

Abstract
Tailor‐made: Discussed herein is the ability to adapt biology's mechanisms for innovation and optimization to solving problems in chemistry and engineering. The evolution of nature's enzymes can lead to the discovery of new reactivity, transformations not known in biology, and reactivity inaccessible by small‐molecule catalysts.
Keywords: biocatalysis, enzymes, heme proteins, protein engineering, synthetic methods
Survival of the Fittest
In this competitive age, when new industries sprout and decay in the span of a decade, we should reflect on how a company survives to celebrate its 350th anniversary. A prerequisite for survival in business is the ability to adapt to changing environments and tastes, and to sense, anticipate, and meet needs faster and better than the competition. This requires constant innovation as well as focused attention to execution. A company that continues to provide meaningful and profitable solutions to human problems has a chance to survive, even thrive, in a rapidly changing and highly competitive world.
Biology has a brilliant algorithm for solving the problem of survival over time: evolution. Those who adapt and (re)produce outcompete the less agile and less fertile. Over the last 30 years—which seems a long time but is less than one‐tenth the time Merck KGaA, Darmstadt, Germany has been in business—I have tried to adapt biology's mechanisms for innovation and optimization to solving problems in chemistry and engineering. It turns out that evolution is a powerful forward‐engineering process, whose widespread adoption in enzyme engineering and synthetic biology has been made possible through advances in molecular biology and high‐throughput screening.
Expanding Nature's Catalytic Repertoire for a Sustainable Chemical Industry
Nature, the best chemist of all time, solves the difficult problem of being alive and enduring for billions of years, under an astonishing range of conditions. Most of the marvelous chemistry that makes life possible is the work of nature's macromolecular protein catalysts, the enzymes. By using enzymes, nature can extract materials and energy from the environment and convert them into self‐replicating, self‐repairing, mobile, adaptable, and sometimes even thinking biochemical systems. These systems are good models for a sustainable chemical industry that uses renewable resources and recycles a good fraction of its products. And biology is not just a model from which to draw inspiration: living organisms or their components can be efficient production platforms. In fact, I predict that DNA‐programmable microorganisms will be producing many of our chemicals in the not‐so‐distant future.
That most chemicals are made using synthetic processes starting from petroleum‐based feedstocks reflects the remarkable creativity of synthetic chemists in developing reaction schemes and catalysts that nature never discovered. Synthetic chemistry has given us an explosion of products, which feed, clothe, house, entertain, and cure us. Synthetic chemistry, however, struggles to match the efficiency and selectivity that biology achieves with enzymes. In many cases, synthetic processes rely on precious metals, toxic reagents and solvents, and extreme conditions, and they generate substantial amounts of unwanted byproducts. DNA‐programmable chemical synthesis using enzymes promises to improve on synthetic chemistry, particularly if we are able to expand biology's catalytic repertoire to include some of the most synthetically useful reactions, under physiological conditions and with earth‐abundant resources. Such clean, green chemistry might sound like pie in the sky, but enzymes already show how a protein can orient substrates for reaction, exclude water from an active site, activate a metal or simple organic cofactor, or suppress competing reactions to draw out new and admirable synthetic capabilities. Synthetic chemists have been drawing inspiration from biology for decades, and now is the time for protein engineers to use inspiration from synthetic chemistry to generate new enzymes that will improve on and replace synthetic catalysts and reaction pathways.1
Unfortunately, our understanding of the link between sequence and function lags well behind our desire for new enzymes. Given that our ability to predict protein sequences, or even just changes to a sequence, which reliably give rise to whole new, finely tuned catalytic activities is rudimentary at best, creating new enzymes capable of improving on current synthetic processes is a pretty tall order. We also dream of going beyond known chemistry to create enzymes that catalyze reactions or make products that are simply not possible with any known method, synthetic or otherwise. Requiring that these new enzymes assemble and function in cells, where they can be made at low cost and incorporated into synthetic metabolic pathways to generate a broader array of products, represents an even greater set of engineering constraints and challenges.
Nature's enzymes are the products of evolution, not design. By using generations of mutation and selection for fitness advantages, evolution allows organisms to continuously update and optimize their enzyme repertoires. New enzymes even appear in real time in response to challenges (e.g. the need to resist antibiotics or pesticides) or opportunities (e.g. the chance to occupy a new food niche by degrading recently introduced, manmade substances). I argue that the process that gave rise to all the remarkable biological catalysts in nature should be able to produce yet more. In the laboratory. Quickly. Advances in molecular biology over the past few decades—the ability to write, cut, and paste DNA and to have that DNA read and translated into proteins in recombinant organisms—have given us the ability to breed enzymes much like we breed sheep or sake yeast. We can direct the evolution of enzymes in the laboratory by requiring them to perform in ways that may not be useful to a bacterium but are useful to us. Directed evolution achieves these desirable functional outcomes while circumventing our deep ignorance of how sequence encodes them.
Directed evolution mimics evolution by artificial selection, and is accelerated in the laboratory setting by focusing on individual genes expressed in fast‐growing microorganisms. We start with existing proteins (sourced from nature or engineered), introduce mutations, and then screen for the progeny proteins with enhanced activity (or another desirable trait). We use the improved enzymes as parents for the next round of mutation and screening, recombining beneficial mutations as needed, and continuing until we reach the target level of performance.
Engineering enzymes in the 1980s and 1990s, I learned the hard way that there was no reliable method to predict performance‐enhancing mutations. Turning instead to random mutagenesis and screening, I quickly realized that such mutations were easy to find and accumulate with the right evolutionary optimization strategy. My students and I observed that proteins, the products of evolution, are themselves readily evolvable. Properties we and others targeted in the early days of directed evolution (the mid‐1990s) included recovering activity in unusual environments (e.g. organic solvents), improving activity on non‐native substrates, enhancing thermostability, and changing enantioselectivity. We learned the then‐surprising fact that beneficial mutations could be far from an active site, and often appeared on the protein surface (which in those days was generally deemed insensitive to mutation and functionally neutral). To this day, no one can explain satisfactorily how such mutations exert their effects, much less predict them.
Evolution of Novelty: Enzymes that Catalyze Reactions Invented by Synthetic Chemists
Although we could enhance activity (and many other properties) by accumulating beneficial mutations over generations of random mutagenesis and screening, evolving a whole new catalytic activity seemed a much more difficult problem. After all, evolution is not good for problems that require multiple, simultaneous, low‐probability events,2 and the active sites of enzymes are so beautifully and precisely configured that it was hard to imagine how the stepwise accumulation of beneficial mutations could create a new one.
Evolution's innovation mechanisms, however, are more simple than they might appear: evolution works best when it does not need to generate a whole new active site from scratch. Instead, evolution can generate a new enzyme from one that is “close”, that is, shares elements of mechanism or machinery from which the new activity can be built. Nature co‐opts old machinery to do new jobs. And sometimes the ability to do the new job is already there, at least at a low level. The biological world is replete with proteins whose capabilities extend well beyond what may be used at any given time. Thus new enzymes are built from promiscuous or side activities that become advantageous in a new biological context, such as when a new food source becomes available.3 Thus a conservative process of accumulating beneficial mutations can innovate because the innovation is already there! The magnificent diversity of the biological world provides the fuel for further innovations.
For directed evolution to be a reliable approach to creating new enzymes, we the breeders of proteins must first identify potential catalytic novelty in the form of starting proteins which have at least low levels of a new activity. We therefore look for activities that are known to synthetic chemistry, but perhaps not explored in nature. Cytochrome P450s, whose native functions include a variety of extremely challenging transformations such as hydroxylation, epoxidation, heteroatom oxidations, nitration and more, looked to me like a promising place to start hunting for new activities. Nature had already exploited this evolvable heme‐protein assembly and the various reactive intermediates in the catalytic cycle to create all the natural P450 functions. We quickly discovered that many more new, non‐natural functions were possible. In the last few years we have engineered P450s and other heme proteins to carry out a plethora of reactions known to synthetic chemists, but not found in biology.4
For example, olefin cyclopropanation by carbene transfer is a reaction well known in the area of transition‐metal catalysis, but not known to be catalyzed by an enzyme. In 2012, inspired by much older reports of heme mimics performing such reactions in organic solvents, we discovered that heme proteins catalyze cyclopropanation when provided with diazo carbene precursors and a suitable olefin, in water.5 This promiscuous activity is manifested when the protein encounters the diazo reagent, forms the reactive carbene, and then transfers it to the olefin. Our lab took advantage of this inherent ability of a bacterial cytochrome P450 to evolve a highly efficient enzyme for production of the chiral cis‐cyclopropane precursor to the antidepressant medication levomilnacipran.6 Our group and that of Rudi Fasan have since pushed a variety of heme proteins to synthesize other chiral cyclopropane pharmaceutical precursors, including one used in the synthesis of ticagrelor, a medication used to prevent the reoccurrence of heart attacks.7 In our case, we identified a truncated globin from Bacillus subtilis, which catalyzes the reaction at low levels and also showed some selectivity for producing the single, desired diastereomer of the ticagrelor cyclopropane precursor from ethyl diazoacetate and 3,4‐difluorostyrene (Figure 1). Just a few generations of directed evolution improved the activity and selectivity of the enzyme so that, of the four possible stereoisomers, it produces the ticagrelor cyclopropane almost exclusively. Because the reaction proceeds in whole Escherichia coli cells which express the evolved enzyme, producing the catalyst is as simple as growing bacteria.
Figure 1.
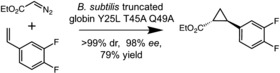
A B. subtilis globin variant, engineered by directed evolution, catalyzes the cyclopropanation of 3,4‐difluorostyrene to make the desired stereoisomer of a ticagrelor precursor with high selectivity and yield.7a
While we were investigating carbene‐transfer reactions catalyzed by heme proteins, we also looked into the possibility of evolving enzymes for nitrene‐transfer reactions. Inspired by a hint in the literature from the 1980′s to attempt intramolecular C−H amination, we were delighted to find that a cytochrome “P411” exhibited a low level of promiscuous activity with an aryl sulfonyl azide nitrene precursor, and that activity could be improved by directed evolution.8 We purposefully engineered the P411 by replacing the completely conserved cytochrome P450 cysteine ligand, which is bound to the iron center, with serine, a ligand not found in any known natural heme protein. This change shifts the characteristic peak in the CO difference spectrum from λ=450 to 411 nm and abolishes the native monooxygenase activity. It also greatly promotes carbene‐transfer and nitrene‐transfer activities.
After demonstrating that the P411 derived from Bacillus megaterium cytochrome P450 could be engineered for intramolecular C−H amination, and intermolecular aziridination and sulfimidation activities unknown in biological systems, our efforts culminated in cytochrome P411CHA, which catalyzes intermolecular benzylic C−H amination.9 Efficient and highly enantioselective intermolecular amination of C(sp3)−H bonds has long been a challenge in chemical catalysis. Despite screening many different heme proteins and protein variants, however, we never found one with the desired activity until postdoctoral fellow Chris Prier discovered that the P411 variant “P4”, evolved for an intermolecular sulfimidation and rearrangement reaction, had acquired promiscuous activity for benzylic C−H amination. Chris Prier and doctoral student Kelly Zhang then directed the evolution of P4 to create P411CHA, which exhibits hundreds of turnovers for the amination of benzylic C−H bonds with excellent enantioselectivities (>99 % ee).9
Free heme does not catalyze any of these nitrene‐transfer reactions, and small‐molecule catalysts for direct C−H amination rely heavily on precious metals which are not sustainable. The protein, however, can impart this new reactivity to earth‐abundant iron in its porphyrin cofactor, and it is evolvable. Evolution enabled P411CHA to promote nitrenoid formation and transfer to a second substrate over the competing nitrene reduction heavily favored in the parent enzyme,9 a property that would be extremely challenging, if not impossible, to design. In fact, we think of these proteins as chiral, self‐assembling, DNA‐encoded macromolecular transition‐metal complexes whose steric and electronic properties are readily tuned by directed evolution to achieve desired activities and selectivities.
Recently we have been exploring enzymes that open yet more chemical space for biocatalysis, including enzymes that form chemical bonds unknown in biology. In the last year we described heme enzymes that catalyze carbene insertion into Si−H and B−H bonds, thus giving living systems their first carbon–silicon10 and carbon–boron11 bond‐forming activities. C−Si bonds are useful in medicinal chemistry, imaging agents, elastomers, and a wide variety of consumer products, but they have never been found in biological systems. Until now, the only methods to create these bonds enantioselectively involved multistep syntheses just to prepare chiral reagents or chiral transition‐metal complexes. The resulting catalysts are often only poorly active, and an iron‐based catalyst had never been reported for this carbene‐insertion reaction. Upon screening a collection of heme proteins, postdoctoral fellow Jennifer Kan and her team discovered that a small (124 aa), highly stable cytochrome c from Rhodothermus marinus (Rma cyt c) could catalyze the reaction between ethyl 2‐diazopropanoate and phenyldimethylsilane to form the chiral organosilicon product with high enantioselectivity (Figure 2 A). Directed evolution discovered three mutations that enable the enzyme to form C−Si bonds with up to 8200 total turnovers (based on Rma cyt c concentration) and enantioselectivities with greater than 99 % ee for a wide range of silicon‐containing substrates. Doctoral student Kai Chen used the engineered enzyme to make 20 organosilicon products, most of which were obtained as single enantiomers. The evolved enzyme also became highly chemoselective for Si−H insertion using substrates having other potentially reactive functional groups (alkene, alkyne, N−H, O−H; Figure 2 B).
Figure 2.
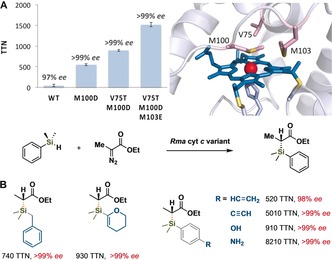
A) Chiral Si−C bond formation catalyzed by a laboratory‐evolved variant of Rhodothermus marinus cytochrome c.10 The three amino acid residues that were mutated to increase this abiological activity include the methionine axial ligand (M100). B) The enzyme catalyzes formation of different organosilane products with high enantiomeric excess from silane and diazo substrates.
We also asked whether an enzyme could catalyze carbene insertion into B−H bonds. No C−B bonds are known in the biological world, and the few natural products that include boron in different forms are thought to incorporate boric acid, available from the environment, non‐enzymatically. Postdoctoral fellows Jennifer Kan and Xiongyi Huang proposed that B−H insertion could be genetically encoded, thus giving living cells the ability to make chiral organoborane products from appropriate, biocompatible carbene precursors and borylating agents. Starting with the Rma cytochrome c, they evolved a very efficient biocatalyst: bacteria expressing evolved Rma cyt c provided access to 16 chiral organoboranes, which had never been made previously, from borane‐Lewis base complexes and various carbene precursors.11 Suitable for gram‐scale biosynthesis, the catalyst offered up to 15 300 turnovers (based on cytochrome c concentration), a 99:1 enantiomeric ratio (e.r.), and 100 % chemoselectivity (Figure 3). These catalyst turnovers are more than 400 times greater than those for known chiral catalysts for the same class of transformation. Furthermore, the enzyme's enantio‐preference could be switched to make either product enantiomer. Fully genetically encoded and functional for hours, these new enzymes open a new world of silicon and boron chemistry in living systems.
Figure 3.
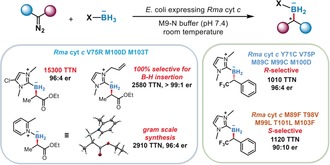
Production of chiral organoboranes by E. coli expressing Rhodothermus marinus cytochrome c.11 The bacterial catalyst uses borane‐Lewis base complexes and diazo reagents to construct boron‐containing carbon stereocenters efficiently and selectively in cells by carbene B−H insertion. The bioconversion can be conducted readily on gram scale, and the enantio‐preference of borylation was switched to give either enantiomer of the organoborane products.
It has been fascinating to see that at least some of nature's vast catalogue of proteins can be evolved in the laboratory, often with only a few mutations, to catalyze abiological reactions (cyclopropanation, N−H insertion, S−H insertion, amination, aziridination, and more). The new enzymes can even create chemical bonds not known in biology (C−Si, C−B). These new reactivities were made possible by 1) use of reagents not found naturally (carbene and nitrene precursors), 2) the promiscuous reactivities of proteins in the presence of these synthetic reagents, 3) directed evolution to enhance and tune the new activities, and 4) chemical intuition and know‐how to identify the right conditions and reactions to test with the right enzymes.
Enzymes to Conquer New Chemistry
The next big challenge is to create enzymes for reactions that neither biology nor synthetic chemistry has conquered. As macromolecular catalysts, for example, enzymes can stabilize transition states and promote reactions through pathways that would be difficult, if not impossible, to access with small‐molecule catalysts because of competition with other, lower‐energy reaction pathways. In a powerful example of how an enzyme active site can be engineered to promote one reaction pathway over another, postdoctoral fellow Stephan Hammer directed the evolution of an alkene anti‐Markovnikov oxygenase (aMOx), which catalyzes the conversion of alkenes into the anti‐Markovnikov carbonyl compounds.12 Intrigued by a report that the cytochrome P450 from Labrenzia aggregata made some phenyacetaldehyde as a side product when it oxidized styrene to the epoxide, Hammer looked more deeply and discovered that this promiscuous reactivity did not involve epoxidation followed by isomerization to the aldehyde, as had been proposed.13 He correctly surmised that it instead went through a competing, stepwise mechanism involving radical/cation intermediates and a 1,2‐hydride migration (Figure 4 A).14 He then exploited this side activity to direct the evolution of by far the most active, and the first enantioselective, direct aMOx catalyst.12 Using earth‐abundant iron, dioxgen, and a recyclable cofactor (NADPH), the laboratory‐evolved P450 enzyme catalyzes thousands of turnovers for anti‐Markovnikov oxidation of different substituted styrenes, including hindered substrates such as internal and 1,1‐disubstituted alkenes.
Figure 4.
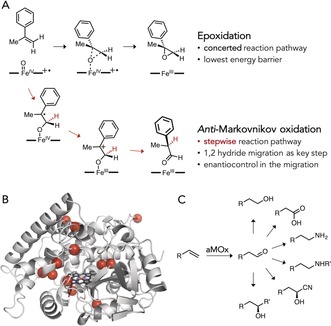
A cytochrome P450 anti‐Markovnikov oxygenase.12 A) Competing reaction pathways for P450‐catalyzed oxo transfer to alkenes. The concerted epoxidation pathway is favored over the stepwise anti‐Markovnikov oxidation consisting of oxo transfer followed by an (enantioselective) 1,2‐hydride migration. B) Ten rounds of directed evolution accumulated 12 amino acid mutations, many of which are distant from the active site. C) aMOx can be combined with established (bio)catalysts for various challenging anti‐Markovnikov alkene functionalization reactions.
Remarkably, the aMOx enzyme reacts with 1,1‐disubstituted alkenes and generates chiral centers by controlling the enantioselectivity of the 1,2‐hydride migration. It is difficult to envision how this could be done outside of a macromolecular active site. The 12 mutations that confer this impressive reactivity and control over substrate orientation during the 1,2‐hydride migration are distributed all over the protein (Figure 4 B). Their specific effects on the enzyme's structure and mechanism that led to this remarkable activity are difficult to rationalize, much less predict.
Synthetic catalysts for anti‐Markovnikov oxidation have not been able to exploit the aMOx mechanism because of competition from the highly favored concerted epoxidation pathway. Instead, synthetic approaches use alternative strategies that require precious metals and/or stoichiometric oxidants such as iodosobenzene. Their very limited activities cannot compete with the laboratory‐evolved enzyme. No catalytic, enantioselective method to convert prochiral alkenes into their chiral anti‐Markovnikov carbonyl compounds was known before this enzyme was engineered.
To enhance utility for production of fine chemicals, aMOx can be coupled with other catalysts, such as an alcohol dehydrogenase, to enable anti‐Markovnikov redox hydration of alkenes.12 This coupled enzyme system yields valuable chiral alcohols with high enantiomeric excess and has the added benefit of recycling the NADPH cofactor. The anti‐Markovnikov alkene functionalization is an important step in the production of many valuable chemicals, which are now in principle approachable through the combination of an evolved aMOx with other enzymes or chemical catalysts (Figure 4 C). Knowing that such a direct aMOx catalyst is possible may inspire creation of even better enzymes or even invention of smaller‐molecule mimics.
Closing Thoughts
I have focused this essay on the efforts of my own group to re‐purpose heme proteins and to use them to demonstrate that directed evolution (and a little design) can readily create new genetically encoded enzyme catalysts for reactions first invented by synthetic chemists and even for reactions which have eluded synthetic approaches. The active sites of the new enzymes can confer high activities and chemo‐, regio‐, and enantioselectivies, as well as product selectivities, which are difficult or impossible to achieve with small‐molecule catalysts. Furthermore, laboratory‐evolved enzymes can stabilize and direct the fates of highly reactive intermediates to promote reactions that are disfavored without the precise control of the enzyme. We hope that these demonstrations will soon be accompanied by deeper insights into the mechanisms of the new enzyme‐catalyzed reactions gained from ongoing structural, spectroscopic, and computational studies.
Other excellent examples of directed evolution for non‐natural chemistry have been produced by other groups, including designed enzymes and enzymes with artificial cofactors, some of which are described in our recent review.15 The designed enzymes don't yet have the sophistication of nature's products, and design struggles with the metals and other cofactors that drive so much interesting chemistry. But when that changes, directed evolution will be there to draw out the new capabilities and fine‐tune the designs, just as it can do with the promiscuous activities of natural proteins.
With the power of evolution realized for engineering, we now have a whole new way to look at the diverse products of natural evolution. Instead of simply asking what enzymes do in nature, we can now ask the question, “what CAN they do?” It will turn out that they can do a lot more than we ever imagined, especially when we use evolution to unleash their latent potentials. What is clear is that evolution is an innovation machine, and nature's products are ready to be let loose to take on new functions, under the discerning eye of the breeder of molecules. A treasure trove of new enzymes is just waiting to be discovered and used for chemistry that we could only have dreamed of just a few years ago.
Conflict of interest
The author declares no conflict of interest.
Biographical Information
Frances Arnold is the Linus Pauling Professor of Chemical Engineering, Bioengineering, and Biochemistry at the California Institute of Technology, where her research focuses on enzyme engineering by directed evolution, with applications in sustainable fuels and chemicals. She uses evolution's innovation mechanisms to bring new chemical reactions to biology. Her honors include the Millennium Technology Prize (2016). She has been elected to the US National Academies of Science, Medicine, and Engineering.

Acknowledgements
I thank Sabine Brinkmann‐Chen, Kai Chen, Stephan Hammer, Kari Hernandez, Xiongyi Huang, Jennifer Kan, Rusty Lewis, Chris Prier and Ruijie Zhang for the ideas, inspiration, data, and hard work that went into the examples presented here. I thank Kim Mayer for excellent editorial assistance. This work was supported by the National Science Foundation, Division of Molecular and Cellular Biosciences (grant MCB‐1513007), the Resnick Sustainability Institute, and the Caltech CI2 innovation program.
F. H. Arnold, Angew. Chem. Int. Ed. 2018, 57, 4143.
References
- 1. Prier C. K., Arnold F. H., J. Am. Chem. Soc. 2015, 137, 13992. [DOI] [PubMed] [Google Scholar]
- 2. Romero P., Arnold F. H., Nat. Rev. Mol. Cell Biol. 2009, 10, 866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. O'Brien P. J., Herschlag D., Chem. Biol. 1999, 6, R91. [DOI] [PubMed] [Google Scholar]
- 4. Brandenberg O., Fasan R., Arnold F. H., Curr. Opin. Biotechnol. 2017, 47, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Coelho P. S., Brustad E. M., Kannan A., Arnold F. H., Science 2013, 339, 307. [DOI] [PubMed] [Google Scholar]
- 6. Wang Z. J., Renata H., Peck N. E., Farwell C. C., Coelho P. S., Arnold F. H., Angew. Chem. Int. Ed. 2014, 53, 6810; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 6928. [Google Scholar]
- 7.
- 7a. Hernandez K. E., Renata H., Lewis R. D., Kan S. B. J., Zhang C., Forte J., Rozzell D., McIntosh J. A., Arnold F. H., ACS Catal. 2016, 6, 7810; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Bajaj P., Sreenilayam G., Tyagi V., Fasan R., Angew. Chem. Int. Ed. 2016, 55, 16110; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 16344. [Google Scholar]
- 8. McIntosh J. A., Coelho P. S., Farwell C. C., Wang Z. J., Lewis J. C., Brown T. R., Arnold F. H., Angew. Chem. Int. Ed. 2013, 52, 9309; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 9479; see also [Google Scholar]; Singh R., Bordeaux M., Fasan R., ACS Catal. 2014, 4, 546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Prier C. K., Zhang R. K., Arnold F. H., Nat. Chem. 2017, 9, 629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kan S. B. J., Lewis R. D., Chen K., Arnold F. H., Science 2016, 354, 1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.S. B. J. Kan, X. Huang, Y. Gumulya, K. Chen, F. H. Arnold, Nature 2017, DOI: https://doi.org/10.1038/nature24996. [DOI] [PMC free article] [PubMed]
- 12. Hammer S. C., Kubik G., Watkins E., Huang S., Minges H., Arnold F. H., Science 2017, 358, 215. [DOI] [PubMed] [Google Scholar]
- 13. Yin Y. C., Yu H. L., Luan Z. J., Li R. J., Ouyang P. F., Liu J., Xu J. H., ChemBioChem 2014, 15, 2443. [DOI] [PubMed] [Google Scholar]
- 14. Groves J. T., Myers R. S., J. Am. Chem. Soc. 1983, 105, 5791. [Google Scholar]
- 15. Hammer S. C., Knight A. M., Arnold F. H., Curr. Opin. Green Sustainable Chem. 2017, 7, 23. [Google Scholar]