

Abstract
Objective
Focal cortical dysplasias (FCDs) are an important cause of drug‐resistant epilepsy. In this work, we aimed to investigate whether abnormal gene regulation, mediated by microRNA, could be involved in FCD type II.
Methods
We used total RNA from the brain tissue of 16 patients with FCD type II and 28 controls. MicroRNA expression was initially assessed by microarray. Quantitative polymerase chain reaction, in situ hybridization, luciferase reporter assays, and deep sequencing for genes in the mTOR pathway were performed to validate and further explore our initial study.
Results
hsa‐let‐7f (p = 0.039), hsa‐miR‐31 (p = 0.0078), and hsa‐miR34a (p = 0.021) were downregulated in FCD type II, whereas a transcription factor involved in neuronal and glial fate specification, NEUROG2 (p < 0.05), was upregulated. We also found that the RND2 gene, a NEUROG2‐target, is upregulated (p < 0.001). In vitro experiments showed that hsa‐miR‐34a downregulates NEUROG2 by binding to its 5′‐untranslated region. Moreover, we observed strong nuclear expression of NEUROG2 in balloon cells and dysmorphic neurons and found that 28.5% of our patients presented brain somatic mutations in genes of the mTOR pathway.
Interpretation
Our findings suggest a new molecular mechanism, in which NEUROG2 has a pivotal and central role in the pathogenesis of FCD type II. In this way, we found that the downregulation of hsa‐miR‐34a leads to upregulation of NEUROG2, and consequently to overexpression of the RND2 gene. These findings indicate that a faulty coupling in neuronal differentiation and migration mechanisms may explain the presence of aberrant cells and complete dyslamination in FCD type II. Ann Neurol 2018;83:623–635
The development of the cerebral cortex involves complex steps, requiring tightly regulated molecular mechanisms for the efficient and precise control of gene expression.1 Some of the molecular pathways that control gene expression are mediated by microRNAs, a class of noncoding RNAs that regulate gene expression at the posttranscriptional level.2 Gene regulation mediated by microRNAs is involved in a large number of key processes in the nervous system,3, 4 especially in neurodevelopment.
Malformations of cortical development (MCDs) are an important cause of disorders in the central nervous system. MCDs are usually highly epileptogenic, which may result from major changes in the pattern of gene expression involved in neuronal excitability.5 Focal cortical dysplasias (FCDs) are a subtype of MCD affecting >25% of all patients undergoing surgery for the treatment of refractory epilepsy.6 FCDs are associated with dysmorphic neurons and occasionally with balloon cells,7 being an important cause of severe drug‐resistant epilepsy. The current classification of FCDs is based upon neuropathological examination of surgical specimens.8 In the present study, we examined FCD type II, which presents as an isolated lesion characterized by cortical dyslamination and dysmorphic neurons without (type IIa) or with balloon cells (type IIb).
Although recent literature points to the involvement of the mTOR pathway in FCD type II,9, 10, 11, 12 the exact molecular mechanism leading to this type of cortical malformation, especially to the presence of aberrant cells, remains undetermined.13, 14 As the development of the cerebral cortex is a sophisticated process, requiring the fine‐tuning of gene expression, we decided to analyze the expression of microRNAs as a shortcut to gain a better understanding of the mechanisms involved in the pathogenesis of FCD type II. Thus, we aimed to investigate whether abnormal gene regulation, mediated by microRNA, could be involved in FCD type II. In addition, we explored the role of putative candidate genes regulated by the abnormally expressed microRNAs in the mechanisms underlying FCD type II.
Subjects and Methods
Patients
We analyzed the brain tissue obtained from 16 patients with FCD type II (6 patients with FCD type IIa and 10 with FCD type IIb) who had undergone selective resection of cortical structures for the treatment of clinically refractory seizures. Adult patients signed a written informed consent approved by the Research Ethics Committee of University of Campinas (UNICAMP), Campinas, Brazil; patients younger than age 18 years gave their assent, and parents signed the consent on their behalf. All patients were recruited at the epilepsy service of UNICAMP. All patients had been diagnosed with drug‐resistant epilepsy and presented magnetic resonance imaging (MRI) abnormalities suggestive of FCD type II (Fig 1A, B). Patients underwent epilepsy surgery with the aim of removal of the epileptogenic zone after extensive clinical assessment, routine electroencephalograms (EEGs), ictal recordings under long‐term video‐EEG monitoring, and structural and functional neuroimaging investigation. In all patients, the epileptogenic zone included the area suggestive of FCD, as observed in the MRI study and intraoperative EEG. We also used tissue from patients with tuberous sclerosis complex (TSC; n = 3). After surgery, FCD type II and TSC were confirmed by 2 neuropathologists according to the International League against Epilepsy classification (see Fig 1C–E).7, 8 Clinical and pathological characteristics are summarized in Table 1. According to the Engel classification, 73.7% (n = 14/19) of patients presented a good postoperative seizure outcome, Engel class I and II. Control samples (n = 28) were obtained from autopsies of individuals whose cause of death was nonneurological, as well as from the normal temporal cortex obtained during anterior temporal lobectomy performed in patients with mesial temporal sclerosis. All control samples were evaluated by histopathological examination, and normal cytoarchitecture of the cerebral cortex was confirmed. Clinical characteristics of control individuals are summarized in Table 2.
Figure 1.
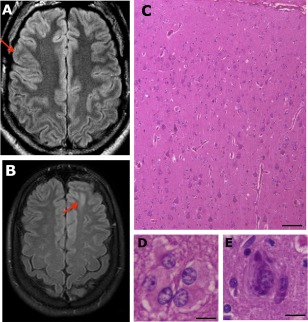
Fluid‐attenuated inversion recovery magnetic resonance imaging and histopathological findings in focal cortical dysplasia (FCD) type II. (A) A 29‐year‐old man with FCD type IIa and drug‐resistant seizures from 4 years of age; the arrow indicates the region of cortex thickening and abnormal sulcation. (B) A 21‐year‐old woman with type FCD IIb and drug‐resistant seizures from 11 years of age; the arrow indicates loss of definition of the limits between cortical and subcortical areas and hyperintense signal at the bottom of the sulcus in the left frontal region. (C–E) Hematoxylin and eosin stain of a specimen of the patient shown in B, exhibiting cortical dyslamination throughout (C), and presence of balloon cells (D) and dysmorphic neuron (E). Scale bar = 100 µm (C), 25 µm (D, E).
Table 1.
Clinical and Neuropathological Findings in 16 Patients with Focal Cortical Dysplasia Type II and 3 Patients with TSC Included in the Present Study
| ID | Sex | Age at Surgery, yr | Site of Surgery | ILAE | Age at Seizure Onset, yr | Seizure Frequency per Daya | Type of Seizure | Engelb | mTOR Mutations, Gene; Position; Allelic Frequency in Brain Tissue |
|---|---|---|---|---|---|---|---|---|---|
| P1 | M | 40 | T P O/R | IIb | 6 | 4–5 | CFS/SFS/GTC | I | Nothing found |
| P2 | M | 26 | T/L | IIa | 7 | 1 | SFS/CFS | I | Nothing found |
| P3 | M | 6 | T O/R | IIb | 0 | 6 | CFS/GTC | IV | NP |
| P4 | F | 33 | O/R | IIb | 8 | 4–5 | CFS/GTC | I | Nothing found |
| P5 | M | 52 | Fr/R | IIb | 18 | 4–5 | SFS/CFS/GTC | I | NP |
| P6 | M | 3 | Fr P/L | IIb | 0 | 7–8 | CFS | I | NP |
| P7 | M | 15 | Fr/L | IIb | 4 | 30 | SFS/GTC | II | NP |
| P8 | M | 10 | Fr P/R | IIa | 6 | 20 | CFS/SFS/GTC | IV | NP |
| P9 | M | 9 | Fr/R | IIb | 0 | 15–20 | CFS | I | NP |
| P10 | F | 15 | Fr/L | IIa | 4 | 10–15 | CFS/SFS/GTC | II | NP |
| P11 | M | 12 | Fr/L | IIa | 2 | 6 | SFS/CFS/GTC | IV | NP |
| P13 | M | 29 | Fr/R | IIa | 4 | 3 | GTC | IV | Nothing found |
| P15 | F | 23 | Fr/R | IIa | 9 | 1 | CFS/GTC | IV | NP |
| P16 | M | 18 | Fr/L | IIb | 0 | 3 | CFS/GTC | I | Nothing found |
| P20 | M | 32 | Fr/L | IIb | 3 | 1 | CFS | I | MTOR; c.4379T>C; 1.8% |
| P21 | F | 21 | Fr/L | IIb | 11 | 2–3 | CFS | II | TSC2; c.3781G>A; 1.7% and AKT1; c.349_351del; 2.2% |
| P14 | F | 29 | Fr/R | TSC | 7 | 1 | SFS/CFS/GTC | II | Nothing found |
| P22 | M | 1 | Fr/R | TSC | 0 | 20‐30 | CFS/GTC | II | NP |
| P23 | F | 4 | Fr/R | TSC | 4 | 10 | CFS/GTC | I | NP |
We used fresh‐frozen tissue specimens in P1–P4, P13–P16, and P20–P23 and formalin‐fixed paraffin‐embedded tissue specimens in patients P5–P11.
Seizure frequency before surgery.
Engel classification scale for surgical outcome.
CFS = complex focal seizures; F = female; Fr = frontal; GTC = generalized tonic–clonic seizures; ILAE = International League against Epilepsy; L = left; M = male; NP = deep sequencing was not performed; O = occipital; P = parietal; R = right; SFS = simple focal seizures; T = temporal; TSC = tuberous sclerosis complex.
Table 2.
Clinical Characteristics of Individuals from Whom Control Samples Were Obtained
| ID | Age at Death/Surgery, yr | Sex | Cause of Death/Surgery | Tissue Type | Site |
|---|---|---|---|---|---|
| C1 | 58 | M | Cardiogenic shock | FF | Fr |
| C2 | 33 | F | Respiratory failure, edema, pulmonary congestion, and diffuse lymphoma | FFPE/FF | Fr |
| C3 | 22 | M | Respiratory failure | FFPE | Fr |
| C4 | 30 | M | Shock lung | FFPE | Fr |
| C5 | 50 | M | Hypovolemic shock and gastrointestinal hemorrhage | FFPE/FF | Fr |
| C7 | 29 | M | Infective endocarditis | FFPE | Fr |
| C9 | 29 | M | Bilateral bronchopneumonia | FFPE | Fr |
| C12 | 29 | F | Pulmonary hypoxia | FFPE | Fr |
| C14 | 42 | M | Septic shock | FFPE | Fr |
| C15 | 44 | F | Acute fulminant hepatitis | FFPE | Fr |
| C16 | 20 | F | Septic shock | FFPE | Fr |
| C18 | 45 | M | Acute hepatitis | FFPE | Fr |
| C19 | 47 | M | Respiratory failure/bilateral pneumonia and tuberculosis | FFPE | Fr |
| C20 | 55 | M | Septic shock | FF | Fr |
| C21 | 37 | F | Septic shock/bronchopneumonia | FF | Fr |
| C22 | 42 | F | Septic shock | FF | Fr |
| C23 | 48 | M | Pulmonary embolism | FF | Fr |
| C24 | 62 | M | Hepatic impairment | FF | Fr |
| C25 | 47 | M | Septic shock | FF | Fr |
| C26 | 50 | M | Anterior temporal lobectomy | FF | Fr |
| C27 | 38 | M | Anterior temporal lobectomy | FF | T |
| C28 | 44 | F | Anterior temporal lobectomy | FF | T |
| C29 | 40 | M | Anterior temporal lobectomy | FF | T |
| C30 | 49 | F | Anterior temporal lobectomy | FF | T |
| C31 | 45 | F | Anterior temporal lobectomy | FF | T |
| C32 | 51 | M | Anterior temporal lobectomy | FF | T |
| C33 | 28 | F | Anterior temporal lobectomy | FF | T |
| C34 | 54 | F | Anterior temporal lobectomy | FF | T |
There were 19 samples from autopsy and 9 samples of temporal neocortex resected as part of selective anterior temporal lobectomy performed in patients with mesial temporal sclerosis.
F = female; FF = fresh‐frozen; FFPE = formalin‐fixed paraffin‐embedded; Fr = frontal lobe; M = male; T = temporal lobe.
Tissue Collection and RNA Extraction
Tissue specimens were divided into 2 groups: fresh‐frozen (FF) and formalin‐fixed paraffin‐embedded (FFPE) tissue that had been preserved between 2003 and 2010. For both FF and FFPE tissues, we selected samples containing representative neuropathological findings of FCD type II that were evaluated on hematoxylin and eosin, anti‐NeuN (MAB377, 1:1,000; Millipore, Darmstadt, Germany), anti‐MAP‐2 (M13, 1:500; Invitrogen, Waltham, MA), and antinestin (AB5922, 1:500, Millipore) stained sections. For FPPE tissue, extraction of total RNA was performed using the Recover All kit (Ambion, Austin, TX), following the manufacturer's recommendations with amendments proposed by Goswani et al.15 For FF tissue, total RNA was extracted using Trizol (Invitrogen) following the manufacturer's recommended protocol. The total RNA yield was determined using an Epoch spectrophotometer (BioTek Instruments, Winooski, VT). RNA integrity was assessed using on‐chip capillary electrophoresis (RNA 6000 Pico and RNA 6000 Nano Chip Kit; Agilent Technologies, Santa Clara, CA) and a 2100 Bio‐Analyzer (Agilent Technologies).
MicroRNA Microarray Analysis
Microarray profiling of microRNA was performed on FFPE tissue using 300ng of total RNA and the GeneChip 1.0 miRNA array (Affymetrix, Santa Clara, CA), which interrogates a total of 847 human microRNAs. In the microarray experiments, we included 9 samples from patients with FCD type II (type IIa, n = 4; type IIb, n = 5) and 5 controls. All experiments were performed according to the manufacturer's recommended protocol. In addition, we performed background correction, summarization, and normalization using the RMA function in the Bioconductor package.16 We compared microRNA expression in tissues with FCD type II and controls using RankProd (Bioconductor package) software and false discovery rate correction for multiple comparisons. MicroRNAs were considered differentially expressed when the fold change (FC) was greater than the absolute value of 1.5 with an adjusted p value of < 0.05. Candidate microRNAs for subsequent validation were prioritized based on the highest absolute values of FC, smallest q values, and a putative biological role that could demonstrate any association with candidate mechanisms underlying FCD.
Bioinformatics Prediction of Human mRNA Targets
We identified candidate mRNAs whose expression was likely to be modulated by the differentially expressed microRNAs found in the microarray experiments using the miRGen algorithm (version 2.0).17 We selected candidate mRNAs by searching for the keywords neurogenesis, cell differentiation, cell migration, and cell proliferation. We included mRNAs previously reported to be potentially associated with FCD. Finally, to refine the list of potential mRNA targets, we searched for favorable binding interactions between microRNAs and mRNA transcripts using RNAhybrid.18 We prioritized lower values of minimal free energy (MFE) to select the strongest microRNA–mRNA interactions.
Quantitative Real‐Time Polymerase Chain Reaction Experiments
We obtained mature microRNAs using 10ng of total RNA from FFPE tissue as the substrate in a reverse transcription reaction. For the synthesis of each microRNA‐specific cDNA, small RNA enrichment was reverse transcribed using the TaqMan microRNA reverse transcription kit (Life Technologies, Foster City, CA) with microRNA‐specific stem‐loop primers, following the manufacturer's instructions. The primers used were: hsa‐miR‐486‐5p (MIMAT0002177, ID1278), hsa‐miR‐106a (MIMAT0000103, ID2169), hsa‐miR‐886‐5p (ID2193), hsa‐miR‐31 (MIMAT0000089, ID2279), hsa‐let‐7f (MIMAT0000067, ID382), hsa‐miR‐1274a (ID2883), hsa‐miR‐34a (MIMAT0000255, ID426), hsa‐miR‐23a (MIMAT0000078, ID399), hsa‐miR‐379 (ID568), and hsa‐miR‐1182 (MIMAT0005827, ID2830) as candidate microRNAs in FCD type II and RNU24 (ID 1001) and RNU48 (ID1006) as endogenous controls (Life Technologies). We used the TaqMan (Life Technologies) assay for the quantification of mature microRNA transcripts. These experiments were performed in triplicate with a final volume of 12 μl, containing 6.25 μl of Master Mix, 0.625 μl of probe (×20), 1.125 μl of RNase‐free water, and 4 μl of cDNA. Reaction conditions were 95 °C for 10 minutes, and 40 cycles at 95 °C for 15 seconds and 60 °C for 1 minute, carried out in a 7500 thermocycler (Life Technologies). Data were analyzed using the 7500 software (Life Technologies, version 2.05) by determining the threshold cycle (Ct). Relative quantities of microRNAs were calculated using the 2−ΔΔCt method19 after normalization to RNU24 and RNU48.
We obtained human mRNA from 1200ng of total RNA, extracted from FF tissue, using the SuperScript III reverse transcriptase kit and random hexamer primers (Life Technologies). To quantify mRNA expression, we used TaqMan polymerase chain reaction (PCR) probes: MAP4K3 (Hs00269284_m1), SEMA4C (Hs00215035_m1), GAS7 (Hs00932959_m1), JAG1 (Hs01070032_m1), PAFAH1B1 (Hs00181182_m1), SYT1 (Hs00194572_m1), NOTCH1 (Hs01062014_m1), SATB2 (Hs00392652_m1), SLC1A2 (Hs01102423_m1), NEUROG2 (Hs00702774_s1), NEUROD1 (Hs01922995_s1), and RND2 (Hs00183269_m1). As endogenous controls, we used glyceraldehyde‐3‐phosphate dehydrogenase (Hs02758991_g1) and β‐actin (Hs01060665_g1). PCR reactions were performed in triplicate, and reaction conditions were the same as those used to quantify mature microRNAs, as described above. We also used SYBR Green–based detection, with primer pairs for the genes NEUROG2 and RND2 that were designed using the National Center for Biotechnology Information primer design tool (http://www.ncbi.nlm.nih.gov/tools/primer-blast/). PCR reactions were also performed in triplicate and Sybr Power Plus Master Mix kit (Life Technologies) was used following the manufacturer's instructions. Following PCR amplifications, a melting curve was analyzed for each gene, and a single melting temperature peak was observed.
Cotransfection Experiments and microRNA Dual Luciferase Assays
Luciferase assays were used to verify whether the 5′‐untranslated region (UTR) and/or 3′‐UTR regions of NEUROG2 bind in vitro with hsa‐miR‐31 and/or hsa‐miR‐34a. Fragments of NEUROG2 5′‐UTR (around 350bp in length, with 2 putative binding sites for hsa‐miR‐34a) and NEUROG2 3′‐UTR (around 400bp in length, with 3 putative binding sites for hsa‐miR‐31) were used. Oligonucleotide sequences were obtained from Integrated DNA Technologies (Coralville, IA; gBlock Gene Fragments) and are described in Supplementary Table 1. In addition, we used a seed sequence‐mutant of both fragments. We first cloned NEUROG2 3′‐UTR and NEUROG2 5′‐UTR downstream of the Renilla luciferase reporter gene in the psiCHECK2 vector (Promega, Madison, WI), using the XhoI/NotI sites of the vector. U87 cells (1 × 105 cells/ml) were grown in 24‐well plates in Dulbecco modified Eagle medium (Sigma Aldrich, Saint Louis, MO) supplemented with 10% fetal bovine serum (Invitrogen), at 37 °C in a 5% CO2 humidified chamber. Cotransfection was carried out using 50nM of chemically synthesized RNA (Life Technologies): hsa‐miR‐34a mimic (Cat.# MC11030)/ hsa‐miR‐31‐5p mimic (Cat.# MC11465), negative mimic (Cat.# 4464058), and positive control (Cat.# 4464080), as well as with 3,900ng of psiCheck2‐5′‐UTR or psiCheck2‐3′‐UTR plasmids. A FuGENE transfection reaction (Promega) was performed according to the manufacturer's instructions. After 48 hours of transfection, luciferase activity was assessed according to the Dual‐Luciferase Reporter Assay protocol (Promega). Relative luciferase levels were expressed as Renilla luciferase (r‐luc) normalized against firefly luciferase signals. Each experiment was performed in hexaplicate and repeated for 3 separate transfection assays carried out on different cell culture passages.
In Situ Hybridization
We performed in situ hybridization using locked nucleic acid (LNA)‐modified DNA, which was 5′‐ and 3′‐digoxigenin labeled. The probe 5′ATCATAAGAGACAGATGGCA3′ was used for the detection of NEUROG2 mRNA (Cat. # 266172‐1), U6 snRNA (Cat. # 99002‐01) as the positive control, and a scramble LNA probe (Cat. # 99004‐15) as the negative control. All probes were obtained from Exiqon (Vedbaek, Denmark). Hybridizations were performed on 4 µm FFPE sections of tissue with FCD type II and control tissue, following the manufacturer's protocol, with amendments as follows: proteinase k digestion for 7 minutes, 40nM of hybridization probe, a hybridization temperature of 56 °C, and an overnight incubation with alkaline phosphatase substrate. Cells were counterstained with nuclear fast red for 1 minute. Qualitative analysis was performed using digitalized images obtained using the PALM MicroBeam System (Zeiss, Jena, Germany).
Sequencing Analysis of mTOR Pathway Genes
To evaluate whether brain somatic mutations in genes belonging to the mTOR pathway could be associated with FCD type II, we performed deep next generation sequencing using a customized panel containing 60 genes of the mTOR pathway. The genes for the panel were selected based on literature information as well as whole exome sequencing performed previously in the same samples. For the sequencing experiments, we used genomic DNA extracted from brain tissue resected by surgery (BTRS) and compared to these sequences obtained from blood samples from 7 patients. We performed capturing and enrichment with SeqCap EZ Choice Library (NimbelGen; Roche Molecular Systems, Pleasanton, CA). Samples were sequenced following a 150bp paired‐end protocol in a Miseq (Illumina, San Diego, CA). Sequences were aligned using BWA‐MEM (0.7.5a‐r405), and variant calling was performed using the Genome Analysis Toolkit (3.6‐0). We evaluated mosaicism using Mutect2.20 Variants were classified as mosaic mutations when <10% of reads were not aligned to the human genome reference (GRCh38/hg38) and were present only in BTRS. Deleterious effect of mutations was evaluated using SIFT, Polyphen2, and SNPs&GO.
Statistical Analysis
R environment (https://www.r-project.org/) was used for statistical analyses. The nonparametric Wilcoxon–Mann–Whitney test with Bonferroni correction was used to compare the log‐transformed relative expression (RQ) of microRNAs among FCD type IIa, FCD type IIb, and controls. We also used the log‐transformed RQ of mRNAs on a coefficient test for the adjusted model via a generalized estimating equation (Geepack, version 1.2.0, Wald test) between FCD type IIa, FCD type IIb, and controls, corrected for multiple comparisons. We performed Student t test to compare relative luciferase activity. The level of significance, alpha, was set to ≤0.05.
Results
Microarray Analysis
Selected samples of FFPE tissue were submitted to microRNA microarray analysis to compare microRNA expression in tissues with FCD type II (type IIa, n = 4; type IIb, n = 5) with that of normal cortical tissue (n = 5). A total of 23 microRNAs were found to be differentially expressed when comparing patients and controls. Twenty‐two of these were significantly downregulated, and only 1 was upregulated in patients (data not shown).
Validation of microRNA Microarray Data by Quantitative PCR
Of the 23 microRNAs identified to be abnormally expressed in FCD type II, we selected 10 (Supplementary Table 2) for validation by quantitative PCR (qPCR) based on the highest absolute values of FC and adjusted p value. Moreover, we used data available in the literature that could indicate a potential functional relationship with FCD type II. The qPCR results confirmed the reduced expression of 3 microRNAs—hsa‐let‐7f (p = 0.039), hsa‐miR‐31 (p = 0.0078), and hsa‐miR34a (p = 0.021)—in patients (type IIa, n = 4; type IIb, n = 7) in comparison with controls (n = 12; Fig 2). We also found that hsa‐miR‐31 was downregulated when comparing FCD type IIb and controls (p = 0.018). No differences were observed in the expression values of other selected microRNAs (data not shown).
Figure 2.
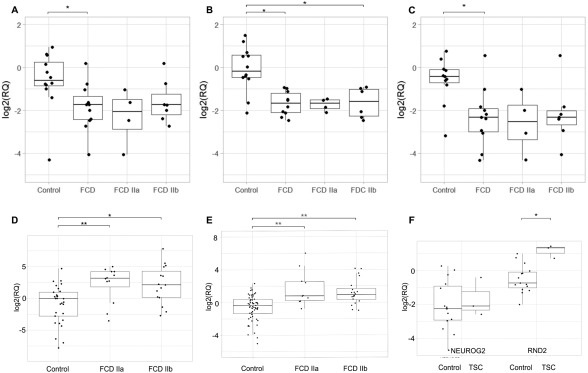
Expression levels of microRNAs, and NEUROG2 and RND2 genes in human brain tissue with focal cortical dysplasia (FCD) type II and tuberous sclerosis complex (TSC). (A–C) Boxplots depicting log‐transformed relative expression values (RQ) of hsa‐let‐7f (A), hsa‐miR‐31 (B), and hsa‐miR‐34a (C), in formalin‐fixed paraffin‐embedded brain tissue from controls (n = 12) and patients with FCD type II (type IIa, n = 4; type IIb, n = 7), as well as only type IIa and only type IIb. Expression levels were normalized to RNU24 and RNU48. Comparisons with statistically significant differences, determined by the Wilcoxon test corrected by Bonferroni, are marked with asterisks. (D) Boxplot depicting log‐transformed RQ of NEUROG2 gene in fresh‐frozen brain tissue from controls (n = 18), and patients with FCD type IIa (n = 3) and FCD type IIb (n = 4). Expression levels were normalized to glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH). (E) Boxplot depicting log‐transformed RQ of RND2 gene in fresh‐frozen brain tissue from controls (n = 15), and patients with FCD type IIa (n = 3) and FCD type IIb (n = 5). Expression levels were normalized to B‐actin. (F) Boxplot depicting log‐transformed RQ of NEUROG2 and RND2 genes in fresh‐frozen brain tissue from controls (n = 15) and patients with TSC (n = 3). Expression levels were quantified by SYBR Green–based detection and normalized to GAPDH. Comparisons with statistically significant differences, obtained by generalized estimating equation comparison corrected for multiple comparisons, are marked with asterisks. *p < 0.05, **p < 0.001.
Selection and Validation of mRNA Targets
To identify putative candidate genes regulated by these abnormally expressed microRNAs, we used the miRGen17 algorithm for in silico prediction of human mRNA targets. We identified 10 potential mRNA targets that could be negatively regulated by the 3 microRNAs (see Supplementary Table 2), which were then evaluated by qPCR. Comparison of tissue from patients with FCD type II (type IIa, n = 4; type IIb, n = 4) with normal cortical tissue (n = 18) revealed the upregulation of a transcription factor involved in mammalian neurogenesis, NEUROG2 (see Fig 2D). We noticed an increase in expression of NEUROG2 in FCD type IIa (p = 0.0002) and in FCD type IIb (p = 0.0075) when compared to control tissue. No significant difference in NEUROG2 expression was observed when comparing FCD type IIa and FCD type IIb (p = 1). Based on these findings, we considered whether NEUROG2 dysregulation could have a direct effect in downstream genes. To test this, we performed qPCR in 2 NEUROG2 targets: NEUROD1 and RND2. We found an increased expression of RND2 in FCD type IIa (p = 0.00013) and in FCD type IIb (p = 2.16e‐07) when compared to control tissues (see Fig 2E). No significant difference in RND2 expression was observed when comparing FCD type IIa and FCD type IIb (p = 0.89), as well as in NEUROD1 expression when the same comparisons listed above were performed.
The elevated levels of expression of NEUROG2 and RND2 led us to investigate whether this dysregulation could also be present in other similar lesions. We then analyzed these transcripts in 3 patients with TSC and noticed overexpression of the RND2 gene (p = 0.002; see Fig 2F) when TSC tissue was compared to the control group. However, no significant difference in NEUROG2 expression was observed when comparing TSC and the control group.
In Silico Binding Prediction and Luciferase Binding Assays
Initially, we found a single putative binding site for hsa‐miR‐31 on the 3′‐UTR of NEUROG2 based on the lowest MFE algorithm (MFE = −26.1kcal/mol; Fig 3). Because a single mRNA can be regulated by several microRNAs, we broadened our initial search for other binding sites to include hsa‐let‐7f and hsa‐miR‐34a in the 3′‐UTR and 5′‐UTR of NEUROG2, leading to the identification of an additional predicted hsa‐miR‐34a binding site in the 5′‐UTR region of NEUROG2 with MFE = −30.4kcal/mol. This last identified interaction is predicted to be more stable than that of hsa‐miR‐31:3′‐UTR NEUROG2. A schematic drawing of NEUROG2 mRNA and the 2 predicted binding sites is presented in Figure 3A.
Figure 3.
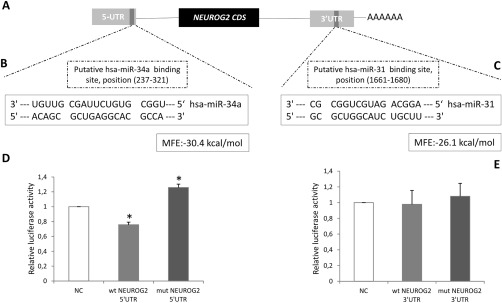
(A) Schematic representation of human NEUROG2 mRNA depicting the putative hsa‐miR‐34a binding site in the 5′‐untranslated region (UTR) and the putative hsa‐miR‐31 binding site in the 3′UTR. We searched for favorable binding interactions between microRNAs and mRNA transcripts, based on lower values of minimal free energy (MFE). (B) Sequence complementarity between hsa‐miR‐34a and the NEUROG2 5′UTR target site. (C) Sequence complementarity between hsa‐miR‐31 and the NEUROG2 3′UTR target site. (D, E) Relative luciferase activity in U87‐MG cells when cotransfected with 50nM of hsa‐miR‐34a mimic (D) and 50nM of hsa‐miR‐31 mimic (E). Forty‐eight hours after transfection, luciferase activity was measured. Firefly luciferase activity was normalized to Renilla luciferase expression from 3 independent experiments performed in hexaplicate, and was repeated in 3 separate transfections carried out on different cell passages. Error bars represent standard deviation; statistical significance, using Student t test: *p < 0.05 compared to controls. mut = mutant sequence; NC = normal control; wt = wild type.
Subsequently, to confirm the interaction of NEUROG2 with hsa‐miR‐31 and/or hsa‐miR‐34a, we performed a luciferase reporter assay. We used glioma U87‐MG cells to assess transcriptional regulation of NEUROG2 through its 3′‐UTR by hsa‐miR‐31 mimic and the 5′‐UTR by hsa‐miR‐34a mimic. Luciferase activity was reduced when cells were cotransfected with hsa‐miR‐34a mimic and NEUROG2 5′‐UTR wild type (see Fig 3D), showing a reduction of 32% relative to control levels (p = 0.037). We also observed that NEUROG2 5′‐UTR mutant significantly reversed the reduction induced by mimic‐34a (p = 0.031), which corroborates the initial finding using the wild‐type sequence. In contrast, there was no significant difference in luciferase activity when hsa‐miR‐31 mimic and NEUROG2 3′‐UTR were cotransfected, or when the mutant sequence was evaluated in the same experimental conditions (see Fig 3E). Taken together, these results indicate that NEUROG2 is directly regulated by hsa‐miR‐34a interaction with its 5′‐UTR region.
In Situ Hybridization Assay
Results of in situ hybridization of NEUROG2 mRNA in selected FFPE samples of patients with FCD type II (type IIa, n = 3; type IIb, n = 3) and controls (n = 3) showed strong NEUROG2 expression in balloon cells with significant nuclear staining as well as nuclear expression in dysmorphic neurons (Fig 4). The expression of NEUROG2 was also observed in neurons from normal brain tissue as well as in glial cells, both with less intense staining. No positive staining was observed using the scrambled probe in normal brain sections.
Figure 4.
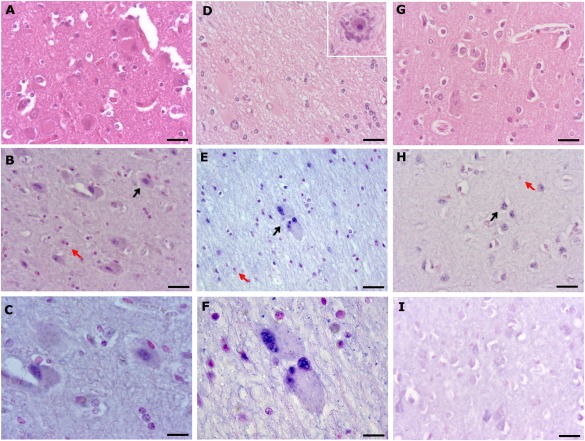
Hematoxylin and eosin (H&E) stain and representative distribution of NEUROG2 determined by in situ hybridization analysis with specific antisense probe in slides of selected formalin‐fixed paraffin‐embedded human brain tissue with focal cortical dysplasia (FCD) type II (type IIa, n = 3; type IIb, n = 3) and controls (n = 3). (A–H) Histopathological findings in patients with FCD type IIa (A–C), patients with FCD type IIb (D–F), and a control individual (G, H). (A, D, G) H&E staining of a frontal lobe section with FCD type IIa of a 15‐year‐old female with drug‐resistant seizures from 4 years age (A), a temporal‐parieto‐occipital section of FCD type IIb of a 40‐year‐old man with refractory seizures from 6 years of age (D; insert: dysmorphic neuron), and a frontal lobe section of a 55‐year‐old male control (G). (B) Representative example of the distribution of NEUROG2 mRNA (5′‐ and 3′‐digoxigenin labeled) after in situ hybridization analysis with an locked nucleic acid–specific antisense probe of brain section with FCD type IIa. Note the nuclear staining in dysmorphic neurons. (C) Higher magnification of B. (E) Brain section with FCD type IIb. Note the strong nuclear staining in balloon cells. (F) Higher magnification of E. (H) Normal brain section. Note staining in neurons. (I) Scrambled probe in normal brain tissue. Black arrows indicate cells with detectable NEUROG2 staining, and red arrows indicate cells without. Scale bar = 10 µm (C), 25 µm (F), 20 µm (others).
Sequencing Analysis of mTOR Pathway Genes
Analysis using the Mutect2 algorithm identified brain somatic variants in genes belonging to the mTOR pathway in 28.5% (n = 2/7) of patients with FCD type II studied by DNA sequencing (see Table 1). Mutations were identified in the following genes: MTOR, c.4379T>C/p.Leu1460Pro (rs1057519779; NM_004958.3) with allele frequency of 9/503 reads (1.8 %) in BTRS and 0/438 (0%) in blood; TSC2, c.3781G>A/p.Ala1261Thr (not reported previously; NM_000548.3) with allele frequency of 7/409 reads (1.7%) in BTRS and 0/373 (0%) reads in blood; and AKT1, c.349_351del/p.Glu117del (rs768025881; NM_005163.2) with allele frequency of 10/454 (2.2%) in BTRS and 1/350 reads (0.29%) in blood. The somatic missense mutations identified in MTOR and TSC2 are probably deleterious, because they are classified as damaging, by 2 (Polyphen2 and SNPs&GO) of the 3 mutation prediction programs used. In addition, rs1057519779 in MTOR has been previously reported in patients with FCD type II.21 Variation identified in AKT1 is a glutamic acid in‐frame deletion. Neither mutation was found in the Exome Aggregation Consortium database or in a publicly available genomic database of Brazilian samples (http://www.BIPMed.org).
Discussion
Our results revealed that 3 microRNAs (hsa‐let‐7f, hsa‐miR‐31, and hsa‐miR‐34a) are downregulated in the brain tissue of patients with FCD type II. In addition, we showed an upregulation of NEUROG2, a gene involved in mammalian neurogenesis,22 suggesting that NEUROG2 plays a central role in the pathogenesis of FCD type II. In addition, we localized NEUROG2 expression in balloon cells and dysmorphic neurons. Furthermore, we also identified the dysregulation of RND2, a NEUROG2 target,23 suggesting an involvement of the radial migration process in the pathogenesis of FCD type II as well. Finally, we demonstrated that overexpression of NEUROG2 is likely to be due to a posttranscriptional regulation mechanism mediated by the previous dysregulation of hsa‐miR‐34a binding to the 5′‐UTR of NEUROG2.
NEUROG2 is a member of the family of transcription factors bHLH (basic helix loop‐helix), which plays a central role in cell fate specification and neuronal differentiation in many regions of the central nervous system.22, 24 During brain development, NEUROG2 has a specific spatiotemporal expression pattern, because it is exclusively detected in the ventricular zone and only during neurogenesis (Fig 5A).25 It has been demonstrated that a negative feedback mechanism is essential for the correct transition between the inhibition of neurogenesis and the induction of early gliogenesis.26 Furthermore, the disruption of proneuronal gene activity, such as that of NEUROG2, may result in loss of commitment in specific neural progenitor cells27 and the generation of ectopic neurons.28 Thus, one may suggest that the transition from neurogenesis to gliogenesis could be hampered due to abnormally high expression of NEUROG2, as identified here in brain tissue of patients with FCD type II, leading to the formation of aberrant cells, such as dysmorphic neurons and others that display characteristics of transition cells, between neurons and glial cells, such as balloon cells (see Fig 5B). The latter cells are known to express markers of neuronal or glial lineage or both, indicating a mixed lineage.29 They also express phospho‐vimentin, Pax6, and BLBP, proteins that are normally expressed by cells within the embryonic ventricular zone,30 suggesting that balloon cells retain a stem cell phenotype.29 Therefore, taken together the results of the expression and in situ hybridization performed in the present study clearly show an overexpression of NEUROG2, indicating that this neurogenic transcription factor plays a role in the mechanisms leading to FCD type II, as well as in the origin of balloon cells.
Figure 5.
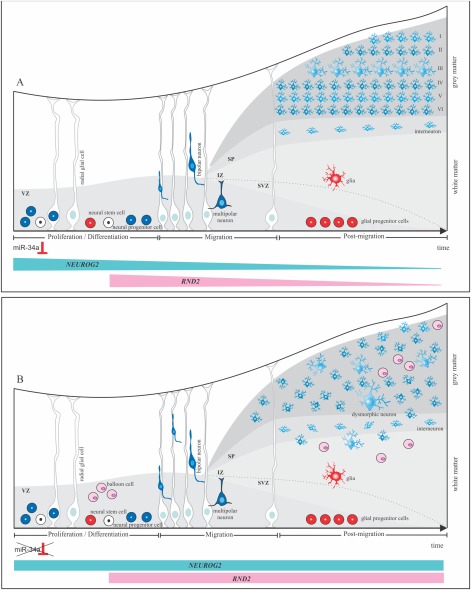
Schematic representation of hypothetical molecular mechanisms involved in failure of cell fate specification and disruption of neuronal migration in focal cortical dysplasia (FCD) type II. (A) Stages of normal cortical development: proliferation of neuroglial precursors, neuron migration, and postmigration. Differentiation of neuroglial precursors in the proliferation stage is potentially regulated by miR‐34a, which inhibits expression of NEUROG2. Thereafter, RND2, whose expression is controlled by NEUROG2, is downregulated and successive waves of newborn neurons undergo sequential steps of radial migration, along radial glial cells, leading to the organization of cortical layers. (B) Model proposed with formation of aberrant cells, such as dysmorphic neurons and balloon cells in FCD type II. Disruption in the interaction between miR‐34a and NEUROG2 leads to an upregulation in the expression of NEUROG2 and RND2. Overexpression of NEUROG2 affects the inhibition of neurogenesis, leading to the formation of undifferentiated cells, such as balloon cells. In addition, dysregulation of RND2 expression could impair the cytoskeleton modulation, resulting in heterotopic and/or dysmorphic neurons and cortical dyslamination due to failure in the organization and orientation of radially migrating neurons. IZ: intermediate zone; SP: subplate; SVZ: subventricular zone and VZ: ventricular zone.
NEUROG2 has also been associated with other important functions modulated by interactions with transcriptional regulators.23, 30, 31, 32 Based on this, we speculated that NEUROG2 dysregulation could affect downstream genes; this hypothesis was confirmed by identification of an overexpression of RND2 in FCD type II. RND2 is also transiently expressed in the ventricular zone; during embryonic cerebral cortex development,23 it has a role in the regulation of actin cytoskeleton organization33 and in radial migration of newborn excitatory neurons.31 Rnd2‐deficient neurons in mouse fail to transit from the multipolar to the bipolar stage,32 suggesting that the excessive levels of RND2 in FCD type II are detrimental to the organization and orientation of radially migrating neurons, which could ultimately be implicated in the complete dyslamination observed in the dysplastic tissue. Furthermore, abnormal cell migration speed could be consistent with the presence of heterotopic clusters of neurons located within the white matter (see Fig 5B),8 as well as an increase in the number of neurons found in the upper cortical layers, mainly in the molecular layer, as noticed in pediatric patients with FCD.34
Our results also provide some clues to explain the intrinsic epileptogenicity observed in FCD type II tissue, as a failure in the proper positioning of migrating cortical neurons during brain development, due to abnormal expression of RND2, can result in the formation of abnormal neural circuits,35 owing to abnormal cortical synaptic connectivity. Furthermore, because NEUROG2 is required for the specification of glutamatergic neurotransmitter identity,36 an increased synaptic excitatory glutamatergic neurotransmission, due to an excessive expression of NEUROG2, may alter the excitation–inhibition balance.37 Therefore, these findings taken together indicate that epileptogenesis in FCD type II may result from a faulty coupling in the neuronal differentiation and migration process, leading to what is called the “dysmaturity hypothesis” of cortical development in FCD.37
Recently, somatic mutations have been associated with hyperactivation of the mTOR pathway in FCD type II.9, 10, 11, 12 In our cohort, 28.5% of the tested patients harbored brain somatic mutations in genes belonging to the mTOR pathway. Few reports have investigated the relationship between the mTOR pathway and NEUROG2 expression.38, 39 One of these studies demonstrated that GSK3, a component of the Wnt signaling pathways, negatively regulates activity of NEUROG2 (an increase in GSK3 activity results in a decline in NEUROG2 expression) in a transient manner during neurogenesis.38 In addition, inhibition of the mTOR signaling by rapamycin leads to the reduced expression of Neurog2 and fewer neurons in the chick prenatal tube.39 However, the exact relationship between somatic mutations in mTOR genes and the proposed mechanism mediated by NEUROG2 remains to be better elucidated.
It is known that microRNAs are fine‐tuning regulators in embryonic brain development and abnormal changes in their expression could disrupt their mechanism of action.40 We found downregulation of hsa‐miR‐34a in FCD type II and hypothesized that the downregulation of hsa‐miR‐34a could lead to the less efficient repression of NEUROG2, resulting in the high expression of this gene in FCD type II (see Fig 5B). In addition, our results show the first evidence that NEUROG2 expression is regulated by hsa‐miR‐34a in the 5′‐UTR region. This type of microRNA interaction is not common, but has been reported previously.41, 42
Furthermore, hsa‐miR‐34a has been implicated directly in embryonic brain development. Chang and coworkers43 reported that the decreased expression of miR‐34a is crucial for the precise deployment of neural precursors to their correct destination. Similarly, Mollinari and coauthors44 identified DCX, a gene involved in neuronal migration disorders, as a new target of miR‐34a; they also observed stage‐specific effects of the overexpression of miR‐34a, detecting an increase in cell proliferation, and a delay in neuroblast migration. Thus, it could be hypothesized that the laminar disorganization and loss of radial orientation45 typically found in FCD type II may also be related to the dysregulation in the expression of hsa‐miR‐34a.
Our results also revealed the downregulation of 2 other microRNAs in FCD type II. First, hsa‐let‐7f has been involved in the mechanisms leading to the transition from neurogenesis to gliogenesis.46, 47 Several studies have provided evidence that the hsa‐let‐7 family is highly expressed in neural stem cells and neural progenitor cells,46 as well as in cell fate determination during human embryonic development. In this way, Patterson and coworkers47 demonstrated that the let‐7 family, including let‐7f, plays a role in the commitment of human neuronal progenitor cells, by repressing HMGA2 and LIN28 expression. Furthermore, we have also shown that hsa‐miR‐31 is significantly downregulated in FCD type II. The decreased expression of miR‐31 was detected in glioblastoma,48 and it is believed that miR‐31 may be an oncogene, because it represses tumor suppressor genes, and acts in cell migration and motility by regulating the gene radixin.49
Nonetheless, there are some caveats to the interpretation of our results. It is noteworthy that we did not perform our experiments using resected tissue from human fetal brain with FCD type II but rather the final product of brain development, adult tissue, to create a hypothesis about the actual pathogenic mechanisms occurring during development in this cortical malformation. Moreover, the dysregulation of NEUROG2 and RND2 may not be specific for FCD type II. Because the cortical lesions of FCD type II share similar histopathological features with those of TSC,10 we performed an expression study in TSC tissue, and our initial results suggest the involvement of the RND2 gene in TSC (see Fig 2F); however, additional experiments in TSC as well as in hemimegaloencephaly and other types of FCD should be performed to further investigate the issue.
In conclusion, our data strongly support the hypothesis that an earlier failure in neuronal–glial differentiation and migration may have taken place during neocortical development, causing the abnormalities seen in FCD type II. This hypothesis was first proposed by Englund et al,50 but has not been proven to date. Furthermore, our data revealed that hsa‐miR‐34a interacts with the 5′‐UTR of NEUROG2, indicating that the observed reduction in hsa‐miR‐34a expression may lead to the less efficient repression of NEUROG2. In turn, the high expression of NEUROG2 and one of its targets, RND2, at a crucial moment in cortical development may result in the presence of aberrant cells and complete dyslamination in FCD type II.
Author Contributions
S.H.A., F.R.T., A.S.V., D.B.D., R.S., and I.L.‐C. participated in the study concept and design. All authors performed data acquisition and analysis. S.H.A., F.R.T., A.S.V., R.S., F.R., F.C., and I.L.‐C. contributed to drafting the manuscript and the figures.
Potential Conflicts of Interest
Nothing to report.
Supporting information
Additional supporting information can be found in the online version of this article.
Supporting Information
Acknowledgment
This study was sponsored by grants from Fundação de Amparo a Pesquisa do Estado de São Paulo, Brazil (2013/07559‐3, 2013/00099‐7); Conselho Nacional de Pesquisa, Brazil; and Coordenação de Pessoal de Ensino Superior, Brazil.
We thank all patients and their families for their helpful cooperation; Drs H. Tedeschi and E. Ghizoni for performing FCD surgeries; L. Reis, A. C. Piaza, M. R. Gomes da Cruz, Dr M. A. Montenegro, Dr Ana Costa, Dr André Costa, Dr A. Riccardi, and Dr V. Paschoal for their technical assistance and helpful discussions; and M. de Fátima Santos for technical assistance with the artwork.
References
- 1. Martynoga B, Drechsel D, Guillemot F. Molecular control of neurogenesis: a view from the mammalian cerebral cortex. Cold Spring Harb Perspect Biol 2012;4:a008359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shen Q, Temple S. Fine control: microRNA regulation of adult neurogenesis. Nat Neurosci 2009;12:369–370. [DOI] [PubMed] [Google Scholar]
- 3. Dogini DB, Ribeiro PA, Rocha C, et al. MicroRNA expression profile in murine central nervous system development. J Mol Neurosci 2008;35:331–337. [DOI] [PubMed] [Google Scholar]
- 4. Olsen L, Klausen M, Helboe L, et al. MicroRNAs show mutually exclusive expression patterns in the brain of adult male rats. PLoS One 2009;4:e7225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Barkovich AJ, Guerrini R, Kuzniecky RI, et al. A developmental and genetic classification for malformations of cortical development: update 2012. Brain 2012;135:1348–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bast T, Ramantani G, Seitz A, Rating D. Focal cortical dysplasia: prevalence, clinical presentation and epilepsy in children and adults. Acta Neurol Scand 2006;113:72–81. [DOI] [PubMed] [Google Scholar]
- 7. Palmini A, Najm I, Avanzini G, et al. Terminology and classification of the cortical dysplasias. Neurology 2004;62:S2–S8. [DOI] [PubMed] [Google Scholar]
- 8. Blümcke I, Thom M, Aronica E, et al. The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc Task Force of the ILAE Diagnostic Methods Commission. Epilepsia 2011;52:158–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Baybis M, Yu J, Lee A, et al. mTOR cascade activation distinguishes tubers from focal cortical dysplasia. Ann Neurol 2004;56:478–487. [DOI] [PubMed] [Google Scholar]
- 10. Crino PB. mTOR: a pathogenic signaling pathway in developmental brain malformations. Trends Mol Med 2011;17:734–742. [DOI] [PubMed] [Google Scholar]
- 11. Marin‐Valencia I, Guerrini R, Gleeson JG. Pathogenetic mechanisms of focal cortical dysplasia. Epilepsia 2014;55:970–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nakashima M, Saitsu H, Takei N, et al. Somatic mutations in the MTOR gene cause focal cortical dysplasia type IIb. Ann Neurol 2015;78:375–386. [DOI] [PubMed] [Google Scholar]
- 13. Lim JS, Kim WI, Kang HC, et al. Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat Med 2015;21:395–400. [DOI] [PubMed] [Google Scholar]
- 14. Schwartzkroin PA, Wenzel HJ. Are developmental dysplastic lesions epileptogenic? Epilepsia 2012;53:35–44. [DOI] [PubMed] [Google Scholar]
- 15. Goswami RS, Waldron L, Machado J, et al. Optimization and analysis of a quantitative real‐time PCR‐based technique to determine microRNA expression in formalin‐fixed paraffin‐embedded samples. BMC Biotechnol 2010;10:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gautier L, Cope L, Bolstad BM, Irizarry RA. affy—analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2004;20:307–315. [DOI] [PubMed] [Google Scholar]
- 17. Alexiou P, Vergoulis T, Gleditzsch M, et al. miRGen 2.0: a database of microRNA genomic information and regulation. Nucleic Acids Res 2010;38:D137–D141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Krüger J, Rehmsmeier M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res 2006;34:W451–W454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods 2001;25:402–408. [DOI] [PubMed] [Google Scholar]
- 20. Cibulskis K, Lawrence MS, Carter SL, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013;31:213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nakashima M, Saitsu H, Takei N, et al. Somatic mutations in the MTOR gene cause focal cortical dysplasia type IIb. Ann Neurol 2015;78:375–386. [DOI] [PubMed] [Google Scholar]
- 22. Guillemot F, Molnár Z, Tarabykin V, Stoykova A. Molecular mechanisms of cortical differentiation. Eur J Neurosci 2006;23:857–868. [DOI] [PubMed] [Google Scholar]
- 23. Heng JI, Nguyen L, Castro DS, et al. Neurogenin 2 controls cortical neuron migration through regulation of Rnd2. Nature 2008;455:114–118. [DOI] [PubMed] [Google Scholar]
- 24. Hirabayashi Y, Gotoh Y. Epigenetic control of neural precursor cell fate during development. Nat Rev Neurosci 2010;11:377–388. [DOI] [PubMed] [Google Scholar]
- 25. Sun Y, Nadal‐Vicens M, Misono S, et al. Neurogenin promotes neurogenesis and inhibits glial differentiation by independent mechanisms. Cell 2001;104:365–376. [DOI] [PubMed] [Google Scholar]
- 26. Bertrand N, Castro DS, Guillemot F. Proneural genes and the specification of neural cell types. Nat Rev Neurosci 2002;3:517–530. [DOI] [PubMed] [Google Scholar]
- 27. Kessaris N, Pringle N, Richardson WD. Ventral neurogenesis and the neuron‐glial switch. Neuron 2001;31:677–680. [DOI] [PubMed] [Google Scholar]
- 28. Mizuguchi R, Sugimori M, Takebayashi H, et al. Combinatorial roles of olig2 and neurogenin2 in the coordinated induction of pan‐neuronal and subtype‐specific properties of motoneurons. Neuron 2001;31:757–771. [DOI] [PubMed] [Google Scholar]
- 29. Yasin SA, Latak K, Becherini F, et al. Balloon cells in human cortical dysplasia and tuberous sclerosis: isolation of a pathological progenitor‐like cell. Acta Neuropathol 2010;120:85–96. [DOI] [PubMed] [Google Scholar]
- 30. Lamparello P, Baybis M, Pollard J, et al. Developmental lineage of cell types in cortical dysplasia with balloon cells. Brain 2007;130:2267–2276. [DOI] [PubMed] [Google Scholar]
- 31. Ge W, He F, Kim KJ, et al. Coupling of cell migration with neurogenesis by proneural bHLH factors. Proc Natl Acad Sci U S A 2006;103:1319–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hand R, Bortone D, Mattar P, et al. Phosphorylation of Neurogenin2 specifies the migration properties and the dendritic morphology of pyramidal neurons in the neocortex. Neuron 2005;48:45–62. [DOI] [PubMed] [Google Scholar]
- 33. Heasman SJ, Ridley AJ. Mammalian rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol 2008;9:690–701. [DOI] [PubMed] [Google Scholar]
- 34. Andres M, Andre VM, Nguyen S, et al. Human cortical dysplasia and epilepsy: an ontogenetic hypothesis based on volumetric MRI and NeuN neuronal density and size measurements. Cereb Cortex 2005;15:194–210. [DOI] [PubMed] [Google Scholar]
- 35. Leventer RJ, Guerrini R, Dobyns WB. Malformations of cortical development and epilepsy. Dialogues Clin Neurosci 2008;10:47–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schuurmans C, Armant O, Nieto M, et al. Sequential phases of cortical specification involve Neurogenin‐dependent and ‐independent pathways. EMBO J 2004;23:2892–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cepeda C, Andre VM, Levine MS, et al. Epileptogenesis in pediatric cortical dysplasia: the dysmature cerebral developmental hypothesis. Epilepsy Behav 2006;9:219–235. [DOI] [PubMed] [Google Scholar]
- 38. Li S, Mattar P, Zinyk D, et al. GSK3 temporally regulates neurogenin 2 proneural activity in the neocortex. J Neurosci 2012;32:7791–7805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fishwick KJ, Li RA, Halley P, et al. Dev Biol 2010;338:215–225. [DOI] [PubMed] [Google Scholar]
- 40. Schratt G. Fine‐tuning neural gene expression with microRNAs. Curr Opin Neurobiol 2009;19:213–219. [DOI] [PubMed] [Google Scholar]
- 41. Lytle JR, Yario TA, Steitz JA. Target mRNAs are repressed as efficiently by microRNA‐binding sites in the 5′ UTR as in the 3′ UTR. Proc Natl Acad Sci U S A 2007;104:9667–9972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhou H, Rigoutsos I. MiR‐103a‐3p targets the 5′ UTR of GPRC5A in pancreatic cells. RNA 2014;20:1431–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chang SJ, Weng SL, Hsieh JY, et al. MicroRNA‐34a modulates genes involved in cellular motility and oxidative phosphorylation in neural precursors derived from human umbilical cord mesenchymal stem cells. BMC Med Genomics 2011;4:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mollinari C, Racaniello M, Berry A, et al. miR‐34a regulates cell proliferation, morphology and function of newborn neurons resulting in improved behavioural outcomes. Cell Death Dis 2015;6:e1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sisodiya SM, Fauser S, Cross JH, Thom M. Focal cortical dysplasia type II: biological features and clinical perspectives. Lancet Neurol 2009;8:830–843. [DOI] [PubMed] [Google Scholar]
- 46. Zhao C, Sun G, Li S, et al. MicroRNA let‐7b regulates neural stem cell proliferation and differentiation by targeting nuclear receptor TLX signaling. Proc Natl Acad Sci U S A 2010;107:1876–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Patterson M, Gaeta X, Loo K, et al. let‐7 miRNAs can act through notch to regulate human gliogenesis. Stem Cell Reports 2014;3:758–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Visani M, de Biase D, Marucci G, et al. Expression of 19 microRNAs in glioblastoma and comparison with other brain neoplasia of grades I‐III. Mol Oncol 2014;8:417–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hua D, Ding D, Han X, et al. Human miR‐31 targets radixin and inhibits migration and invasion of glioma cells. Oncol Rep 2012;27:700–706. [DOI] [PubMed] [Google Scholar]
- 50. Englund C, Folkerth RD, Born D, et al. Aberrant neuronal‐glial differentiation in Taylor‐type focal cortical dysplasia (type IIA/B). Acta Neuropathol 2005;109:519–533. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information can be found in the online version of this article.
Supporting Information