

Abstract
Delafloxacin, a fluoroquinolone, has activity against gram‐positive organisms including methicillin‐resistant Staphylococcus aureus and fluoroquinolone‐susceptible and –resistant gram‐negative organisms. This study was conducted to determine delafloxacin pharmacokinetics after a single intravenous infusion or oral dose administration in subjects with varying degrees of renal function. The study was an open‐label, parallel‐group crossover study in subjects with normal renal function or with mild, moderate, or severe renal impairment. Subjects received 300 mg delafloxacin intravenously, placebo intravenously, and 400 mg delafloxacin orally in 3 periods separated by ≥14‐day washouts. Blood and urine pharmacokinetic parameters were calculated using noncompartmental methods. Delafloxacin total clearance decreased with decreasing renal function, with a corresponding increase in AUC0–∞. After intravenous administration, mean total clearance was 13.7 and 7.07 L/h, and mean AUC0–∞ was 22.6 and 45.0 μg·h/mL in normal and severe renal subjects, respectively. Mean renal clearance as determined by urinary excretion was 6.03 and 0.44 L/h in normal and severe renal impairment subjects, respectively. Total clearance exhibited linear relationships to eGFR and CLCR. Similar observations were found after oral administration of delafloxacin. Single doses of delafloxacin 300 mg intravenously and 400 mg orally were well tolerated in all groups. In conclusion, renal insufficiency has an effect on delafloxacin clearance; a dosing adjustment for intravenous dosing is warranted for patients with severe renal impairment (eGFR < 30 mL/min).
Keywords: delafloxacin, fluoroquinolones, pharmacokinetics, renal dysfunction
Antibiotic resistance among gram‐positive bacteria, including Staphylococcus, has led to the search for new antibiotics with better activity against these resistant organisms.1, 2, 3 Delafloxacin is an anionic fluoroquinolone antibiotic with a broad spectrum of antibacterial activity that includes gram‐positive organisms (methicillin‐resistant Staphylococcus aureus, methicillin‐susceptible S. aureus, Streptococcus pyogenes), gram‐negative organisms (Escherichia coli, Klebsiella spp., and Pseudomonas aeruginosa), atypical bacteria, and anaerobic organisms (Bacteroides and Clostridium spp.).4 Delafloxacin was recently approved by the U.S. Food and Drug Administration (FDA) for the treatment of acute bacterial skin and skin structure infections (ABSSSI)5, 6 and currently is being evaluated for the treatment of community‐acquired bacterial pneumonia.7
The pharmacokinetics and absorption, distribution, metabolism, and excretion (ADME) characteristics of delafloxacin have been assessed in multiple phase 1 studies.8, 9, 10 Delafloxacin distribution and disposition are characterized in healthy volunteers by a steady‐state volume of distribution of ∼40 L and plasma protein binding of 84% in humans (data on file at Melinta Therapeutics). Total clearance is approximately 13 L/h, and renal clearance of the parent compound accounts for about 35%–40% of total clearance. The liver is also involved in elimination both through the production of water‐soluble glucuronide conjugates cleared by the kidney and in the fecal elimination of about 30% of an intravenous dose as the unchanged parent compound.
Given the extent of renal clearance, it was hypothesized that systemic exposure to delafloxacin would be greater in patients with impaired renal function than in those with normal renal function. Thus, a study in patients with renal impairment was conducted, in which the primary objective was to evaluate the pharmacokinetics of delafloxacin after single intravenous or oral doses of delafloxacin in subjects with mild, moderate, and severe renal impairment compared with subjects with normal renal function (estimated glomerular function rate [eGFR] > 80 mL/min). A secondary objective was to assess the safety and tolerability of delafloxacin in these populations.
Methods
Study Design
This study was conducted at DaVita Clinical Research, Minneapolis, Minnesota, and approval was obtained from the principal investigator's institutional review board (Western Institutional Review Boards, Olympia, Washington) prior to the beginning of the study. All subjects voluntarily signed informed consent prior to admission into the study. The study was conducted according to the International Conference on Harmonization guideline, Good Clinical Practice: Consolidated Guideline, and the United States Code of Federal Regulations.
This was a phase 1 open‐label, parallel‐group, single‐dose, crossover, single‐site study assessing the pharmacokinetics and toleration of delafloxacin in subjects with varying degrees of renal function. A total of 32 subjects were planned (4 groups of 8 subjects each) as follows:
Group A: healthy subjects (eGFR > 80 mL/min/1.73 m2);
Group B: subjects with mild renal impairment (eGFR > 50–80 mL/min/1.73 m2);
Group C: subjects with moderate renal impairment (eGFR > 30–50 mL/min/1.73 m2);
Group D: subjects with severe renal impairment (eGFR ≤ 30 mL/min/1.73 m2).
At screening, subjects were assigned to a study group based on their eGFR, calculated using the isotope dilution mass spectrometry‐traceable Modification of Diet in Renal Disease (MDRD)11 formula as follows:
eGFR (mL/min/1.73 m2) = 175 × serum creatinine (SCr; mg/dL)−1.154 × age (years)−0.203 × (0.742 if female) × (1.212 if African American).
Subject creatinine clearance (CLCR) was also calculated using the Cockcroft‐Gault formula12 as follows:
CLCR (mL/min) = ([140 – age (years)] × weight [kg])/(72 × SCr [mg/dL]) × 0.85 (if female).
For inclusion in the study, each subject was required to meet all the following criteria: male or female between 18 and 80 years of age; body mass index between 18.5 and 40 kg/m2; baseline laboratory values within reference ranges or deemed not clinically significant by the investigator; subjects with renal impairment with laboratory values consistent with their disease; not childbearing, rendered not childbearing (eg, vasectomy, hysterectomy), or using an adequate method of contraception from screening through 12 weeks after the last dose of study drug. Renally impaired subjects were accepted if they had been taking medications that did not affect study drug ADME.
Subjects were excluded if the subject had a clinically or laboratory significant abnormality in the medical history or at screening (excluding renal insufficiency) that in the investigator's opinion may have placed the subject at risk; any surgical or medical condition or medication history (active or chronic) that may have interfered with the ADME of delafloxacin or production and/or excretion of SCr; a functioning renal transplant; abnormal vital signs (systolic and diastolic blood pressure < 90 or > 200 and < 45 or > 110 mm Hg, respectively, heart rate < 50 or > 120 bpm); hemoglobin < 10 g/dL, blood/plasma donation within 30 days before dosing, or had lost >1200 mL of blood within 4 months prior to first dose of study drug; a history of or current hepatitis, or subject was a carrier of hepatitis B surface antigen and/or hepatitis C antibodies (a subject with hepatitis C who had normal liver function test results was allowable with investigator approval); a history or evidence of drug abuse within 6 months of screening, positive serum drug screen or alcohol breathalyzer result at screening or admission to study center; HIV/AIDS; a history of an investigational drug within 30 days or 5 half‐lives, whichever was longer; or a baseline QTc interval ≥ 480 or ≥ 500 milliseconds for men and women, respectively.
Study Medication
Delafloxacin for injection, 300 mg/vial, is formulated as a sterile, nonpyrogenic, lyophilized powder. Each vial contains the following ingredients: 433 mg delafloxacin meglumine equivalent of 300 mg free acid, 58.56 mg meglumine, 2.4 g sulfobutylether‐β‐cyclodextrin (SBECD; Captisol), 3.4 mg ethylenediaminetetraacetate disodium (equivalent to 2.6 mg EDTA). Delafloxacin vials were stored at controlled room temperatures (68°F–77°F) and protected from light. Placebo for intravenous delafloxacin was supplied as a 12.4‐mL frozen solution in 20‐mL vials that contained the same amounts of excipients per vial as the active vials. Oral delafloxacin was supplied as four 100‐mg capsules containing neat compound for a total dose of 400 mg.
Study Drug Administration
Subjects participated in 3 treatment periods separated by washout periods of ≥14 days:
Period 1: delafloxacin 300 mg intravenous infusion over 1 hour;
Period 2: placebo containing 2.4 g of an SBECD intravenous infusion over 1 hour;
Period 3: oral delafloxacin 400 mg.
Subjects fasted for 4 hours before and after dosing (or after the end of the infusion for the intravenous treatments). For oral dosing, the study drug was administered with a minimum of 240 mL of room‐temperature water. Water was allowed ad libitum throughout the study period, but was restricted to the water provided for dosing between 1 hour before and 2 hours after dosing of study medication.
Pharmacokinetic Samples
For the intravenous treatments (periods 1 and 2), blood for plasma samples was collected before dosing and 0.33, 0.66, 1 (end of infusion), 1.083, 1.167, 1.33, 1.5, 2, 3, 4, 6, 8, 12, 24, 36, and 48 hours after the start of the infusion. For the oral treatment (period 3), subjects had blood drawn before dosing and 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 16, 20, 24, 30, 36, and 48 hours after dosing. Urine samples were collected in intervals from ‐2 to 0 hours and after dosing from 0 to 12, 12 to 24, 24 to 36, and 36 to 48 hours. For all subjects, a 10‐mL blood sample was collected before dosing in period 1 for the in vitro determination of plasma protein binding of delafloxacin.
Both plasma and urine samples were analyzed for delafloxacin using validated methods for each matrix utilizing liquid chromatography coupled with detection by tandem mass spectrometry (LC‐MS/MS). Sample analyses were performed by Alta Analytical Laboratory (Intertek Analytical Services, El Dorado Hills, California). Delafloxacin was quantitated in plasma samples (dipotassium EDTA) using a validated LC‐MS/MS method with a nominal concentration range of 5 to 5000 ng/mL. Sample preparation was performed by supported liquid phase extraction on Biotage Isolute 96‐well SLE+ plates. Analysis of the final extract was performed with high‐pressure liquid chromatography using an XBridge C18 column and MS/MS detection using positive ion electrospray. The method demonstrated acceptable linearity, accuracy, and precision. Delafloxacin stability was demonstrated in standard freeze/thaw and room‐temperature tests and in samples frozen at less than or equal to ‐20°C for up to 484 days (data on file at Melinta Therapeutics).
Pharmacokinetic Analyses
Individual plasma concentration–versus–actual time data were used to derive noncompartmental pharmacokinetic parameters using Phoenix WinNonlin versions 6.2.1 and 6.4 (Certara, Princeton, New Jersey). The following pharmacokinetic parameters were calculated: area under the plasma concentration–time curve (AUC) by the trapezoidal rule, including AUC0–∞ (time zero extrapolated to infinity) and AUC0–t (time zero to time of last quantifiable concentration); maximum observed plasma concentration (Cmax) and time to reach Cmax (Tmax); apparent terminal elimination rate constant and terminal half‐life (λz and t½, respectively); total body clearance (CLtot) from the ratio of dose to AUC0–∞; area under the intravenous plasma concentration–time moment curve (AUMC); intravenous mean residence time (MRT) from the ratio of AUMC to AUC0–∞, corrected for the infusion time; and intravenous volume of distribution at steady state (Vss) calculated by the product of CLtot and MRT.
The following pharmacokinetic parameters were calculated from urine concentrations of delafloxacin: amount of delafloxacin excreted in urine (Ae) over various periods from time zero to 48 hours after dosing; fraction of the dose excreted in urine (Fe%) from time zero to 48 hours; and renal clearance (CLr) from the ratio of Ae0–48 to AUC0–t.
Safety Assessments
Safety assessments included evaluation of treatment‐emergent adverse events (TEAEs), clinical laboratory results (hematology [including coagulation], serum chemistry [including liver function tests], and urinalysis), vital signs, 12‐lead electrocardiogram measurements, and physical examination findings.
Statistical Analysis
Summary statistics were prepared with WinNonlin and SAS software version 9.1.3 (SAS Institute, Cary, North Carolina). In general, continuous data are summarized by presenting the number of subjects, mean, standard deviation (SD), median, minimum, and maximum. Categorical data are summarized by presenting the number (frequency) and percentage of subjects at each varying degree of renal function. Linear regression was conducted using GraphPad Prism version 5.01 (GraphPad Software, La Jolla, California). Linear regression of the intravenous pharmacokinetic parameters was performed without the implausible data from the severe impairment subject indicated below. There were 2 analysis populations: the pharmacokinetic population, which included all subjects who had sufficient concentration data to calculate reliable estimates of pharmacokinetic parameters; and the safety population, which included all subjects who received at least 1 dose of study drug (delafloxacin or placebo).
Results
Study Population Demographics
Thirty‐four subjects were enrolled in the study, but 2 subjects were discontinued early because of TEAEs. Both subjects were replaced and were not included in the pharmacokinetic population, but all 34 subjects were included in the safety population. Thirty‐two subjects who received delafloxacin were considered evaluable and included in the pharmacokinetic population.
Subject demographic and baseline characteristics are summarized in Table 1. With the exception of renal status, overall mean demographic characteristics were similar across groups. As observed in Table 1, mean values of eGFR were consistently lower than those calculated by the Cockcroft‐Gault method (CLCR).
Table 1.
Subject Demographics and Baseline Characteristics (Mean ± SD)
| Normal | Mild Impairment | Moderate Impairment | Severe Impairment | |
|---|---|---|---|---|
| n (completed) | 8 | 8 | 8 | 8 |
| n (discontinued) | 1 | 0 | 0 | 1 |
| Age (y) | 52 ± 4 | 56 ± 10 | 57 ± 9 | 54 ± 9 |
| Weight (kg) | 84 ± 13 | 94 ± 15 | 98 ± 25 | 92 ± 18 |
| Height (cm) | 174 ± 13 | 174 ± 8 | 175 ± 9 | 170 ± 9 |
| BMI (kg/m2) | 28 ± 2 | 31 ± 3 | 32 ± 7 | 32 ± 5 |
| eGFR (mL/min/1.73 m2) | 92 ± 11 | 63 ± 8 | 39 ± 5 | 22 ± 6 |
| CLCR (mL/min) | 121 ± 19 | 87 ± 16 | 58 ± 15 | 35 ± 10 |
Delafloxacin Pharmacokinetics
For 1 subject in the severe impairment group (subject 1404), concentration data from the intravenous infusion period (0.33, 0.66, and 1 hour) appeared to be pharmacokinetically implausible (data not shown). Outlier tests (extreme studentized deviate) of the plasma concentrations at each point in the severe impairment group indicated the implausible concentrations were statistical outliers. Mean noncompartmental parameters for intravenous dosing of the severe impairment group were calculated excluding the data from the subject with the outlier results. The mean plasma profile in Figure 1 for the intravenous severe impairment group excluded the statistical outlier data.
Figure 1.
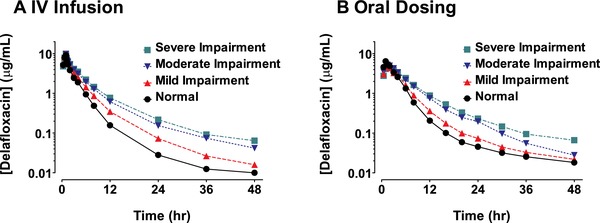
Mean plasma concentration–time profiles in subjects with varying degrees of renal function after either intravenous infusion of 300 mg of delafloxacin (A) or oral administration of 400 mg of delafloxacin (B).
After the end of the 1‐hour 300‐mg delafloxacin intravenous infusions, plasma concentrations declined in a multiphasic manner in patients from all renal function categories (Figure 1A), and delafloxacin total exposure increased consistently as the degree of renal impairment worsened (Table 2). Compared with the normal renal function group, mean AUC0–t and AUC0–∞ values for the severe renal impairment group were 1.8‐fold and 2‐fold greater, respectively (43.4 vs 23.6 and 45.0 vs 22.6 μg·h/mL). Mean peak exposures (Cmax) were similar across all subjects with varying degrees of renal impairment. Apparent t½ was meaningfully increased only for the severe renal impairment group (14.9 hours) compared with other study groups. Mean Vss values increased somewhat from 45.6 to 57.2 L as baseline renal function worsened. The fraction of the dose of delafloxacin excreted in urine during the 48 hours after dosing declined from 45.3% for the normal group to 5.8% for the severe renal impairment group (Table 2). The majority of the urinary excretion of delafloxacin occurred in the first 12 hours after dosing (Figure 2A). Consistent with the loss of renal function, mean CLr decreased from 6.03 L/h for the normal group to 0.44 L/h for the severe renal impairment group.
Table 2.
Mean (%CV) Pharmacokinetic Parameters Following Delafloxacin 300 mg Intravenously in Subjects With Varying Degrees of Renal Function
| Normal | Mild Impairment | Moderate Impairment | Severe Impairment* | |
|---|---|---|---|---|
| Plasma pharmacokinetics | ||||
| Cmax (μg/mL) | 9.28 (25.3) | 9.80 (11.1) | 9.86 (25.6) | 9.89 (22.3) |
| t½ (h) | 9.3 (46.7) | 10.7 (22.9) | 8.9 (33.5) | 14.9 (41.3) |
| AUC0–t (μg·h/mL) | 23.6 (23.2) | 31.0 (19.3) | 39.3 (26.3) | 43.4 (26.6) |
| AUC0‐∞ (μg·h/mL) | 22.6 (20.0) | 31.3 (19.0) | 38.4 (27.9) | 45.0 (28.7) |
| CL (L/h) | 13.7 (19.1) | 9.92 (20.3) | 8.25 (22.9) | 7.07 (23.8) |
| Vss (L) | 45.6 (23.3) | 48.2 (33.7) | 47.2 (26.7) | 57.2 (32.0) |
| Urinary pharmacokinetics | ||||
| Ae0–48 (mg) | 136 (16.8) | 84.2 (58.6) | 49.2 (40.3) | 17.5 (76.0) |
| Fe0–48 (%) | 45.3 (16.8) | 28.0 (58.6) | 16.4 (40.3) | 5.83 (75.6) |
| CLr (L/h) | 6.03 (26.8) | 2.96 (72.7) | 1.30 (49.0) | 0.44 (83.6) |
Noncompartmental analysis performed with 1 subject (1404) excluded for implausible plasma concentrations (refer to text).
Figure 2.
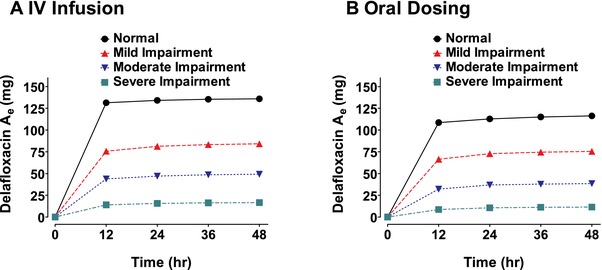
Cumulative mean urinary excretion (Ae) of delafloxacin in subjects with varying degrees of renal function after either intravenous infusion of 300 mg of delafloxacin (A) or oral administration of 400 mg of delafloxacin (B).
Following the 400‐mg oral dose of delafloxacin, plasma concentrations exhibited a median Tmax of 1 to 1.5 hours, followed by a multiphasic decline in patients from all renal function categories (Figure 1B). The normal renal function group had total exposure similar to that observed following the 300‐mg delafloxacin intravenous infusion (Table 3). In the group with normal renal function, mean AUC0–t values were 23.6 μg·h/mL after intravenous dosing and 25.8 μg·h/mL after oral dosing. Mean AUC0–t values for the moderate and severe renal impairment groups were approximately 1.5‐fold higher than the corresponding value for the normal group; however, mean Cmax varied little across the various renal function groups after oral dosing, with a slightly higher mean Cmax of 7.2 μg/mL for the normal group compared with 5.4 μg/mL for those with severe renal impairment. Mean CLr values after oral dosing were similar to the values and pattern observed for intravenous dosing (Figure 2B) and decreased from 5.09 L/h for the normal group to 0.29 L/h for the severe renal impairment group (Table 3).
Table 3.
Mean (%CV) Pharmacokinetic Parameters Following Oral Administration of 400 mg of Delafloxacin in Subjects With Varying Degrees of Renal Function
| Normal | Mild Impairment | Moderate Impairment | Severe Impairment | |
|---|---|---|---|---|
| Plasma pharmacokinetics | ||||
| Cmax (μg/mL) | 7.16 (34.9) | 5.67 (34.2) | 6.00 (29.7) | 5.35 (24.9) |
| Tmax (h)a | 1 (0.5–1.5) | 1 (0.5–2) | 1 (0.5–3) | 1.5 (0.5–6) |
| t½ (h) | 15.4 (43.3) | 12.5 (21.9) | 10.5 (40.5) | 15.5 (33.8) |
| AUC0–t (μg·h/mL) | 25.8 (29.3) | 26.8 (31.4) | 36.9 (18.8) | 37.8 (26.9) |
| AUC0–∞ (μg·h/mL) | 25.4 (31.6) | 28.3 (28.9) | 37.3 (18.8) | 39.5 (27.9) |
| CLtot/F (L/h) | 17.6 (40.2) | 15.9 (47.5) | 11.0 (18.4) | 10.8 (25.7) |
| Urinary pharmacokinetics | ||||
| Ae0–48 (mg) | 116 (21.8) | 75.5 (70.3) | 38.5 (64.6) | 11.5 (56.0) |
| Fe0–48 (%) | 29.1 (21.8) | 18.9 (70.3) | 9.62 (64.6) | 2.88 (56.0) |
| CLr (L/h) | 5.09 (46.3) | 2.95 (54.2) | 1.03 (57.4) | 0.29 (41.4) |
Tmax values are median (range).
Plasma protein binding of delafloxacin showed an overall trend to decrease slightly with decreasing renal function. Mean (%CV) protein binding in the normal group was 84.1% (10.2%) and decreased to 81.2% (20.0%), 82.7% (10.9%), and 79.8% (17.4%) in those subjects with mild, moderate, and severe renal impairment, respectively.
Relationship Between Renal Function and Pharmacokinetic Parameters
Analysis of the relationships between renal function and plasma pharmacokinetic parameters of delafloxacin were performed by linear regression. Evaluations were made using renal function as measured by eGFR and CLCR. The regression results are presented in Table 4 for intravenous administration and in Table 5 for oral administration. Plots of CLtot versus eGFR for intravenous and oral delafloxacin are shown in Figure 3 together with linear regression lines and 95% confidence intervals of the estimated regression.
Table 4.
Linear Regression of Pharmacokinetic Parameter Estimates Versus eGFR or CLCR in Subjects After Intravenous Infusion Administration of 300 mg of Delafloxacin
| PK Parameter | Renal Function Measure | n | Slope Estimate | 95%CI of Slope Estimate | r 2 | P |
|---|---|---|---|---|---|---|
| AUC0–∞ (μg·h/mL) | eGFR | 28 | −0.307 | (−0.453 to ‐0.161) | 0.419 | .0002 |
| CLCR | 28 | −0.228 | (−0.319 to ‐0.137) | 0.505 | <.0001 | |
| CL (L/h) | eGFR | 28 | 0.0934 | (0.0609–0.126) | 0.573 | <.0001 |
| CLCR | 28 | 0.0687 | (0.0496–0.0878) | 0.678 | <.0001 | |
| CLr (L/h) | eGFR | 31 | 0.0786 | (0.0602–0.0970) | 0.724 | <.0001 |
| CLCR | 31 | 0.0576 | (0.0437–0.0715) | 0.713 | <.0001 | |
| Cmax (μg/mL) | eGFR | 31 | −0.00693 | (−0.0346 to 0.0208) | 0.00893 | .613 |
| CLCR | 31 | −0.0141 | (−0.0339 to 0.00576) | 0.0677 | .158 | |
| Vss (L) | eGFR | 28 | −0.128 | (−0.360 to 0.104) | 0.0468 | .269 |
| CLCR | 28 | −0.0254 | (−0.186 to 0.135) | 0.00407 | .747 |
Table 5.
Linear Regression of Pharmacokinetic Parameter Estimates Versus eGFR or CLCR in Subjects After Oral Administration of 400 mg of Delafloxacin
| PK Parameter | Renal Function Measure | n | Slope Estimate | 95%CI of Slope Estimate | r 2 | P |
|---|---|---|---|---|---|---|
| AUC0–∞ (μg·h/mL) | eGFR | 30 | −0.189 | (−0.311 to ‐0.0661) | 0.262 | .0038 |
| CLCR | 30 | −0.168 | (−0.257 to ‐0.0800) | 0.353 | .0005 | |
| CLtot/F (L/h) | eGFR | 30 | 0.0985 | (0.0263–0.171) | 0.218 | .0093 |
| CLCR | 30 | 0.0918 | (0.0399–0.144) | 0.319 | .0011 | |
| CLr (L/h) | eGFR | 32 | 0.0654 | (0.0455–0.0852) | 0.601 | <.0001 |
| CLCR | 32 | 0.0511 | (0.0376–0.0647) | 0.664 | <.0001 | |
| Cmax (μg/mL) | eGFR | 32 | 0.0233 | (−0.00155 to 0.0482) | 0.109 | .0651 |
| CLCR | 32 | 0.00940 | (−0.00991 to 0.0287) | 0.032 | .328 |
Figure 3.
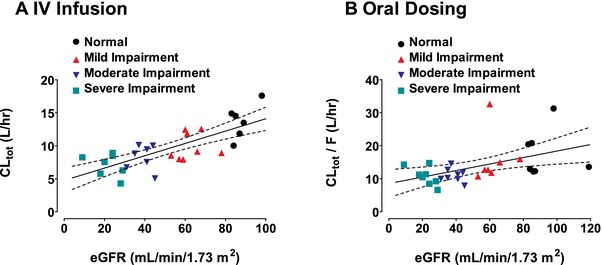
Linear regression of total clearance (/F) versus eGFR after either 300 mg intravenous delafloxacin (A) or 400 mg delafloxacin orally (B). Solid lines are linear regressions of total clearance versus eGFR; dashed lines are the 95% confidence intervals of the linear regressions.
Following a single dose of delafloxacin intravenously, statistically significant slopes were obtained for the AUC0–∞, CLtot, and CLr parameters, with P < .001 for the slope estimates. For the linear relationships between CLtot and CLr versus eGFR or CLCR, r 2 was >0.55, whereas for the relationship between AUC0–∞ and renal function, r 2 was somewhat less, at 0.4 or greater. Because the major excretion route of delafloxacin is renally mediated, a strong correlation between renal clearance and renal function was observed, with r 2 > 0.7. The relationships between Cmax and Vss versus eGFR or CLCR were not significant, with P > .15. A similar pattern was observed for the oral pharmacokinetic parameters (Table 5), with significant relationships between AUC0–∞, CLtot/F, and CLr versus eGFR or CLCR, but not for Cmax.
Safety
Overall, 56 TEAEs were reported, and 18 of 34 subjects (52.9%) experienced at least 1 TEAE after receiving the study drug. To summarize, TEAEs were reported by 2 of 9 subjects (22.2%) in the normal renal function group, 5 of 8 subjects (62.5%) in the mild renal impairment group, 3 of 8 subjects (37.5%) in the moderate renal impairment group, and 5 of 9 subjects (55.6%) in the severe renal impairment group after the 300‐mg intravenous delafloxacin infusion. TEAEs were reported by no subjects in the normal and mild renal impairment groups, 3 of 8 subjects (37.5%) in the moderate renal impairment group, and 2 of 8 subjects (25.0%) in the severe renal impairment group after the 400‐mg oral delafloxacin dose. Following the placebo intravenous infusion, TEAEs were reported by 1 of 9 subjects (11.1%) in the normal group, 4 of 8 subjects (50.0%) in the mild renal impairment group, 3 of 8 subjects (37.5%) in the moderate renal impairment group, and no subjects in the severe renal impairment group.
The most commonly reported TEAEs overall in those subjects receiving delafloxacin were classified as gastrointestinal disorders (6 of 34 subjects [17.6%]), with diarrhea the most commonly reported gastrointestinal disorder TEAE (4 subjects [66.7%]). Of the total number of TEAEs, 14 TEAEs in 10 subjects were considered possibly or probably related to delafloxacin treatment; no TEAE was considered definitely related to treatment. After receiving the 300 mg of intravenous delafloxacin in period 1 and placebo intravenously in period 2, 1 female subject in the healthy group (group A) was discontinued on day 4 of period 2 because of a severe TEAE of C. difficile, considered unrelated to the study drug by the investigator. This 52‐year‐old subject did not meet all inclusion/exclusion criteria, as she received amoxicillin within the 30‐day restriction period. Another subject, a 52‐year‐old man, in the severe renal impairment group (group D) received a single dose of delafloxacin in period 1 and discontinued from the study because of a TEAE of moderate severity of bursitis in the left shoulder, which was considered unrelated to the study drug by the investigator. No deaths or treatment‐emergent serious adverse events occurred during the study.
Discussion
We studied the pharmacokinetics and safety of delafloxacin administered as a single 300‐mg intravenous dose infused over 1 hour, a single 400‐mg oral dose, and a 1‐hour infusion of placebo containing SBECD in normal healthy subjects and otherwise healthy subjects with mild, moderate, and severe renal impairment. After the intravenous infusion of delafloxacin, the mean total exposure of delafloxacin increased consistently as the degree of renal impairment worsened, with mean AUC0–∞ for the severe renal impairment group about 2‐fold higher than in the healthy group. In contrast, decreasing renal function did not have a significant effect on Cmax or Vss. Delafloxacin urinary pharmacokinetics corroborated the plasma pharmacokinetics with decreasing Ae0‐48 and Fe0–48 values in association with declining renal clearance. Similarly, after a single oral dose of delafloxacin, declining renal function was associated with increased total exposure of delafloxacin. Oral Cmax values were slightly lower for the renal impairment groups compared with the healthy group, but the difference was not clinically relevant.
These data are consistent with studies indicating that delafloxacin elimination is primarily renal, whether as intact drug or as glucuronide conjugates. This study indicates that, independent of the route of administration, delafloxacin elimination was highly correlated with renal function, whether assessed by the eGFR, calculated by the MDRD formula, or estimated by the Cockcroft‐Gault equation (CLCR). A previous report on delafloxacin oral pharmacokinetics in elderly subjects similarly suggested decreased delafloxacin clearance was correlated with decreased creatinine clearance.9 Although delafloxacin is also subject to a degree of hepatic elimination, a previous study showed a lack of change in delafloxacin clearance in patients with hepatic impairment, including those with severe (Child‐Pugh class C) hepatic impairment.11
Fluoroquinolone effectiveness is based on the ratio of the AUC to the minimum inhibitory concentration (MIC) of the target bacteria.12 This study showed that AUC values did not decrease, but rather increased with reduced renal function, and that Cmax values remained largely unchanged, so the AUC/MIC ratio would not be diminished but rather might increase in the presence of renal impairment because of increased plasma delafloxacin concentrations. However, some drugs are associated with increased incidence of toxicity with increasing exposures. In this study we did not find any trend toward increased adverse events correlating with increased exposure in the renally‐impaired subjects. In general, a single dose of delafloxacin was as well tolerated in the renally impaired population as in those with normal renal function.
This study also evaluated the MDRD formula13 and the established Cockcroft‐Gault formula.14 Although there may be limitations in the Cockcroft‐Gault equation, it has been validated by the measurement of the glomerular filtration rate by inulin clearance, which, however, is an impractical method of assessing renal function in clinical practice.15 Nonetheless, delafloxacin clearance was correlated with both estimations of renal clearance, which may simplify decision‐making on dosing.
The FDA‐approved intravenous delafloxacin dose in patients with normal renal function or mild to moderate renal impairment (eGFR, 30–89 mL/min/1.73 m2) is 300 mg given every 12 hours.16 Given the approximately 2‐fold increase in AUC seen in severe renal impairment compared with normal renal function, a dose adjustment to 200 mg given intravenously every 12 hours is warranted in patients with severe renal impairment (eGFR, 15–29 mL/min/1.73 m2). This dose should still provide an efficacious AUC while reducing unnecessary exposure to excess drug. No dose adjustment for oral delafloxacin is warranted in severe renal impairment. The phase 3 studies in ABSSSI did enroll patients with primarily mild or moderate renal impairment. Overall, delafloxacin was effective and well tolerated in these patients.17
Conclusions
In conclusion, increased degrees of renal impairment correlate with decreased delafloxacin clearance and consequent increased exposure. In addition, delafloxacin was well tolerated in the 4 groups of renal function spanning CLcr > 80 to < 30 mL/min.
Declaration of Conflicting Interests
Three authors (L.L., M.Q., S.K.C.) are employed by Melinta Therapeutics, Inc.
Funding
All research was funded by Melinta Therapeutics, Inc.
These data were presented in part at ICAAC 2013 in Denver CO (poster #A‐107e).
References
- 1. Spellberg B, Brass E, Bradley J, et al. White Paper: Recommendations on the conduct of superiority and organism‐specific clinical trials of antibacterial agents for the treatment of infections caused by drug‐resistant bacterial pathogens. Clin Infect Dis. 2012;55(8):1031–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Munita J, Bayer A, Arias C. Evolving resistance among Gram‐positive pathogens. Clin Infect Dis. 2015;61(suppl 2):S48–S57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Draenert R, Seybold U, Grutzner E, Bogner J. Novel antibiotics: Are we still in the pre ‐ post ‐ antibiotic era? Infection. 2015;43:145–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Van Bambeke F. Delafloxacin, a non‐zwitterionic fluoroquinolone in Phase III of clinical development: evaluation of its pharmacology, pharmacokinetics, pharmacodynamics and clinical efficacy. Future Microbiol. 2015;10(7):1111–1123. [DOI] [PubMed] [Google Scholar]
- 5. Cammarata S, Gardovskis J, Farley B, et al. Results of a global phase 3 study of delafloxacin (DLX) compared to vancomycin with aztreonam (VAN) in acute bacterial skin and skin structure infections (ABSSSI). 2015. http://melinta.com/wp-content/uploads/2016/03/IDWeek2015-complete_302_ABSSSI_study_results.pdf. [DOI] [PMC free article] [PubMed]
- 6. O'Riordan W, McManus A, Teras J, et al. A global phase 3 study of delafloxacin (DLX) compared to vancomycin/ aztreonam (VAN/AZ) in patients with acute bacterial skin and skin structure infections (ABSSSI). 2016. http://melinta.com/wp-content/uploads/2016/10/IDWEEK-1347-Baxdela-vs-VAN-AZ-302-Ph3-Results.pdf.
- 7. Study to compare delafloxacin to moxifloxacin for the treatment of adults with community‐acquired bacterial pneumonia (DEFINE‐CABP). https://clinicaltrials.gov/ct2/show/NCT02679573?term=delafloxacin&rank=3. Accessed August 23, 2017. [DOI] [PMC free article] [PubMed]
- 8. Hoover R, Hunt T, Benedict M, et al. Safety, tolerability, and pharmacokinetic properties of intravenous delafloxacin after single and multiple doses in healthy volunteers. Clin Ther. 2016;38(1):53–65. [DOI] [PubMed] [Google Scholar]
- 9. Hoover R, Hunt T, Benedict M, et al. Single and multiple ascending‐dose studies of oral delafloxacin: effects of food, sex, and age. Clin Ther. 2016;38(1):39–52. [DOI] [PubMed] [Google Scholar]
- 10. McEwen A, Lawrence L, Hoover R, et al. Disposition, metabolism and mass balance of delafloxacin in healthy human volunteers following intravenous administration. Xenobiotica. 2015;45(12):1054–1062. [DOI] [PubMed] [Google Scholar]
- 11. Hoover R, Marbury T, Preston R, et al. Clinical pharmacology of delafloxacin in patients with hepatic impairment. J Clin Pharmacol. 2017;57(3):328–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Craig W. Dose the dose matter? Clin Infect Dis. 2001;33(suppl 3):S233–S237. [DOI] [PubMed] [Google Scholar]
- 13. Levey A, Bosch J, Lewis J, Greene T, Rogers N, Roth D. A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Modification of Diet in Renal Disease study group. Ann Intern Med. 1999;130:461–470. [DOI] [PubMed] [Google Scholar]
- 14. Cockcroft D, Gault M. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16:31–41. [DOI] [PubMed] [Google Scholar]
- 15. Luke D, Halstenson C, Opsahl J, Matzke G. Validity of creatinine clearance estimates in the assessment of renal function. Clin Pharmacol Ther. 1990;48:503–508. [DOI] [PubMed] [Google Scholar]
- 16. Delafloxacin Prescribing Information. 2017. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208610s000,208611s000lbl.pdf. Accessed August 23, 2017.
- 17. Beasley R, Oguchi G, Liang S, Lawrence L, Cammarata S. Delafloxacin (DLX) is effective and well‐tolerated in treatment of patients with renal impairment with acute bacterial skin and skin structure infections (ABSSSI) versus vancomycin/aztreonam (VAN/AZ). 2017. http://melinta.com/wp-content/uploads/2017/04/ECCMID-2017-Renal.pdf.