

Abstract
Objective
Loss of function of the astrocyte‐specific protein MLC1 leads to the childhood‐onset leukodystrophy “megalencephalic leukoencephalopathy with subcortical cysts” (MLC). Studies on isolated cells show a role for MLC1 in astrocyte volume regulation and suggest that disturbed brain ion and water homeostasis is central to the disease. Excitability of neuronal networks is particularly sensitive to ion and water homeostasis. In line with this, reports of seizures and epilepsy in MLC patients exist. However, systematic assessment and mechanistic understanding of seizures in MLC are lacking.
Methods
We analyzed an MLC patient inventory to study occurrence of seizures in MLC. We used two distinct genetic mouse models of MLC to further study epileptiform activity and seizure threshold through wireless extracellular field potential recordings. Whole‐cell patch‐clamp recordings and K+‐sensitive electrode recordings in mouse brain slices were used to explore the underlying mechanisms of epilepsy in MLC.
Results
An early onset of seizures is common in MLC. Similarly, in MLC mice, we uncovered spontaneous epileptiform brain activity and a lowered threshold for induced seizures. At the cellular level, we found that although passive and active properties of individual pyramidal neurons are unchanged, extracellular K+ dynamics and neuronal network activity are abnormal in MLC mice.
Interpretation
Disturbed astrocyte regulation of ion and water homeostasis in MLC causes hyperexcitability of neuronal networks and seizures. These findings suggest a role for defective astrocyte volume regulation in epilepsy. Ann Neurol 2018;83:636–649
“Megalencephalic leukoencephalopathy with subcortical cysts” (MLC; MIM 604004) is a rare childhood‐onset leukodystrophy,1, 2 caused by homozygous recessive mutations in MLC1 3 or GLIALCAM.4 MLC1 is a membrane protein almost exclusively expressed in brain astrocytes.5 GlialCAM is a chaperone of MLC1, ensuring its localization in the membrane of astrocyte endfeet.4, 6, 7 Patients show very high water content in the brain white matter. Biopsies reveal countless fluid‐filled vacuoles within the outer lamellae of myelin sheaths and, to a lesser degree, in perivascular astrocyte endfeet.8 Mutations affecting MLC1 protein negatively impact volume‐regulated anion channel (VRAC) activity in astrocytes, leading to impaired regulatory volume decrease9 and resulting in chronically swollen astrocytes.10
Because the pathophysiology of MLC has mainly been studied in isolated cells, consequences of astrocyte dysfunction for neuronal network functioning are unknown. The disease is characterized by infantile‐onset macrocephaly, slow deterioration of motor functions, and later cognitive decline. Minor head trauma can lead to temporary worsening, often with seizures, prolonged unconsciousness, and motor deterioration.11 Mild epilepsy, which typically responds well to treatment, has been described in many MLC patients, but status epilepticus has also been described.12 Occurrence of epilepsy has not been studied in a large cohort of genetically confirmed patients, and its cellular basis is not understood.
The panglial syncytium, the glial network consisting of gap‐junction coupled astrocytes and oligodendrocytes, is essential for the uptake and dispersal of extracellular K+ ([K+]o) released from neurons during periods of high neuronal activity.13, 14 Disruption of proteins important for this process, such as Kir4.1 K+ channels,15, 16 glial gap‐junction proteins,17 and the astrocytic water channel aquaporin‐4 (AQP4),18 lead to impaired [K+]o clearance and to epileptiform brain activity or an altered seizure threshold. MLC1 and GlialCAM undergo molecular interactions with several of these proteins.19, 20 Therefore, we hypothesize that defective astrocyte volume regulation in MLC leads to impaired control of [K+]o,8 resulting in hyperexcitability of neuronal networks and epilepsy.
We recently developed an Mlc1‐null mouse recapitulating major pathological features of MLC.10 Additionally, Glialcam‐null mice21 show similar pathological features.7, 22 Mlc1‐ and Glialcam‐null mice (collectively referred to here as MLC mice) are excellent tools to investigate the pathophysiology of MLC. Here, we combine a clinical inventory of MLC patients with in vivo and in vitro electrophysiology in MLC mice to study the cellular pathophysiology of epilepsy in MLC. We confirm that early seizure onset is common in MLC patients. Similarly, MLC mice show spontaneous epileptiform activity and a reduced threshold for seizure induction. At the cellular level, we find that excitability of single neurons is unchanged, whereas stimulation‐induced increases in [K+]o and network excitability are increased. Taken together, we demonstrate that loss of function of the astrocytic protein MLC1 disturbs neuronal network activity while leaving intrinsic neuronal properties intact.
Materials and Methods
Study Approval
Experimental procedures involving mice were in strict compliance with animal welfare policies of the Dutch government and were approved by the Institutional Animal Care and Use Committee of the VU University, Amsterdam. The clinical inventory was approved by the Medical Ethics Committee of the VU University Medical Center.
MLC Patients
We executed a cross‐sectional observational study among 205 genetically proven patients with autosomal‐recessive MLC present in the Amsterdam Leukodystrophy database, containing patients referred to the VUmc Center for Childhood White Matter Disorders for genetic testing. As part of a larger study on the clinical phenotype of MLC,23 standardized clinical questionnaires including items on epilepsy were completed primarily by the patient's physician. If this source was unavailable, information was derived from medical records, supplemented by information provided by families.
Mice
Transgenic mice and their wild‐type littermates had a C57BL/6J background. Generation of both Mlc1 deficient mice (Mlc1‐null)10 and Glialcam deficient mice (Glialcam‐null)21, 22 was previously described. Null animals were compared with wild‐type littermate controls, with the exception of extracellular field potential/seizure threshold recordings. Here, littermate data for both MLC lines were grouped because no differences between littermates were observed.
Behavioral Assays
In vivo experiments were performed on adult (8–12 months old) mice. Forty‐eight‐hour recordings of basal activity were performed on both male and female mice. Kainate‐induced seizures were only studied in male mice because sex strongly influences sensitivity to kainate.24 To score hindlimb clasping, each mouse was suspended for 30 seconds by its tail and kept 10cm above a stable platform. Seizure threshold and severity were assessed using an intraperitoneal injection of kainate (kainic acid; 10mg/kg). Behavioral response was scored using a revised Racine's seizure scale.25 Maximal score during subsequent 10‐minute time bins was assessed by a trained observer unaware of genotype. Mice were sacrificed if seizure severity reached score 5c.
Telemetric Extracellular Field Potential Recordings
Electrocorticogram (ECoG) recordings and hippocampal local field potential (LFP) recordings were performed with wireless ETA‐F10 transmitters (Data Science International, St. Paul, MN). For electrode placement, animals were anesthetized with isoflurane (induction 3%, flow rate 0.8l/min; maintenance 1.5–1.8%, flow rate 0.6l/min). For ECoG recordings, a recording electrode (stereotaxic coordinates relative to bregma: 2.22mm anterior and 1mm lateral, corresponding to motor cortex) was implanted subdurally through a small hole drilled in the skull and held in place with a screw (A2‐70; Jeveka, Almere‐Poort, The Netherlands) made of stainless steel. For hippocampal LFP recordings, an insulated depth‐electrode (W‐electrode; Open Source Instruments, Watertown, MA) was lowered into the brain (stereotaxic coordinates relative to bregma: 2.2mm posterior, 2.0mm lateral, 1.7mm depth, corresponding to the CA1 region of hippocampus). A ground electrode was placed subdurally 6mm posterior and 1mm lateral relative to bregma. All electrodes were sealed with dental cement. The calibrated transmitter was placed subcutaneously along the dorsal flank of the animal. Mice received analgesia (Temgesic; 0.1mg/kg subcutaneously) before surgery and recovered for 7 days before recordings.
ECoG/LFP signal was collected through a radiofrequency receiver (RLA1020; Data Science International) and analog output adapter (Option RO8, Data Science International) using Powerlab 4/10 (AD‐Instruments, Austin, TX; sampling: 1kHz; filtering: 200Hz low pass). Infrequent periods of severe electrical noise were manually removed from the recording before further analysis. Recordings were analyzed using the Neuroarchiver tool (Open Source Instruments; http://www.opensourceinstruments.com/Electronics/A3018/Seizure_Detection.html). For more details on analysis, see the website or publication.26 In brief, data were split into 1‐second epochs. A power threshold was set (5 times baseline power), and putative events were epochs in which this threshold was crossed. Values of six metrices (power (in the 0–200Hz band), coastline, voltage asymmetry, coherence, intermittency, and rhythm) were determined for each putative event, and these were compared with a library of events manually classified as seizure‐like. Events were classified according to their Euclidian distance to events in a manually constructed event library. The library was constructed by an operator who classified events as “no event/baseline” (no obvious electrographic or behavioral event), “interictal event,” or “artifact” (grooming related noise or antenna glitches). In recordings from kainate‐injected animals, additional event types appeared: large single negative peak spike waves (simple spike‐waves; SS); spike waves having one or more shoulders (complex spike‐waves; CS); and runs of intermediate frequency (IF) discharges (5–12Hz) lasting 0.2 to 12 seconds with typically smaller amplitude (Fig 4B and Supplementary Movie 1). Following library construction, all data were analyzed without further manual interference.
Figure 4.
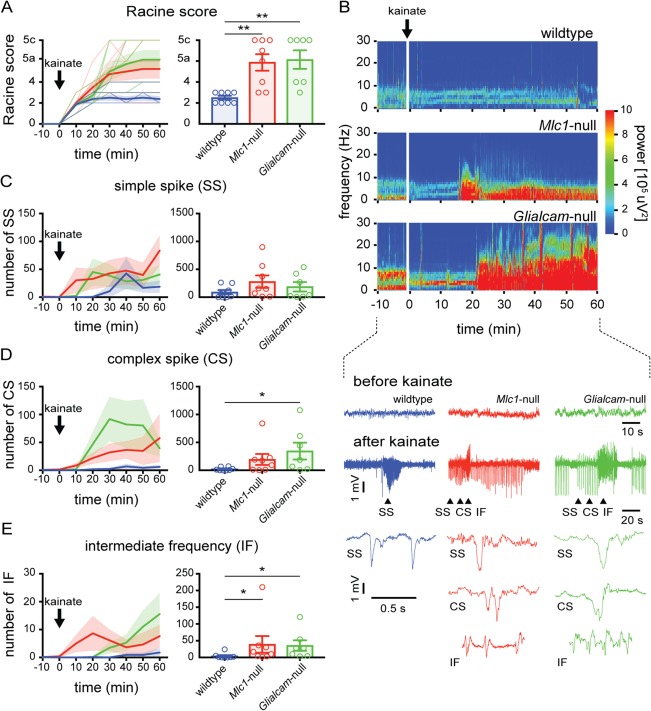
Lowered seizure threshold in MLC mice. (A) Behavioral scoring of seizure activity over time in wild‐type (blue) and MLC mice (Mlc1‐null: red; Glialcam‐null: green; thin lines show individual mouse responses; thick lines show averages) following kainate injection (10mg/kg intraperitoneally). For each 10‐minute interval, the highest level of epileptic activity was scored using the modified Racine seizure scale. The bar graph shows maximum Racine score during 60‐minute trial (open circles indicate individual mice, bars show average). MLC mice reached significantly higher behavioral seizure scores (wildtype: n = 8; Mlc1‐null: n = 8; Glialcam‐null: n = 7; p = 0.0014; wildtype vs Mlc1‐null, p = 0.0060; wildtype vs Glialcam‐null, p = 0.0023). (B) Top: Morlet‐wavelet ECoG spectra from kainate‐injected mice. Bottom: ECoG traces showing simple spike waves (SS), complex spike waves (CS) and runs of intermediate frequency (IF) discharges (expanded below). (C–E) Temporal progression of kainate‐induced SS, CS, and IF events, respectively. Bar graphs show total number of SS, CS, and IF events in 60‐minute trial. A significantly higher number of IF discharges was observed in MLC mice (wildtype: 4.1 ± 2.9 discharges/h; Mlc1‐null: 39.0 ± 24.8; Glialcam‐null: 35.4 ± 15.8; p = 0.0101; wildtype vs Mlc1‐null, p = 0.028; wildtype vs Glialcam‐null, p = 0.012). Error bars and shaded regions indicate SEM. *p < 0.05; **p < 0.01; ***p < 0.001. ECoG = electrocorticogram; MLC = megalencephalic leukoencephalopathy with subcortical cysts.
Slice Preparation
Acute brain slices were prepared from 3‐ to 5‐month‐old mice. After decapitation, the brain was removed in ice‐cold solution containing (mM): 70 sucrose, 70 NaCl, 25 D‐glucose, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4*H2O, 5.0 MgSO4*H2O, 1 CaCl2, 3 sodium pyruvate, and 1 sodium ascorbate (carboxygenated with 5%CO2/95%O2, 300–310mOsm). Coronal hippocampal slices (350 µm) were cut on a microtome (Thermo Fisher Scientific, Waltham, MA), kept in slicing solution at 32 ºC for 20 minutes, followed by 20‐minute recovery at room temperature. Afterward, slices were stored for >1 hour at room temperature in ACSF containing (mM): 125 NaCl, 3 KCl, 1.2 NaH2PO4, 1 MgCl2, 2 CaCl2, 26 NaHCO3, and 10 glucose (carboxygenated with 5%CO2/95%O2, 300–305mOsm).
Patch‐Clamp Recording
Slices were transferred to the setup and perfused with ACSF (5ml/min at 30–31 ºC). Recording ACSF contained CNQX (10 µM), AP‐5 (50 µM), and Gabazine (3 µM). Patch pipettes (3–5MΩ) were filled with intracellular solution containing (mM): 111 K‐gluconate, 8 KCl, 10 HEPES, 4 Mg‐ATP, 0.4 Na‐GTP, 10 K‐phosphocreatine, 0.2 EGTA, and 0.37% biocytin (7.4 pH; 290 ± 10mOsm). Recordings were made using an Axopatch 700B amplifier and Clampex software (sampling: 50kHz; filtering: 30kHz; Axon Instruments, Union City, CA). After determination of resting membrane potential, cells were kept in current clamp at –65mV through steady current injection. Data were analyzed using a custom‐written script in Matlab (version R2012a; The MathWorks, Inc., Natick, MA). Action potentials (APs) were required to exceed a membrane potential threshold of –20mV and a speed threshold of 10mV/ms to be included in analysis. AP threshold was defined by determining the peak of the AP derivative, and measuring the membrane potential at which 10% of this maximum was reached. AP half‐width was calculated as the time difference between the two time points corresponding to 50% of the AP amplitude (AP peak/AP threshold). For most neurons, pyramidal identity was confirmed after recording by biocytin staining, using the avidin‐biotin‐peroxidase method.27
K+‐Sensitive Electrode Recordings
K+‐sensitive microelectrodes were prepared from thin walled nonfilamented glass capillaries (GC150T‐10; Harvard Apparatus, Holliston, MA) pulled to a tip inner diameter in the range of 2 μm.28 Microelectrodes were silanized with gaseous N,N‐dimethyltrimethylsilylamine (41716; Sigma‐Aldrich, St. Louis, MO) at 200 ºC for 8 to 10 hours before being filled with a short column (120–150 μm) of liquid membrane solution (99373, K+ Ionophore I Cocktail B; Sigma‐Aldrich) and 150mM of NaCl, 3mM of KCl as backfilling solution (150–180MΩ). Tips of a K+‐sensitive and a field‐potential electrode (2MΩ, filled with 150mM of NaCl, 3mM of KCl) were placed ∼5 μm (±2 μm) from each other. Voltage signal from both channels to the same reference electrode were recorded using a MultiClamp 700B double amplifier (Axon Instruments), and differential voltage between the two electrodes was calculated by subtraction. K+‐sensitive electrodes were calibrated before and after the experiment (calibration solutions: 3, 6, 12, 24, or 48mM of KCl, 150mM of NaCl). Data obtained with a K+‐sensitive electrode were excluded when calibrations before and after differed by more than 2.5mV for 3, 6, or 12mM of KCl.
For recording [K+]o dynamics in hippocampus, both K+‐sensitive electrode and field‐potential electrode were placed in CA1 stratum radiatum (depth ∼80–100 μm). A concentric bipolar stimulation electrode was placed ∼550 to 600 μm lateral from the recording site onto stratum radiatum. Input‐output curves were constructed by recording field excitatory postsynaptic potentials (fEPSPs) following single stimulation pulses of increasing strength (0–950 μA; 50‐μA steps). Trains of 10‐second stimulation at 5, 10, or 20Hz, 450 μA, were applied twice and the averaged response parameters were calculated. Subsequently, recording electrodes were moved into stratum pyramidale (stimulation electrode remained in the same location). Data were analyzed using custom‐written procedures in IGOR‐pro (Wavemetrics, Lake Oswego, OR). Population spike area in stratum pyramidale was defined as the area between large negative deflections in the measured fEPSP and a fitted diagonal connecting the two neighboring local maxima (Fig. 8B; shaded area).
Figure 8.
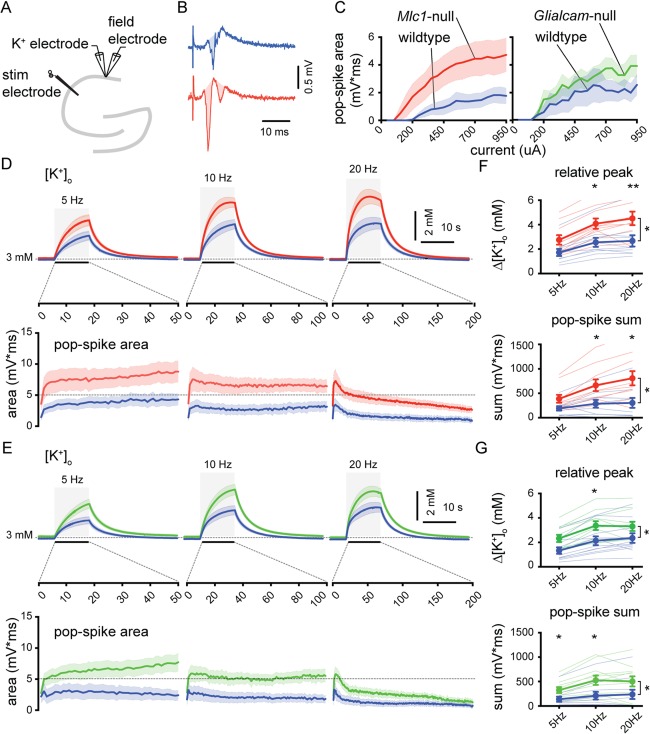
Increased stimulation‐induced [K+]o rises and network excitability in CA1 stratum pyramidale of MLC mice. (A) Schematic drawing of the hippocampus showing recording and stimulating electrodes. (B) Representative field recording upon single Schaffer collateral stimulation (450 µA) in a wildtype mouse (blue) and an Mlc1‐null mouse (red). The pop‐spike area was defined as the shaded region in the traces. (C) Pop‐spike area versus stimulation current. Although a trend towards larger pop‐spike area was observed this did not reach significance (wildtype [blue; n = 10] vs Mlc1‐null [red; n = 10]: p = 0.053; wildtype [blue; n = 11] vs Glialcam‐null [green; n = 11]: p = 0.076). (D,E) Top: averaged trace of [K+]o upon 10‐second stimulation (gray area) at 5, 10, and 20Hz. Bottom: averaged pop‐spike area for each stimulation pulse during 5, 10, and 20Hz stimulation. (F,G) Significant stimulation frequency dependent increase in [K+]o in MLC mice (wildtype vs Mlc1‐null: p = 0.021; wildtype vs Glialcam‐null: p = 0.035). Total summed pop‐spike area was also significantly increased in MLC mice (wildtype vs Mlc1‐null: p = 0.015; wildtype vs Glialcam‐null: p = 0.033). Thin lines show individual experiment. Error bars and shaded regions indicate SEM. *p < 0.05; **p < 0.01. MLC = megalencephalic leukoencephalopathy with subcortical cysts.
Statistical Analysis
Statistical analysis was performed with Prism (version 7; GraphPad Software Inc., La Jolla, CA). For behavioral and ECoG recordings, nonparametric analysis of variance (ANOVA; Kruskal‐Wallis), followed by Dunn's multiple comparisons (wild‐type versus Mlc1‐null and Glialcam‐null) was performed. For patch‐clamp data and basal [K+]o, parameters, we tested for normality using the Shapiro‐Wilk test. Data were compared using regular t test or, in case of deviant normal distribution, Mann–Whitney U test. For input‐output analysis and analysis of stimulation‐induced [K+]o, two‐way ANOVA followed by Sidak's multiple comparison test was performed. Kaplan‐Meier survival curves were compared using log‐rank statistics.
Results
Clinical Seizures in MLC Patients
Information on seizure characteristics was obtained from 141 patients with recessive MLC1 mutations and 17 patients with recessive GLIALCAM mutations (137 families; Supplementary Table 1). Survival analysis showed that 75.8% of patients with MLC1 mutations and 73.4% of patients with GLIALCAM mutations experienced at least one seizure before 25 years of age (Fig 1A,B). Curves were not different for patients with an MLC1 defect and patients with a GLIALCAM defect.
Figure 1.
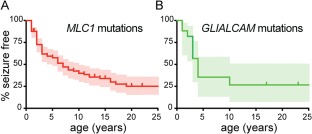
Early‐onset seizures are often observed in MLC patients. (A) Survival analysis (Kaplan‐Meier) of the proportion of patients that are seizure free as a function of age for patients with homozygous recessive MLC1 or (B) GLIALCAM mutations. Censored patients (seizure free at last follow‐up) are indicated by vertical lines. Ninety‐five percent confidence intervals are depicted by shaded areas. Survival curves were not different for patients with an MLC1 defect and patients with a GLIALCAM defect (log‐rank test: p = 0.56). Median age at the occurrence of the first seizure was 3 years, mode 2 years (range from first year of life up to 56 years). MLC = megalencephalic leukoencephalopathy with subcortical cysts. [Color figure can be viewed at http://www.annalsofneurology.org]
Of all patients with seizures, 13% had experienced only one seizure at the time of clinical inventory, often provoked by mild head trauma (8 of 12 patients) or by febrile illness (2 of 12 patients). Seventy‐three percent had epilepsy that was well controlled with medication. Of all patients ≥6 years of age, 63% met the International League against Epilepsy criteria for epilepsy.29 Clinically, generalized tonic‐clonic seizures and focal seizures (sometimes with impaired awareness and/or focal to bilateral tonic‐clonic seizures) were most common. Mild head trauma was an important provoking factor, causing one or more seizures in 55% of patients with seizures. One or more status epilepticus had occurred in 17%, of which most (58%) occurred for the first time a few years after the first seizure. 6 of the 158 patients were deceased. Of these 6, 3 deaths were related to epilepsy.
Taken together, the patient inventory shows that an early onset of epileptic seizures is an integral part of the MLC phenotype. Mild head trauma is an important provoking factor and status epilepticus occurs relatively frequently and early in the disease.
Hindlimb Clasping and Spontaneous Interictal Activity in MLC Mice
To understand the cellular pathophysiology of epilepsy in MLC, we studied two MLC mouse models: the Mlc1‐null mouse and the Glialcam‐null mouse. Previous automated behavioral analysis of Mlc1‐null mice did not uncover behavioral abnormalities.10 For Glialcam‐null mice, behavioral analysis has not been reported. We observed that both Mlc1‐null and Glialcam‐null mice showed hindlimb clasping when suspended from the tail (Fig 2). This is a sign of neurological abnormality, co‐occurring with seizures or increased seizure susceptibility in several mouse models for neurological diseases.30, 31
Figure 2.
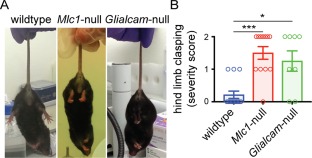
Hindlimb clasping in MLC mice. (A) Representative pictures illustrating the hindlimb clasping phenotype. Scoring was as follows: 0: normal (hindlimbs fully spread and moving); 1: intermediate (intermittent clasping of one hindlimb); 2: full (both hindlimbs fully drawn up to the abdomen). Left: wildtype, scored 0; middle, right: Mlc1‐null and Glialcam‐null mice respectively, both scored 2. (B) Both Mlc1‐null (n = 12) and Glialcam‐null (n = 8) mice exhibited significantly increased hindlimb clasping severity compared to the wild‐type controls (n = 14; p = 0.0003; wild‐type vs Mlc1‐null, p = 0.0003; wild‐type vs Glialcam‐null, p = 0.014). Open circles show individual mice. Bars indicate mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001. MLC = megalencephalic leukoencephalopathy with subcortical cysts. [Color figure can be viewed at http://www.annalsofneurology.org]
To test whether abnormal electrical activity is present in brains of MLC mice, we performed wireless ECoG recordings with an electrode placed above the motor cortex in both MLC mouse models (Fig 3A). Long‐term recordings (48 hours) from mice in their home cage revealed that both MLC lines show a significantly higher occurrence of abnormal discharges than wild‐type mice (Fig 3B–E). These discharges bear resemblance to interictal spikes.32 They did not have a clear behavioral correlate, as assessed from simultaneous video recordings of mouse behavior.
Figure 3.
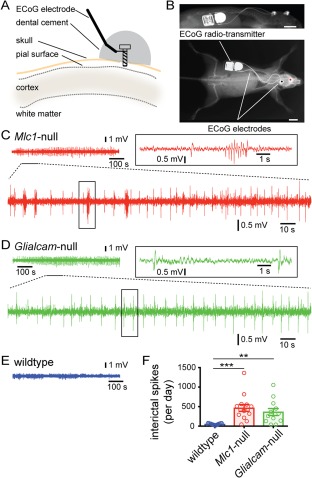
Spontaneous interictal activity in MLC mice. (A) Schematic representation of the implanted electrode for wireless ECoG recording. (B) X‐ray image of a mouse showing subcutaneous placement of the radiotransmitter (bar = 5mm). (C) Top: representative example 12‐minute ECoG recording from a Mlc1‐null mouse. Bottom: Expanded display of 100 seconds from the top trace. Right, boxed: Further expansion (10 seconds) of the boxed region. (D) Same as (C), but for a Glialcam‐null mouse. (E) Same as (B,C), but for a wild‐type mouse. (F) Bar graph showing the number of interictal spikes per day for all three groups. Open circles indicate individual mice, bars show average ± SEM. Both MLC mouse lines had a significant increase in interictal spikes (wildtype: 51.8 ± 5.3 events/day, n = 15; Mlc1‐null: 465.1 ± 86.8 events/day, n = 14; Glialcam‐null: 361.5 ± 89.0, n = 12; p = 0.0001; wildtype vs Mlc1‐null, p < 0.0001; wildtype vs Glialcam‐null, p = 0.0027). *p < 0.05; **p < 0.01; ***p < 0.001. ECoG = electrocorticogram; MLC = megalencephalic leukoencephalopathy with subcortical cysts.
To assess whether interictal spikes also occurred in the hippocampus, we performed wireless LFP recordings with a depth‐electrode placed in the hippocampal CA1 area. Similar to what we observed in motor cortex, MLC mice showed a significantly higher occurrence of interictal discharges compared to wild‐type mice (wild‐type: 15.7 ± 7.1 events/day, n = 3; MLC: 331.2 ± 95.9 events/day, n = 3 Glialcam‐null and 1 Mlc1‐null; p = 0.039).
Taken together, MLC mice show an overt hindlimb clasping phenotype. Although major behavioral seizures did not occur during our recording sessions, and were not observed during previous phenotyping,10 unprovoked interictal spikes are present in MLC mice.
Lowered Seizure Threshold and Increased Seizure Severity in MLC Mice
The presence of unprovoked interictal spikes in MLC mice suggests that they might be more sensitive to evoked seizure activity. To test this, mice received a single intraperitoneal injection with the chemoconvulsant kainate, at a dose which does not induce severe seizures in C57BL/6J mice (10mg/kg),33 followed by ECoG recording and behavioral scoring using the modified Racine score.25 While wild‐type mice only reached Racine score 2 or 3, with time Mlc1‐null and Glialcam‐null mice developed more severe seizures with multiple generalized tonic‐clonic seizure events (Racine score 5a–5c; Fig 4A).
Analysis of ECoG recordings following kainate injection revealed three main types of epileptic events: spike‐wave complexes either with a single negative peak (SS) or featuring one or more shoulders (CS), interspersed with runs of intermediate frequency (IF) discharges (5–12Hz) lasting 0.2 to 12 seconds, which typically had a small amplitude (Fig 4B). IF discharges correlated with severe motor seizures (Supplementary Movie 1), in line with previous data.34 In the 60‐minute period following kainate injection, wild‐type mice showed few simple SS and CS events, with very rare IF discharges (Fig 4C–E). In contrast, both Mlc1‐null and Glialcam‐null mice showed a significantly higher number of IF discharges than wild‐type animals (Fig 4E). No significant differences were observed in the occurrence of SS and CS events, although a trend toward more events in MLC mice was present. Taken together, these findings highlight that MLC mice have a lower threshold for kainate induced seizures than wild‐type mice.
Neuronal Excitability in MLC Mice
Spontaneous interictal activity in motor cortex and hippocampus and a lowered threshold for evoked seizures suggest neuronal hyperexcitability in MLC mice. To test whether this was alternatively to increased intrinsic neuronal excitability, we performed whole‐cell patch‐clamp recordings from CA1 pyramidal neurons in acute hippocampal brain slices (Fig 5 and Supplementary Table 2). Passive electrophysiological properties (membrane potential, input resistance) did not differ between MLC mice and their wild‐type littermates (Fig 5D,E). Furthermore, AP waveforms were indistinguishable between Mlc1‐null mice and littermate controls (Fig 5C,D). Glialcam‐null mice also showed similar AP waveform properties as their wild‐type littermates, apart from a slight increase in AP peak and half‐width (Fig 5E). Importantly, input‐output characteristics and maximal firing frequency were identical in both MLC mouse lines compared to their wild‐type littermate controls (Fig 5F,G).
Figure 5.
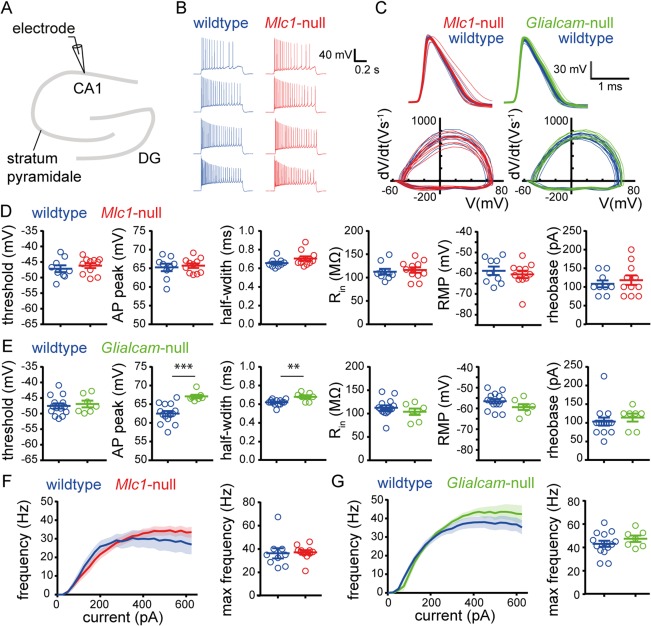
Intrinsic properties of CA1 pyramidal neurons are unaltered in MLC mice. (A) Schematic drawing of the hippocampus showing the patch‐clamp electrode at the region of recording (CA1). (B) Representative AP firing pattern of a wild type (blue) and an Mlc1‐null (red) pyramidal neuron upon 200‐, 300‐, 400‐, and 600‐pA current injection. (C) Top: AP waveform of the first AP fired by single cells (thin lines) and the average AP waveform per genotype (thick lines). Bottom: AP phase plane plot of the first AP fired by single cells. Data of Mlc1‐null (red) and wild‐type littermates (blue) are shown on the left and data of Glialcam‐null (green) and wild‐type littermates (blue) on the right. (D) Active properties (first three panels; threshold, AP peak, and half‐width) and passive properties (last three panels; input resistance [Rin], resting membrane potential [RMP], and rheobase) for wild‐type (blue) and Mlc1‐null mice (red) and (E) for wild‐type (blue) and Glialcam‐null mice (green). AP properties were derived from the first AP fired by a cell. For mean values and statistics, see Supplementary Table 2. (F) Input‐output curve showing AP firing frequency vs stimulation current (left) and scatter plot showing maximum firing frequency (right) for wild‐type (blue) and Mlc1‐null (red) pyramidal neurons and (G) for wild‐type (blue) and Glialcam‐null (green) pyramidal neurons. Input‐output curve was determined using 800‐ms‐long step current injections (25‐pA steps from –100 to 625pA). No significant differences were observed in input‐output curves (wildtype vs Mlc1‐null: p = 0.74; wildtype vs Glialcam‐null: p = 0.39). Error bars and shaded regions indicate SEM. **p < 0.01; ***p < 0.001. AP = action potential; MLC = megalencephalic leukoencephalopathy with subcortical cysts.
We hypothesized that stimulating single neurons to fire APs for a prolonged period might uncover more subtle differences in intrinsic properties. Sustained AP firing was evoked by applying prolonged current injections with step‐wise increments (Fig 6A). Prolonged stimulation altered the AP waveform (Fig 6B), but again this did not differ between MLC mice and their wild‐type littermate controls (Fig 6C–E).
Figure 6.
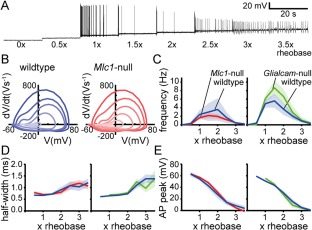
Prolonged stimulation leads to similar action potential (AP) firing in MLC mice and wild‐type littermates. (A) Representative firing response trace of a CA1 neuron during prolonged stimulation. Prolonged stimulation consisted of eight 20‐second long steps with an increment of one half rheobase (rheobase: current at which an AP was first observed). (B) Representative phase plane plots showing the first derivative (V s–1) versus membrane voltage (mV) for representative wild‐type (blue, left) and Mlc1‐null (red, right) neurons. Each line represents the first AP from a 20‐second current step, with the earliest AP as darkest color and latest AP as lightest color. (C–E) Input‐output curves showing the average firing frequency (C), average half‐width (D), and average AP peak (E) per 20‐second current step vs stimulation current (represented as multiplications of the rheobase). Left panels show data of Mlc1‐null mice (red) and wild‐type littermates (blue) and right panels show data of Glialcam‐null mice (green) and wild‐type littermates (blue). No significant differences between MLC mice and littermate controls were observed for any of these parameters (wildtype vs Mlc1‐null: p = 0.55 [frequency], p = 0.95 [half‐width], and p = 0.71 [absolute peak]; wildtype vs Glialcam‐null: p = 0.26 [frequency], p = 0.30 [half‐width], and p = 0.83 [absolute peak]). Shaded regions indicate SEM. AP = action potential; MLC = megalencephalic leukoencephalopathy with subcortical cysts.
To check whether intrinsic neuronal properties in other brain regions are similarly unaffected in MLC mice, we performed an additional set of patch‐clamp recordings from layer 5 pyramidal neurons in primary motor cortex (M1). Like in hippocampus, we observed no differences in passive and active neuronal properties, nor in responses to prolonged current injections, that could explain hyperexcitability in MLC mice (data not shown).
Taken together, these results show that intrinsic excitability of principal neurons in hippocampus and neocortex is largely unchanged in MLC mice, and cannot account for the observed interictal activity and decreased seizure threshold.
Increased Stimulation‐Induced [K+]o Around CA1 Synapses of MLC Mice
Impaired astrocyte functioning can lead to disturbed [K+]o dynamics. Therefore, we measured [K+]o dynamics using calibrated K+‐sensitive electrodes28 in acute brain slices of hippocampal CA1 area (stratum radiatum; Fig 7A). We did not observe differences in basal [K+]o between MLC mice and wild‐type littermates at rest (Fig 7B).
Figure 7.
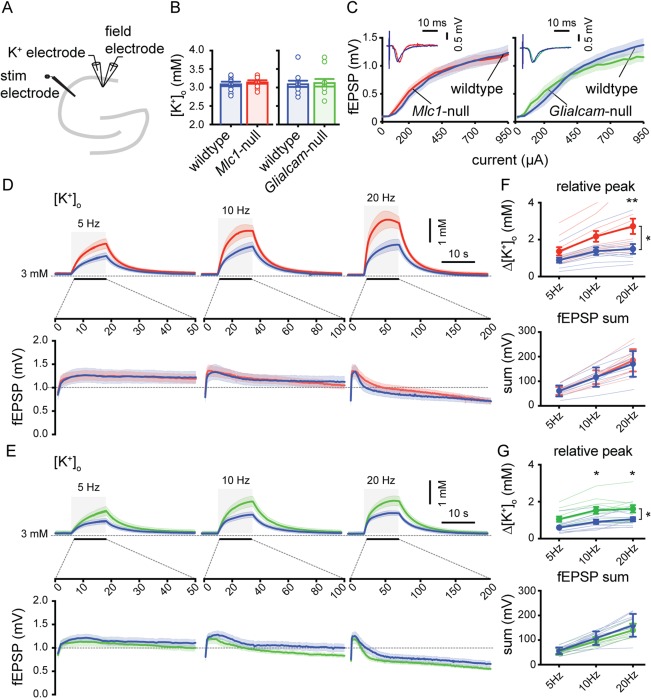
Larger stimulation‐induced rises in [K+]o in CA1 stratum radiatum of MLC mice. (A) Schematic drawing of the hippocampus showing recording and stimulating electrodes. (B) Basal [K+]o in stratum radiatum (wildtype [blue]: 3.0 ± 0.06mM; n = 10 vs Mlc1‐null [red]: 3.1 ± 0.04; n = 10; p = 0.63; wildtype [blue]: 3.1 ± 0.08; n = 11 vs Glialcam‐null [green]: 3.1 ± 0.10; n = 11; p > 0.99). (C) Input‐output curve showing fEPSP amplitude vs stimulation current. Curves did not differ between MLC mice and littermate controls (wildtype vs Mlc1‐null: p = 0.47; wildtype vs Glialcam‐null: p = 0.14) (D,E) Top: averaged trace of [K+]o upon 10‐second stimulation (gray area) at 5, 10, and 20Hz. Bottom: averaged fEPSP amplitude for each stimulation pulse during 5, 10, and 20Hz stimulation. (F,G) Significant stimulation frequency‐dependent increase in [K+]o in MLC mice (wildtype vs Mlc1‐null: p = 0.034; wildtype vs Glialcam‐null: p = 0.013) with no significant change in fEPSP amplitude sum (wildtype vs Mlc1‐null: p = 0.087; wildtype vs Glialcam‐null: p = 0.29). Thin lines show individual experiment. Error bars and shaded regions indicate SEM. *p < 0.05; **p < 0.01. MLC = megalencephalic leukoencephalopathy with subcortical cysts.
Next, we simultaneously recorded [K+]o and field potential responses in stratum radiatum upon extracellular stimulation of Schaffer collaterals (Fig 7A). First, we determined the input‐output curve of the fEPSP amplitude by single stimulations with step‐wise increases in stimulation strength. Notably, no genotype differences were observed in input‐output curves, showing that basal synaptic strength of the Schaffer collaterals is unchanged in MLC mice (Fig 7C).
Increases in [K+]o were induced by sustained repetitive Schaffer collateral stimulation at a fixed stimulation strength for 10 seconds, either at 5, 10, or 20Hz. A stimulation frequency‐dependent increase in [K+]o was observed in all experiments (Fig 7D,E). However, significantly higher stimulation‐induced increases in [K+]o were measured both in Mlc1‐null (Fig 7F) and in Glialcam‐null mice (Fig 7G). In contrast, the sum of all fEPSP amplitudes measured during repetitive stimulation was not different between genotypes (Fig 7F,G). Observed differences in peak [K+]o were stimulation frequency‐dependent, with 20‐Hz stimulation showing the strongest increase in peak [K+]o (Fig 7F,G). Despite the fact that MLC mice showed a significantly higher increase in [K+]o during the stimulation, the poststimulation [K+]o recovery (80–20% decay time) was not different between MLC mice and wild‐type littermates (data not shown).
These findings show that while the strength and efficacy of Schaffer collateral synapses are unchanged in MLC, prolonged high‐frequency stimulation of these synapses leads to a larger increase in [K+]o in MLC mice than in wild‐type littermates.
Increased Network Excitability in CA1 Somatic Layer of MLC Mice
Increased [K+]o in the dendritic layer upon repetitive stimulation could lead to stronger and more sustained depolarization of the dendritic tree and may therefore result in increased network excitability. After finishing recording in stratum radiatum, we moved recording electrodes into the CA1 cell body layer (stratum pyramidale) while keeping the stimulation electrode at the same position (Fig 8A). When determining the input‐output curve in stratum pyramidale with single stimulations, both MLC mice showed a trend toward an increase in population spikes (pop‐spikes; Fig 8B), although this did not reach significance (Fig 8B,C). Similar to what was observed in stratum radiatum, prolonged Schaffer collateral stimulation led to larger increases in [K+]o in both MLC mice than in wild‐type littermates (Fig 8C). Also similar to stratum radiatum, the increase in [K+]o was frequency dependent (Fig 8F,G). Strikingly, the sum of all pop‐spike areas evoked during the train stimulation was also increased in both MLC mouse models (Fig 8C), indicating increased network excitability in MLC mice (Fig 8F, G). Similar to what we observed in stratum radiatum, the poststimulation [K+]o recovery (80–20% decay time) in stratum pyramidale was not different between MLC mice and wild‐type littermates (data not shown).
In conclusion, [K+]o recordings show that, while Schaffer collateral input strength is unchanged, prolonged synaptic stimulation leads to higher rises in [K+]o in CA1, both in stratum radiatum and in stratum pyramidale. Furthermore, there is an increase in stimulation‐induced pop‐spikes in CA1, suggesting increased excitability of neuronal networks in MLC mice.
Discussion
We have shown that an early onset of epileptic seizures is common in MLC patients. We studied the cellular pathophysiology of seizures in two different genetic mouse models for MLC. Both Mlc1‐null and Glialcam‐null mice show hindlimb clasping, unprovoked interictal brain activity, and a lowered threshold for kainate‐induced seizures. While intrinsic excitability of principal neurons is unchanged in MLC mice, increases in [K+]o upon network activation and network excitability are higher in both MLC mouse models. Thus, astrocyte dysfunction in MLC leads to disturbed [K+]o dynamics and network hyperexcitability, and lowers the threshold for seizures.
Seizures in MLC Patients and MLC Mouse Models
This is the first report on seizure characteristics in a large cohort of genetically confirmed patients with recessive MLC. An estimated 75% of all MLC patients experiences at least one seizure and 63% of patients older than 6 years has been diagnosed with epilepsy, which is in line with previous reports,12, 35 and confirms that epilepsy is common in MLC. Epilepsy in MLC patients is mostly well controlled with medication compared to the general epilepsy population (only 14% had moderately to poorly controlled epilepsy versus 20–30% of general epilepsy patients).36, 37 However, occurrence of status epilepticus is high (∼17%) compared to the overall occurrence reported for children and adults with epilepsy during follow‐up (∼10%),38, 39 indicating that once seizures are initiated, restoring neuronal network activity to its normal state is hampered in MLC.
A striking feature of MLC is the high susceptibility to develop seizures immediately following mild head injury (54% of patients with seizures). In patients with epilepsy in general, mild head trauma is only rarely reported as a seizure‐precipitating factor.40 Interestingly, animal studies show that closed head injury leads to a rise in brain [K+]o.41 The high sensitivity to mild head injury of MLC patients could therefore reflect an increased sensitivity to rises in [K+]o, which would be in line with our observations in mouse models for MLC (see below).
Our results in Mlc1‐null and Glialcam‐null mice confirm that epileptiform brain activity and a lowered seizure threshold are an integral part of MLC. However, because overt spontaneous behavioral seizures are absent in both mouse models for MLC, the phenotype of MLC mice is milder than that of MLC patients. This discrepancy between MLC mice and patients has been discussed before10 and might be related to different compensation by mice to loss of MLC1 function compared to humans. Another likely contributing factor is the much shorter life‐span of mice than that of humans, given that MLC patients develop major neurological dysfunction only after a delay of several years to decades.1, 8
Cellular Basis of Epilepsy in MLC
The use of mouse models allowed us to investigate the cellular basis of epilepsy in MLC. Decreased seizure threshold is often associated with increased neuronal excitability. However, we did not observe any change in intrinsic neuronal excitability of either hippocampal CA1 pyramidal neurons or cortical L5 pyramidal neurons in MLC mice that could explain the decreased seizure threshold. We conclude that intrinsic hyperexcitability of principal neurons is not the cause of seizures in MLC.
K+‐sensitive electrode recordings revealed increased peak [K+]o rises in hippocampal CA1 upon trains of Schaffer collateral stimulation in MLC mice. This was not attributed to increased synaptic strength, given that fEPSP amplitudes were unchanged. When recording population responses to the same Schaffer collateral stimulation in stratum pyramidale, we observed an increase in CA1 pyramidal neuron population spiking, indicative of increased network excitability. In line with this, it was previously shown that modest rises in [K+]o increase hippocampal population spiking while leaving levels of synaptic transmission unaffected.42 Therefore, we conclude that although intrinsic excitability of individual neurons is unchanged, MLC mice show altered [K+]o dynamics and increased network excitability.
How could loss of function of the astrocyte‐specific protein MLC1 lead to disturbed [K+]o dynamics and seizures? It is well established that astrocytes are crucial for [K+]o homeostasis.13, 14 Mathematical models show that disturbing glial K+ uptake can lead to seizures,43 and dysfunctional astrocyte [K+]o homeostasis is implicated in epilepsy.44 K+ homeostasis is tightly linked to the homeostasis of other ions, and MLC1 and GlialCAM colocalize and/or interact with several proteins involved in ion and water homeostasis. This includes the dystrophin glycoprotein complex,5, 45 Na+/K+‐ATPase,46 the swelling‐sensitive cation channel, TRPV4,47 and the gap‐junction protein, connexin 43.20 MLC1 and GlialCAM may therefore play a central role in organizing these components necessary for astrocyte ion and water homeostasis.
In the healthy brain, astrocytes counteract increases in [K+]o during periods of high neuronal activity through transporter‐mediated K+ uptake48 and spatial K+ buffering.14 This leads to astrocyte swelling and depolarization, which can be counteracted by activation of VRACs. We and others have previously shown that mutations in MLC1 or GLIALCAM lead to dysfunction of astrocytic VRACs,8, 9, 10 resulting in chronic astrocyte swelling. We previously hypothesized that this could limit the K+ uptake capacity of astrocytes, thereby leading to activity‐dependent white matter vacuolization.8 In line with this hypothesis, our study confirms that [K+]o is disturbed in MLC. We show that this is accompanied by network hyperexcitability and seizures. The fact that MLC patients show a high occurrence of status epilepticus fits in this framework, given that a disturbed uptake of K+ likely hampers restoration of disturbed ion gradients following a seizure.
MLC1 and GlialCAM share their localization in astrocyte endfeet abutting blood vessels with inwardly rectifying Kir4.1 K+ channels and AQP4 water channels. Both these proteins are necessary for [K+]o regulation and have been linked to seizures. Kir4.1 is crucial for K+ spatial buffering49, 50, 51 and for efflux of K+ from astrocytes following active transport.48 Astrocyte‐specific Kir4.1 conditional knockout results in reduced astrocyte K+ uptake, disturbed clearance of [K+]o, white matter vacuolization, ataxia, and stress‐induced seizures,15, 16 thereby resembling the MLC phenotype. However, astrocyte‐specific Kir4.1 knockout mice are much more severely affected and die at 3 to 4 weeks of age.15 Disturbed AQP4 in perivascular endfeet also leads to abnormal [K+]o dynamics and a seizure phenotype,18, 52, 53 although the mechanism by which AQP4 interacts with Kir4.1 and impacts on [K+]o is not fully understood.
Epilepsy was long thought to be primarily caused by malfunction of neurons, but in the last decades many studies have convincingly shown that alterations in astrocyte function play an important role in its pathogenesis.43, 53 In line with this, our study demonstrates that loss of function of the astrocyte‐specific protein MLC1 leads to dysregulation of [K+]o, network hyperexcitability and seizures. These results highlight the importance of astrocytes for neuronal network functioning and form an important next step in our understanding of the cellular pathophysiology of MLC.
Author Contributions
M.D., E.B., H.D.M., M.S.v.d.K., and R.M. contributed to the conception and design of the study. M.D., E.B., E.M.C.H., O.S., M.B., H.K., M.H.P.K., U.B., R.C.W., H.D.M., M.S.v.d.K., and R.M. contributed to the acquisition and analysis of data. M.D., E.B., E.M.C.H., M.S.v.d.K., and R.M. contributed to drafting the text and preparing the figures.
Potential Conflicts of Interest
Nothing to report.
Supporting information
Additional supporting information can be found in the online version of this article.
Supporting Information Movie 1
Supporting Information Tables
Acknowledgment
This study was financially supported by E‐Rare (11‐330‐1024), the Dutch Hersenstichting (2009(2)‐14), an NWO Spinoza award (2008), and an Amsterdam Neuroscience proof‐of‐concept grant (2014).
We thank all families and physicians who contributed to the inventory of clinical characteristics. We thank Prof Juha Voipio for advice on potassium‐sensitive electrode recordings, Dr Thijs Verhoog for help with action potential analysis, Dr Kevan Hashemi for ECoG analysis software, and Anton Pieneman, Dr Tim Heistek, and Hans Lodder for technical assistance.
References
- 1. van der Knaap MS, Barth PG, Stroink H, et al. Leukoencephalopathy with swelling and a discrepantly mild clinical course in eight children. Ann Neurol 1995;37:324–334. [DOI] [PubMed] [Google Scholar]
- 2. Singhal BS, Gursahani RD, Udani VP, Biniwale AA. Megalencephalic leukodystrophy in an Asian Indian ethnic group. Pediatr Neurol 1996;14:291–296. [DOI] [PubMed] [Google Scholar]
- 3. Leegwater PA, Yuan BQ, van der Steen J, et al. Mutations of MLC1 (KIAA0027), encoding a putative membrane protein, cause megalencephalic leukoencephalopathy with subcortical cysts. Am J Hum Genet 2001;68:831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lopez‐Hernandez T, Ridder MC, Montolio M, et al. Mutant GlialCAM causes megalencephalic leukoencephalopathy with subcortical cysts, benign familial macrocephaly, and macrocephaly with retardation and autism. Am J Hum Genet 2011;88:422–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boor I, Nagtegaal M, Kamphorst W, et al. MLC1 is associated with the dystrophin‐glycoprotein complex at astrocytic endfeet. Acta Neuropathol 2007;114:403–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Capdevila‐Nortes X, Lopez‐Hernandez T, Apaja PM, et al. Insights into MLC pathogenesis: GlialCAM is an MLC1 chaperone required for proper activation of volume‐regulated anion currents. Hum Mol Genet 2013;22:4405–4416. [DOI] [PubMed] [Google Scholar]
- 7. Hoegg‐Beiler MB, Sirisi S, Orozco IJ, et al. Disrupting MLC1 and GlialCAM and ClC‐2 interactions in leukodystrophy entails glial chloride channel dysfunction. Nat Commun 2014;5:3475. [DOI] [PubMed] [Google Scholar]
- 8. van der Knaap MS, Boor I, Estevez R. Megalencephalic leukoencephalopathy with subcortical cysts: chronic white matter oedema due to a defect in brain ion and water homoeostasis. Lancet Neurol 2012;11:973–985. [DOI] [PubMed] [Google Scholar]
- 9. Ridder MC, Boor I, Lodder JC, et al. Megalencephalic leucoencephalopathy with cysts: defect in chloride currents and cell volume regulation. Brain 2011;134(pt 11):3342–3354. [DOI] [PubMed] [Google Scholar]
- 10. Dubey M, Bugiani M, Ridder MC, et al. Mice with megalencephalic leukoencephalopathy with cysts: a developmental angle. Ann Neurol 2015;77:114–131. [DOI] [PubMed] [Google Scholar]
- 11. Bugiani M, Moroni I, Bizzi A, et al. Consciousness disturbances in megalencephalic leukoencephalopathy with subcortical cysts. Neuropediatrics 2003;34:211–214. [DOI] [PubMed] [Google Scholar]
- 12. Yalcinkaya C, Yuksel A, Comu S, et al. Epilepsy in vacuolating megalencephalic leukoencephalopathy with subcortical cysts. Seizure 2003;12:388–396. [DOI] [PubMed] [Google Scholar]
- 13. Walz W. Role of astrocytes in the clearance of excess extracellular potassium. Neurochem Int 2000;36:291–300. [DOI] [PubMed] [Google Scholar]
- 14. Kofuji P, Newman EA. Potassium buffering in the central nervous system. Neuroscience 2004;129:1045–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Djukic B, Casper KB, Philpot BD, et al. Conditional knock‐out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short‐term synaptic potentiation. J Neurosci 2007;27:11354–11365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Haj‐Yasein NN, Jensen V, Vindedal GF, et al. Evidence that compromised K+ spatial buffering contributes to the epileptogenic effect of mutations in the human Kir4.1 gene (KCNJ10). Glia 2011;59:1635–1642. [DOI] [PubMed] [Google Scholar]
- 17. Odermatt B, Wellershaus K, Wallraff A, et al. Connexin 47 (Cx47)‐deficient mice with enhanced green fluorescent protein reporter gene reveal predominant oligodendrocytic expression of Cx47 and display vacuolized myelin in the CNS. J Neurosci 2003;23:4549–4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Binder DK, Yao X, Zador Z, et al. Increased seizure duration and slowed potassium kinetics in mice lacking aquaporin‐4 water channels. Glia 2006;53:631–636. [DOI] [PubMed] [Google Scholar]
- 19. Brignone MS, Lanciotti A, Camerini S, et al. MLC1 protein: a likely link between leukodystrophies and brain channelopathies. Front Cell Neurosci 2015;9:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wu M, Moh MC, Schwarz H. HepaCAM associates with connexin 43 and enhances its localization in cellular junctions. Sci Rep 2016;6:36218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Favre‐Kontula L, Rolland A, Bernasconi L, et al. GlialCAM, an immunoglobulin‐like cell adhesion molecule is expressed in glial cells of the central nervous system. Glia 2008;56:633–645. [DOI] [PubMed] [Google Scholar]
- 22. Bugiani M, Dubey M, Breur M, et al. Megalencephalic leukoencephalopathy with cysts: the Glialcam‐null mouse model. Ann Clin Transl Neurol 2017;4:450–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hamilton EMC, Tekturk P, Cialdella F, et al. Megalencephalic leukoencephalopathy with subcortical cysts: characterization of disease variants. Neurology 2018. (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang XM, Zhu SW, Duan RS, et al. Gender differences in susceptibility to kainic acid‐induced neurodegeneration in aged C57BL/6 mice. Neurotoxicology 2008;29:406–412. [DOI] [PubMed] [Google Scholar]
- 25. Luttjohann A, Fabene PF, van LG. A revised Racine's scale for PTZ‐induced seizures in rats. Physiol Behav 2009;98:579–586. [DOI] [PubMed] [Google Scholar]
- 26. Wykes RC, Heeroma JH, Mantoan L, et al. Optogenetic and potassium channel gene therapy in a rodent model of focal neocortical epilepsy. Sci Transl Med 2012;4:161ra52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Horikawa K, Armstrong WE. A versatile means of intracellular labeling: injection of biocytin and its detection with avidin conjugates. J Neurosci Methods 1988;25:1–11. [DOI] [PubMed] [Google Scholar]
- 28. Voipio J, Pasternack M, MacLeod K. Ion‐sensitive microelectrodes In: Ogden D, editor. Microelectrode Techniques: The Plymouth Workshop Handbook, 2nd ed. Cambridge, UK: The Company of Biologists Limited; 1994:275–316. [Google Scholar]
- 29. Perucca E. Introduction to the clinical definition of epilepsy by the ILAE president. Epilepsia 2014;55:473‐474. [DOI] [PubMed] [Google Scholar]
- 30. Novarino G, El‐Fishawy P, Kayserili H, et al. Mutations in BCKD‐kinase lead to a potentially treatable form of autism with epilepsy. Science 2012;338:394–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ivanov SV, Ward JM, Tessarollo L, et al. Cerebellar ataxia, seizures, premature death, and cardiac abnormalities in mice with targeted disruption of the Cacna2d2 gene. Am J Pathol 2004;165:1007–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Glasscock E, Qian J, Yoo JW, Noebels JL. Masking epilepsy by combining two epilepsy genes. Nat Neurosci 2007;10:1554–1558. [DOI] [PubMed] [Google Scholar]
- 33. McCord MC, Lorenzana A, Bloom CS, et al. Effect of age on kainate‐induced seizure severity and cell death. Neuroscience 2008;154:1143–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Katzel D, Nicholson E, Schorge S, et al. Chemical‐genetic attenuation of focal neocortical seizures. Nat Commun 2014;5:3847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Singhal BS, Gorospe JR, Naidu S. Megalencephalic leukoencephalopathy with subcortical cysts. J Child Neurol 2003;18:646–652. [DOI] [PubMed] [Google Scholar]
- 36. Berg AT, Zelko FA, Levy SR, Testa FM. Age at onset of epilepsy, pharmacoresistance, and cognitive outcomes: a prospective cohort study. Neurology 2012;79:1384–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sander JW. Some aspects of prognosis in the epilepsies: a review. Epilepsia 1993;34:1007–1016. [DOI] [PubMed] [Google Scholar]
- 38. Berg AT, Shinnar S, Testa FM, et al. Status epilepticus after the initial diagnosis of epilepsy in children. Neurology 2004;63:1027–1034. [DOI] [PubMed] [Google Scholar]
- 39. Stroink H, Geerts AT, van Donselaar CA, et al. Status epilepticus in children with epilepsy: Dutch study of epilepsy in childhood. Epilepsia 2007;48:1708–1715. [DOI] [PubMed] [Google Scholar]
- 40. Nakken KO, Solaas MH, Kjeldsen MJ, et al. Which seizure‐precipitating factors do patients with epilepsy most frequently report? Epilepsy Behav 2005;6:85–89. [DOI] [PubMed] [Google Scholar]
- 41. Katayama Y, Becker DP, Tamura T, Hovda DA. Massive increases in extracellular potassium and the indiscriminate release of glutamate following concussive brain injury. J Neurosurg 1990;73:889–900. [DOI] [PubMed] [Google Scholar]
- 42. Balestrino M, Aitken PG, Somjen GG. The effects of moderate changes of extracellular K+ and Ca2+ on synaptic and neural function in the CA1 region of the hippocampal slice. Brain Res 1986;377:229–239. [DOI] [PubMed] [Google Scholar]
- 43. Kager H, Wadman WJ, Somjen GG. Simulated seizures and spreading depression in a neuron model incorporating interstitial space and ion concentrations. J Neurophysiol 2000;84:495–512. [DOI] [PubMed] [Google Scholar]
- 44. Coulter DA, Steinhauser C. Role of astrocytes in epilepsy. Cold Spring Harb Perspect Med 2015;5:a022434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ambrosini E, Serafini B, Lanciotti A, et al. Biochemical characterization of MLC1 protein in astrocytes and its association with the dystrophin‐glycoprotein complex. Mol Cell Neurosci 2008;37:480–493. [DOI] [PubMed] [Google Scholar]
- 46. Brignone MS, Lanciotti A, Macioce P, et al. The beta1 subunit of the Na,K‐ATPase pump interacts with megalencephalic leucoencephalopathy with subcortical cysts protein 1 (MLC1) in brain astrocytes: new insights into MLC pathogenesis. Hum Mol Genet 2011;20:90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lanciotti A, Brignone MS, Molinari P, et al. Megalencephalic leukoencephalopathy with subcortical cysts protein 1 functionally cooperates with the TRPV4 cation channel to activate the response of astrocytes to osmotic stress: dysregulation by pathological mutations. Hum Mol Genet 2012;21:2166–2180. [DOI] [PubMed] [Google Scholar]
- 48. Macaulay N, Zeuthen T. Glial K(+) clearance and cell swelling: key roles for cotransporters and pumps. Neurochem Res 2012;37:2299–2309. [DOI] [PubMed] [Google Scholar]
- 49. Newman EA. High potassium conductance in astrocyte endfeet. Science 1986;233:453–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Newman EA, Frambach DA, Odette LL. Control of extracellular potassium levels by retinal glial cell K+ siphoning. Science 1984;225:1174–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Seifert G, Huttmann K, Binder DK, et al. Analysis of astroglial K+ channel expression in the developing hippocampus reveals a predominant role of the Kir4.1 subunit. J Neurosci 2009;29:7474–7488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Strohschein S, Huttmann K, Gabriel S, et al. Impact of aquaporin‐4 channels on K+ buffering and gap junction coupling in the hippocampus. Glia 2011;59:973–980. [DOI] [PubMed] [Google Scholar]
- 53. Haj‐Yasein NN, Bugge CE, Jensen V, et al. Deletion of aquaporin‐4 increases extracellular K(+) concentration during synaptic stimulation in mouse hippocampus. Brain Struct Funct 2015;220:2469–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information can be found in the online version of this article.
Supporting Information Movie 1
Supporting Information Tables