

Abstract
Background and Purpose
Aucubin, the predominant component of Eucommia ulmoides Oliv., has been shown to have profound effects on oxidative stress. As oxidative stress has previously been demonstrated to contribute to acute and chronic myocardial injury, we tested the effects of aucubin on cardiac remodelling and heart failure.
Experimental Approach
Initially, H9c2 cardiomyocytes and neonatal rat cardiomyocytes pretreated with aucubin (1, 3, 10, 25 and 50 μM) were challenged with phenylephrine. Secondly, the transverse aorta was constricted in C57/B6 and neuronal NOS (nNOS)‐knockout mice, then aucubin (1 or 5 mg·kg−1 body weight day−1) was injected i.p. for 25 days. Hypertrophy was evaluated by assessing morphological changes, echocardiographic parameters, histological analyses and hypertrophic markers. Oxidative stress was evaluated by examining ROS generation, oxidase activity and NO generation. NOS expression was determined by Western blotting.
Key Results
Aucubin effectively suppressed cardiac remodelling; in mice, aucubin substantially inhibited pressure overload‐induced cardiac hypertrophy, fibrosis and inflammation, whereas knocking out nNOS abolished these cardioprotective effects of aucubin. Blocking or knocking down the β3‐adrenoceptor abolished the protective effects of aucubin in vitro. Furthermore, aucubin enhanced the protective effects of a β3‐adrenoceptor agonist in vitro by increasing cellular cAMP levels, whereas treatment with an adenylate cyclase (AC) inhibitor abolished the cardioprotective effects of aucubin.
Conclusions and Implications
Aucubin suppresses oxidative stress during cardiac remodelling by increasing the expression of nNOS in a process that requires activation of the β3‐adrenoceptor/AC/cAMP pathway. These findings suggest that aucubin could have potential as a treatment for cardiac remodelling and heart failure.
Abbreviations
- β‐myosin heavy chain
β‐MHC
- atrial natriuretic peptide
ANP
- body weight
BW
- connective tissue growth factor
CTGF
- endoplasmic reticulum
ER
- heart weight
HW
- high dose
HD
- knockout
KO
- left ventricle
LV
- l‐nitro‐arginine methyl ester
l‐NAME
- low dose
LD
- lung weight
LW
- N‐acetyl‐l‐cysteine
NAC
- neonatal rat cardiomyocytes
NRCMs
- Picro‐Sirius Red
PSR
- tibia length
TL
- vinyl‐l‐NIO hydrochloride
l‐VNIO
Introduction
Pathological cardiac remodelling is usually characterized by alterations in cardiac structure, shape and function. Initially, these alterations are adaptive responses to maintain cardiac function; however, under conditions of sustained stress and as time progresses, these changes become maladaptive, and the heart ultimately fails (Bui et al., 2011). Accumulating evidence has suggested that the pathological remodelling response is coordinated by orchestrated cellular modifications, including oxidative stress, endoplasmic reticulum (ER) stress, impairment of autophagy, metabolism (Shimizu and Minamino, 2016) and numerous signalling pathways. For example, the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=30‐http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2352‐http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=284, GPCR Gαq/PLC‐http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1482 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2503/http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=285 pathways comprise a complex network that converges on corresponding transcription factors and promotes abnormal changes associated with pathological cardiac remodelling (Heineke and Molkentin, 2006). However, far less is known about how cardiac remodelling is suppressed. Negative regulation of these signalling pathways could be of significant therapeutic value and could facilitate the development of therapies for the treatment of cardiac remodelling and heart failure.
Eucommia ulmoides Oliv. is a famous traditional Chinese medicine that exhibits antioxidative stress and neuroprotective effects (Andoh et al., 2017). Aucubin, a major compound found in E. ulmoides Oliv., has been shown to regulate oxidative stress (Xue et al., 2009), ER stress (Lee et al., 2013), autophagy and apoptosis (Wang et al., 2017). Previous studies have explored the therapeutic potential of aucubin for many other diseases, such as diabetes mellitus (Jin et al., 2008), skin fibrosis (Ho et al., 2005) and peripheral neuropathy (Andoh et al., 2017). Aucubin has been reported to exert antioxidative effects in neurons (Xue et al., 2009), liver and kidneys (Jin et al., 2008) and to exert anti‐ER stress effects in peripheral Schwann cells (Andoh et al., 2017) and HepG2 cells (Lee et al., 2013). Aucubin also inhibits inflammation in many cell types, such as adipocytes (Lee et al., 2013), articular chondrocytes (Wang et al., 2015) and mast cells (Jeong et al., 2002). These data suggest a potential protective application for aucubin in cardiac pathology, especially in terms of cardiac remodelling. However, data supporting this hypothesis have been largely lacking. In the current study, we demonstrate that aucubin suppresses pathological cardiac remodelling elicited by pressure overload or http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=485 challenge. In addition, our mechanistic investigations reveal a critical role for the β3‐adrenoceptor‐http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1251 (nNOS) cascades in aucubin‐mediated protection against cardiac remodelling.
Methods
Animals and animal models
All of the animal care and experimental procedures conformed to the Animals (Scientific Procedures) Act 1986, of the UK Parliament, Directive 2010/63/EU of the European Parliament, the Guidelines for the Care and Use of Laboratory Animals, published by the United States National Institutes of Health (NIH Publication, revised 2011) and the Guidelines for the Care and Use of Laboratory Animals of the Chinese Animal Welfare Committee and were approved by the Animal Use Committees of our hospital and our institute. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). The current study had no implications for replacement, refinement or reduction. Male C57/B6 mice [8 to 10 weeks old; body weight (BW): 25.5 ± 2 g, NO: 11401300036042] were purchased from the Institute of Laboratory Animal Science, Chinese Academy of Medical Sciences (Beijing, China). nNOS‐knockout (KO) mice (stock no. 002633) were purchased from the Jackson Laboratory (Bar Harbor, USA) through the Model Animal Research Centre of Nanjing University. The animals were housed with a maximum of six other mice in individually‐ventilated cages (with a floor area of 542 cm2 and bedded with corncob). The animals were allowed free access to food and water and were maintained on a 12 h light/dark cycle in a controlled temperature (20–25°C), humidity (50 ± 5%) and specific pathogen‐free environment for a period of 1 week before the study commenced. Then, the mice were grouped according to a random number table to either a sham surgery or an aortic banding (AB) group with or without aucubin treatment. Aucubin was dissolved in sterile saline and administered to the animals by i.p. injection [low dose (LD): 1 mg·kg−1 BW day−1; high dose (HD): 5 mg·kg−1 BW day−1]. Injections began at 3 days post‐surgery and were performed for a further 25 days. Animals in the control group were administered equal volumes of vehicle. The AB surgery and subsequent analyses were performed in a blinded fashion (without knowledge of the operator) for all of the groups. The mice were anaesthetized with an i.p. injection of 3% sodium pentobarbital at a dose of 40 mg·kg−1. When anaesthetized mice ceased to respond to a foot squeeze. The AB or sham procedure was performed according to Jiang et al. (2014) in an aseptic surgery room. To treat post‐operative pain, Temgesic (0.1 mg·kg−1) was applied once daily for 6 days post‐surgery. Doppler analysis, as described previously (Jin et al., 2008), was performed without knowledge of the treatment to confirm that hypertrophy was induced. Four weeks post‐AB surgery, the mice were killed by cervical dislocation, and the hearts and lungs were harvested and weighed to compare the heart weight (HW)/BW (mg·g−1), lung weight (LW)/BW (mg·g−1) and HW/tibia length (TL) (mg·mm−1) ratios between vehicle‐ and aucubin‐treated mice.
Echocardiography and haemodynamics
Echocardiography was performed on anaesthetized (1.5% isoflurane) mice using a MyLab 30CV ultrasound (Biosound Esaote, Florence, Italy) with a 10 MHz linear array ultrasound transducer, as described previously (Wu et al., 2016; Xiao et al., 2017). The left ventricle (LV) was assessed in both parasternal long‐axis and short‐axis views at a frame rate of 120 Hz. End systole and end diastole were defined as the phases in which the smallest and largest areas of the LV were obtained respectively. LV end‐systolic diameter and LV end‐diastolic diameter were measured via LV M‐mode tracing with a sweep speed of 50 mm·s−1 at the midpapillary muscle level.
Haemodynamics were measured in anaesthetized (1.5% isoflurane) mice using cardiac catheterization, as described previously (Wu et al., 2016; Xiao et al., 2017). A microtip catheter transducer (SPR‐839; Millar Instruments, Houston, TX, USA) was inserted into the right carotid artery and advanced into the LV. Fifteen minutes after stabilization, pressure signals and heart rate were continuously recorded with a Millar Pressure‐Volume System (MPVS‐400; Millar Instruments) coupled to a Powerlab/4SP A/D converter and then stored and displayed on a personal computer. The data were processed using PVAN data analysis software.
Histological analysis and immunohistochemistry
As described previously (Wu et al., 2016; Xiao et al., 2017), hearts were excised, placed immediately in 10% potassium chloride solution to ensure that they were stopped in diastole, washed with saline solution, placed in 10% formalin and embedded in paraffin. Hearts were cut transversely close to the apex to visualize the left and right ventricles. Several sections of each heart (4 to 5 μm thick) were prepared and stained with haematoxylin and eosin for histopathology or Picro‐Sirius Red (PSR) for collagen deposition analysis. The sections were then visualized by light microscopy. Single myocytes were measured using a quantitative digital image analysis system (Image‐Pro Plus, version 6.0). Between 100 and 200 LV myocytes were outlined in each group. The LV collagen volume fraction was calculated from the PSR‐stained sections as the area stained by PSR was divided by the total area. For immunohistochemistry, the heart sections were heated using the pressure cooker method for antigen retrieval, incubated with anti‐CD31 (ab28364; Abcam, Cambridge, UK) or 4‐hydroxynonenal (ab46545; Abcam) followed by incubation with goat anti‐rabbit EnVisionTM+/HRP) reagent and stained using a DAB detection kit. For immunofluorescence, heart sections were incubated with anti‐CD68 (ab125212; Abcam) and anti‐CD45 antibody (ab10558; Abcam).
Quantitative real‐time RT‐PCR
To examine the mRNA expression of hypertrophy and fibrosis markers, RNA was collected from LV tissue using TRIzol (15596‐026; Invitrogen) and reverse transcribed into cDNA for PCR analysis using oligo (DT) primers and the Transcriptor First Strand cDNA Synthesis Kit (04896866001; Roche, Basel, Switzerland). cDNA was synthesized from 2 g of total RNA. The PCR amplifications were quantified using a LightCycler 480 SYBR Green 1 Master Mix (04707516001; Roche), and the results were normalized against GAPDH gene expression.
Western blot
Protein amounts from all samples were assessed using the BCA Protein Assay Kit (23227; Thermo Fisher Scientific, Waltham, MA, USA), and protein concentration was normalized prior to all Western blot experiments. Protein samples (50 μg) were separated by SDS‐PAGE and then transferred to Immobilon‐FL transfer membrane (IPFL00010; Millipore, Billerica, MA, USA). The membrane was blocked with 5% skimmed milk in Tris‐buffered saline Tween‐20 for 1 h and then incubated overnight at 4°C with the indicated primary antibodies, including the β3‐adrenoceptor (ab94506; Abcam), nNOS (4321; Cell Signalling Technology), http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1249 (sc‐654; Santa Cruz, CA, USA) and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1250 (ab3523; Abcam). The antibody for GAPDH (sc‐25778) was purchased from Santa Cruz. The secondary antibody used was goat anti‐rabbit IgG (926‐32211; LI‐COR, Lincoln, NE, USA) or goat anti‐mouse IgG (C11026‐03; LI‐COR). The blots were scanned by a two‐colour IR imaging system (Odyssey; LI‐COR). Specific protein expression levels were normalized to GAPDH protein for total cell lysates.
NO production
NO production was determined as the amount of nitrate plus nitrite using the Griess reaction assay (Cayman Chemical, Ann Arbor, MI, USA) according to the manufacturer's instructions (Niu et al., 2012).
Cell culture
H9c2 cells (Cell Bank of the Chinese Academy of Sciences, Shanghai, China) were grown in high‐glucose DMEM (C11995; Gibco, Grand Island, NY, USA) supplemented with 10% FBS (FBS, 10099; Gibco), penicillin (100 U·mL−1) and streptomycin (100 mg·mL−1) (15140; Gibco) in a humidified CO2 incubator (SANYO 18M, Osaka, Japan) with 5% CO2 at 37°C. Cells undergoing exponential growth were dissociated with 0.25% trypsin (25200; Gibco) and seeded in six‐well culture plates at 1 × 106 cells per well then incubated for 24 h. Next, the cells were cultured with serum‐free DMEM for another 12 h. Aucubin was dissolved in PBS at a concentration of 1 mM. Phenylephrine (50 μM), in the presence or absence of different concentrations of aucubin (1, 5, 10 and 50 μM), was added to the medium, and the cells were incubated for 24 h. After incubation, total RNA was extracted from the cells. The mRNA expression levels of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4869 (ANP) and β‐myosin heavy chain (β‐MHC) were examined by qRT‐PCR. The cell surface area was determined by immunofluorescence staining of α‐actinin.
Neonatal rat cardiomyocyte culture
One‐ to two‐day‐old Sprague–Dawley rats were killed by cervical dislocation. Hearts were quickly removed, and ventricles were washed with PBS three times and incubated with 0.125% trypsin–EDTA (2520‐072; Gibco) for 15 min. Ventricles were then enzymatically digested four times for 15 min each in 0.125% trypsin–EDTA in PBS. Digestion was stopped by the addition of FBS at a final concentration of 10%. The cells were then centrifuged at 250 × g for 8 min and were resuspended in DMEM/F12 (C11330; Gibco) supplemented with 10% FBS. Resuspended cells were incubated for 1–2 h in a 100 mm dish to allow non‐cardiac myocytes (mainly cardiac fibroblasts) to adhere to plastic. Cells were then plated in six‐well plates at a density of 5 × 105 cells per well with 1% bromodeoxyuridine for 48 h.
Recombinant adenoviral vectors and infection
To silence β3‐adrenoceptor expression, replication‐defective adenoviral vectors under the control of the U6 promoter were used. Three shβ3‐adrenoceptor constructs were obtained from Santa Cruz (sc‐39869) for sc‐39869A (sense: GCAACCUGCUGGUAAUCAUTT; antisense: AUGAUUACCAGCAGGUUGCTT), sc‐39869B (sense: GAGUGUUCGUUGUGGCUAATT; antisense: UUAGCCACAACGAACACUCTT) and sc‐39869C (sense: GAACCCAGGCAUCUCUAUATT; antisense: UAUAGAGAUGCCUGGGUUCTT). Three shβ3‐adrenoceptor vectors were generated using one adenoviral vector by Vigene Bioscience (Rockville, MD, USA). Ad‐shRNA was used as a non‐targeting control. The culture medium was changed to serum‐free DMEM/F12 for 8 h before the experiment, after which neonatal rat cardiac myocytes (NRCM) were infected with Ad‐shβ3‐adrenoceptors.
Detection of ROS in cardiomyocytes
Myocytes were cultured in nine‐well plates and pretreated with aucubin and phenylephrine for the indicated times. ROS were then detected by dichlorofluorescein diacetate assay (DCFH‐DA). The cells were incubated with DCFH‐DA (10 μM) for 60 min at 37°C, and immunofluorescence was detected using a fluorescence microplate reader normalized to the vehicle‐PBS group to control for unwanted sources of variation (excitation wavelength/emission wavelength: 485/525 nm), by light microscopy (BX51TRF; Olympus Corporation, Tokyo, Japan) and by flow cytometry (BD Biosciences, San Jose, CA, USA) (Ma et al., 2016).
Cell viability assay
Cell viability was evaluated using the MTT assay, in accordance with the manufacturer's instructions. Briefly, 20 μL of MTT (5 mg·mL−1) was added to each well of a 96‐well plate and incubated at 37°C for 4 h. After incubation, the solution was discarded, and 100 μL of DMSO was added. Cell viability was determined by measuring absorbance at 495 nm using an elisa reader (Synergy HT; Bio‐tek, Winooski, VT, USA) (Zhao et al., 2017). The effect of aucubin on cell viability was determined by calculating the percentage cell viability of aucubin‐treated cells compared with that of vehicle‐treated cells, which was set at 100%.
Immunofluorescence
The cells were washed three times with PBS and then fixed with RCL2 (RCL2‐CS100; ALPHELYS, Plaisir, France), permeabilized in 0.1% Triton X‐100 (Amresco, Atlanta, GA, 1050 Satellite Blvd, USA) in PBS and stained with anti‐α‐actinin (05‐384; Millipore) at a dilution of 1:100 in 1% goat serum (GTX27481; GeneTex, Sanantonio, Texas, USA) overnight. Cells were then incubated with the secondary antibody goat anti‐mouse IRdye 800CW (926‐32210; LI‐COR) for 60 min. Glass slides were used to mount the cells on coverslips with SlowFade Gold anti‐fade reagent with DAPI (S36939; Invitrogen, Carlsbad, CA, USA). Surface areas of single cells were determined by measuring α‐actinin staining with a quantitative digital image analysis system (Image‐Pro Plus, version 6.0). We traced the outlines of 40 cells for each group, as described previously (Wu et al., 2016).
cAMP measurement
The cAMP complete enzyme immunoassay kit from Assay Designs was used as directed by the manufacturer (#ab65355; Abcam).
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). The results are expressed as the means ± SD. SPSS software, version 13.0, was used for data analysis. The assignment of mice to different groups was randomized via a random number table. The raw data were assessed independently by two authors to ensure the correctness of the conclusions. All measurements were obtained while blinded using a random number. The group sizes of the in vivo experiments were estimated based on power analysis of HW/BW with an α error of 5% and a power of 80%, consistent with a previously published article (Ma et al., 2016). To detect a 10% change in HW/BW with an expected SD of 5%, we needed five animals per group. In our study, n = 8 fulfilled the statistical requirements. Groups were compared by one‐way ANOVA followed by the post hoc least significant difference test when ANOVA found a significant value of F and no variance in homogeneity; otherwise, Tamhane's T2 post hoc test was used. Comparisons between two groups were performed using Student's unpaired t‐test. All in vivo, in vitro and imaging studies were performed blinded. Statistical significance was assigned at P < 0.05.
Materials
Aucubin (CAS: 479‐98‐1) was obtained from Shanghai Winherb Medical Science Co. (Shanghai, China, http%3A%2F%2Fwww.sh-winherb.com%2FZproduct.aspx%3Ffn%3D2%26amp%3Bkey%3Daucubin). The purity of aucubin was above 98% as determined by HPLC analysis. Phenylephrine (PHR1695; Sigma, St. Louis, MO, USA; D9568‐5G), N‐acetyl‐l‐cysteine (NAC, A7250; Sigma), l‐nitro‐arginine methyl ester (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5213, N5751; Sigma), http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=567 (B169; Sigma), http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=721 (A4474; Sigma), Vinyl‐l‐NIO hydrochloride (l‐VNIO, sc‐205541; Santa Cruz) and SR95230A (0.1 μM) were obtained from the indicated sources. Anti‐α‐actin was purchased from Millipore. DCFH‐DA was obtained from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). Proteins were measured with assay kits obtained from Thermo Scientific™ Pierce™ (23225; Wyman Street, Waltham, MA). All other chemicals were of analytical grade.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b).
Results
Aucubin suppresses phenylephrine‐induced cardiomyocyte hypertrophy
To explore the effects of aucubin on cardiac hypertrophy, we first investigated whether aucubin affects cardiomyocyte hypertrophic responses. We treated H9c2 cardiomyocytes with phenylephrine (50 μM), a hypertrophic agonist, and examined the effect of aucubin on cardiac hypertrophy. Cardiomyocyte viability was not affected by the addition of aucubin at the concentrations tested (1, 3, 10, 25 and 50 μM) (Figure 1A). However, higher concentrations of aucubin (10 and 50 μM) caused a significant decrease in cell surface area and decreased the mRNA expression levels of ANP and β‐MHC (Figure 1B–D).
Figure 1.
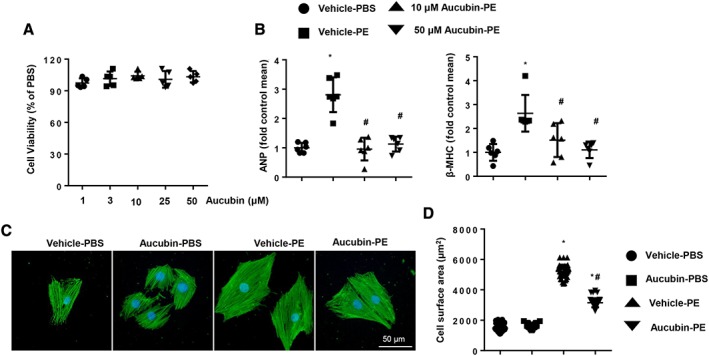
Aucubin suppresses phenylephrine (PE)‐induced cardiomyocyte hypertrophy. (A) H9c2 cardiomyocytes were treated with aucubin (1, 3, 10, 25 and 50 μM) for 24 h. MTT assays were performed to detect cell viability (n = 5 per experiment). (B) H9c2 cardiomyocytes were treated with aucubin (10 and 50 μM) and phenylephrine (50 μM) for 24 h. RT‐PCR analysis of ANP and β‐MHC mRNA expression levels was performed in each group (n = 6 per experiment). (C and D) H9c2 cardiomyocytes were treated with aucubin (50 μM) and phenylephrine (50 μM) for 24 h. Immunofluorescence staining of α‐actinin was used to detect the cell surface area (n = 6 per experiment). (C) Representative images; (D) quantitative results (n = 50+ cells per group). *P < 0.05 versus vehicle‐PBS group; #P < 0.05 versus vehicle‐phenylephrine group. All of the experiments were repeated three independent times.
Aucubin blunts cardiac hypertrophy and improves cardiac function after chronic pressure overload in mice
Mice were subjected to sham or AB surgeries, and extensive examinations were conducted 4 weeks post‐surgery. AB‐induced cardiac hypertrophic responses were markedly blunted in the HD (5 mg·kg−1·day−1)‐treated mice compared with the vehicle‐treated mice, as indicated by decreased HW/BW, HW/TL, LW/BW and LW/TL ratios in the HD group (Figure 2A–D). Conversely, the LD (1 mg·kg−1·day−1)‐treated mice exhibited decreased LW/BW and LW/TL ratios, and their HW/BW and HW/TL ratios were not significantly altered compared with vehicle‐treated mice. Moreover, HD aucubin treatment in mice with the sham surgery did not affect BW, HW/BW, HW/TL, LW/BW or LW/TL ratios, compared with the vehicle‐treated group (Supporting Information Figure S1). Thus, 5 mg·kg−1·day−1 aucubin was used for further study. The AB surgery triggered an increase in cross section area, and mRNA expression levels of hypertrophic markers were also inhibited in the HD‐treated mice compared with vehicle‐treated mice (Figure 2E–G). Four weeks post‐AB surgery, vehicle‐treated mice exhibited decreased heart function, with a dilated left ventricular diameter, reduced ejection fraction and fractional shortening. Conversely, aucubin‐treated mice displayed a marked decrease in AB‐induced cardiac dysfunction, with a 14.5% reduction in LV end‐diastolic diameter and a 31.8% improvement in left ventricular ejection fraction. Moreover, the aucubin‐treated mice also exhibited reduced interventricular septal thickness compared with the vehicle‐treated group (Table 1). As demonstrated by pressure–volume analysis, aucubin improved systolic and diastolic functions after AB surgery (Table 1).
Figure 2.
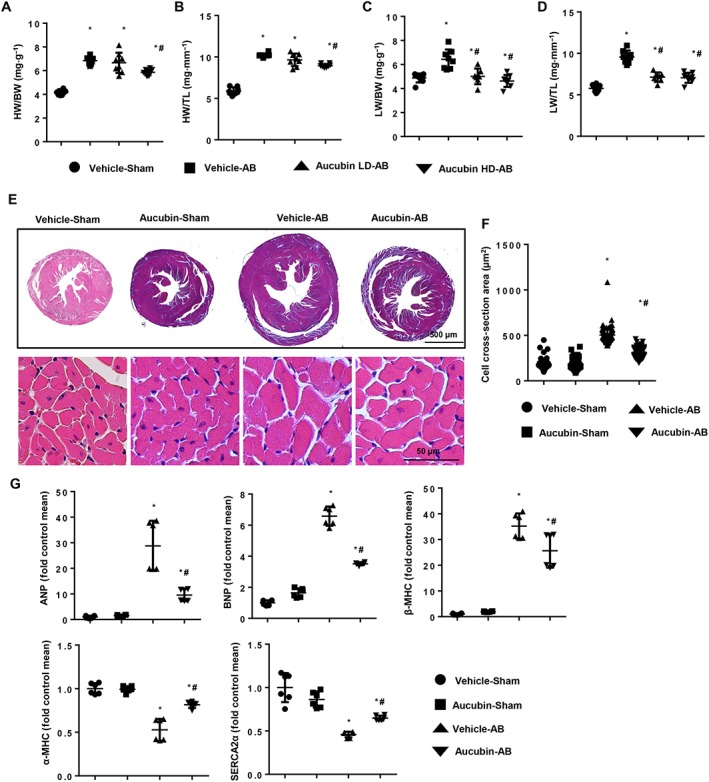
Aucubin blunts cardiac hypertrophy after chronic pressure overload in mice. (A–D) Statistical results for the ratios of HW/BW, HW/TL, LW/BW and LW/TL in aucubin‐ and vehicle‐treated mice 4 weeks post‐AB surgery (n = 8, LD, 1 mg·kg−1·day−1; HD, 5 mg·kg−1·day−1). (E and F) Histological analyses of the haematoxylin and eosin staining of aucubin‐treated and vehicle‐treated mice 4 weeks post‐AB surgery (n = 6 mice per experimental group). (E) Haematoxylin and eosin staining; (F) statistical results for the CSA (n = 100+ cells per group). (G) RT‐PCR analyses of hypertrophic markers (ANP, BNP, β‐MHC, α‐MHC and http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=159#841) induced by AB surgery in the indicated mice (n = 6 per experimental group). *P < 0.05 versus vehicle‐sham; #P < 0.05 versus vehicle‐AB.
Table 1.
Echocardiography and haemodynamic parameters in mice treated with aucubin or vehicle 4 weeks post‐AB surgery
| Parameter | Sham | Sham | AB | AB |
|---|---|---|---|---|
| Vehicle | Aucubin | Vehicle | Aucubin | |
| n = 8 | n = 8 | n = 8 | n = 8 | |
| LVEDd (mm) | 3.34 ± 0.07 | 3.31 ± 0.04 | 4.81 ± 0.04* | 4.11 ± 0.05* , # |
| LVESd (mm) | 2.71 ± 0.07 | 2.61 ± 0.06 | 3.9 ± 0.05* | 3.26 ± 0.05* , # |
| LVPWd (mm) | 0.78 ± 0.02 | 0.77 ± 0.02 | 1.23 ± 0.03* | 0.95 ± 0.02* , # |
| LVEF (%) | 69 ± 1.8 | 69 ± 1.6 | 41.7 ± 1.5* | 55 ± 1.7* , # |
| LVFS (%) | 33.1 ± 0.9 | 33.6 ± 0.8 | 21.6 ± 0.6* | 27.5 ± 0.8* , # |
| ESP (mmHg) | 99.1 ± 2.4 | 95.7 ± 4.2 | 145 ± 1.1* | 148 ± 1.8* |
| EDP (mmHg) | 11.9 ± 0.8 | 12.1 ± 0.69 | 22.5 ± 0.4* | 16.8 ± 0.4* , # |
| CO (μL·min−1) | 8675 ± 368 | 8486 ± 358 | 4530 ± 105* | 5615 ± 99* , # |
| dp/dtmax (mmHg·s−1) | 9566 ± 138 | 9376 ± 135 | 6351 ± 126* | 7272 ± 98* , # |
| dp/dtmin (mmHg·s−1) | −9541 ± 91 | −9826 ± 249 | −6079 ± 248* | −7455 ± 214* , # |
LVEDd, left ventricular end‐diastolic diameter; LVESd, left ventricular end‐systolic diameter; LVPWd, left ventricular end‐diastolic posterior wall dimension; LVEF, left ventricular ejection fraction; LVFS, left ventricular fractional shortening; ESP, end‐systolic pressure; EDP, end‐diastolic pressure; CO, cardiac output; dp/dtmax, maximal rate of pressure development; dp/dtmin, maximal rate of pressure decay.
P < 0.05 for difference from the corresponding sham group.
P < 0.05 versus vehicle‐AB group after AB.
Aucubin attenuates cardiac fibrosis and inflammation in vivo
As shown in Figure 3A–C, aucubin‐treated mice exhibited a significant decrease in average collagen volume after AB surgery compared with vehicle‐treated mice. Transcript levels of collagen I, collagen III, fibronectin and connective tissue growth factor (CTGF) were dramatically decreased in aucubin‐treated mice. However, aucubin treatment did not affect the expression of fibrotic genes under basal conditions. Whereas decreased microvascular density is involved in hypertrophic responses, we found that aucubin treatment increased microvascular density in hypertrophic hearts as assessed by CD31 staining (Figure 3D, E). Cardiac hypertrophy is also associated with an inflammatory response, and we found that aucubin treatment decreased CD45‐labelled leukocytes and CD68‐labelled macrophage infiltration into mouse hearts 4 weeks post‐AB surgery (Figure 3F–H).
Figure 3.
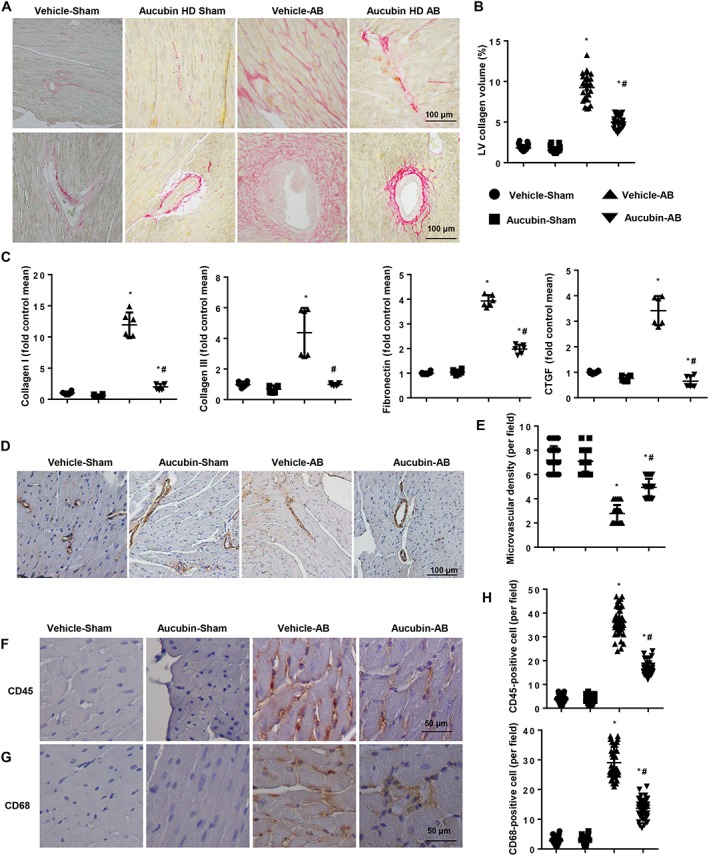
Aucubin attenuates cardiac fibrosis and inflammation induced by pressure overload in mice. (A and B) PSR staining of histological sections of the LV in the indicated groups 4 weeks post‐AB surgery (n = 6 per experimental group). (A) Representative image; (B) quantification of the total collagen volume in aucubin‐ and vehicle‐treated mice 4 weeks post‐AB surgery (n = 25+ fields per experimental group). (C) Real‐time PCR analyses of fibrotic markers (collagen I, collagen III, fibronectin and CTGF) in the indicated groups (n = 6). (D and E) Detection of microvessel density by immunohistochemical staining of CD31 in the indicated mice 4 weeks post‐AB surgery (n = 6 per experimental group). (D) Representative image; (E) quantitative result (n = 10+ fields per experimental group). Immunohistochemical staining of (F) CD45 and (G) CD68 in mouse hearts in the indicated groups (n = 6 per experimental group). (H) Quantification of CD45‐ and CD68‐positive cells in mouse hearts in the indicated groups (n = 10+ fields per experimental group). *P < 0.05 versus vehicle‐sham; #P < 0.05 versus vehicle‐AB.
Aucubin inhibits oxidative stress in vitro and in vivo
Because aucubin plays an indispensable role in regulating oxidative stress, we next explored the effect of aucubin on cellular ROS generation. Aucubin treatment inhibited phenylephrine‐induced ROS generation as assessed by DCFH‐DA assay using an elisa reader, light microscopy and flow cytometry (Figure 4A–C). Aucubin treatment also increased the mRNA levels of SOD and glutathione (GSH) peroxidase and decreased the mRNA levels of NADPH P67 phox in cardiomyocytes (Figure 4D). NO production was increased by aucubin treatment (Figure 4E), and oxidative stress was also detected in our in vivo study. Consistently, decreased levels of the lipid peroxidation product 4‐hydroxynonenal were observed in aucubin‐treated mice compared with vehicle‐treated mice (Figure 4F). Aucubin treatment increased the mRNA levels of SOD and GPx and decreased the mRNA levels of P67 phox in mouse hearts (Figure 4G), and we also observed increased activity of SOD and NO production in aucubin‐treated mouse hearts (Figure 4H).
Figure 4.
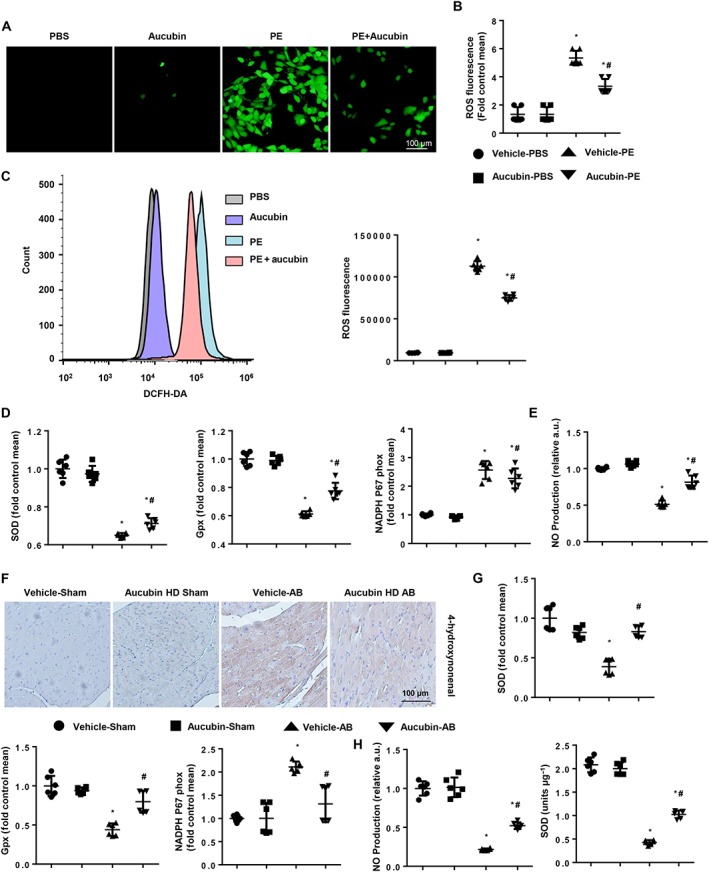
Aucubin inhibits oxidative stress and increases NO production in hypertrophic hearts and cardiomyocytes. (A–E) H9c2 cardiomyocytes were treated with aucubin (50 μM) and phenylephrine (PE; 50 μM) for 24 h. ROS were detected by DCFH‐DA with (A) light microscopy (n = 6 per experiment), (B) an elisa reader (n = 6 per experiment) and (C) flow cytometry (n = 6 per experiment) in the indicated groups. (D) Real‐time PCR analyses of oxidative markers (SOD, GPx and NADPH p67 phox, n = 6 per experiment). (E) NO production in the indicated groups (n = 6 per experiment). *P < 0.05 versus vehicle‐PBS group; #P < 0.05 versus vehicle‐phenylephrine group. All experiments were performed three independent times. (F) Immunohistochemical staining of 4‐hydroxynonenal in the indicated groups 4 weeks post‐AB surgery (n = 6 per experimental group). (G) RT‐PCR analyses of oxidative markers (SOD, GPx and NADPH p67 phox) in the indicated groups 4 weeks post‐AB surgery (n = 6 per experimental group). (H) NO production and SOD activation in the indicated groups 4 weeks post‐AB surgery (n = 6). *P < 0.05 versus vehicle‐sham; #P < 0.05 versus vehicle‐AB.
Aucubin increased the expression of nNOS in vivo and in vitro
Since aucubin increased NO production, we next explored whether aucubin affected NOS expression. Three isoforms of NOS contribute to the production of NO: the neuronal (nNOS), inducible (iNOS) and endothelial (eNOS) isoforms. We found increased mRNA and protein expression levels of iNOS and nNOS at 4 weeks post‐AB surgery in vivo and post‐phenylephrine stimulus in vitro, whereas the expression of eNOS was unchanged (Figure 5A–F). Aucubin treatment increased nNOS expression after both AB surgery in vivo and phenylephrine stimulus in vitro (Figure 5A–F). We also found that nNOS expression was unchanged at 1 week and up‐regulated at 2 weeks post‐AB surgery and that this up‐regulation lasted for 4 weeks post‐surgery (Supporting Information Figure S2A–C).
Figure 5.
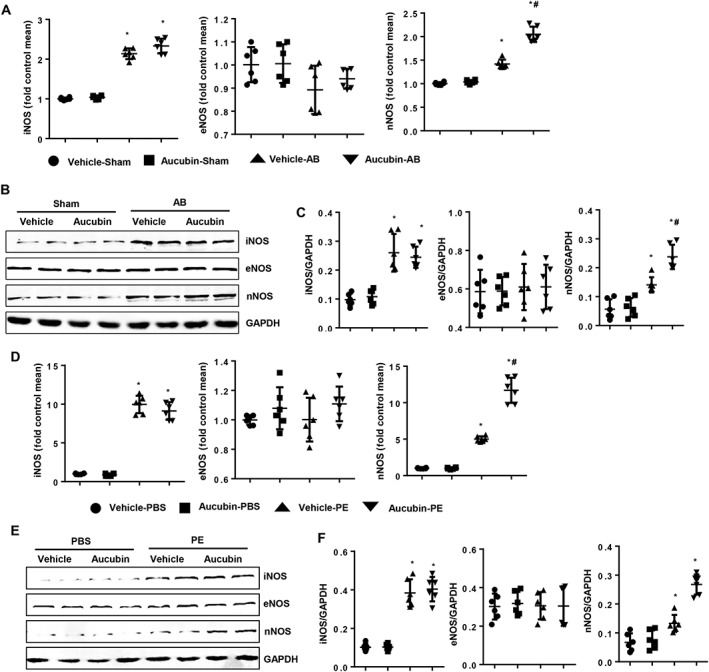
The effects of nNOS on aucubin‐mediated cardioprotection in vivo and in vitro. (A–C) The mRNA and protein expression levels of iNOS, eNOS and nNOS in the indicated groups 4 weeks post‐AB surgery. (A) Real‐time PCR results (n = 6 per experimental group), (B) Western blot image and (C) quantitative results of Western blots (n = 6 per experimental group). *P < 0.05 versus vehicle‐sham; #P < 0.05 versus vehicle‐AB. (D–F) H9c2 cardiomyocytes were treated with aucubin (50 μM) and phenylephrine (PE; 50 μM) for 24 h. The mRNA and protein expression levels of iNOS, eNOS and nNOS in the indicated groups. (D) RT‐PCR results (n = 6 per experimental group), (E) Western blot images and (F) quantitative results of Western blots (n = 6 per experimental group). *P < 0.05 versus vehicle‐PBS group; #P < 0.05 versus vehicle‐phenylephrine group. All of the experiments are repeated three independent times.
To further evaluate whether the effects of aucubin on cardiomyocyte hypertrophy were related to suppression of oxidative stress, cardiomyocytes were treated with NAC (2 mM). NAC treatment suppressed the phenylephrine‐induced hypertrophic response, and aucubin treatment (50 μM) did not augment this improvement, as evidenced by both treatments resulting in the same degree of decrease in transcription of hypertrophic markers (Supporting Information Figure S2D) and ROS generation (Supporting Information Figure S2F, G). To evaluate the necessity of nNOS in the effect of aucubin on cardiac hypertrophy, we treated cardiomyocytes with the non‐isoform‐selective NOS inhibitor l‐NAME (100 μM), which is partially selective for eNOS and nNOS over iNOS (Yang et al., 2015). Interestingly, the aucubin‐mediated amelioration of phenylephrine‐induced hypertrophy in cardiomyocytes was effectively prevented by l‐NAME treatment (Supporting Information Figure S2E–G). We next used l‐VNIO (10 μM), a specific inhibitor of nNOS. Notably, l‐VNIO prevented the anti‐hypertrophic effects of aucubin in cardiomyocytes following phenylephrine stimulus (Supporting Information Figure S2E–G). Together, these data robustly verified the critical involvement of nNOS in aucubin‐mediated anti‐cardiac hypertrophy effects.
Aucubin‐mediated cardioprotection depends on increased nNOS
Next, we used nNOS‐KO mice to determine the role of nNOS in aucubin‐mediated protection against cardiac remodelling. In agreement with our in vitro results, the ameliorated phenotypes of pressure overload‐induced hypertrophy identified in the aucubin‐treated wild‐type mice were effectively prevented by nNOS deficiency, as evidenced by both the AB‐operated aucubin‐treated mice and the vehicle‐treated mice exhibiting the same extent of increased HW/BW, LW/BW and HW/TL, LW/TL ratios (Figure 6A), worsened cardiac function (Table 2), larger cardiomyocyte size (Figure 6B, C), aggravated fibrosis (Figure 6D, E), increased mRNA levels of hypertrophic and fibrotic cardiac genes (Supporting Information Figure S3A, B) and augmented oxidative stress (Supporting Information Figure S3C, D). No significant phenotypic differences were identified between the aucubin‐ and vehicle‐treated mice, suggesting that nNOS ablation completely abolished the protective effects of aucubin treatment. These data indicated that increased expression of nNOS in aucubin‐treated mice contributed to its protective effect against cardiac hypertrophy and heart failure. Thus, we concluded that aucubin‐regulated amelioration of cardiac remodelling is dependent on its regulation of nNOS expression.
Figure 6.

nNOS‐KO counteracts the protective effects of aucubin in vivo. nNOS‐KO mice were subjected to AB surgery and treated with 5 mg·kg−1·day−1 aucubin for 25 days. (A) Statistical results for the ratios of HW/BW, HW/TL, LW/BW and LW/TL in aucubin‐ and vehicle‐treated mice 4 weeks post‐AB surgery AB (n = 8). (B and C) Histological analyses of the haematoxylin and eosin staining of aucubin‐ and vehicle‐treated mice 4 weeks post‐AB surgery (n = 6 mice per experimental group). (B) Haematoxylin and eosin staining; (C) statistical results for CSA (n = 100+ cells per group). (D and E) PSR staining of histological sections of the LV in the indicated groups 4 weeks post‐AB surgery (n = 6 per experimental group). (D) Representative images; (E) quantification of the total collagen volume in aucubin‐ and vehicle‐treated mice 4 weeks post‐AB surgery (n = 25+ fields per experimental group). *P < 0.05 versus vehicle‐sham; #P < 0.05 versus vehicle‐AB.
Table 2.
Echocardiography and haemodynamic parameters in nNOS‐KO mice treated with aucubin or vehicle 4 weeks post‐AB surgery
| Parameter | Sham | Sham | AB | AB |
|---|---|---|---|---|
| Vehicle | Aucubin | Vehicle | Aucubin | |
| n = 8 | n = 8 | n = 8 | n = 8 | |
| LVEDd (mm) | 3.68 ± 0.09 | 3.63 ± 0.13 | 4.95 ± 0.06* | 5.0 ± 0.07* |
| LVESd (mm) | 2.63 ± 0.12 | 2.73 ± 0.06 | 4.08 ± 0.04* | 4.10 ± 0.07* |
| LVPWd (mm) | 0.82 ± 0.02 | 0.81 ± 0.03 | 1.34 ± 0.02* | 1.33 ± 0.04* |
| LVEF (%) | 64 ± 1.9 | 65 ± 1.9 | 39.6 ± 1.1* | 40 ± 1.8* |
| LVFS (%) | 30.6 ± 0.7 | 29.3 ± 0.9 | 18.5 ± 0.8* | 18.9 ± 1.0* |
| ESP (mmHg) | 103 ± 1.8 | 98.3 ± 1.35 | 148 ± 2.11* | 149 ± 1.3* |
| EDP (mmHg) | 11.8 ± 1.3 | 11.2 ± 0.68 | 24.7 ± 1.7* | 23.5 ± 0.7* |
| CO (μL·min−1) | 7609 ± 177 | 8089 ± 192 | 3693 ± 101* | 3658 ± 333* |
| dp/dtmax (mmHg·s−1) | 8018 ± 157 | 7954 ± 145 | 4921 ± 306* | 4826 ± 179* |
| dp/dtmin (mmHg·s−1) | −7525 ± 107 | −7808 ± 166 | −4121 ± 232* | −4036 ± 201* |
LVEDd, left ventricular end‐diastolic diameter; LVESd, left ventricular end‐systolic diameter; LVPWd, left ventricular end‐diastolic posterior wall dimension; LVEF, left ventricular ejection fraction; LVFS, left ventricular fractional shortening; ESP, end‐systolic pressure; EDP, end‐diastolic pressure; CO, cardiac output; dp/dtmax, maximal rate of pressure development; dp/dtmin, maximal rate of pressure decay.
P < 0.05 for difference from corresponding sham group.
P < 0.05 versus Vehicle‐AB group after AB.
Aucubin regulates nNOS by activating β3‐adrenoceptors
The requirement of nNOS for the protective function of aucubin against cardiac remodelling raised another important question: how does aucubin regulate nNOS expression? Previous studies have indicated that the β3‐adrenoceptor is closely associated with nNOS activation and NO production (Trappanese et al., 2015). However, whether the involvement of β3‐adrenoceptors in this process contributes to aucubin's protective effects was unknown. Therefore, we investigated the levels of β3‐adrenoceptors in hypertrophic mouse hearts after AB surgery and in cardiomyocytes after phenylephrine stimulus. Figure 7A–C shows that hypertrophic stimuli dramatically enhanced the expression levels of β3‐adrenoceptors, whereas aucubin treatment did not affect β3‐adrenoceptor expression. In the presence of the β3‐adrenoceptor antagonist SR95230A (0.1 μM), the phenylephrine‐induced hypertrophic response was accelerated, and aucubin treatment (50 μM) was unable to ameliorate the augmented hypertrophic response, as evidenced by the increased cell surface area and hypertrophic markers (Figure 7D–F). As expected, treatment with a β3‐adrenoceptor agonist (BRL37344, 10 μM) exerted anti‐hypertrophic effects, and aucubin treatment (50 μM) enhanced the β3‐adrenoceptor agonist‐induced anti‐hypertrophic effects compared with treatment with BRL37344 alone (Figure 7G). To detect whether increased NO is the ultimate effect of aucubin treatment, l‐arginine, the substrate for NO production, was used. l‐arginine (2 mM) mimicked the effect of aucubin and even reversed the phenotype induced by l‐VNIO (Figure 7G). These data suggest that increased NO is the ultimate effect of aucubin treatment and that this increase relies on the activation of β3‐adrenoceptors.
Figure 7.
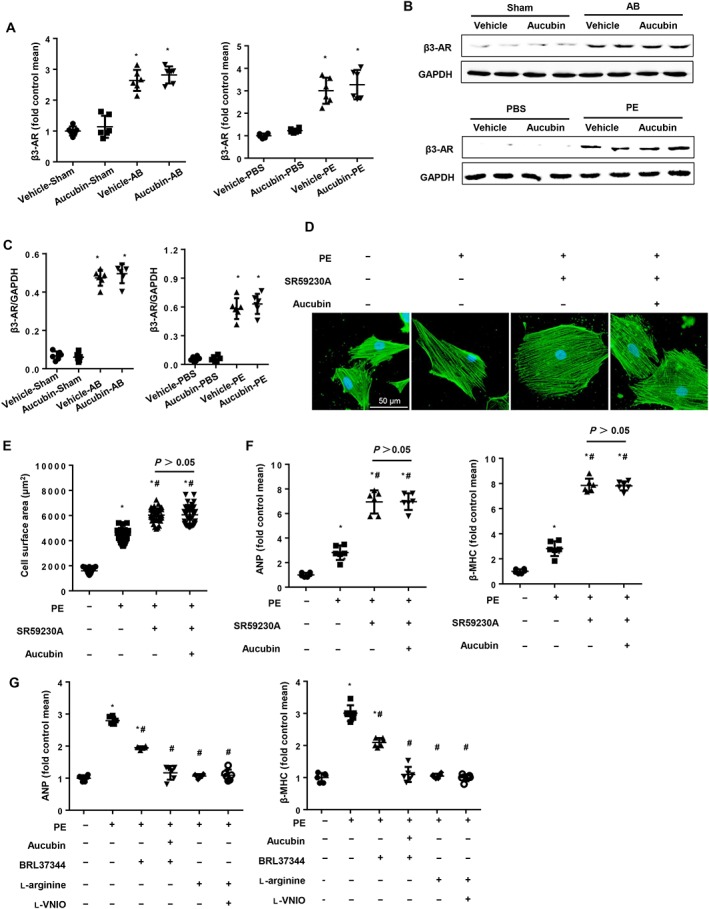
Aucubin regulates nNOS by activating β3‐adrenoceptors. The (A) mRNA and (B and C) protein expression levels of β3‐adrenoceptors in the indicated groups (n = 6). *P < 0.05 versus vehicle‐sham/PBS; #P < 0.05 versus vehicle‐AB/phenylephrine (PE). (D–G) H9c2 cardiomyocytes were treated with aucubin (50 μM), the β3‐adrenoceptor antagonist (SR59230A, 0.1 μM), β3‐adrenoceptor agonist (BRL37344, 10 μM) or l‐arginine (2 mM) and stimulated with phenylephrine (50 μM) for 24 h. (D and E) Detection of cell surface area by immunofluorescence staining of α‐actinin (n = 6). (D) Representative images; (E) quantitative results (n = 50+ cells per group). (F and G) RT‐PCR analysis of ANP and β‐MHC mRNA expression levels in each group (n = 6). *P < 0.05 versus vehicle‐PBS group; #P < 0.05 versus vehicle‐phenylephrine group. All of the experiments were repeated three independent times.
Anti‐hypertrophic effects of aucubin on neonatal rat cardiomyocytes
To further evaluate the requirement for β3‐adrenoceptors in the protective effect of aucubin against cardiomyocyte hypertrophy, we isolated neonatal rat cardiomyocytes (NRCMs). Consistent with the results obtained from H9c2 cells, different concentrations of aucubin (1, 3, 10, 25 and 50 μM) did not affect cardiomyocyte viability compared with the control group (Figure 8A). However, aucubin treatment at 50 μM caused a significant decrease in cell surface area and decreased mRNA expression of ANP and β‐MHC (Figure 8B–D).
Figure 8.
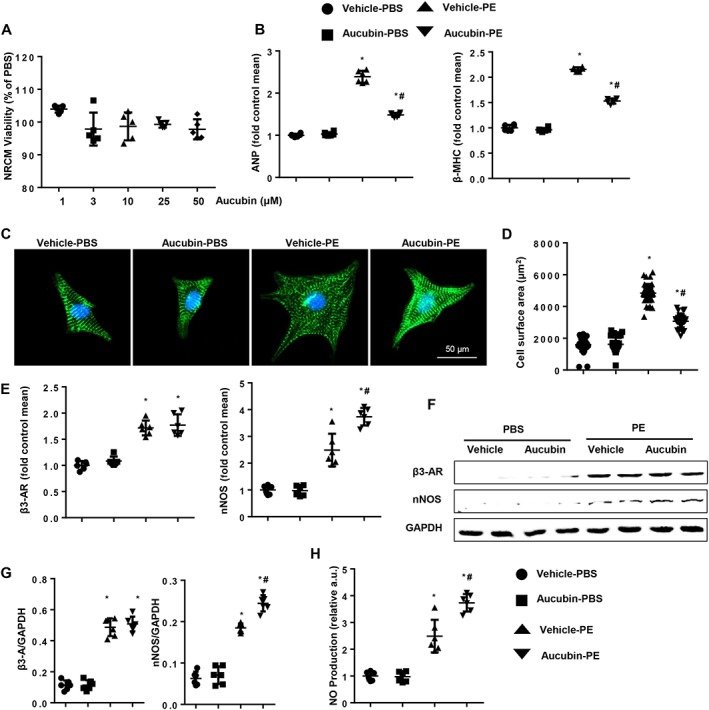
The effects of aucubin on neonatal rat cardiomyocytes (NRCMs). (A) NRCMs were treated with aucubin (1, 3, 10, 25 and 50 μM) for 24 h. MTT assays were performed to detect cell viability (n = 5 per experimental group). (B–D) NRCMs were treated with aucubin (50 μM) and phenylephrine (PE; 50 μM) for 24 h. (B) RT‐PCR analysis of ANP and β‐MHC mRNA expression levels in each group (n = 6 per experimental group). (C and D) Detection of cell surface area by immunofluorescence staining of α‐actinin (n = 6 per experimental group). (C) Representative images; (D) quantitative results (n = 50+ cells per group). *P < 0.05 versus vehicle‐PBS group; #P < 0.05 versus vehicle‐phenylephrine group. The (E) mRNA and (F and G) protein expression levels of β3‐adrenoceptors (AR) and nNOS in the indicated groups (n = 6 per experimental group). (H) NO production in the indicated groups (n = 6 per experimental group). *P < 0.05 versus vehicle‐PBS; #P < 0.05 versus vehicle‐phenylephrine. All of the experiments were repeated three independent times.
The expression of β3‐adrenoceptors in NRCMs was increased after stimulation with phenylephrine for 24 h. Aucubin treatment (50 μM) did not affect β3‐adrenoceptor expression (Figure 8E–G), but it increased the expression of nNOS and the production of NO (Figure 8H). We infected NRCMs with Ad‐shRNA or Ad‐shβ3‐adrenoceptor alone (Supporting Information Figure S4A) or in combination with aucubin treatment. The infected cells were treated with phenylephrine for 24 h. Inhibition of the β3‐adrenoceptor impaired the protective functions of aucubin and resulted in a further enlarged cardiomyocyte size (Supporting Information Figure S4B, C), higher mRNA levels of fetal cardiac genes (Supporting Information Figure S4D) and augmented oxidative stress (Supporting Information Figure S4E, F). Taken together, these in vitro data demonstrated that the β3‐adrenoceptor/nNOS signalling is essential for aucubin‐mediated inhibition of cardiac remodelling.
The effect of aucubin on β3‐adrenoceptor signalling in neonatal rat cardiomyocytes
To evaluate the mechanism through which aucubin affects β3‐adrenoceptor/NO signalling in NRCMs, we treated NRCMs with the β3‐adrenoceptor antagonist SR95230A (0.1 μM), the β3‐adrenoceptor agonist BRL37344 (10 μM) and l‐arginine (2 mM). As expected, the β3‐adrenoceptor antagonist SR95230A accelerated phenylephrine‐induced cardiac hypertrophy, and aucubin treatment was unable to ameliorate the augmented hypertrophic response (Figure 9A–C). Importantly, the β3‐adrenoceptor agonist BRL37344 exerted anti‐hypertrophic effects, and aucubin treatment (50 μM) enhanced these protective effects (Figure 9A–C). l‐arginine mimicked the protective effects of aucubin and even reversed the adverse phenotype induced by l‐VNIO (Figure 9A–C). To investigate the mechanism through which aucubin alters β3‐adrenoceptor signalling, we measured cAMP levels and found that cAMP levels were increased 4 weeks post‐AB surgery. Aucubin treatment did not alter baseline cAMP levels, but cAMP levels in heart tissue were increased 4 weeks post‐surgery in the aucubin‐treated group compared with the vehicle‐treated group (Figure 9D). Aucubin also increased cAMP levels in cardiomyocytes after phenylephrine stimulus (Figure 9E). An http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=257) inhibitor (2′5‐dd‐Ado, 200 μM) was used to investigate the possible role of cAMP. 2′5‐dd‐Ado treatment decreased the elevated cAMP levels and nNOS expression induced by phenylephrine stimulation (Figure 9E, F). 2′5‐dd‐Ado also exaggerated the phenylephrine‐induced hypertrophic response, and aucubin treatment was unable to exert its protective effect (Figure 9E–G). These data indicated that aucubin regulated the β3‐adrenoceptor/AC/cAMP pathway to activate nNOS/NO signalling.
Figure 9.
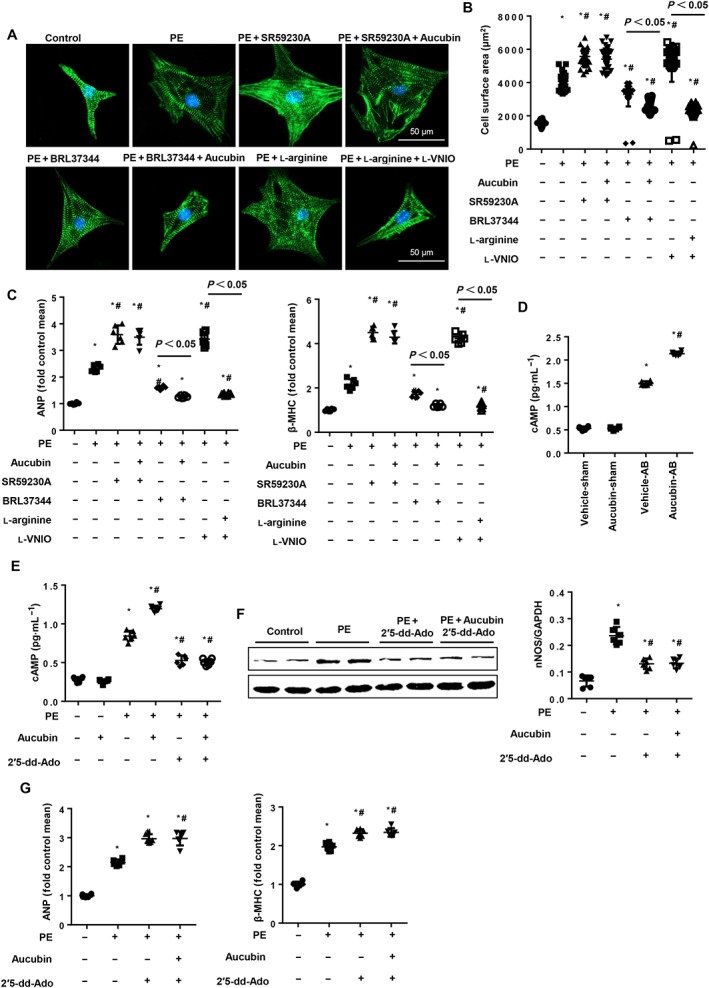
The effect of aucubin on β3‐adrenoceptor signalling in neonatal rat cardiomyocytes (NRCMs). NRCMs were treated with aucubin (50 μM), a β3‐adrenoceptor antagonist (SR59230A, 0.1 μM), β3‐adrenoceptor agonist (BRL37344, 10 μM), l‐arginine (2 mM) or l‐VNIO (1 μM) and stimulated with phenylephrine (PE; 50 μM) for 24 h. (A and B) Detection of cell surface area by immunofluorescence staining of α‐actinin (n = 6). (A) Representative images; (B) quantitative results (n = 50+ cells per group). (C) RT‐PCR analysis of ANP and β‐MHC mRNA expression levels (n = 6) in each group. *P < 0.05 versus vehicle‐PBS group; #P < 0.05 versus vehicle‐phenylephrine group. (D) cAMP levels in hypertrophic mice hearts in the indicated groups (n = 6, *P < 0.05 vs. vehicle‐sham group; #P < 0.05 vs. vehicle‐AB group). (E–G) NRCMs were treated with aucubin (50 μM) or 2′5‐dd‐Ado (200 μM) and stimulated with phenylephrine (50 μM) for 24 h. (E) cAMP levels in cardiomyocytes in the indicated groups (n = 6). (F) nNOS protein expression in the indicated cardiomyocytes (n = 6). (G) RT‐PCR analysis of ANP and β‐MHC mRNA expression levels (n = 6) in each group. *P < 0.05 versus vehicle‐PBS group; #P < 0.05 versus vehicle‐phenylephrine group. All of the experiments were repeated three independent times.
Discussion
Aucubin, a compound isolated from traditional Korean medicinal herbs, such as Aucuba japonica, Eucommia ulmoides, Plantago asiatica and Melitaea cinxia, was originally identified as an antioxidant (Kim et al., 2014). Aucubin has since been shown to have multiple pharmacological effects, including BP reduction and liver protection as well as anti‐inflammatory (Kim et al., 2014) and anti‐apoptotic effects and regulation of autophagy (Wang et al., 2017). The profound effects of aucubin on oxidative stress, inflammation and apoptosis suggests a potential application for aucubin in cardiovascular disease as well. However, the relevance of aucubin in cardiac remodelling and heart failure was previously unknown. In the present study, aucubin dramatically inhibited pathological cardiomyocyte enlargement, cardiac dysfunction, inflammation and fibrosis induced by pro‐hypertrophic stimuli.
Over the past several decades, clinical and experimental studies have provided substantial evidence that oxidative stress, defined as excess production of ROS relative to antioxidant defences, is enhanced in heart failure (Seddon et al., 2007). Excessive ROS cause cellular dysfunction, protein and lipid peroxidation and DNA damage and can lead to irreversible cell damage and death (Seddon et al., 2007). Moreover, ROS activate a broad variety of signalling kinases and transcription factors and mediate apoptosis, all of which are involved in the development and progression of maladaptive myocardial remodelling and heart failure (Tsutsui et al., 2011). During our investigations into the mechanism responsible for aucubin‐mediated protection against pathological cardiac remodelling, we demonstrated a dramatic suppression of oxidative stress by aucubin following hypertrophic stress. Furthermore, the necessity of oxidative stress for the protective effects of aucubin was demonstrated by the ability of NAC to alleviate cardiomyocyte hypertrophy, and aucubin could not further augment its improvements.
NO is a labile (half‐life ~5 s) and reactive free radical. It is generated by NOS, all three isoforms of which are expressed in the heart (Tang and Ziolo, 2014; Barouch et al., 2002). nNOS and eNOS are constitutively expressed and regulated by Ca2+, whereas iNOS is not Ca22+ regulated and is induced in almost any cell type with appropriate stimulation (Badorff, 2003). eNOS localizes to the caveolae, whereas nNOS is present in the sarcoplasmic reticulum. Although nNOS and eNOS can have opposing effects on contractility, genetically engineered mice have been used to link both anti‐hypertrophic responses (Booz, 2005). It has been established that NO produced from constitutive nNOS provides an intrinsic regulatory mechanism for myocardial contraction and relaxation in both healthy and diseased hearts (Zhang, 2017). nNOS‐derived NO controls the intracellular oxidative status and ROS‐dependent downstream effects in the myocardium by targeting cardiac oxidases, such as xanthine oxidoreductase and NADPH oxidase, as well as mitochondrial ROS production (Zhang, 2017). nNOS exerts its cardioprotection through the regulation of ion channels, modulating abnormal Ca2+ homeostasis, mitochondrial function and signalling pathways during pathological progression (Zhang and Casadei, 2012; Omar and Webb, 2014). Importantly, nNOS protein expression and activity have been shown to be increased in the myocardium in diseased hearts (Zhang et al., 2014), such as with ischaemia–reperfusion injury, infarct, hypertrophy and heart failure, and nNOS has been shown to exert manifold beneficial effects: nNOS‐derived NO prevented diastolic dysfunction and increased β‐adrenergic reserves, reduced left ventricular hypertrophy/dilatation/infarct size and protected the myocardium from arrhythmogenesis (Niu et al., 2012; Zhang et al., 2014). During our investigation into the signalling pathways responsible for aucubin‐mediated protection against pathological cardiac remodelling, we demonstrated that NO production was increased by aucubin treatment. After hypertrophic stimulus, iNOS and nNOS were increased, eNOS was unchanged and aucubin treatment increased only nNOS expression without altering expression of iNOS or eNOS. The necessity of nNOS signalling for the cardioprotective effects of aucubin was evidenced by the ability of an artificial nNOS inhibitor to reverse the beneficial effects of aucubin treatment on cardiomyocyte enlargement and ROS generation. Moreover, the essential function of nNOS in aucubin‐mediated suppression of cardiac remodelling was verified by rescue experiments using nNOS‐KO mice. Assessment of the protective capacity of aucubin in nNOS‐KO mice led to the very interesting observation that aucubin did not counteract the pro‐remodelling effects of nNOS deficiency. Thus, the regulation of nNOS signalling by aucubin treatment is the primary mechanism leading to limitation of the remodelling response.
There is accumulating evidence that the β3‐adrenoceptor plays an important role in the modulation of cardiovascular function in heart failure via its effect on nNOS. In contrast to the well‐characterized β1/β2‐adrenoceptor, the β3‐adrenoceptor has been found to be up‐regulated in failing hearts in both humans (Moniotte et al., 2001) and animal models (Zhao et al., 2007). The β3‐adrenoceptor has also been shown to exert protective effects in LV hypertrophy (Moens et al., 2009; Belge et al., 2014) and to induce NO‐mediated cardioprotection in various animal models of cardiac pressure overload (Aragon et al., 2011; Niu et al., 2012; Watts et al., 2013; Belge et al., 2014). The β3‐adrenoceptor‐stimulated nNOS‐NO‐soluble guanylyl cyclase signalling pathway triggered cGMP production and preserved cardiac function in volume‐overloaded hearts (Trappanese et al., 2015). Moreover, the anti‐hypertrophic and antioxidant effects of β3‐adrenoceptor activation in myocytes require differential nNOS phosphorylation (Watts et al., 2013). The data in our study suggested that aucubin enhanced the activation of β3‐adrenoceptors by BRL37344. The β3‐adrenoceptor antagonist (SR59230A) or shRNA abolished the anti‐hypertrophic effects of aucubin. These data suggest that aucubin sensitizes the β3‐adrenoceptor and promotes the activation of nNOS. Moreover, the NO donor l‐arginine mimicked the protective effects of aucubin on cardiomyocytes and even reversed the deteriorating phenotype induced by an nNOS inhibitor, suggesting that nNOS‐triggered NO was the ultimate effect of aucubin treatment. The β3‐adrenoceptor is a GPCR, which can couple to either Gs/AC or Gi/MAPK (Dessy, 2010). In our study, aucubin seemed to affect Gs/AC‐mediated effects by increasing cAMP levels. The aucubin‐induced increase in cAMP was blocked by an AC inhibitor, which also abolished the anti‐hypertrophic effects of aucubin triggered by increased nNOS expression. Thus, aucubin sensitized the β3‐adrenoceptor/Gs/AC pathway, and its protective effects were mediated through nNOS/NO. Interestingly, aucubin treatment did not affect baseline cAMP levels, perhaps due to low expression levels of the β3‐adrenoceptor in the baseline state.
Several limitations of the present study should be considered. Firstly, since aucubin shows multiple pharmacological effects, including BP reduction, liver protection and anti‐inflammatory effects, the contribution of potential systemic effects to its anti‐hypertrophic effects requires further study. Secondly, only two dosages were tested in our in vivo study, so whether a lower or higher dosage might exert the same effects is not clear. Further studies concerning the tissue distribution, concentration and pharmacokinetics of aucubin will be needed to choose the best dosage and delivery method for aucubin.
In summary, we found that aucubin inhibited cardiac remodelling via a β3‐adrenoceptor‐dependent regulation of the nNOS/NO pathway. Our study could have implications for the future treatment of cardiac hypertrophy through the application of aucubin.
Author contributions
Q.‐Q.W. and Q.Z.T. contributed to the conception and design of the experiments; Q.‐Q.W., Y.X., M.‐X.D. and Y.Y. carried out the experiments; X.‐H.J. and W.D. analysed the experimental results and revised the manuscript; Z.Y. and H.‐H.L. wrote and revised the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Aucubin treatment not affect body weight in vivo. Statistical results for body weight, the ratios of HW/BW, HW/TL, LW/BW,andLW/TL in aucubin‐(5 mg kg−1d−1)and vehicle‐treated mice in the sham group(n = 8).
Figure S2 The effects ofnNOS on aucubin mediated cardio‐protection. A‐C.nNOS expression levels at0–4 weeks after AB surgery. A. RT‐PCR results(n = 6). B and C western blot analysis (n = 6). D‐G. H9c2 cardiomyocytes aretreated with aucubin (50 μM) and/or NAC (2 mM), L‐NAME (100 μM), and L‐VNIO (1 μM) and stimulated with PE (50 μM) for 24 h. D, E.RT‐PCR analysis of ANP and β‐MHC mRNA expressionlevels (n = 6per experimental group) in each group. F, G.ROS aredetected byDCFH‐DA with flow cytometry in the indicated groups (n = 6per experimental group, F. representative image; G. quantified result). *P < 0.05 vs vehicle‐PBS group; #P < 0.05 vs vehicle‐PE group; §P < 0.05 vs aucubin‐PE group.All of the experiments are repeated 3times independently.
Figure S3 nNOS knock out counteracts the protective effects of aucubin in vivo. nNOS‐KO mice aresubjected to AB surgery and treated with 5 mg kg−1d−1aucubin for 25 days. A‐C.RT‐PCR analyses of hypertrophic markers (A, ANP,BNP, β‐MHC, α‐MHC), fibrosis markers (B, collagen I, collagen III, fibronectin) and oxidative stress markers (C, SOD, Gpx, NADPH p67 phox) induced by AB in the indicated mice (n = 6 per experimental group). *P < 0.05 vs vehicle‐Sham; #P < 0.05 vs vehicle‐AB. D. Immunohistochemistry staining of 4‐hydroxynonenal in the indicated groups 4 weeks after AB (n = 6 per experimental group).
Figure S4 The effects of aucubin on neonatal rat cardiomyocytes A. β3AR expression levels in NRCMs after infectionwith Ad‐shβ3‐AR orscrambleshRNA(n = 6per experimental group). B‐F. Neonatal rat cardiomyocytesareinfected with Ad‐shβ3‐AR orscrambleshRNA and are treatedwith aucubin (50 μM) and stimulated with PE (50 μM) for 24 h. B and C.Detectionof the cell surface area by immunofluorescence staining of α‐actinin(n = 6per experimental group). B, representative images; C, quantitative results (n = 50+ cells per group). Dand E.RT‐PCR analysis of mRNA expressionlevels of hypertrophic markers (D, ANP and β‐MHC) and oxidative markers (E, SOD, Gpx, NADPH p67 phox) (n = 6 per experimental group) in each group. F.ROS aredetected by DCFH‐DA with an ELISA reader in the indicated groups(n = 6per experimental group). *P < 0.05 vs vehicle‐PBS group; #P < 0.05 vs vehicle‐PE group.All of the experiments are repeated 3times independently.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (nos. 81470516, 81530012 and 81700353), Hubei Province's Outstanding Medical Academic Leader programme, and Fundamental Research Funds of the Central Universities (2042017kf0060).
Wu, Q.‐Q. , Xiao, Y. , Duan, M.‐X. , Yuan, Y. , Jiang, X.‐H. , Yang, Z. , Liao, H.‐H. , Deng, W. , and Tang, Q.‐Z. (2018) Aucubin protects against pressure overload‐induced cardiac remodelling via the β3‐adrenoceptor–neuronal NOS cascades. British Journal of Pharmacology, 175: 1548–1566. doi: 10.1111/bph.14164.
References
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andoh T, Uta D, Kato M, Toume K, Komatsu K, Kuraishi Y (2017). Prophylactic administration of aucubin inhibits paclitaxel‐induced mechanical allodynia via the inhibition of endoplasmic reticulum stress in peripheral Schwann cells. Biol Pharm Bull 40: 473–478. [DOI] [PubMed] [Google Scholar]
- Aragon JP, Condit ME, Bhushan S, Predmore BL, Patel SS, Grinsfelder DB et al (2011). Beta3‐adrenoreceptor stimulation ameliorates myocardial ischemia‐reperfusion injury via endothelial nitric oxide synthase and neuronal nitric oxide synthase activation. J Am Coll Cardiol 58: 2683–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badorff C, DS (2003). NO balance: regulation of the cytoskeleton in congestive heart failure by nitric oxide. Circulation 107: 1348–1349. [DOI] [PubMed] [Google Scholar]
- Barouch LA, Harrison RW, Skaf MW, Rosas GO, Cappola TP, Kobeissi ZA et al (2002). Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature 416: 337–339. [DOI] [PubMed] [Google Scholar]
- Belge C, Hammond J, Dubois‐Deruy E, Manoury B, Hamelet J, Beauloye C et al (2014). Enhanced expression of β3‐adrenoceptors in cardiac myocytes attenuates neurohormone‐induced hypertrophic remodeling through nitric oxide synthase. Circulation 129: 451–462. [DOI] [PubMed] [Google Scholar]
- Booz GW (2005). Putting the brakes on cardiac hypertrophy: exploiting the NO‐cGMP counter‐regulatory system. Hypertension 45: 341–346. [DOI] [PubMed] [Google Scholar]
- Bui AL, Horwich TB, Fonarow GC (2011). Epidemiology and risk profile of heart failure. Nat Rev Cardiol 8: 30–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessy C, Balligand JL (2010). Beta3‐adrenergic receptors in cardiac and vascular tissues emerging concepts and therapeutic perspectives. Adv Pharmacol 59: 135–163. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heineke J, Molkentin JD (2006). Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol 7: 589–600. [DOI] [PubMed] [Google Scholar]
- Ho JN, Lee YH, Lee YD, Jun WJ, Kim HK, Hong BS et al (2005). Inhibitory effect of aucubin isolated from Eucommia ulmoides against UVB‐induced matrix metalloproteinase‐1 production in human skin fibroblasts. Biosci Biotechnol Biochem 69: 2227–2231. [DOI] [PubMed] [Google Scholar]
- Jeong HJ, Koo KH, Na HJ, Kim MS, Hong SH, Eom JW et al (2002). Inhibition of TNF‐alpha and IL‐6 production by aucubin through blockade of NF‐kappaB activation RBL‐2H3 mast cells. Cytokine 18: 252–259. [DOI] [PubMed] [Google Scholar]
- Jiang DS, Wei X, Zhang XF, Liu Y, Zhang Y, Chen K et al (2014). IRF8 suppresses pathological cardiac remodelling by inhibiting calcineurin signalling. Nat Commun 5: 3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Xue HY, Jin LJ, Li SY, Xu YP (2008). Antioxidant and pancreas‐protective effect of aucubin on rats with streptozotocin‐induced diabetes. Eur J Pharmacol 582: 162–167. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG, NC3RS Reporting Guidelines Working Group (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YM, Sim UC, Shin Y, Kim Kwon Y (2014). Aucubin promotes neurite outgrowth in neural stem cells and axonal regeneration in sciatic nerves. Exp Neurobiol 23: 238–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HY, Lee GH, Lee MR, Kim HK, Kim NY, Kim SH et al (2013). Eucommia ulmoides Oliver extract, aucubin, and geniposide enhance lysosomal activity to regulate ER stress and hepatic lipid accumulation. PLoS One 8: e81349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma ZG, Dai J, Zhang WB, Yuan Y, Liao HH, Zhang N et al (2016). Protection against cardiac hypertrophy by geniposide involves the GLP‐1 receptor/AMPKα signalling pathway. Br J Pharmacol 173: 1502–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moens AL, Leyton‐Mange JS, Niu X, Yang R, Cingolani O, Arkenbout EK et al (2009). Adverse ventricular remodeling and exacerbated NOS uncoupling from pressure‐overload in mice lacking the beta3‐adrenoreceptor. J Mol Cell Cardiol 47: 576–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moniotte S, Kobzik L, Feron O, Trochu JN, Gauthier C, Balligand JL (2001). Upregulation of beta(3)‐adrenoceptors and altered contractile response to inotropic amines in human failing myocardium. Circulation 103: 1649–1655. [DOI] [PubMed] [Google Scholar]
- Niu X, Watts VL, Cingolani OH, Sivakumaran V, Leyton‐Mange JS, Ellis CL et al (2012). Cardioprotective effect of beta‐3 adrenergic receptor agonism: role of neuronal nitric oxide synthase. J Am Coll Cardiol 59: 1979–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omar SA, Webb AJ (2014). Nitrite reduction and cardiovascular protection. J Mol Cell Cardiol 73: 57–69. [DOI] [PubMed] [Google Scholar]
- Seddon M, Looi YH, Shah AM (2007). Oxidative stress and redox signalling in cardiac hypertrophy and heart failure. Heart 93: 903–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu I, Minamino T (2016). Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol 97: 245–262. [DOI] [PubMed] [Google Scholar]
- Tang L, Wang H, Ziolo MT (2014). Targeting NOS as a therapeutic approach for heart failure. Pharmacol Ther 142: 306–315. [DOI] [PubMed] [Google Scholar]
- Trappanese DM, Liu Y, McCormick RC, Cannavo A, Nanayakkara G, Baskharoun MM et al (2015). Chronic β1‐adrenergic blockade enhances myocardial β3‐adrenergic coupling with nitric oxide‐cGMP signaling in a canine model of chronic volume overload: new insight into mechanisms of cardiac benefit with selective β1‐blocker therapy. Basic Res Cardiol 110: 456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsui H, Kinugawa S, Matsushima S (2011). Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol 301: H2181–H2190. [DOI] [PubMed] [Google Scholar]
- Wang J, Li Y, Huang WH, Zeng XC, Li XH, Li J et al (2017). The protective effect of aucubin from Eucommia ulmoides against status epilepticus by inducing autophagy and inhibiting necroptosis. Am J Chin Med 45: 557–573. [DOI] [PubMed] [Google Scholar]
- Wang SN, Xie GP, Qin CH, Chen YR, Zhang KR, Li X et al (2015). Aucubin prevents interleukin‐1 beta induced inflammation and cartilage matrix degradation via inhibition of NF‐kappaB signaling pathway in rat articular chondrocytes. Int Immunopharmacol 24: 408–415. [DOI] [PubMed] [Google Scholar]
- Watts VL, Sepulveda FM, Cingolani OH, Ho AS, Niu X, Kim R et al (2013). Anti‐hypertrophic and anti‐oxidant effect of beta3‐adrenergic stimulation in myocytes requires differential neuronal NOS phosphorylation. J Mol Cell Cardiol 62: 8–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu QQ, Yuan Y, Jiang XH, Xiao Y, Yang Z, Ma ZG et al (2016). OX40 regulates pressure overload‐induced cardiac hypertrophy and remodelling via CD4+ T‐cells. Clin Sci (Lond) 130: 2061–2071. [DOI] [PubMed] [Google Scholar]
- Xiao Y, Yang Z, Wu QQ, Jiang XH, Yuan Y, Chang W et al (2017). Cucurbitacin B protects against pressure overload induced cardiac hypertrophy. J Cell Biochem 18: 3899–3910. [DOI] [PubMed] [Google Scholar]
- Xue HY, Jin L, Jin LJ, Li XY, Zhang P, Ma YS et al (2009). Aucubin prevents loss of hippocampal neurons and regulates antioxidative activity in diabetic encephalopathy rats. Phytother Res 23: 980–986. [DOI] [PubMed] [Google Scholar]
- Yang Y, Yu T, Lian YJ, Ma R, Yang S, Cho JY (2015). Nitric oxide synthase inhibitors: a review of patents from 2011 to the present. Expert Opin Ther Pat 25: 49–68. [DOI] [PubMed] [Google Scholar]
- Zhang YH (2017). Nitric oxide signalling and neuronal nitric oxide synthase in the heart under stress. F1000Res 6: 742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YH, Casadei B (2012). Sub‐cellular targeting of constitutive NOS in health and disease. J Mol Cell Cardiol 52: 341–350. [DOI] [PubMed] [Google Scholar]
- Zhang YH, Jin CZ, Jang JH, Wang Y (2014). Molecular mechanisms of neuronal nitric oxide synthase in cardiac function and pathophysiology. J Physiol 592: 3189–3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Wu TG, Jiang ZF, Chen GW, Lin Y, Wang LX (2007). Effect of beta‐blockers on beta3‐adrenoceptor expression in chronic heart failure. Cardiovasc Drugs Ther 21: 85–90. [DOI] [PubMed] [Google Scholar]
- Zhao X, Kong F, Wang L, Zhang H (2017). c‐FLIP and the NOXA/Mcl‐1 axis participate in the synergistic effect of pemetrexed plus cisplatin in human choroidal melanoma cells. PLoS One 12: e0184135. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Aucubin treatment not affect body weight in vivo. Statistical results for body weight, the ratios of HW/BW, HW/TL, LW/BW,andLW/TL in aucubin‐(5 mg kg−1d−1)and vehicle‐treated mice in the sham group(n = 8).
Figure S2 The effects ofnNOS on aucubin mediated cardio‐protection. A‐C.nNOS expression levels at0–4 weeks after AB surgery. A. RT‐PCR results(n = 6). B and C western blot analysis (n = 6). D‐G. H9c2 cardiomyocytes aretreated with aucubin (50 μM) and/or NAC (2 mM), L‐NAME (100 μM), and L‐VNIO (1 μM) and stimulated with PE (50 μM) for 24 h. D, E.RT‐PCR analysis of ANP and β‐MHC mRNA expressionlevels (n = 6per experimental group) in each group. F, G.ROS aredetected byDCFH‐DA with flow cytometry in the indicated groups (n = 6per experimental group, F. representative image; G. quantified result). *P < 0.05 vs vehicle‐PBS group; #P < 0.05 vs vehicle‐PE group; §P < 0.05 vs aucubin‐PE group.All of the experiments are repeated 3times independently.
Figure S3 nNOS knock out counteracts the protective effects of aucubin in vivo. nNOS‐KO mice aresubjected to AB surgery and treated with 5 mg kg−1d−1aucubin for 25 days. A‐C.RT‐PCR analyses of hypertrophic markers (A, ANP,BNP, β‐MHC, α‐MHC), fibrosis markers (B, collagen I, collagen III, fibronectin) and oxidative stress markers (C, SOD, Gpx, NADPH p67 phox) induced by AB in the indicated mice (n = 6 per experimental group). *P < 0.05 vs vehicle‐Sham; #P < 0.05 vs vehicle‐AB. D. Immunohistochemistry staining of 4‐hydroxynonenal in the indicated groups 4 weeks after AB (n = 6 per experimental group).
Figure S4 The effects of aucubin on neonatal rat cardiomyocytes A. β3AR expression levels in NRCMs after infectionwith Ad‐shβ3‐AR orscrambleshRNA(n = 6per experimental group). B‐F. Neonatal rat cardiomyocytesareinfected with Ad‐shβ3‐AR orscrambleshRNA and are treatedwith aucubin (50 μM) and stimulated with PE (50 μM) for 24 h. B and C.Detectionof the cell surface area by immunofluorescence staining of α‐actinin(n = 6per experimental group). B, representative images; C, quantitative results (n = 50+ cells per group). Dand E.RT‐PCR analysis of mRNA expressionlevels of hypertrophic markers (D, ANP and β‐MHC) and oxidative markers (E, SOD, Gpx, NADPH p67 phox) (n = 6 per experimental group) in each group. F.ROS aredetected by DCFH‐DA with an ELISA reader in the indicated groups(n = 6per experimental group). *P < 0.05 vs vehicle‐PBS group; #P < 0.05 vs vehicle‐PE group.All of the experiments are repeated 3times independently.