

Abstract
A double‐blind, 4‐period crossover study (NCT01327066) was conducted to assess the effect of the novel norepinephrine prodrug droxidopa on the QT interval in in healthy subjects. Subjects were randomized to receive a single dose of droxidopa 600 mg (maximal dose) and 2000 mg (supratherapeutic dose) compared with the positive control, moxifloxacin 400 mg, and placebo, each separated by a 3‐day washout period. Patients were monitored by continuous Holter monitoring, and electrocardiograms (ECGs) were extracted 0.5–23 hours after dosing. Blood samples for pharmacokinetic analysis were collected before dosing and after ECG data collection. The primary end point was the time‐matched placebo‐adjusted change from baseline in the individually corrected QT (QTcI). The time‐averaged QTcI mean placebo‐corrected changes from baseline for droxidopa 600 and 2000 mg were 0.1 milliseconds (90%CI, ‐0.9 to 1.0 milliseconds) and 0.3 milliseconds (90%CI, ‐0.6 to 1.3 milliseconds), respectively, and 9 milliseconds (90%CI, 8.4–10.3 milliseconds) for moxifloxacin. This study found no effect of either dose of droxidopa on cardiac repolarization using QTcI. Analysis of the pharmacokinetic/pharmacodynamic relationship and cardiac repolarization showed no association with droxidopa exposure. There were no clinically relevant effects of droxidopa on heart rate, atrioventricular conduction, or cardiac depolarization identified. No morphologic ECG changes were observed.
Keywords: droxidopa, neurogenic orthostatic hypotension, QTc interval, cardiovascular safety, cardiac repolarization, pharmacokinetics, pharmacodynamics
Patients with neurodegenerative diseases may have impaired norepinephrine release causing neurogenic orthostatic hypotension (nOH), defined as an inadequate blood pressure (BP) and compensatory heart rate response during orthostatic change.1 Therefore, patients with nOH may experience dizziness, lightheadedness, or syncope on standing because of cerebral hypoperfusion.1 Droxidopa (l‐threo‐dihydroxyphenylserine) is a norepinephrine prodrug approved by the US Food and Drug Administration (FDA) for the treatment of orthostatic dizziness and lightheadedness or presyncope in adult patients with symptomatic nOH caused by autonomic failure in Parkinson's disease, multiple system atrophy, pure autonomic failure, dopamine β‐hydroxylase deficiency, or nondiabetic autonomic neuropathy.2, 3 The efficacy and safety of droxidopa have been investigated in 3 randomized, controlled trials in patients with nOH.4, 5, 6, 7
Droxidopa is converted by dopa decarboxylase to norepinephrine.2, 8, 9 In single‐dose studies in healthy subjects or patients with autonomic failure conditions, the peak plasma concentrations of droxidopa and norepinephrine occurred 3 to 5 and 3 to 8 hours, respectively, after oral administration.10, 11, 12 In patients with autonomic failure, pharmacodynamic effects (increases in blood pressure) were generally coincident with the pharmacokinetic profile.11, 12 In preclinical safety studies, potential cardiovascular effects of droxidopa were assessed in human ether‐a‐go‐go‐related gene (hERG)–transfected HEK293 cells (see Supplemental Methods) and isolated spontaneous beating rat atria preparations, as well as in conscious and anesthetized rats, anesthetized cats, and conscious and anesthetized dogs. Droxidopa (10, 30, and 90 μg/mL) did not inhibit hERG tail current in HEK293 cells stably transfected with hERG cDNA, whereas the reference compound, E‐4031 (100 nM), decreased hERG tail current by 88% to a steady‐state level.
New drugs introduced into clinical practice have the rare potential for significant QT prolongation that can herald the development of proarrhythmia effects, including torsades de pointes.13, 14, 15, 16 Examples of newly approved drugs that are associated with QT prolongation include ribociclib17 and valbenazine.18
The risks associated with prolongation of the QT interval include presyncope, syncope, and sudden cardiac death. General demographic and clinical characteristics of patients with nOH (eg, advanced age, existence of comorbidities, and use of polypharmacy) indicate that the proarrhythmia effect assessment is of importance; thus, the impact of droxidopa on QT interval was evaluated at both the maximum approved clinical dose and a supratherapeutic dose.19 The effect of droxidopa on cardiac repolarization (ie, QT interval) was studied in healthy volunteers during clinical development, in accordance with International Conference on Harmonisation (ICH) E14 guidance.20 Specifically, the effects of single therapeutic and supratherapeutic doses of droxidopa (600 and 2000 mg, respectively) were compared with moxifloxacin (positive control) and placebo on cardiac conduction parameters in healthy subjects.21 During the study, pharmacokinetic and BP variables were also examined.
Methods
Study Design
The study was approved by the institutional review board of the study center (IntegraReview Ltd, Austin, Texas) and conducted in accordance with the ICH Good Clinical Practice guidelines and all national, state, and local laws and regulations. Subjects provided written informed consent before any study procedure began.
The thorough QT interval study in healthy volunteers was performed at a single site (PPD Phase I Clinic, Austin, Texas) using a double‐blind, 4‐period crossover design. Subjects were recruited by advertisement and mailings to healthy subjects who had previously participated in studies conducted in the region. Eligible enrolled subjects were randomly assigned to a treatment sequence consisting of 4 periods (each separated by at least a 3‐day washout period) in which they received a single dose of each study drug (droxidopa at the maximum therapeutic dose [600 mg] and at a supratherapeutic dose [2000 mg], overencapsulated moxifloxacin [400 mg, a positive control], and matching placebo). For each treatment, electrocardiogram (ECG) parameter changes, pharmacokinetics, and safety parameters were evaluated. The trial is registered at ClinicalTrials.gov (NCT01327066).
Study Participants
Healthy men and women aged 18 to 45 years with a body mass index of 18.0 to 30.0 kg/m2 without any clinically important disorders, including serious medical illness typically excluded for thorough QT studies, such as cardiovascular disorders, cancer, uncontrolled diabetes or hypertension, and liver and kidney diseases (based on medical history, vital signs, physical examination, clinical laboratory tests, or 12‐lead ECG), were eligible to participate. Individuals were excluded if they had risk factors for torsades de pointes (eg, unexplained syncope, known long QT syndrome, heart failure, myocardial infarction, angina, or clinically significant hypokalemia, hypercalcemia, or hypomagnesemia), abnormal ECG at screening (eg, second‐ or third‐degree atrioventricular block, QRS interval > 110 milliseconds, QT interval corrected for heart rate by Fridericia's formula [QTcF] > 450 milliseconds, PR interval > 200 milliseconds, or other clinically significant rhythm abnormality as determined by the investigator), family history of long QT syndrome or Brugada syndrome, or other clinically significant allergic, hematologic, endocrine, pulmonary, gastrointestinal, cardiovascular, hepatic, psychiatric, or neurologic disease. Individuals with sustained (defined as 2 assessments) supine hypertension (systolic BP [SBP] > 140 mm Hg or diastolic BP [DBP] > 95 mm Hg) and individuals with sustained hypotension (systolic BP < 90 mm Hg or diastolic BP < 50 mm Hg) during the screening or check‐in examination were also excluded.
Use of all prescription or over‐the‐counter medication (including herbal remedies and drugs known to prolong the QT/QTc interval) was prohibited 1 week before initial study dosing and throughout the study. Female subjects who were pregnant or lactating were excluded, and those of childbearing potential were required to use appropriate birth control methods (including oral, injectable, or implantable contraceptives) during the study. Subjects using drugs that affect platelet function (eg, aspirin) or anticoagulant agents (current use or within 3 months of check‐in) were excluded because of the need for multiple venipunctures in the pharmacokinetic study. Subjects consuming ≥ 500 mg/day of caffeine (eg, 3 to 5 cups of tea/coffee, ∼100 ounces of cola), nicotine‐containing products (2 weeks before study start), alcohol, or xanthine‐containing products (72 hours before study start for both) were also excluded.
Study Treatments
In this double‐blind, double‐dummy study, subjects received a maximal therapeutic dose of droxidopa (600 mg), a supratherapeutic dose of droxidopa (2000 mg), placebo, and a positive control (moxifloxacin 400 mg) in single oral doses and were randomly assigned to 1 of 8 possible treatment sequences (Figure 1). Each study drug dose was separated by a washout period of at least 3 days. On the morning of each treatment day, subjects received the assigned treatment in the fasted state (at least 8 hours) with water (240 mL). Subjects remained fasting for approximately 4 hours after dosing.
Figure 1.
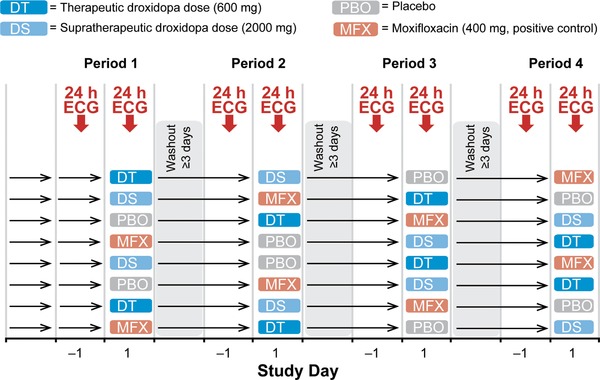
Study design. ECG, electrocardiogram.
Electrocardiogram Assessments and Outcomes
On the day before dosing day and on each dosing day, subjects were monitored for 23 hours using a continuous 12‐lead ECG digital Holter monitor (H12+; Mortara Instrument, Milwaukee, Wisconsin). The ECGs were extracted in quadruplicate 0.5, 1, 2, 3, 4, 5, 6, 8, 10, 12, 18, and 23 hours after dosing by the ECG central laboratory (eResearch Technology, Inc., Philadelphia, Pennsylvania) and analyzed using a high‐resolution manual on‐screen caliper method by a central cardiologist who was blinded to study treatment. Manual on‐screen measurements of the RR, PR, QRS, and QT intervals were performed. Before each ECG extraction time, subjects maintained a 10‐minute supine rest period. The primary study end point was the time‐matched, placebo‐adjusted change from baseline in QT interval using an individual correction method (QTcI) derived from within‐subject data.20, 22, 23 Secondary ECG end points included analyses of changes from baseline in QTcF, QT interval corrected for heart rate by Bazett's formula (QTcB), heart rate, PR and QRS intervals, and ECG changes (second‐ and third‐degree heart block, complete right or left bundle branch block, ST segment elevation or depression, T‐wave abnormalities, abnormal U waves, and myocardial infarction pattern). QTcF and QTcB were calculated using the following formulas: QTcF = QT/(RR)1/3 and QTcB = QT/(RR)1/2.
Pharmacokinetic Assessments
On each dosing day, blood samples for pharmacokinetic analysis were taken before dosing and at the times for the ECG data collections, as described previously. Blood sampling for pharmacokinetic studies was done after the time‐matched ECG extractions to avoid changes in autonomic tone/blood volume associated with blood collection. The concentration of droxidopa was measured from dipotassium ethylenediaminetetraacetic acid human plasma samples treated with ethylene glycol‐bis(2‐aminoethylether)‐N,N,Nʹ,Nʹ‐tetraacetic acid and reduced l‐glutathione. Underivatized samples were prepared using alumina solid‐phase extraction and analyzed by liquid chromatography (Primesep 200 column; water/acetonitrile/formate mobile phase) tandem mass spectrometry (MS/MS) methods with detection by MS/MS monitoring of positive ions produced in the TurboIonspray ion source of the API 4000 (AB Sciex, Ontario, Canada). The precursor ion 214 (Q1 m/z) and the product ion 152.1 (Q3 m/z) were monitored. The internal standard was [13C7]‐3,4‐dihydroxyphenylserine hydrochloride. The lower limit of quantitation was 15.0 ng/mL, the quantification range was 5 to 3000 ng/mL, and the between‐day variation was ≤4%. Data acquisition was performed using Analyst version 1.4.1 (Applied Biosystems, Foster City, California). Quantitation was performed using weighted (1/concentration2) least‐squares linear regression from the plasma calibration curves prepared with each run using Thermo Scientific™ Watson LIMS version 7.3.0.0.1 (Thermo Fisher Scientific, Waltham, Massachusetts).
Area under the plasma concentration–time curve (AUC) from time 0 to 23 hours (AUC0–23 h), AUC extrapolated to infinity (AUC0–inf), maximum observed plasma concentration (Cmax), apparent terminal half‐life (t½), and time to achieve Cmax (tmax) were calculated using noncompartmental analysis with the actual times of blood collection using WinNonlin version 6.0.
Safety
Safety was assessed by reported adverse events (AEs), vital signs, and physical examination findings, results of laboratory tests (hematology, serum chemistry, and urinalysis), and safety 12‐lead ECG results. Vital signs (measured before dosing and 1, 2, 3, 4, 6, 8, 12, and 23 hours after dosing) and safety 12‐lead ECG assessments were done after a 5‐minute supine rest period.
Statistical Analyses
A time‐matched analysis of mean change from the average baseline before each treatment and then QTcI change (placebo‐corrected) for baseline (day ‐1) versus day 1 was performed to determine if the upper 2‐sided 90% confidence interval (90%CI) exceeded 10 milliseconds, per ICH E14 guidance.20 Assay sensitivity would be established if the lower CI of the mean difference of moxifloxacin and placebo was less than 5 milliseconds at 1 or more times. A clinically significant QTcI was defined as > 5 milliseconds based on the threshold of regulatory concern ICH E14 guidance.
Outlier analyses for QTcI, heart rate, PR interval, and QRS interval were based on the most extreme values (minimal and maximal) across all times for the subject. The QTcI outlier values included those that were new (defined as not present at baseline and observed after baseline) measurements > 500 and > 480 milliseconds, and changes from baseline of 31 to 60 and >60 milliseconds.20 A bradycardic outlier was defined as a heart rate < 50 bpm and a ≥25% decrease from baseline mean heart rate. A tachycardic outlier was defined as a heart rate > 100 bpm and a ≥25% increase from baseline mean heart rate. A PR interval outlier was defined as >200 milliseconds and a ≥25% increase from the baseline mean PR interval. A QRS interval outlier was defined as >100 milliseconds and a ≥25% increase from the baseline mean QRS interval.
The pharmacokinetic/pharmacodynamic relationship between the placebo‐adjusted change from baseline in QTcI and droxidopa plasma concentration was examined using linear mixed‐effects modeling.
All analyses were performed using version 9.2 or higher of SAS software (SAS Institute, Inc, Cary, North Carolina).
Sample size
Based on the assumption of a true difference of 3 milliseconds between the time‐matched change from baseline in QTcI for droxidopa versus placebo (a commonly used estimate for drugs that have a negative cardiac risk in preclinical studies) and an SD of 9 milliseconds (a conservative null hypothesis estimate of the upper limit of the 2‐sided 90%CI for the time‐matched placebo‐adjusted QTcI change from baseline), a sample size of 44 evaluable subjects would provide at least 80% power to show that the upper limit of the 2‐sided 90%CI for the comparison of droxidopa with placebo would be below 10 milliseconds. A total of 52 healthy subjects (approximately 26 women and 26 men) were planned for enrollment to allow for the potential that some patients may not complete the study.
Results
Demographic Characteristics
A total of 52 subjects were randomized into the study; all participants completed the study and were included in all analyses. The study population consisted of similar numbers of men (n = 27; 51.9%) and women (n = 25; 48.1%). Subject age ranged from 19 to 45 years (mean ± SD, 28.9 ± 7.3 years). The cohort consisted of 69.2% white and 44.2% Hispanic subjects (Supplemental Table S1).
QTc Interval
The time‐matched placebo‐corrected change in QTcI from baseline for droxidopa (600 and 2000 mg) and moxifloxacin versus time is shown in Figure 2. The upper bound of the 2‐sided 90%CI did not approach 10 milliseconds, indicating no clinically significant effect of droxidopa at therapeutic or supratherapeutic doses on cardiac repolarization. The estimates of the time‐averaged placebo‐corrected change from baseline for droxidopa 600 and 2000 mg were 0.1 milliseconds (90%CI, ‐0.9 to 1.0 milliseconds) and 0.3 milliseconds (90%CI, ‐0.6 to 1.3 milliseconds), respectively. The lower bound of the 2‐sided 90%CI for moxifloxacin was less than 5 milliseconds at multiple points, demonstrating the sensitivity of the assay. The estimate of the time‐averaged placebo‐corrected change from baseline for moxifloxacin was 9.3 milliseconds (90%CI, 8.4–10.3 milliseconds).
Figure 2.
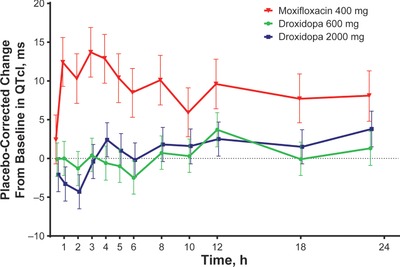
Mean placebo‐corrected change from baseline in QTcI versus time. Error bars represent 90% confidence intervals. QTcI, QT interval using an individual correction method.
No clinical signal for QTc prolongation (eg, presyncope, syncope, seizure, or sudden cardiac death) in either men or women was found in any subgroup analysis.
Categorical Changes in QTcI, Outlier Analyses, and Morphologic Changes
Categorical changes in QTcI, QTcF, and QTcB parameters are shown in Table 1. No patient experienced a new QTcI, QTcF, or QTcB interval > 500 or > 480 milliseconds. Minimal changes from baseline in heart rate were demonstrated at either dose of droxidopa (mean decreases of 1 to 2 bpm). Outlier values for bradycardia were observed for 1 subject at the therapeutic dose and 2 subjects at the supratherapeutic dose of droxidopa. No subjects had outlier values for bradycardia while receiving placebo. No tachycardic outliers were noted at either dose of droxidopa. At either dose of droxidopa, placebo‐corrected changes from baseline for PR or QRS interval were small and not clinically relevant (less than 1 millisecond for all; Table 1). One subject met the PR outlier criteria at both doses, and there were no outliers for QRS values. No clinically relevant signals were identified from analysis of morphologic changes. On the supratherapeutic dose of droxidopa (2000 mg), 1 subject developed a new T‐wave inversion. A single subject developed second‐degree atypical Mobitz I heart block at both droxidopa doses. No subject developed new ST‐segment changes (depression or elevation), left or right bundle branch block, Q waves, or high‐grade heart block.
Table 1.
Electrocardiogram Parameter Analyses
| Parameter | Droxidopa 600 mg (n = 52) | Droxidopa 2000 mg (n = 52) | Moxifloxacin 400 mg (n = 52) | Placebo (n = 52) |
|---|---|---|---|---|
| QTcI | ||||
| Mean change from baseline, ms | −2.9 | −2.7 | 6.3 | −3.0 |
| New > 500 ms, n (%) | 0 | 0 | 0 | 0 |
| New > 480 ms, n (%) | 0 | 0 | 0 | 0 |
| 31–60 ms, n (%) | 0 | 1 (2) | 1 (2) | 0 |
| >60 ms, n (%) | 0 | 1 (2) | 0 | 0 |
| QTcF | ||||
| Mean change from baseline, ms | −2.8 | −2.6 | 6.1 | −3.1 |
| New > 500 ms, n (%) | 0 | 0 | 0 | 0 |
| New > 480 ms, n (%) | 0 | 0 | 0 | 0 |
| 31–60 ms, n (%) | 0 | 1 (2) | 1 (2) | 0 |
| >60 ms, n (%) | 0 | 1 (2) | 0 | 0 |
| QTcB | ||||
| Mean change from baseline, ms | −4.2 | −4.2 | 7.4 | −3.1 |
| New > 500 ms, n (%) | 0 | 0 | 0 | 0 |
| New > 480 ms, n (%) | 0 | 0 | 0 | 0 |
| 31–60 ms, n (%) | 0 | 3 (6) | 5 (10) | 0 |
| >60 ms, n (%) | 0 | 1 (2) | 0 | 0 |
| Heart rate | ||||
| Mean change from baseline, bpm | −1.3 | −1.5 | 1.1 | 0.0 |
| Tachycardic outliers,a n (%) | 0 | 0 | 0 | 0 |
| Bradycardic outliers,b n (%) | 1 (2) | 2 (4) | 0 | 0 |
| PR interval | ||||
| Mean change from baseline, ms | 0.4 | 0.7 | –1.6 | −0.3 |
| Outliers,c n (%) | 1 (2) | 1 (2) | 0 | 0 |
| QRS interval | ||||
| Mean change from baseline, ms | −0.1 | −0.5 | −0.3 | 0.0 |
| Outliers,d n (%) | 0 | 0 | 0 | 0 |
| Morphologic analyses, n (%) | ||||
| New T‐wave inversion | 0 | 1 (2) | 0 | 0 |
| Mobitz I second‐degree heart block | 1 (2) | 1 (2) | 0 | 0 |
QTcB, QT interval corrected for heart rate using Bazett's formula; QTcF, QT interval corrected for heart rate using Fridericia's formula; QTcI, QT interval using an individual correction method.
Defined as heart rate > 100 bpm and ≥25% increase from baseline mean heart rate.
Defined as heart rate < 50 bpm and ≥25% decrease from baseline mean heart rate.
Defined as >200 ms and ≥25% increase from baseline mean PR interval.
Defined as >100 ms and ≥25% increase from baseline mean QRS interval.
Pharmacokinetic/Pharmacodynamic Relationship
The pharmacokinetics of droxidopa are shown in Figure 3 and Table 2. The 3.3‐fold increase for therapeutic versus supratherapeutic droxidopa dose resulted in a 2.3‐fold increase in mean total exposure (AUC0–23 h, 16 589 versus 37 510 ng·h/mL; AUC0–inf, 16 637 versus 37 711 ng·h/mL) and a 2‐fold increase in Cmax (3966 versus 7923 ng/mL). Median tmax and mean t½ were similar for both droxidopa doses (approximately 2 and 3 hours, respectively). The relationship between placebo‐corrected change in QTcI from baseline and plasma concentration is shown in Figure 4. The slope (SE) of the plasma‐concentration effect on placebo‐corrected change in QTcI from baseline for droxidopa was ‐0.0002 (0.0001). For the therapeutic and supratherapeutic doses of droxidopa, the predicted placebo‐ and baseline‐corrected value at the maximum plasma concentration was ‐0.079 milliseconds (upper 95%CI, 0.827 milliseconds) and ‐0.756 milliseconds (upper 95%CI, 0.578 milliseconds), respectively.
Figure 3.
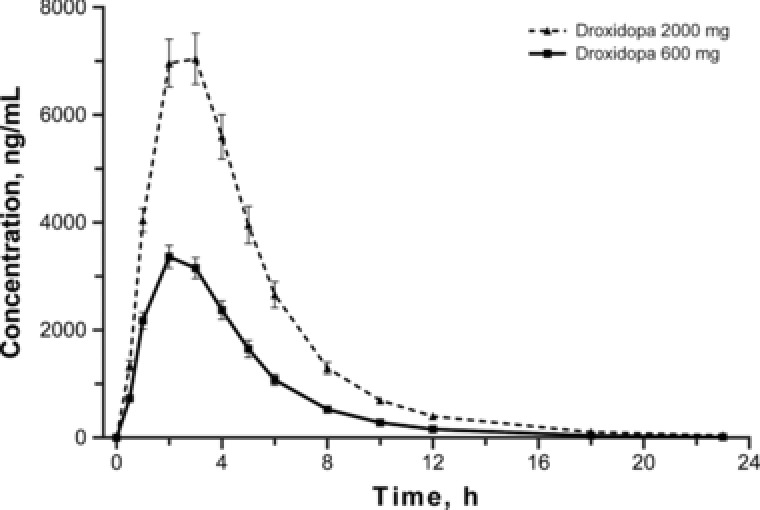
Mean plasma concentrations of droxidopa. Error bars represent standard error of the mean.
Table 2.
Pharmacokinetic Parameters of Droxidopa
| Parameter | Droxidopa 600 mg (n = 52) | Droxidopa 2000 mg (n = 52) |
|---|---|---|
| AUC0–23h, ng·h/mL | ||
| Mean (SD) | 16 589 (6231) | 37 510 (17,500) |
| AUC0–inf, ng·h/mL | ||
| Mean (SD) | 16 637 (6252) | 37 711 (17,534) |
| Cmax, ng/mL | ||
| Mean (SD) | 3966 (1280) | 7923 (3295) |
| tmax, h | ||
| Median | 2.05 | 2.06 |
| t½, h | ||
| Mean (SD) | 2.85 (0.32) | 3.35 (0.53) |
AUC0–23 h, area under the plasma concentration–time curve from time zero to 23 hours; AUC0–inf, area under the plasma concentration–time curve extrapolated to infinity; Cmax, maximum observed plasma concentration; t½, half‐life; tmax, time to Cmax.
Figure 4.
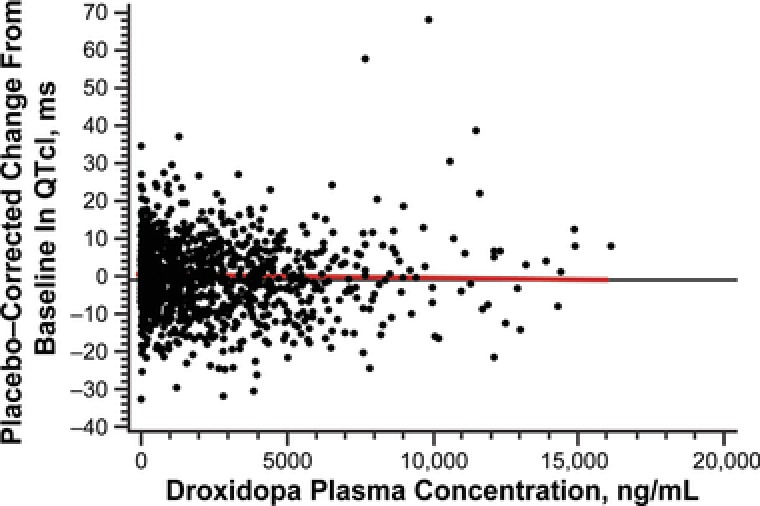
Scatterplot of placebo‐corrected change from baseline in QTcI versus plasma concentration of droxidopa. The red line represents the slope of the plasma concentration effect of droxidopa on the placebo‐corrected change from baseline in QTcI. QTcI, QT interval using an individual correction method.
Blood Pressure Effects
Treatment‐related placebo‐corrected increases in BP were observed in the supine position. At 2, 3, and 4 hours after administration of the therapeutic dose of droxidopa (600 mg), mean increases from baseline in SBP were 8.2, 8.3, and 5.5 mm Hg, respectively, and mean increases from baseline in DBP were 7.9, 7.6, and 5.7 mm Hg, respectively. With the supratherapeutic dose of droxidopa (2000 mg) at the same points, mean increases from baseline in SBP were 19.2, 19.9, and 13.3 mm Hg, respectively, and mean increases from baseline in DBP were 14.1, 14.4, and 10.7 mm Hg, respectively. BP levels returned to baseline by 6 hours after droxidopa dosing with both doses of droxidopa.
Safety
Similar rates of treatment‐emergent AEs (TEAEs) were observed in the therapeutic droxidopa dose, moxifloxacin, and placebo arms (26.9%–30.8%); the TEAE rate at the supratherapeutic dose of droxidopa was 53.8% (Supplemental Table S2). Other than contact dermatitis (all instances of which were judged by the investigator to be related to the ECG electrodes), the most frequent individual TEAEs in either droxidopa arm were abdominal pain (9.6%–30.8%), headache (9.6%–15.4%), nausea (7.7%–21.2%), and vomiting (1.9%–13.5%). Each of these TEAEs was reported more frequently with the supratherapeutic than with the maximum therapeutic dose of droxidopa. At the supratherapeutic dose of droxidopa, 4 subjects (7.7%) reported a sensation of increased heart rate; all were considered by the investigator to be possibly related to study drug. No deaths, serious AEs, or discontinuations due to an AE were reported. No clinically significant concerns or treatment‐related trends were identified in other vital sign measurements (ie, other than BP changes previously described), physical examination, safety 12‐lead ECG results, hematology, serum chemistry, and urinalysis findings.
Discussion
The results of this study showed no signal on cardiac repolarization using the QTcI at either the therapeutic dose (600 mg) or a supratherapeutic dose (2000 mg). In addition, there was no signal for any effect of droxidopa on heart rate, atrioventricular conduction (PR interval), or ventricular depolarization (QRS interval), and no new clinically relevant ECG morphologic changes were observed. Analysis of the pharmacokinetic/pharmacodynamic relationship and cardiac repolarization effects using the QTcI also showed no signal associated with droxidopa exposure. The validity of this study was demonstrated by the expected change from baseline in QTcI for the positive control, moxifloxacin, and lack of QTcI change from baseline for placebo.
Droxidopa at therapeutic doses was generally well tolerated in this study. Reported TEAE rates were similar for the therapeutic dose of droxidopa and placebo, although there was a greater reported incidence of TEAEs with the supratherapeutic dose of droxidopa. Other than a treatment‐related trend for the BP increases, which were consistent with both the mechanism of action of the drug and the pharmacokinetic profile of droxidopa, no safety signals were identified from other evaluations (ie, other vital sign assessments, physical examination, safety 12‐lead ECG results, or laboratory results).24
The findings of this phase 1 study support the cardiac safety profile of droxidopa found during clinical trials in patients with nOH. In 3 short‐term double‐blind clinical trials of droxidopa versus placebo (in which a total of 666 patients with nOH were included), no clinically significant effects were noted in safety ECGs, and cardiac AE rates were low.4, 5, 6, 7 Similarly, low rates of cardiac AEs were found in 2 long‐term open‐label extension studies of droxidopa treatment.25, 26
High rates of concomitant medication use occur in patients with nOH.25, 27 In patients receiving polypharmacy, metabolic effects resulting from drug combinations and the potential for drug–drug interaction effects on QT interval and proarrhythmia risk are a concern.28 The cytochrome P450 system is the predominant route of metabolism for the majority of marketed drugs, and the polypharmacy effects on P450 metabolism and the potential for drug–drug interactions are well documented.13, 29, 30 However, droxidopa is metabolized via the catecholamine pathway, and involvement of cytochrome P450 enzymes has not been identified in its metabolic fate.2, 24 Thus, it is anticipated that there is a low likelihood of negative pharmacokinetic interactions with concomitantly administered medications metabolized by cytochrome P450 enzymes.
Theoretically, concomitant catechol‐O‐methyltransferase inhibitor (COMT) use could affect the metabolism of droxidopa. In a single‐dose coadministration study of droxidopa (400 mg) and the COMT inhibitor entacapone (200 mg), there were no significant differences in peak levels of droxidopa or norepinephrine or in the pharmacodynamic effects (ie, systolic BP response) compared with droxidopa monotherapy.8 However, no dedicated drug–drug interaction studies regarding these effects have been done.
Only 2 drugs have been approved by the FDA for the treatment of nOH (droxidopa) or orthostatic hypotension of any origin (midodrine); to our knowledge, our study is the only report of a thorough QTc study for an approved pharmacologic orthostatic hypotension treatment option. Our study is limited in that it evaluated healthy volunteers and not patients with nOH. Thus, the effects of characteristics associated with a diagnosis of nOH (older age, comorbidities, polypharmacy), which might potentially affect the overall proarrhythmia profile, were not evaluated in this study. In addition, effects of droxidopa in individuals with a predisposition to the development of drug‐induced long QT syndrome because of genetic polymorphisms were not determined.31
The QT effects with repeated dosing of droxidopa were not evaluated. Dosing to steady state of the investigational drug is not a regulatory requirement for QTc studies unless there are concerns about achieving adequate exposure (parent drug or metabolites) with single‐dose administration.32 Based on the known pharmacokinetic profile of droxidopa and other data (preclinical and cardiovascular safety studies, lack of QTc effect with a supratherapeutic dose of droxidopa), adequate exposure was demonstrated using a supratherapeutic dose of 2000 mg of droxidopa. This dose showed no effects on cardiac conduction, but induced larger increases in supine blood pressure than the maximal droxidopa clinical dose of 600 mg.
Conclusion
This study indicated that droxidopa did not have a meaningful impact on cardiac repolarization in healthy volunteers. In addition, no effects on heart rate, atrioventricular conduction, or cardiac depolarization were found, and no new ECG morphologic changes were identified that led to any clinical concern.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Declaration of Conflicting Interests
W.B. White is on the droxidopa data safety committee for Lundbeck and has no other consultant roles with the company. L.A. Hewitt is an employee of Lundbeck. A.A. Mehdirad is on speakers’ bureaus for Bristol‐Myers Squibb, Pfizer, and Lundbeck.
Funding
The data reported were derived from clinical trials funded by Chelsea Therapeutics, now Lundbeck. The authors received editorial assistance from CHC Group (North Wales, Pennsylvania), which was supported by Lundbeck.
Fellows of the American College of Clinical Pharmacology: None
References
- 1. Freeman R, Wieling W, Axelrod FB, et al. Consensus statement on the definition of orthostatic hypotension, neurally mediated syncope and the postural tachycardia syndrome. Clin Auton Res. 2011;21(2):69–72. [DOI] [PubMed] [Google Scholar]
- 2. Kaufmann H, Norcliffe‐Kaufmann L, Palma JA. Droxidopa in neurogenic orthostatic hypotension. Expert Rev Cardiovasc Ther. 2015;13(8):875–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gibbons CH, Schmidt P, Biaggioni I, et al. The recommendations of a consensus panel for the screening, diagnosis, and treatment of neurogenic orthostatic hypotension and associated supine hypertension. J Neurol. 2017;264(8):1567–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Biaggioni I, Freeman R, Mathias CJ, et al. Randomized withdrawal study of patients with symptomatic neurogenic orthostatic hypotension responsive to droxidopa. Hypertension. 2015;65(1):101–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hauser RA, Hewitt LA, Isaacson S. Droxidopa in patients with neurogenic orthostatic hypotension associated with Parkinson's disease (NOH306A). J Parkinsons Dis. 2014;4(1):57–65. [DOI] [PubMed] [Google Scholar]
- 6. Hauser RA, Isaacson S, Lisk JP, Hewitt LA, Rowse G. Droxidopa for the short‐term treatment of symptomatic neurogenic orthostatic hypotension in Parkinson's disease (NOH306B). Mov Disord. 2015;30(5):646–654. [DOI] [PubMed] [Google Scholar]
- 7. Kaufmann H, Freeman R, Biaggioni I, et al. Droxidopa for neurogenic orthostatic hypotension: a randomized, placebo‐controlled, phase 3 trial. Neurology. 2014;83(4):328–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Goldstein DS, Holmes C, Sewell L, Pechnik S, Kopin IJ. Effects of carbidopa and entacapone on the metabolic fate of the norepinephrine prodrug L‐DOPS. J Clin Pharmacol. 2011;51(1):66–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Biaggioni I. New developments in the management of neurogenic orthostatic hypotension. Curr Cardiol Rep. 2014;16(11):542. [DOI] [PubMed] [Google Scholar]
- 10. Goldstein DS, Holmes C, Kaufmann H, Freeman R. Clinical pharmacokinetics of the norepinephrine precursor L‐threo‐DOPS in primary chronic autonomic failure. Clin Auton Res. 2004;14(6):363–368. [DOI] [PubMed] [Google Scholar]
- 11. Kaufmann H, Saadia D, Voustianiouk A, et al. Norepinephrine precursor therapy in neurogenic orthostatic hypotension. Circulation. 2003;108(6):724–728. [DOI] [PubMed] [Google Scholar]
- 12. Suzuki T, Higa S, Sakoda S, et al. Pharmacokinetic studies of oral L‐threo‐3,4‐dihydroxyphenylserine in normal subjects and patients with familial amyloid polyneuropathy. Eur J Clin Pharmacol. 1982;23(5):463–468. [DOI] [PubMed] [Google Scholar]
- 13. van Noord C, Eijgelsheim M, Stricker BH. Drug‐ and non‐drug‐associated QT interval prolongation. Br J Clin Pharmacol. 2010;70(1):16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yap YG, Camm AJ. Drug induced QT prolongation and torsades de pointes. Heart. 2003;89(11):1363–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Roden DM. Drug‐induced prolongation of the QT interval. N Engl J Med. 2004;350(10):1013–1022. [DOI] [PubMed] [Google Scholar]
- 16. Kannankeril P, Roden DM, Darbar D. Drug‐induced long QT syndrome. Pharmacol Rev. 2010;62(4):760–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Geoerger B, Bourdeaut F, DuBois SG, et al. A phase I study of the CDK4/6 inhibitor ribociclib (LEE011) in pediatric patients with malignant rhabdoid tumors, neuroblastoma, and other solid tumors. Clin Cancer Res. 2017;23(10):2433–2441. [DOI] [PubMed] [Google Scholar]
- 18. Davis MC, Miller BJ, Kalsi JK, Birkner T, Mathis MV. Efficient trial design — FDA approval of valbenazine for tardive dyskinesia. N Engl J Med. 2017;376(26):2503–2506. [DOI] [PubMed] [Google Scholar]
- 19. Maule S, Milazzo V, Maule MM, et al. Mortality and prognosis in patients with neurogenic orthostatic hypotension. Funct Neurol. 2012;27(2):101–106. [PMC free article] [PubMed] [Google Scholar]
- 20. US Department of Health and Human Services . Guidance for Industry: E14 clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non‐antiarrhythmic drugs, October 2005. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm073153.pdf. Accessed August11, 2017.
- 21. Demolis JL, Kubitza D, Tenneze L, Funck‐Brentano C. Effect of a single oral dose of moxifloxacin (400 mg and 800 mg) on ventricular repolarization in healthy subjects. Clin Pharmacol Ther. 2000;68(6):658–666. [DOI] [PubMed] [Google Scholar]
- 22. Malik M, Farbom P, Batchvarov V, Hnatkova K, Camm AJ. Relation between QT and RR intervals is highly individual among healthy subjects: implications for heart rate correction of the QT interval. Heart. 2002;87(3):220–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Desai M, Li L, Desta Z, Malik M, Flockhart D. Variability of heart rate correction methods for the QT interval. Br J Clin Pharmacol. 2003;55(6):511–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Goldstein DS. L‐dihydroxyphenylserine (L‐DOPS): a norepinephrine prodrug. Cardiovasc Drug Rev. 2006;24(3–4):189–203. [DOI] [PubMed] [Google Scholar]
- 25. Isaacson S, Shill HA, Vernino S, Ziemann A, Rowse GJ. Safety and durability of effect with long‐term, open‐label droxidopa treatment in patients with symptomatic neurogenic orthostatic hypotension (NOH303). J Parkinsons Dis. 2016;6(4):751–759. [DOI] [PubMed] [Google Scholar]
- 26. Isaacson S, Vernino S, Ziemann A, et al. Long‐term safety of droxidopa in patients with symptomatic neurogenic orthostatic hypotension. J Am Soc Hypertens. 2016;10(10):755–762. [DOI] [PubMed] [Google Scholar]
- 27. LeWitt P, Gorny S. Analysis of efficacy and safety outcomes in patients treated with droxidopa in combination with other drug classes. Mov Disord. 2012;27:S425–S426. [Google Scholar]
- 28. Wisniowska B, Tylutki Z, Wyszogrodzka G, Polak S. Drug‐drug interactions and QT prolongation as a commonly assessed cardiac effect ‐ comprehensive overview of clinical trials. BMC Pharmacol Toxicol. 2016;17:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lynch T, Price A. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am Fam Physician. 2007;76(3):391–396. [PubMed] [Google Scholar]
- 30. Guengerich FP. Cytochrome p450 and chemical toxicology. Chem Res Toxicol. 2008;21(1):70–83. [DOI] [PubMed] [Google Scholar]
- 31. Behr ER, Roden D. Drug‐induced arrhythmia: pharmacogenomic prescribing? Eur Heart J. 2013;34(2):89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Darpo B. The thorough QT/QTc study 4 years after the implementation of the ICH E14 guidance. Br J Pharmacol. 2010;159(1):49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information