4). CXCL11P2G also showed a 15‐fold improved binding to CXCR3 compared with CXCL11WT. However, in contrast to ACKR3, this chemokine showed a drastic reduction in its ability to induce β‐arrestin‐2 recruitment to CXCR3 (Figure 4D). More surprisingly, the introduction of the P2G mutation in CXCL11 only led to modest reduction of its agonist activity (Figure 4E, F, Table 4), which may be related to its higher affinity.
Effect of amino‐terminal truncation on binding and activity of chemokine‐derived peptides and modified CXCL11
N‐terminal truncations were also introduced in peptides derived from both CXCL12 and CXCL11, and their binding and activity towards the receptors was compared. For CXCR4, N‐terminal residue truncation (CXCL122–17) drastically affected the agonist properties of the peptide, turning it into an antagonist in the calcium assay (IC50 = 31.5 μM, pIC50 = 4.50 ± 0.12), while the binding was only slightly reduced compared with CXCL121–17. A similar change of activity towards CXCR3 was observed with CXCL112–17 lacking the amino‐terminal phenylalanine (IC50 = 7.36 μM, pIC50 = 5.32 ± 0.15) (Figures 5E‐inset, Tables 1 and 2).
Figure 5.
β‐arrestin‐2 recruitment and G protein signalling induced by N‐terminally truncated CXCL11 variants towards ACKR3 and CXCR3. (A and B) Comparison of the effects of N‐terminal residue truncation in CXCL12‐ and CXCL11‐derived peptides on (A) binding to ACKR3 assessed by competition studies with Alexa Fluor 647‐coupled CXCL12 and (B) β‐arrestin‐2 recruitment to ACKR3 monitored by NanoLuc complementation in U87 cells. (C) Impact of progressive N‐terminal truncation on the ability of CXCL11 (100 nM) to recruit β‐arrestin‐2 to ACKR3 and CXCR3. Values are expressed as percentage of the maximum β‐arrestin‐2 recruitment monitored with saturating concentrations of CXCL11WT. (D and F) Comparison of β‐arrestin‐2 recruitment to ACKR3 (D) and CXCR3 (F) induced by CXCL11WT and variants lacking the first two residues (CXCL113–73) monitored by BRET. (E) Comparison of maximum CXCR3 mediated intracellular calcium release induced by CXCL11WT and CXCL113–73 variant in HEK cells using FLIPR 4 Calcium Flux kit dye. (E‐inset) Antagonist properties of peptide CXCL112–17 monitored in calcium assay. Each experiment was performed five times, and the data shown are means ± SEM.
Interestingly, truncation of the first residue of the CXCL12 and CXCL11 peptides (Lys1 and Phe1, respectively) had different effects on their binding to ACKR3, with no effect on the IC50 for CXCL122–17 and an over 10‐fold increase for that of CXCL112–17 (Figures 5A). Although it substantially affected the potency in CXCL11, the N‐terminal deletion in CXCL12 and CXCL11 peptides did not convert them to ACKR3 antagonists, as observed for CXCR4 and CXCR3 (Figures 5B).
The effects of N‐terminal truncation were further investigated using full‐length chemokines. A series of CXCL11 variants lacking 2, 4 or 6 N‐terminal residues were generated, and their ability to bind and activate CXCR3 and ACKR3 was measured (Figure 1E, Table 4). Truncated CXCL11 proteins (CXCL113–73, CXCL115–73 and CXCL117–73) all showed diminished maximal β‐arrestin‐2 recruitment compared with CXCL11WT for both CXCR3 and ACKR3 (Figure 5C). However, CXCL113–73 retained substantially more agonist efficacy at ACKR3 (Emax = 81%) than CXCR3 (Emax = 22%) (Figure 5D, Table 4). The EC50 value for CXCL113–73‐induced β‐arrestin‐2 recruitment to ACKR3 was also only two‐fold higher than that of CXCL11WT (Figure 5C, Table 4). This result indicates that neither ACKR3 agonist potency nor efficacy is substantially altered by removal of the two N‐terminal amino acids of CXCL11.
The analysis of the CXCL113–73–CXCR3 interaction showed that N‐terminal truncation had a significant effect on receptor binding and activation. CXCL113–73 bound to CXCR3 with an IC50 of 123.3 nM (pIC50 = 6.90 ± 0.09) as compared with the 3.2 nM (pIC50 = 8.48 ± 0.09) for CXCL11WT (Table 4), which is indicative of a substantial loss of binding affinity. In addition, its potency and efficacy in G protein activation and β‐arrestin‐2 recruitment were drastically reduced (Figure 5E, F, Table 4).
For the two receptors, the longer N‐terminal deletions (CXCL115–73 and CXCL117–73) markedly reduced both binding and β‐arrestin‐2 recruitment efficacy and potency. However, the shorter truncation (CXCL113–73) maintained a higher portion of ACKR3 activity than CXCR3 activity, suggesting a higher tolerance for N‐terminal processing.
Taken together, these results confirmed the previously demonstrated importance of the chemokine proximal N‐terminal residues in CXCR3 and CXCR4 for both binding and activation and showed, for the first time, that ACKR3 ligand binding and activation was less dependent on the presence of the first amino acids and N loop residues. Moreover, this study suggested that ACKR3 could also act as a scavenger receptor for N terminally processed chemokines.
Discussion
Chemokine‐receptor selectivity is dictated by numerous interactions along the receptor extracellular surface and transmembrane segments (TM; Steen et al., 2014; Burg et al., 2015 ; Qin et al., 2015). Recent structural and mechanistic breakthroughs demonstrated that chemokines make extensive contacts with their receptors in a 1:1 stoichiometry through at least three major recognition sites (Veldkamp et al., 2008; Szpakowska et al., 2012; Kufareva et al., 2014; Steen et al., 2014; Burg et al., 2015; Qin et al., 2015; Zheng et al., 2017). Despite these advances, and in the absence of comparative structural data, the roles of the conserved features present in chemokines, and especially in their N‐terminal regions, remain obscure. Although a growing body of evidence indicates that the two‐site/two‐step binding mode proposed for chemokines and their receptors is oversimplified, the essential role of chemokine N termini in mediating receptor affinity and activity is well demonstrated, as their truncation or modification drastically affects chemokine function (Crump et al., 1997; Qin et al., 2015; Kleist et al., 2016). While studies have documented the role of chemokine N‐terminal interactions with CXCR4 for CXCL12 and vCCL2 (Heveker et al., 1998; Loetscher et al., 1998; Zhou et al., 2000; Chevigne et al., 2011), none has compared the role of chemokine N termini between multiple receptor targets, limiting our understanding of the complex regulatory principles in promiscuous chemokine systems.
In this study, we examined the importance of different features present in the N‐terminal regions of CXCL12, CXCL11 and vCCL2 for binding and activation of a trio of receptors: two conventional receptors, CXCR4 and CXCR3, and the ACKR3. Using this subset of interconnected receptors, we also investigated how widespread the binding capacity of D‐stereoisomers and the improved binding of dimeric ligands are, these properties being commonly recognized for CXCR4 (Heveker et al., 2001; Zhou et al., 2002; Choi et al., 2012) (Figure 7).
Figure 7.
Contributions of chemokine and receptor regions to binding and activation of ACKR3, CXCR3 and CXCR4. (A–C) Importance of structural determinants of chemokines CXCL12 (A), CXCL11 (B) and vCCL2 (C) for binding and activity towards the receptors. Upper diagram: effects of the different modifications of chemokine N‐terminal domains on receptor binding and/or activity: ≈ no effect or <5‐fold change; ↑ or ↓ increase or decrease by >5 fold; ↑↑ or ↓↓ increase or decrease by >50 fold. Lower diagram: activity (shown as green circles) and lack of activity (shown as red circles) in G protein signalling (Gpro) and β‐arrestin recruitment (βarr) of peptides comprising the flexible N terminus and the N‐loop (1–17/21), their D stereoisomers and proximally modified variants (P2G, 2–17). (D) Comparison of ACKR3 and CXCR3/4 binding pockets. The activation determinants of ACKR3 are localized closer to the surface compared with CXCR3 and CXCR4. Ligand binding to ACKR3 pocket triggers a limited signalling repertoire (i.e. β‐arrestin‐2 recruitment). In contrast, CXCR4 and CXCR3 activation determinants are localized deeper in the ligand binding pocket with chemokine triggering G protein signalling and, subsequently, arrestin recruitment.
The results from binding and activity analyses of peptides derived from CXCL12 and vCCL2 re‐examined in this study were consistent with previous reports (Crump et al., 1997; Heveker et al., 1998; Loetscher et al., 1998; Luo et al., 2000; Zhou et al., 2000, 2002; Chevigne et al., 2011). The investigation of a larger set of modifications of chemokine‐derived peptides, not only on CXCR4 but also on CXCR3 and ACKR3, provided new insights into the importance of different chemokine N‐terminal features for receptor binding and activation. Notably, our study showed that the use of peptides derived from chemokine N termini to probe the ligand‐binding pocket or to evaluate the importance of specific modifications on binding and signalling is not only restricted to CXCR4 but can also be extended to other receptors in the CXC and ACKR subfamilies.
Different roles of chemokine N‐loop and proximal N‐terminal residues in binding and activation of CXCR3 and CXCR4, compared with ACKR3
The importance of the chemokine N‐loop for tight receptor binding initially observed for the CXCR4‐CXCL12 pair was conserved in CXCR3 (Figure 7A, B). N‐loop truncation in peptides derived from CXCL12 and CXCL11 reduced drastically their binding to CXCR4 and CXCR3. Moreover, exchange of the N‐loop of CXCL11 for that of CXCL10 had only a minimal effect on CXCR3 activation, consistent with the ability of CXCL10 to bind CXCR3 with a somewhat lower affinity compared to CXCL11 (Clark‐Lewis et al., 2003; Heise et al., 2005). In contrast, substitution with the N‐loop of CXCL12 decreased CXCR3 binding and reduced the potency of both G protein and β‐arrestin‐2 signalling by approximately 10‐fold. This finding was expected, as CXCL12 is not a ligand for CXCR3, thus confirming the important role of the N‐loop for chemokine binding to CRS1 of canonical chemokine receptors, CXCR3 and CXCR4.
In contrast, N‐loop truncation in peptides derived from CXCL12 or CXCL11 only slightly affects ACKR3 binding and signalling. ACKR3 binding and activation by full‐length CXCL11 was also insensitive to N‐loop substitutions with those of CXCL10 or CXCL12. Taken together, the combined results from the N‐loop chimeras and truncated peptides suggest that N‐loop contacts, which are important for CXCR4 and CXCR3 recognition, are not essential for ACKR3‐chemokine interactions (Figure 7A).
As previously been reported for CXCR4, dimeric peptides derived from CXCL11 were also more potent in CXCR3 binding and activation than their monomeric counterparts (Figure 7B). This improvement in affinity and consequentially in potency may result from the presence of the cysteine bridge linking the two monomers possibly mimicking the first disulfide bridge of the parental chemokines, which, as suggested by the first crystal structures of chemokine‐receptor complexes, could be crucial for tight receptor binding (Burg et al., 2015; Qin et al., 2015). Alternatively, better binding of bivalent peptides may arise from an avidity effect through binding of receptor homodimers. Indeed, as described for CXCR4, CXCR3 may also form homodimers, possibly through a TM5 and TM6 dimer interface, bringing the two chemokine N terminus‐binding pockets (CRS2) into close proximity (Wu et al., 2010; Chevigne et al., 2014). Finally, we cannot exclude the possibility that the improvement in binding and potency results from allosteric contacts of the second protomer with extracellular non‐CRS2 determinants of the same receptor.
In our system, dimeric peptides derived from the flexible N termini of CXCL12 and CXCL11 bound to ACKR3 with higher affinities and induced β‐arrestin‐2 recruitment to the receptor with increased potency. These data indicate that the first disulfide bridge of chemokines may also be an important factor in ACKR3 recognition. Interestingly, dimerization of the vCCL2‐derived peptide had little effect on ACKR3 binding suggesting that the vCCL2 N terminus may occupy the receptor's sub‐pockets differently or bind more deeply in the TM region of ACKR3 than the N termini of either CXCL12 or CXCL11. Furthermore, although the full‐length chemokines CXCL12 and CXCL11 display a higher affinity for ACKR3 than does vCCL2, the N‐terminal vCCL2 peptide retained a higher proportion of the parental chemokine binding capacity than the peptides derived from the endogenous chemokines. A similar observation was made for CXCR4 (Figure 7C). This higher affinity may simply result from the larger size of vCCL2 peptide (21 residues versus 17 for the CXC chemokines) or may be related to the ability of vCCL2 to bind a broad spectrum of receptors of all the four classes (XC, CC, CXC and CX3C), implying that its core may have evolved less tight and more promiscuous binding capacity, while the N terminus plays a more important role in specific binding and modulation of downstream activity (Luo et al., 2000; Szpakowska and Chevigne, 2015).
Furthermore, peptides bearing simple modifications such as amino‐terminal residue deletion, P2G mutation or D‐amino acid replacement retained their ability to bind to both CXCR3 and CXCR4, but their agonist activity was drastically reduced or changed to antagonism. These results illustrate the crucial role of the proximal N‐terminal region of chemokines and the stringency of the contacts at CRS2 that are required for classical CXC receptor activation (Figure 7).
Strikingly, although some modifications introduced in the proximal part of the peptides derived from the three chemokines affected their binding to ACKR3, none of them resulted in a loss of receptor activation. For instance, in contrast to the results with CXCR4 and CXCR3, proximal N‐terminal truncation, P2G mutation, and even complete D‐amino acid replacement in CXCL12‐ and CXCL11‐derived peptides did not change their activity towards ACKR3 (Figure 7).
Collectively, these data suggest that the molecular interaction network within the binding pocket required for ACKR3 activation is different and less stringent than that for CXCR4 and CXCR3 and relies less on the proximal residues of the chemokine (Figures 6A and 7D).
Mechanistic interpretation of ACKR3 permissiveness to chemokine N‐terminal modifications
Our results as well as numerous previous studies conducted with non‐chemokine ligands demonstrated that in contrast to classical chemokine receptors, such as CXCR4 or CXCR3, ACKR3 is an ‘activation‐prone’ receptor (Figure 6B) and is able to accommodate a diverse set of ligands including small molecules, peptides and several chemokines, in each case resulting in receptor activation and ultimately arrestin recruitment (Burns et al., 2006; Wijtmans et al., 2012; Ikeda et al., 2013; Montpas et al., 2015; Oishi et al., 2015; Szpakowska et al., 2016; Benredjem et al., 2017; Gustavsson et al., 2017). Numerous CXCR4 antagonists, including http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=844, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2902 and vCCL2, act as agonists towards ACKR3. Recent screening by phage display of N‐terminally randomized CXCL12 also led to the selection of variants all displaying ACKR3 agonist properties (Hanes et al., 2015). Jointly, these results suggest that ACKR3 has a highly plastic binding pocket which funnels the diverse binding modes and unique contacts of its many ligands at the extracellular half of the receptor into a limited signalling repertoire (i.e. arrestin recruitment) arising from its intracellular half (Figure 7D). As such, it is tempting to speculate that the extracellular half of ACKR3 and its intracellular half may operate semi‐autonomously, save for the ability of all ACKR3 ligands to set off an easily triggered ‘tripwire’ leading to arrestin recruitment (Rajagopal et al., 2013).
ACKR3 was the only receptor studied for which activation was insensitive to changes in the N‐loop, D‐stereoisomer replacement, mutations and truncations at the proximal N termini of its chemokine ligands. Although recent structural data suggest that ACKR3 activation relies on a mechanism similar to classical receptors (Gustavsson et al., 2017), the higher propensity of ACKR3 towards activation appears to depend on a less sophisticated mechanism, involving a simple, highly accessible molecular switch leading to the observed ‘agonism bias’. Indeed, our findings suggest that the principal determinants for chemokine affinity and activation of ACKR3 are located between residues 2 to 9 of CXCL12 and CXCL11 with no or little contribution of the N‐loop and the N‐terminal residues. Therefore, different sub‐pocket occupancy or structural changes may be required for ACKR3 as compared with CXCR4 and CXCR3, with activation determinants located not only at the bottom but also more on the outside of the orthosteric ligand binding pocket (Figure 7) (Montpas et al., 2015). To support this hypothesis, it has been demonstrated that charged residues located at the top of TM4, TM6 and in extracellular loop (ECL) 2 [e.g. R197ECL2, D1794.60 and D2756.58; Ballesteros‐Weinstein nomenclature in superscript; Ballesteros and Weinstein, (Academic Press, 1995)] are crucial for activation by TC14012 and its derivatives. Due to the localization of these residues to the upper part of the TM domains and the roots of the ECLs in ACKR3, it was speculated that ACKR3‐mediated β‐arrestin‐2 recruitment may not rely on a classical CRS2 (Montpas et al., 2015), characterized by activation toggle interactions located exclusively at the bottom of the ligand binding pocket. Recent studies on ACKR3 further support this hypothesis. First, it was shown that CXCL11 and CXCL12 depend on many of the same residues as TC14012 in and adjacent to the ECLs of ACKR3 for binding and/or activation, as well as some newly described sites (e.g. E1143.22 and K206ECL2). Surprisingly, a mutation at the top of TM3 adjacent to ECL2 (K1183.26A) caused constitutive β‐arrestin recruitment giving credence to the notion that arrestin signalling could also be triggered by the top of the ligand binding pocket (Benredjem et al., 2017). Conversely, mutation of a tryptophan at the top of the cavity at the ECL2‐TM5 junction (W2085.34A) caused a decrease in β‐arrestin recruitment for CXCL12 and a small molecule partial agonist binding at the bottom of ACKR3 ligand binding pocket (Gustavsson et al., 2017).
These mechanistic considerations aside, the tendency of ACKR3 towards activation poses challenges to the discovery of efficient and specific antagonists. So far, no small molecules have been shown to decrease arrestin recruitment at ACKR3, illustrating the importance of understanding the interactions and mechanisms underlying its activation. Encouragingly, however, the recent discovery of two chemokine receptor antagonists targeting the intracellular domain of CCR2 and CCR9 (Oswald et al., 2016; Zheng et al., 2016) supports the feasibility of developing similar antagonists at ACKR3.
Implications of N‐terminal modifications on chemokine scavenging function of ACKR3
The higher permissivity of ACKR3 to ligand modifications, which at first sight may appear surprising, may be of functional importance with respect to its scavenging functions. Indeed, in addition to scavenging native chemokines to limit their agonist activity on CXCR4 and CXCR3, our results suggest that ACKR3 may also bind and clear N‐terminally processed chemokine species, such as those resulting from the action of proteases, including http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1612. Cleavage of CXCL11 and CXCL12 is an efficient post‐translational processing event that inactivates agonist activity towards CXCR3 and CXCR4 and leads to detectable serum levels of CXCL113–73 and CXCL123–73 (Proost et al., 1998, 2007). Although CXCL113–73 showed ~5‐fold reduction in binding to ACKR3 compared to CXCL11WT, the deletion of the first two residues of CXCL11 decreased binding to CXCR3 to a larger extent (~40 fold) and, more importantly, had only a modest effect on ACKR3 activation (Proost et al., 2001, 2007). Our results are in agreement with data from a recent study on proteolyzed CXCL12 and its interactions with CXCR4 and ACKR3 (Janssens et al., 2017).
In conclusion, this study represents the first comparative, in‐depth, structure–function analysis of the importance of the chemokine N‐terminal features for the binding to and activation of conventional and ACKRs. Our results revealed unexpected contrasts between the structural motifs required for conventional and atypical receptors with CRS1 (binding to the N‐loop) or CRS2 (binding to the chemokine N terminus) interactions important for CXCR3 and CXCR4 binding and activation but dispensable for ACKR3 (Figure 7). These data demonstrate that in addition to distinct functional roles, ACKR3 also presents an activation mechanism different from that of CXCR4 and CXCR3, with which it shares chemokine ligands, supporting its recent classification as an ACKR (Bachelerie et al., 2014b). However, whether the observations reported in this study are characteristic of all ACKRs and CKRs remains to be investigated.
Author contributions
B.F.V., N.H. and A.C. supervised the study. M.S., A.M.N., D.R., T.D., F.G.V., N.D., P.‐A.G., M.C., M.M. and G.S.O. performed the experiments. M.S., A.M.N., J.H., T.D., A.K., M.M., D.S., B.F.V., N.H. and A.C. analysed and interpreted the data. M.S., A.N., A.K., B.F.V., N.H. and A.C. wrote the paper. All authors contributed to the editing.
Conflict of interest
B.F.V. is a co‐founder and has significant financial interest in Protein Foundry, LLC.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This study was supported by the Luxembourg Institute of Health (MESR) grants 20160116 and 20170113, Fonds De La Recherche Scientifique ‐ FNRS ‐ Télévie grants 7456814 and 7461515, the programme financing of the KU Leuven (PF/10/018), F.R.S.‐FNRS incentive grant for scientific research (MIS‐F.4510.14) as well as National Institutes of Health (NIH) grants F30CA196040 (A.K.) and R01 AI058072 (B.F.V.) and grant MOP123421 from the Canadian Institutes of Health Research (CIHR) (N.H.). M.S., M.M. and P.‐A.G. are the Luxembourg National Research Fund (‘Fonds National de la Recherche ‐ Aides à la Formation‐Recherche’, FNR‐AFR) PhD fellows (grants AFR‐3004509, AFR‐11274579 and AFR‐5907281) and INTER/FWO/15/10358798. J.H. is a ‘Fonds de la Recherche Scientifique’ (Fonds de la Recherche Scientifique ‐ FNRS, Belgium) research associate. N.D. is a ‘Fonds pour la Recherche dans l'Industrie et l'Agriculture (FRIA)’ research fellow. A.K. is a member of the NIH‐supported (T32 GM080202) Medical Scientist Training Program at MCW. F.G.V. acknowledges a CIHR scholarship. The authors wish to thank Mathias Plourde in the Heveker Lab for his contributions to the BRET measurements.
Szpakowska, M. , Nevins, A. M. , Meyrath, M. , Rhainds, D. , D'huys, T. , Guité‐Vinet, F. , Dupuis, N. , Gauthier, P.‐A. , Counson, M. , Kleist, A. , St‐Onge, G. , Hanson, J. , Schols, D. , Volkman, B. F. , Heveker, N. , and Chevigné, A. (2018) Different contributions of chemokine N‐terminal features attest to a different ligand binding mode and a bias towards activation of ACKR3/CXCR7 compared with CXCR4 and CXCR3. British Journal of Pharmacology, 175: 1419–1438. doi: 10.1111/bph.14132.
References
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen SJ, Crown SE, Handel TM (2007). Chemokine: receptor structure, interactions, and antagonism. Annu Rev Immunol 25: 787–820. [DOI] [PubMed] [Google Scholar]
- Bachelerie F, Ben‐Baruch A, Burkhardt AM, Combadiere C, Farber JM, Graham GJ et al (2014a). International Union of Basic and Clinical Pharmacology. [corrected]. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol Rev 66: 1–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachelerie F, Graham GJ, Locati M, Mantovani A, Murphy PM, Nibbs R et al (2014b). New nomenclature for atypical chemokine receptors. Nat Immunol 15: 207–208. [DOI] [PubMed] [Google Scholar]
- Balabanian K, Lagane B, Infantino S, Chow KY, Harriague J, Moepps B et al (2005). The chemokine SDF‐1/CXCL12 binds to and signals through the orphan receptor RDC1 in T lymphocytes. J Biol Chem 280: 35760–35766. [DOI] [PubMed] [Google Scholar]
- Balkwill F (2004). Cancer and the chemokine network. Nat Rev Cancer 4: 540–550. [DOI] [PubMed] [Google Scholar]
- Ballesteros JA, Weinstein H (1995). Integrated methods for the construction of three‐dimensional models and computational probing of structure–function relations in G protein‐coupled receptors In: Sealfon SC Conn PM, (eds.) Methods in Neurosciences, Vol. 25. Academic Press: San Diego, CA: pp. 366–428. [Google Scholar]
- Benredjem B, Girard M, Rhainds D, St‐Onge G, Heveker N (2017). Mutational analysis of atypical chemokine receptor 3 (ACKR3/CXCR7) interaction with its chemokine ligands CXCL11 and CXCL12. J Biol Chem 292: 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berahovich RD, Zabel BA, Lewen S, Walters MJ, Ebsworth K, Wang Y et al (2014). Endothelial expression of CXCR7 and the regulation of systemic CXCL12 levels. Immunology 141: 111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berchiche YA, Gravel S, Pelletier ME, St‐Onge G, Heveker N (2011). Different effects of the different natural CC chemokine receptor 2b ligands on beta‐arrestin recruitment, Galphai signaling, and receptor internalization. Mol Pharmacol 79: 488–498. [DOI] [PubMed] [Google Scholar]
- Bjorndal A, Deng H, Jansson M, Fiore JR, Colognesi C, Karlsson A et al (1997). Coreceptor usage of primary human immunodeficiency virus type 1 isolates varies according to biological phenotype. J Virol 71: 7478–7487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldajipour B, Mahabaleshwar H, Kardash E, Reichman‐Fried M, Blaser H, Minina S et al (2008). Control of chemokine‐guided cell migration by ligand sequestration. Cell 132: 463–473. [DOI] [PubMed] [Google Scholar]
- Burg JS, Ingram JR, Venkatakrishnan AJ, Jude KM, Dukkipati A, Feinberg EN et al (2015). Structural biology. Structural basis for chemokine recognition and activation of a viral G protein‐coupled receptor. Science 347: 1113–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns JM, Summers BC, Wang Y, Melikian A, Berahovich R, Miao Z et al (2006). A novel chemokine receptor for SDF‐1 and I‐TAC involved in cell survival, cell adhesion, and tumor development. J Exp Med 203: 2201–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevigne A, Fievez V, Schmit JC, Deroo S (2011). Engineering and screening the N‐terminus of chemokines for drug discovery. Biochem Pharmacol 82: 1438–1456. [DOI] [PubMed] [Google Scholar]
- Chevigne A, Fievez V, Szpakowska M, Fischer A, Counson M, Plesseria JM et al (2014). Neutralising properties of peptides derived from CXCR4 extracellular loops towards CXCL12 binding and HIV‐1 infection. Biochim Biophys Acta 1843: 1031–1041. [DOI] [PubMed] [Google Scholar]
- Choi WT, Kumar S, Madani N, Han X, Tian S, Dong CZ et al (2012). A novel synthetic bivalent ligand to probe chemokine receptor CXCR4 dimerization and inhibit HIV‐1 entry. Biochemistry 51: 7078–7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark‐Lewis I, Mattioli I, Gong JH, Loetscher P (2003). Structure‐function relationship between the human chemokine receptor CXCR3 and its ligands. J Biol Chem 278: 289–295. [DOI] [PubMed] [Google Scholar]
- Cole KE, Strick CA, Paradis TJ, Ogborne KT, Loetscher M, Gladue RP et al (1998). Interferon‐inducible T cell alpha chemoattractant 0(I‐TAC): a novel non‐ELR CXC chemokine with potent activity on activated T cells through selective high affinity binding to CXCR3. J Exp Med 187: 2009–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crump MP, Elisseeva E, Gong J, Clark‐Lewis I, Sykes BD (2001). Structure/function of human herpesvirus‐8 MIP‐II (1‐71) and the antagonist N‐terminal segment (1‐10). FEBS Lett 489: 171–175. [DOI] [PubMed] [Google Scholar]
- Crump MP, Gong JH, Loetscher P, Rajarathnam K, Amara A, Arenzana‐Seisdedos F et al (1997). Solution structure and basis for functional activity of stromal cell‐derived factor‐1; dissociation of CXCR4 activation from binding and inhibition of HIV‐1. EMBO J 16: 6996–7007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon AS, Schwinn MK, Hall MP, Zimmerman K, Otto P, Lubben TH et al (2016). NanoLuc Complementation Reporter Optimized for Accurate Measurement of Protein Interactions in Cells. ACS Chem Biol 11: 400–408. [DOI] [PubMed] [Google Scholar]
- Dyer DP, Salanga CL, Volkman BF, Kawamura T, Handel TM (2016). The dependence of chemokine‐glycosaminoglycan interactions on chemokine oligomerization. Glycobiology 26: 312–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez EJ, Lolis E (2002). Structure, function, and inhibition of chemokines. Annu Rev Pharmacol Toxicol 42: 469–499. [DOI] [PubMed] [Google Scholar]
- Ganju RK, Brubaker SA, Meyer J, Dutt P, Yang Y, Qin S et al (1998). The alpha‐chemokine, stromal cell‐derived factor‐1alpha, binds to the transmembrane G‐protein‐coupled CXCR‐4 receptor and activates multiple signal transduction pathways. J Biol Chem 273: 23169–23175. [DOI] [PubMed] [Google Scholar]
- Gilissen J, Geubelle P, Dupuis N, Laschet C, Pirotte B, Hanson J (2015). Forskolin‐free cAMP assay for Gi‐coupled receptors. Biochem Pharmacol 98: 381–391. [DOI] [PubMed] [Google Scholar]
- Gustavsson M, Wang L, van Gils N, Stephens BS, Zhang P, Schall TJ et al (2017). Structural basis of ligand interaction with atypical chemokine receptor 3. Nat Commun 8: 14135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanes MS, Salanga CL, Chowdry AB, Comerford I, McColl SR, Kufareva I et al (2015). Dual targeting of the chemokine receptors CXCR4 and ACKR3 with novel engineered chemokines. J Biol Chem 290: 22385–22397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al. (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091‐D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heise CE, Pahuja A, Hudson SC, Mistry MS, Putnam AL, Gross MM et al (2005). Pharmacological characterization of CXC chemokine receptor 3 ligands and a small molecule antagonist. J Pharmacol Exp Ther 313: 1263–1271. [DOI] [PubMed] [Google Scholar]
- Heveker N, Montes M, Germeroth L, Amara A, Trautmann A, Alizon M et al (1998). Dissociation of the signalling and antiviral properties of SDF‐1‐derived small peptides. Curr Biol 8: 369–376. [DOI] [PubMed] [Google Scholar]
- Heveker N, Tissot M, Thuret A, Schneider‐Mergener J, Alizon M, Roch M et al (2001). Pharmacological properties of peptides derived from stromal cell‐derived factor 1: study on human polymorphonuclear cells. Mol Pharmacol 59: 1418–1425. [DOI] [PubMed] [Google Scholar]
- Ikeda Y, Kumagai H, Skach A, Sato M, Yanagisawa M (2013). Modulation of circadian glucocorticoid oscillation via adrenal opioid‐CXCR7 signaling alters emotional behavior. Cell 155: 1323–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens R, Mortier A, Boff D, Ruytinx P, Gouwy M, Vantilt B et al (2017). Truncation of CXCL12 by CD26 reduces its CXC chemokine receptor 4‐ and atypical chemokine receptor 3‐dependent activity on endothelial cells and lymphocytes. Biochem Pharmacol 132: 92–101. [DOI] [PubMed] [Google Scholar]
- Kalatskaya I, Berchiche YA, Gravel S, Limberg BJ, Rosenbaum JS, Heveker N (2009). AMD3100 is a CXCR7 ligand with allosteric agonist properties. Mol Pharmacol 75: 1240–1247. [DOI] [PubMed] [Google Scholar]
- Kaya AI, Onaran HO, Ozcan G, Ambrosio C, Costa T, Balli S et al (2012). Cell contact‐dependent functional selectivity of beta2‐adrenergic receptor ligands in stimulating cAMP accumulation and extracellular signal‐regulated kinase phosphorylation. J Biol Chem 287: 6362–6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kledal TN, Rosenkilde MM, Coulin F, Simmons G, Johnsen AH, Alouani S et al (1997). A broad‐spectrum chemokine antagonist encoded by Kaposi's sarcoma‐associated herpesvirus. Science 277: 1656–1659. [DOI] [PubMed] [Google Scholar]
- Kleist AB, Getschman AE, Ziarek JJ, Nevins AM, Gauthier PA, Chevigne A et al (2016). New paradigms in chemokine receptor signal transduction: moving beyond the two‐site model. Biochem Pharmacol 114: 53–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kufareva I, Stephens BS, Holden LG, Qin L, Zhao C, Kawamura T et al (2014). Stoichiometry and geometry of the CXC chemokine receptor 4 complex with CXC ligand 12: molecular modeling and experimental validation. Proc Natl Acad Sci U S A 111: E5363–E5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leduc M, Breton B, Gales C, Le Gouill C, Bouvier M, Chemtob S et al (2009). Functional selectivity of natural and synthetic prostaglandin EP4 receptor ligands. J Pharmacol Exp Ther 331: 297–307. [DOI] [PubMed] [Google Scholar]
- Levoye A, Balabanian K, Baleux F, Bachelerie F, Lagane B (2009). CXCR7 heterodimerizes with CXCR4 and regulates CXCL12‐mediated G protein signaling. Blood 113: 6085–6093. [DOI] [PubMed] [Google Scholar]
- Loetscher M, Gerber B, Loetscher P, Jones SA, Piali L, Clark‐Lewis I et al (1996). Chemokine receptor specific for IP10 and mig: structure, function, and expression in activated T‐lymphocytes. J Exp Med 184: 963–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loetscher P, Gong JH, Dewald B, Baggiolini M, Clark‐Lewis I (1998). N‐terminal peptides of stromal cell‐derived factor‐1 with CXC chemokine receptor 4 agonist and antagonist activities. J Biol Chem 273: 22279–22283. [DOI] [PubMed] [Google Scholar]
- Ludwig A, Schiemann F, Mentlein R, Lindner B, Brandt E (2002). Dipeptidyl peptidase IV (CD26) on T cells cleaves the CXC chemokine CXCL11 (I‐TAC) and abolishes the stimulating but not the desensitizing potential of the chemokine. J Leukoc Biol 72: 183–191. [PubMed] [Google Scholar]
- Luker KE, Steele JM, Mihalko LA, Ray P, Luker GD (2010). Constitutive and chemokine‐dependent internalization and recycling of CXCR7 in breast cancer cells to degrade chemokine ligands. Oncogene 29: 4599–4610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z, Fan X, Zhou N, Hiraoka M, Luo J, Kaji H et al (2000). Structure‐function study and anti‐HIV activity of synthetic peptide analogues derived from viral chemokine vMIP‐II. Biochemistry 39: 13545–13550. [DOI] [PubMed] [Google Scholar]
- Montpas N, Cabana J, St‐Onge G, Gravel S, Morin G, Kuroyanagi T et al (2015). Mode of binding of the cyclic agonist peptide TC14012 to CXCR7: identification of receptor and compound determinants. Biochemistry 54: 1505–1515. [DOI] [PubMed] [Google Scholar]
- Naumann U, Cameroni E, Pruenster M, Mahabaleshwar H, Raz E, Zerwes HG et al (2010). CXCR7 functions as a scavenger for CXCL12 and CXCL11. PloS one 5: e9175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oishi S, Kuroyanagi T, Kubo T, Montpas N, Yoshikawa Y, Misu R et al (2015). Development of novel CXC chemokine receptor 7 (CXCR7) ligands: selectivity switch from CXCR4 antagonists with a cyclic pentapeptide scaffold. J Med Chem 58: 5218–5225. [DOI] [PubMed] [Google Scholar]
- Oswald C, Rappas M, Kean J, Dore AS, Errey JC, Bennett K et al (2016). Intracellular allosteric antagonism of the CCR9 receptor. Nature 540: 462–465. [DOI] [PubMed] [Google Scholar]
- Princen K, Hatse S, Vermeire K, De Clercq E, Schols D (2003). Evaluation of SDF‐1/CXCR4‐induced Ca2+ signaling by fluorometric imaging plate reader (FLIPR) and flow cytometry. Cytometry A 51: 35–45. [DOI] [PubMed] [Google Scholar]
- Proost P, Mortier A, Loos T, Vandercappellen J, Gouwy M, Ronsse I et al (2007). Proteolytic processing of CXCL11 by CD13/aminopeptidase N impairs CXCR3 and CXCR7 binding and signaling and reduces lymphocyte and endothelial cell migration. Blood 110: 37–44. [DOI] [PubMed] [Google Scholar]
- Proost P, Schutyser E, Menten P, Struyf S, Wuyts A, Opdenakker G et al (2001). Amino‐terminal truncation of CXCR3 agonists impairs receptor signaling and lymphocyte chemotaxis, while preserving antiangiogenic properties. Blood 98: 3554–3561. [DOI] [PubMed] [Google Scholar]
- Proost P, Struyf S, Schols D, Durinx C, Wuyts A, Lenaerts JP et al (1998). Processing by CD26/dipeptidyl‐peptidase IV reduces the chemotactic and anti‐HIV‐1 activity of stromal‐cell‐derived factor‐1alpha. FEBS Lett 432: 73–76. [DOI] [PubMed] [Google Scholar]
- Qin L, Kufareva I, Holden LG, Wang C, Zheng Y, Zhao C et al (2015). Structural biology. Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science 347: 1117–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal S, Bassoni DL, Campbell JJ, Gerard NP, Gerard C, Wehrman TS (2013). Biased agonism as a mechanism for differential signaling by chemokine receptors. J Biol Chem 288: 35039–35048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal S, Kim J, Ahn S, Craig S, Lam CM, Gerard NP et al (2010). Beta‐arrestin‐ but not G protein‐mediated signaling by the ‘decoy’ receptor CXCR7. Proc Natl Acad Sci U S A 107: 628–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romagnani P, Lasagni L, Annunziato F, Serio M, Romagnani S (2004). CXC chemokines: the regulatory link between inflammation and angiogenesis. Trends Immunol 25: 201–209. [DOI] [PubMed] [Google Scholar]
- Rossi D, Zlotnik A (2000). The biology of chemokines and their receptors. Annu Rev Immunol 18: 217–242. [DOI] [PubMed] [Google Scholar]
- Steen A, Larsen O, Thiele S, Rosenkilde MM (2014). Biased and g protein‐independent signaling of chemokine receptors. Front Immunol 5: 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens B, Handel TM (2013). Chemokine receptor oligomerization and allostery. Prog Mol Biol Transl Sci 115: 375–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Cheng Z, Ma L, Pei G (2002). Beta‐arrestin2 is critically involved in CXCR4‐mediated chemotaxis, and this is mediated by its enhancement of p38 MAPK activation. J Biol Chem 277: 49212–49219. [DOI] [PubMed] [Google Scholar]
- Szpakowska M, Chevigne A (2015). vCCL2/vMIP‐II, the viral master KEYmokine. J Leukoc Biol 99: 893–900. [DOI] [PubMed] [Google Scholar]
- Szpakowska M, Dupuis N, Baragli A, Counson M, Hanson J, Piette J et al (2016). Human herpesvirus 8‐encoded chemokine vCCL2/vMIP‐II is an agonist of the atypical chemokine receptor ACKR3/CXCR7. Biochem Pharmacol 114: 14–21. [DOI] [PubMed] [Google Scholar]
- Szpakowska M, Fievez V, Arumugan K, van Nuland N, Schmit JC, Chevigne A (2012). Function, diversity and therapeutic potential of the N‐terminal domain of human chemokine receptors. Biochem Pharmacol 84: 1366–1380. [DOI] [PubMed] [Google Scholar]
- Takekoshi T, Ziarek JJ, Volkman BF, Hwang ST (2012). A locked, dimeric CXCL12 variant effectively inhibits pulmonary metastasis of CXCR4‐expressing melanoma cells due to enhanced serum stability. Mol Cancer Ther 11: 2516–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldkamp CT, Seibert C, Peterson FC, De la Cruz NB, Haugner JC 3rd, Basnet H et al (2008). Structural basis of CXCR4 sulfotyrosine recognition by the chemokine SDF‐1/CXCL12. Sci Signal 1: ra4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts AO, van Lipzig MM, Jaeger WC, Seeber RM, van Zwam M, Vinet J et al (2013). Identification and profiling of CXCR3‐CXCR4 chemokine receptor heteromer complexes. Br J Pharmacol 168: 1662–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijtmans M, Maussang D, Sirci F, Scholten DJ, Canals M, Mujic‐Delic A et al (2012). Synthesis, modeling and functional activity of substituted styrene‐amides as small‐molecule CXCR7 agonists. Eur J Med Chem 51: 184–192. [DOI] [PubMed] [Google Scholar]
- Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V et al (2010). Structures of the CXCR4 chemokine GPCR with small‐molecule and cyclic peptide antagonists. Science 330: 1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Han GW, Abagyan R, Wu B, Stevens RC, Cherezov V et al (2017). Structure of CC chemokine receptor 5 with a potent chemokine antagonist reveals mechanisms of chemokine recognition and molecular mimicry by HIV. Immunity 46: 1005, `e1005–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Qin L, Zacarias NV, de Vries H, Han GW, Gustavsson M et al (2016). Structure of CC chemokine receptor 2 with orthosteric and allosteric antagonists. Nature 540: 458–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou N, Luo Z, Luo J, Fan X, Cayabyab M, Hiraoka M et al (2002). Exploring the stereochemistry of CXCR4‐peptide recognition and inhibiting HIV‐1 entry with D‐peptides derived from chemokines. J Biol Chem 277: 17476–17485. [DOI] [PubMed] [Google Scholar]
- Zhou N, Luo Z, Luo J, Hall JW, Huang Z (2000). A novel peptide antagonist of CXCR4 derived from the N‐terminus of viral chemokine vMIP‐II. Biochemistry 39: 3782–3787. [DOI] [PubMed] [Google Scholar]
- Zlotnik A, Yoshie O (2012). The chemokine superfamily revisited. Immunity 36: 705–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou YR, Kottmann AH, Kuroda M, Taniuchi I, Littman DR (1998). Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature 393: 595–599. [DOI] [PubMed] [Google Scholar]