

Abstract
Over the course of most common neurodegenerative diseases the amygdala accumulates pathologically misfolded proteins. Misfolding of 1 protein in aged brains often is accompanied by the misfolding of other proteins, suggesting synergistic mechanisms. The multiplicity of pathogenic processes in human amygdalae has potentially important implications for the pathogenesis of Alzheimer disease, Lewy body diseases, chronic traumatic encephalopathy, primary age-related tauopathy, and hippocampal sclerosis, and for the biomarkers used to diagnose those diseases. Converging data indicate that the amygdala may represent a preferential locus for a pivotal transition from a relatively benign clinical condition to a more aggressive disease wherein multiple protein species are misfolded. Thus, understanding of amygdalar pathobiology may yield insights relevant to diagnoses and therapies; it is, however, a complex and imperfectly defined brain region. Here, we review aspects of amygdalar anatomy, connectivity, vasculature, and pathologic involvement in neurodegenerative diseases with supporting data from the University of Kentucky Alzheimer’s Disease Center autopsy cohort. Immunohistochemical staining of amygdalae for Aβ, Tau, α-synuclein, and TDP-43 highlight the often-coexisting pathologies. We suggest that the amygdala may represent an “incubator” for misfolded proteins and that it is possible that misfolded amygdalar protein species are yet to be discovered.
Keywords: Amyloid, Entorhinal, Hippocampus, Neuropathology, Proteomics, SNAP, Subpial
INTRODUCTION
Protein misfolding is a recurring theme in age-related neurodegenerative diseases: the brains of most elderly persons, with or without clinical dementia, contain >1 species of insoluble proteinaceous aggregates, often involving the amygdala (1–3). Here we review the literature on the pathobiology of the amygdala in common neurodegenerative diseases, with particular focus on protein aggregates. Excellent previously published articles have focused on related topics (4–13), including molecular mechanisms, which are not addressed in detail here. This review is oriented toward a broad readership interested in human brain pathology, and is organized as follows: (i) definition of key terms and conceptual overview; (ii) presentation of a hypothesis that has emerged from analyses of human studies, implicating pathologic synergies (as defined below) in the amygdala; (iii) review of the anatomy, connectivity, and vascularization of the amygdala; (iv) discussion of prior studies related to pathologic synergy in the amygdala, along with some relevant data from the University of Kentucky Alzheimer’s Disease Center (UK-ADC) autopsy cohort; (v) presentation of neuropathologic data on 6 representative persons’ amygdalae, including immunohistochemical stains for β-amyloid (Aβ), Tau, α-synuclein (α-SN), and TDP-43; and (vi) conclusions and possible future directions.
A primary assumption of this review article is that studies of autopsied human brains provide key contributions in the worldwide effort to advance the field of neurodegenerative disease research. At the very least, the exploration of human neuropathologies offers insights into phenomena that merit follow-up in other experimental contexts. Moreover, recent analyses of data from large autopsy series have enabled important conceptual breakthroughs. These studies (some described below) have revealed hitherto unexpected complexity and underscored that no other experimental system can fully recapitulate the unique milieu of the aged human brain. As such, research focusing on the neuropathologies of aged humans has exposed limitations of simplistic models while suggesting new opportunities for diagnostic and therapeutic strategies.
DEFINITIONS AND OVERVIEW
A “pathologic marker” refers to a microscopic structure interpretable to diagnose the presence of a disease, and possibly, as an indicator of disease severity (14). In neurodegenerative diseases, most pathologic markers are proteinaceous aggregates comprising peptide polymers and/or protein adducts (15). The proteinaceous aggregates of common neurodegenerative diseases include molecular fragments derived from the following genes (polypeptides): APP (Αβ), MAPT (Tau), SNCA (α-SN), and TARDBP (TAR-DNA binding protein 43 [TDP-43]). Over the course of neurodegenerative diseases, proteinaceous aggregates are deposited in predictable temporal and neuroanatomic patterns that may mirror the clinical features of disease. In other words, the presence and density of these pathologic markers correlate with decreased function in the affected regions (16, 17). However, the implications of each pathologic marker are context-specific and hence they are usually not pathognomonic. For example, a neurofibrillary tangle (NFT), composed of polymerized Tau protein, may be present in the brain as a result of infectious (18–20), neoplastic (21, 22), metabolic/developmental (23, 24), autoimmune (25), toxic (24, 26), or brain trauma-induced (27) conditions, in addition to the archetypal neurodegenerative diseases (such as Alzheimer disease [AD]), in which NFTs are seen (28, 29).
Nor are pathologic markers usually seen in isolation. In persons >80 years old, with or without frank dementia, it is the rule and not the exception for multiple pathologic markers to coexist in the same brain (2, 3, 30). One of the hypotheses underlying this review, which has been articulated previously, is that the pathologic aggregation of 1 protein can work synergistically to initiate or otherwise promote the aggregation of different protein species (7, 9, 31–34). This process is what we refer to with the term “pathologic synergy” (Fig. 1A). Pathologic synergy seems to be deleterious and is associated with accelerated cognitive impairment in dementing disorders (Fig. 1B, C) (12, 35–7).
FIGURE 1.
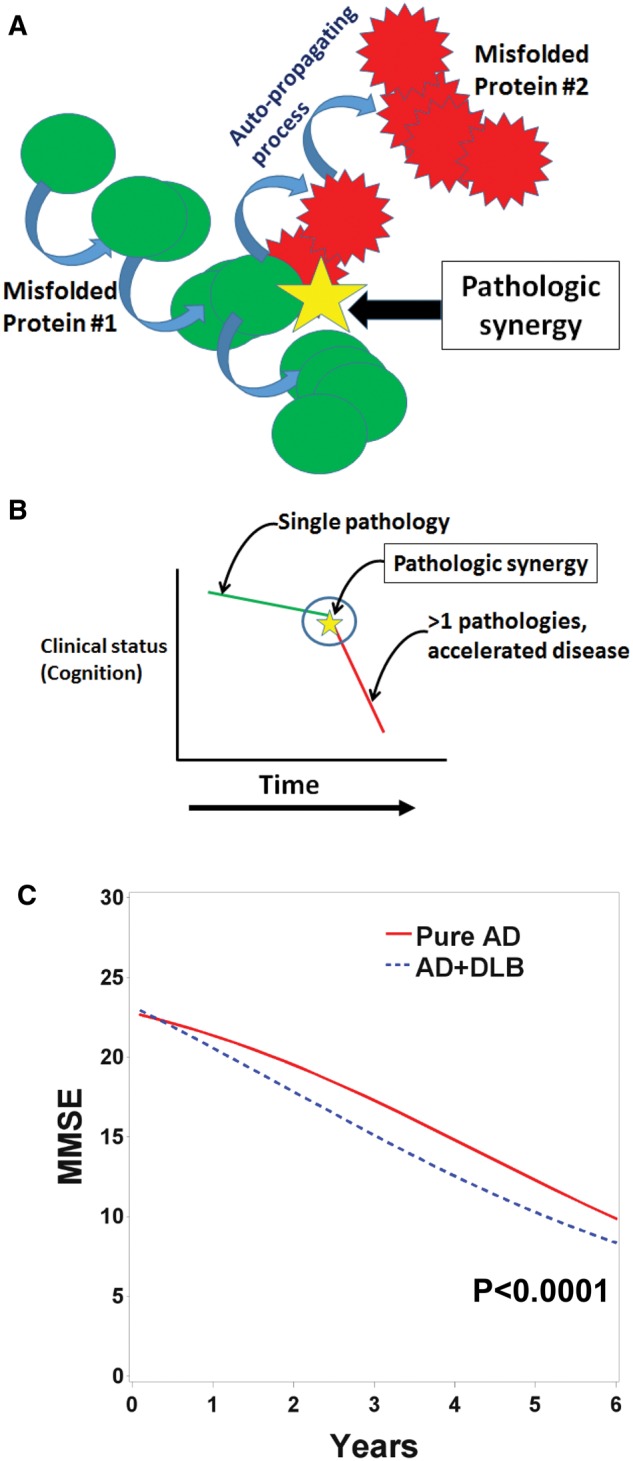
A combination of schematic illustration (A, B) and previously published data (C) help describe how pathologic synergies are associated with worsened clinical outcomes in neurodegenerative disease brains. (A) Cartoon depicts how one pathologic protein may interact with and promote the pathologic aggregation of another protein via mechanisms that are still not well understood; this is the process we are terming “pathologic synergy.” (B) The significance of pathologic synergies is that 1 misfolded protein can induce a conformational change in a separate protein species that may lead to altered (worsened) clinical course of the brain disease. (C) Data from the UK-ADC autopsy cohort provides support of this concept. The change of Mini-Mental State Exam (MMSE) scores over time, prior to death, is depicted using a statistical model of data from age-matched patients who died with Pure Alzheimer disease (AD) (n = 45) or AD with comorbid Lewy body disease, (AD + DLB, n = 20) pathologies, after having been followed longitudinally at the UK-ADC for >4 years before death. Note that the trajectory of cognitive impairment is more severe for AD + DLB patients (all with amygdala LBs) than Pure AD patients. The p value is derived from the “midpoint” parameter of the logistic model. Curves were generated using a 3-parameter logistic regression model (243) to allow between-cohort comparisons, as described in previously in detail (36).
The histomorphologic appearance of a pathologic marker may provide clues about its genesis. An example of a phenomenon that we interpret to indicate pathologic synergy is the neuritic amyloid plaque of AD, which contains both Αβ and Tau (Fig. 2A). More specifically, neuritic amyloid plaques are made up of extracellular Αβ deposits in close proximity with axons and dendrites that are both morphologically disfigured (38), and also packed with pathologic Tau filaments (39, 40). Because it has been demonstrated that axons from different extrinsic sources can show Tau-immunoreactive dystrophic features within a single neuritic amyloid plaque (41, 42), there are intuitive implications: apparently, a pathologic synergy existed in which some agent(s) in the extracellular plaque triggered neurochemical pathway(s) that promoted intracellular Tau misfolding. Details of this process remain incompletely understood; articles with proposed mechanisms are numerous (43–54), but a discussion of them is beyond the scope of this review. The physical nearness of misfolded Αβ and Tau within the plaque does not necessarily indicate that Αβ itself has a direct role in Tau pathology because many other potentially toxic molecules are also present in amyloid plaques (e.g. apolipoprotein E; Fig. 2B, C) (55–57). Nevertheless, even if Αβ in plaques is a proxy for other toxic agent(s), the evocative histomorphologic features of the neuritic plaque appear to support the conclusion of Barcikowska et al in regard to Tau pathology, i.e. it “may represent a nonspecific response … to different kinds of injuries, like the deposition of amyloid in Alzheimer disease” (58).
FIGURE 2.
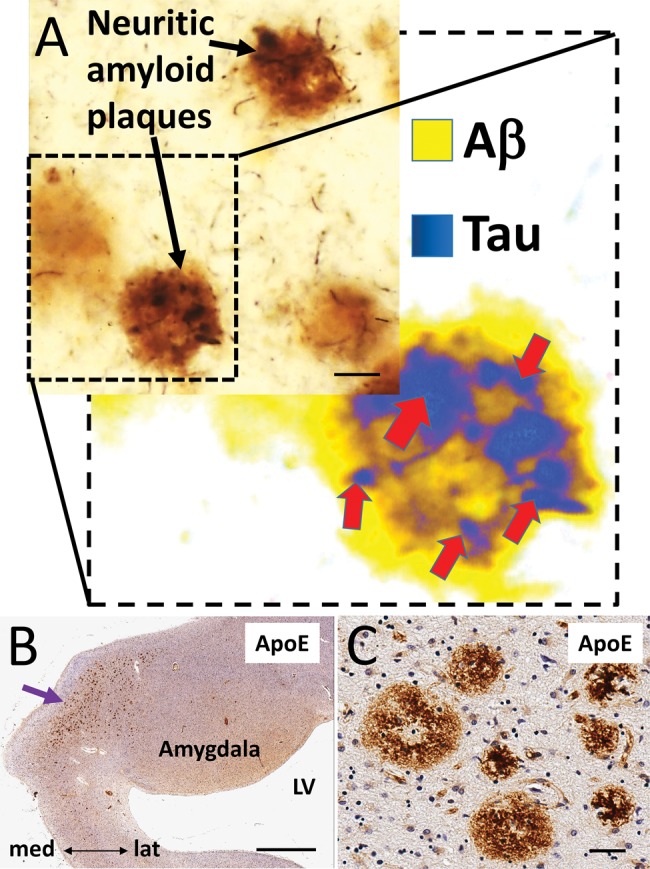
Neuritic amyloid plaques have histomorphologic features that suggest the existence of pathologic synergy. (A) A photomicrograph of an AD brain section immunolabeled for both Αβ (brown chromogen; R1280) and Tau (black chromogen; Alz-50). The inset shows a false-colored rendering to highlight how the intracellular Tau (blue/black) pathology is entangled with the extracellular Αβ (yellow) of the plaque. Whereas the close proximity of Αβ and Tau in the neuritic amyloid plaques conveys the potential for pathologic synergies between extracellular toxic substance(s) and Tau pathology, it does not prove direct mechanistic interactions. (B, C) There are numerous complicating variables including the many biologically active molecules other than Αβ that are also found in neuritic amyloid plaques. For example, Apolipoprotein E is visualized here, stained immunohistochemically in a human amygdala (panel B is low-power, C is high-magnification from the same section). The appearance of Apolipoprotein E-immunopositive plaques (clustered near the purple arrow) in this amygdala (B) resembles the pattern demonstrated by Unger et al (139) using a Tau antibody to visualize neuritic amyloid plaques. Scale bars: A = 100 µm; B = 500 µm; C = 70 µm.
Other phenomena observed in aged brains suggest additional pathologic synergies. For example, investigators have documented the colocalization of α-SN and Tau aggregates (59–64), TDP-43 and Tau aggregates (64–67), and TDP-43 and α-SN aggregates (64, 65, 68) within the same cells, although in some brain regions, the colocalization of comorbid pathologies appears only infrequently (69–72). The development of a finite set of protein aggregates across different brain diseases indicates that diverse upstream factors can lead to common downstream reactions; these reactions may overlap with adaptive (neuroplasticity and/or inflammatory) pathways (73–76), but may ultimately evolve to become harmful (34, 77–79). The deleterious influences of any proteinaceous aggregate may include its propensity to cause other proteins to misfold. Further, since some proteinopathic processes are hypothesized to spread through the brain in a “prion-like” manner (10, 11, 80), a logical focal-point is how the prion-like agent(s) were introduced. In summary, pathologic synergies represent a potential common mechanism for the initiation and expansion of misfolded protein species in the aged human brain.
HYPOTHESIS: PATHOLOGIC SYNERGY IN THE AMYGDALA
Cross-sectional data from large autopsy cohorts indicate that neurodegenerative diseases develop in the brain over decades; these findings are increasingly confirmed via clinical biomarker studies (81–84). From these studies, evidence has also been gathered in support of the hypothesis that particularly important pathological synergies occur in the amygdala. Before more detailed discussion below, we consider this hypothesis in the context of the 2 most commonly observed neurodegenerative conditions: primary age-related tauopathy (PART) and AD (Fig. 3).
FIGURE 3.

(A–C) Cartoons depicting how pathology develops in the brain during the course of 2 of the most common known neurodegenerative conditions: primary age-related tauopathy (PART) (A) and Alzheimer disease (AD) (B, C). In the course of PART (i.e. lacking Αβ plaques) there is expanding distribution of Tau/NFT pathology, starting from the brainstem and evolving to exist also in medial temporal lobe structures including the amygdala. (B) In early AD, amyloid plaques occur first in neocortical regions and evolve in a rostral to caudal (toward the brainstem) direction. For reasons currently not well understood, the presence of Αβ amyloid plaques correlates with a significant transition such that Tau/NFT pathology occurs in the neocortex. (C) In advanced AD, both plaques and tangles are widely distributed throughout many regions of the CNS, and apparent secondary/tertiary misfolding (α-SN and TDP-43 pathologies) occur preferentially in the amygdala.
In the absence of comorbid Aβ, α-SN, or TDP-43 pathologies, Tau/NFT pathology is thought to develop during the course of aging in all humans, evolving in a brainstem-toward-cortex direction, being first seen in locus coeruleus, and later in medial temporal lobes (85, 86). This pathology underlies a disease that is now referred to as PART (87). The maximum severity of PART pathology is limited to Braak NFT Stage IV (87, 88), due to pathophysiologic factors that are not currently understood. Relatively advanced PART pathology (Braak NFT Stage III/IV) is associated with clinical subjective memory complaints (89) and mild cognitive impairment (90).
The presence of Aβ plaques constitutes a sine qua non for the expanded distribution of NFTs to merit the designation of isocortical Braak NFT stages (V/VI), the pathologic substrates for most AD-type dementia (30, 88, 91). Pathologic synergy that occurs in the amygdala between amyloid plaques and Tau/NFT may facilitate the transition to more severe clinical disease (Fig. 3). Further, in brains with severe AD pathology by consensus-based definition (92), both Aβ plaque and NFT pathologies are widely distributed, but the amygdala has a special propensity to develop apparent secondary misfolding of TDP-43 and/or α-SN (Fig. 3C).
These observations and hypotheses raise additional questions: Where exactly in the amygdala or in the peri-amygdaloid regions (e.g. anterior hippocampus, basal forebrain, entorhinal cortex, and ambient gyrus) does the pathology occur? How is the amygdala involved during the course of other diseases, such as Lewy body diseases (LBDs), TDP-43 diseases, and non-AD tauopathies? What features of the amygdala make it prone to be involved in pathologic synergies? Are Aβ, Tau, α-SN, and TDP-43 pathologies the only proteinaceous aggregates that occur in the amygdala? Before discussing these topics, we will first address a more fundamental question: what is the amygdala?
THE HUMAN AMYGDALA: ANATOMY, CONNECTIVITY, AND VASCULARIZATION
The amygdala is a centrally located brain region that plays fundamental roles in human emotion, memory, and various homeostatic responses (93–97). Situated in the rostral part of the temporal lobe and abutting the basal forebrain, the human amygdala is closely connected with olfactory structures, the hippocampal formation, basal ganglia, basal forebrain components including ventral striatum and nucleus basalis, insula, claustrum, hypothalamus, and various thalamic nuclei. The amygdala contributes to white matter tracts that stream nearby, including the anterior commissure, inferior longitudinal fasciculus, stria terminalis, and uncinate fasciculus. Excellent descriptions of the developmental, phylogenetic, neurobehavioral, and neuroimaging aspects of the amygdala are available (94–96, 98–105).
Studies of the amygdala attest to the complexity of this imperfectly defined anatomic region. Morphologic variability of the amygdala is high between primate species (106), and even among individual humans, possibly in association with their life experiences (107, 108). There also have been inconsistencies in published studies of the human amygdala. For example, some prior studies have reported that the human amygdala averages ∼1.1 cm3 in volume (96, 102), whereas other studies reported that the volume of human amygdala averages ∼1.7 cm3 (103, 109), or even larger (110). These divergent results are presumably due to substantial discrepancies in how its boundaries were defined.
Regardless of its peripheral boundaries, what we are referring to as “amygdala” comprises separate cell groups according to multiple criteria of distinction (Supplementary DataFig. S1). It has been suggested that a more appropriate term would be “amygdaloid nuclear complex” (96, 111), and, even more provocatively (because “the amygdala is neither a structural nor a functional unit”), it could be described as an “arbitrarily defined set of cell groups” (93, 111, 112). Extension of amygdala-like neurons into the basal forebrain (so-called “extended amygdala,” briefly described below) further complicates the idea of a single, well-defined anatomic structure (113).
The amygdala can be parsed into between 4 and 32 different subnuclei, as defined by various investigators (93, 95, 96, 100, 101, 114). Six regions are generally acknowledged: the cortical/transitional zone, medial nucleus, central nucleus, lateral nucleus, basal nucleus, and accessory basal nucleus. To economize on detail, 3 main regions of the amygdala, similar to de Olmos et al (94), are discussed here (Fig. 4). They are the cortical/transitional zone, the anteromedial area (including medial and central nuclei), and the basolateral complex (comprising lateral, basal, and accessory basal nuclei).
FIGURE 4.

The amygdala is an approximately 1.5-cubic-centimeter amalgamation of cell groups, with complex connections that include regions of the brain that are vulnerable to neurodegenerative disease pathologies. (A) A schematically rendered axial/horizontal oriented profile of the amygdala to depict some surrounding structures, as well as the 3 main regions of the amygdala: the basolateral complex (green), the anteriomedial area (red), and the medial cortex-like zones (yellow), roughly following de Olmos et al (94); this is not an accurate depiction because not all of these structures are in the exact same plane. (B) The medial cortex-like zone of the amygdala is the least well understood in terms of connectivity in human brains, but has strong reciprocal relationships with the hippocampal formation, entorhinal cortex, and olfactory structures. (C, D) Inputs (C) and outputs (D) of the basolateral complex and anteriomedial region are better understood from prior studies performed in rodents and nonhuman primates. Within the amygdala, information flows from the basolateral and accessory nuclei, toward the anteromedial nuclei, from which derives the main amygdalar output to the diencephalon and brainstem. (E) Panel depicts a right hemibrain to show some of the many brain regions that are directly connected with the amygdala and have been strongly implicated in the earliest phases of various neurodegenerative diseases. More information on amygdala connectivity may be found in (93, 95, 96, 100, 101, 114). Acc. Bas.n., accessory basal nucleus; ag, ambient gyrus; AnCing, anterior cingulate cortex; ARAS, ascending reticular activating system; Bas.n., basal nucleus; Bsl. Forbr., basal forebrain, including magnocellular neurons (nucleus basalis); BNST, bed nucleus of the stria terminalis; Cau, tail of the caudate; Cent.Med.n.g., centromedian nuclear group; Claus, claustrum; Co.n., cortical nucleus; Co.A.Tr., cortico-amygdaloid transition area; cs, collateral sulcus; DMThal, dorsomedial thalamus; d.m.V., dorsal motor nucleus of the vagus nerve; Ectx, Entorhinal cortex; ers, endorhinal sulcus; fg, fusiform gyrus; Ins, insula; Lat.n., lateral nucleus; LC, locus coeruleus; NbM, nucleus basalis of Meynert; PAG, periaqueductal grey matter; PFC, prefrontal cortex; phg, parahippocampal gyrus; sg, semilunar gyrus; Str., striatum/ventral extension of putamen; TEctx, transentorhinal cortex.
The cortical/transitional zone of the amygdala seems to be affected relatively early in the course of multiple diseases (115, 116). Anatomically it consists of a “superficial cortex-like region” (96) along the medial aspect of the amygdala as well as an ill-defined region near the entorhinal cortex, which is termed the cortical-amygdaloid transition zone. Physical connection between this region and the entorhinal cortex is extensive; according to Insausti and Amaral, the entorhinal cortex exists both anterior and posterior to the amygdala (117). The cortical/transition zone has been referred to as the “olfactory amygdala” (94), but with strong connections with the olfactory bulbs, hippocampal formation and parahippocampal cortices, it also serves memory and cognitive functions (94, 96, 100). There remains much to be learned about the medial region of the human amygdala, including the exact demarcation of borders. As stated by Yilmazer-Hanke, this area “is the most controversial amygdaloid region with regard to classification … and delineation of its sectors” (100).
Better characterized are the anteromedial subnuclei of the amygdala, sometimes referred to as the “centromedial nuclear group” (Supplementary DataFig. S1). Neurons in these subnuclei have strong connectivity with autonomic, visceral, and sensory input-related structures. Within the anteromedial region, the medial nuclei of the amygdala also receive strong olfactory input (93). The anteromedial nuclei are continuous structurally and functionally with the sublenticular region and the bed nucleus of the stria terminalis, collectively termed the “extended amygdala” (96, 118). Considered the amygdala’s main output source, the central (particularly) and medial nuclei relay integrated stimuli to autonomic effector regions in the hypothalamus, forebrain, and brainstem, and thus the anteromedial region has been described as a striatum-like “somatomotor” center (100).
By volume, the basolateral nuclear complex comprises much of what is collectively referred to as amygdala, particularly the regions of amygdala overlying the temporal/inferior horn of the lateral ventricle. The lateral nucleus is the largest subnucleus of the human amygdala and is usually asymmetric, i.e. with the right side larger than the left (96, 119). Neurons in the basolateral complex of the amygdala receive widespread input from “higher-order association” cortex and hippocampus, in addition to sensory cortex and thalamus. Notably strong bidirectional communication links the basolateral nuclear complex with the prefrontal cortex and the dorsomedial thalamus. Because of these connections, this area has been termed the amygdala’s “frontotemporal system” (93).
Synaptic connectivity within the amygdala has been described through studies of nonhuman species. Connections flow from the basolateral complex into the anteromedial nuclei, modulating the main outputs to the diencephalon and brainstem that help control bodily functions that may manifest as emotion (93, 95, 96, 100, 101, 114). These pathways incorporate multimodal stimuli, while helping to integrate the circuits of memory and emotion. Remarkably, many of the brain regions that project to and/or from the amygdala are highly prone to develop pathology in neurodegenerative diseases (Fig. 4E).
It currently is not known why specific brain areas are most vulnerable in neurodegenerative diseases. With regard to special anatomic features of the human amygdala, we highlight the brain’s internal (ependyma) and external (pia mater) limiting layers (Fig. 5). The tissues directly internal to these layers (subependymal and subpial compartments) often stain positively for misfolded proteins in neurodegenerative diseases of aging. For example, TDP-43 pathology has been observed in the subependymal and subpial regions in the periamygdaloid region of individuals with neuropsychiatric conditions (120) and subpial and subependymal Tau pathology are characteristic of aging-related tau astrogliopathy (ARTAG) (121). These observations suggest the possibilities that these compartments are susceptible to protein misfolding pathology, or alternatively, the tissue may be in direct contact with disease-stimulating agent(s). The area immediately ventromedial to the amygdala is noteworthy for the close proximity of the ependyma and the pia, separated only by a layer of gray matter (Fig. 5).
FIGURE 5.

Select features of the anatomy and histology of the human amygdala. (A) Low-power photomicrograph of medial human amygdala to visualize how the lateral ventricle (LV) and medial pia mater (Pia) are juxtaposed, separated only by a ∼2.5-mm-band of peri-amygdaloid cortical gray matter. Hematoxylin and eosin. (B) This is shown in higher power. (C, D) Inset regions from (B), depicting the ependyma (Ep) (C) and the pia mater (D). (E–G) In the brains of older persons, the regions of the superficial (subpial) medial amygdala tend to show degenerative features, including gliosis and corpora amylacea (E); corpora amylacea are indicated with green arrows. Immunohistochemistry with anti-phospho-TDP-43 antibody (red arrowheads) highlights the subpial TDP-43 pathology (F, G); the TDP-43 pathology is in close proximity to a corpus amylacea. Scale bars: A = 2 mm; B = 800 µm; C = 100 µm; D = 150 µm; E = 60 µm; F = 125 µm; G = 25 µm.
There is increasing awareness that the “vascular” and “neurodegenerative” pathogenic processes may interact (122–124); therefore, an overview of the amygdala’s blood supply is germane to this discussion. The amygdala’s blood supply is complex, deriving from multiple arterial territories (96, 125, 126), with substantial interindividual variation (125). Further, this is a region rich in vascular anastomoses (125). Primary arterial input to the amygdala derives from the middle cerebral artery (96), and/or the anterior choroidal artery (127), but the amygdala often also receives arterial branches from the internal carotid artery and/or the posterior cerebral artery (125, 126). Huther et al concluded that “… the anterior choroidal artery supplies the posteromedial part of the amygdala and … the middle cerebral artery irrigates the anterolateral part…” (125). TDP-43 pathology in the amygdala may appear in a peculiar distribution around small blood vessels, suggesting a potential link between vascular pathology and misfolded proteins (128). Di Marino et al emphasized that the amygdala’s vascular supply and drainage are aspects of human biology that merit additional study: “The vascularization of the amygdala is much less studied than the vascularization of the neighboring hippocampus…” (96). The need for further relevant studies is particularly pressing in the context of brain aging, in which small vessel pathologies tend to be widespread and are associated with cognitive impairment (123, 129–132).
NEURODEGENERATIVE DISEASE PATHOLOGIES IN THE HUMAN AMYGDALA
Whereas the amygdala harbors misfolded proteins relatively early in multiple neurodegenerative diseases, many questions remain. As summarized by McDonald and Mott: “There are discrepancies in different reports as to which amygdalar nuclei are most affected, which may be related to individual variability and/or stages of the disease” (133). The involvement of each brain region in each disease can be registered in terms of the staging systems that have been created through careful study of neurodegenerative diseases in large autopsy series. These staging systems are helpful for defining the expected distribution of pathology in specific diseases (87, 134), and for gauging disease severity. The particular stages of neurodegenerative diseases during which the amygdala is first affected are depicted in Table 1. Relatively few recent human studies have described in detail the distribution of pathologic markers within subnuclei of the amygdala, to provide a sense of expected interindividual and interdisease variability. Following those caveats, our review focuses on amygdalar pathologies in AD, in LBDs, in TDP-43 proteinopathies, and in rarer human brain conditions. We also present some primary data from the UK-ADC autopsy cohort based on methods published in prior studies.
TABLE 1.
Amygdala and Selected Anatomical Regions in Neurodegenerative Disease Pathology Staging Systems
| Anatomic Region Reference | Classification Schemes [possible number of stages in each system] |
||||||
|---|---|---|---|---|---|---|---|
| Braak NFT Stages [0–VI] (136) | Thal Aβ Phases [0–5] (241) | AGD [0–3] (242) | Braak PD Stages [0–6] (151) | Amygdala-Predominant LBD*(166) | TDP-43 in AD [0–6] (188) | TDP-43 in Aging [0–3] (195) | |
| Below are the stages each anatomic region is first affected in each staging scheme | |||||||
| Amygdala | I–II | 2–3 | 1† | 4 | Early/Strong | 1 | 1 |
| Locus coeruleus | 0–i | 5 | n/a | 2 | 0/mild | n/a | n/a |
| Entorhinal cortex | I | 2 | 2 | 5 | n/a | 2 | 2 |
| Hippocampus | II | 2 | 2 | 4 | n/a | 2–3 | 2 |
| Neocortex-affected early | V | 1 | 3 | 6 | 0/mild | 6 | 3 |
| Neocortex-affected late | VI | n/a | n/a | 6 | 0 | n/a | 3 |
AGD, argyrophilic grain disease; PD, Parkinson disease; LBD, Lewy body disease; n/a, not applicable.
Amygdala-predominant LBD is not a stage-based scheme, and is currently considered “low-likelihood of dementia with Lewy bodies” (159).
Gyrus ambiens is immediately rostro-dorsal to the amygdala’s cortical/transition region with indistinct boundaries.
In terms of AD-type pathology, the amygdala is first affected by amyloid plaques in Thal Αβ stage 2 (135), and by NFTs in Braak NFT stage I–II (“a few isolated NFTs”) (136, 137). According to the seminal NFT staging paper of Braak and Braak, “…the corticomedial complex of the amygdala reveals the presence of many neuritic plaques, while NFT and neuropil threads predominate in the basolateral nuclei.” (136). Scott et al correspondingly highlighted atrophy in the basal nuclei in AD (138). Unger et al described a distinctive pattern of neuritic amyloid plaques early in the disease in the amygdala cortical transition zone (139), and Tsuchiya and Kosaka reported that the density of NFTs and cell loss was greatest in the corticomedial group in AD (140). Overall, it is generally agreed that neither plaques nor tangles are seen first in the amygdala but in late AD the amygdala harbors extensive Αβ amyloid and Tau pathology (141–148).
Additional insights can be gained if one considers the importance of pathologic synergies. In Figure 6, data from the UK-ADC autopsy cohort show that neocortical regions (dorsal prefrontal, inferior parietal, superior/mid-temporal neocortex, and occipital visual neocortex) are affected early in AD by neuritic amyloid plaques but not by NFTs. In sharp contrast, many hippocampal and entorhinal NFTs are present in the early stages of AD, whereas numerous neuritic amyloid plaques are seen later in those medial temporal lobe areas. The pathologic stages are oriented toward indicating where any pathology is recognized (Table 1). A more quantitative approach provides complementary information, in this case providing support for the hypothesis of early convergence of substantial densities of both neuritic amyloid plaques and NFTs in the amygdala (Fig. 6). This highlights a key transition-point of early AD at which some aspect of amyloid plaque biology correlates with (promotes?) a change from relatively benign PART to the more malignant, and possibly auto-propagating, widespread tauopathy of AD. Given the early concentration of numerous NFTs and neuritic amyloid plaques in the amygdala and its connectivity linking brainstem, allocortical, and neocortical structures, the amygdala is a credible anatomical location for this critical transition to occur.
FIGURE 6.

The amygdala (large red arrows) is distinguished by having relatively high numbers of both neuritic amyloid plaques and neurofibrillary tangles (NFTs), relatively early in the clinical course of Alzheimer disease. (A, B) Neuritic amyloid plaque counts (A) and NFT counts (B) from 7 different brain areas among 603 research subjects. Cases were stratified by final MMSE scores, which provide a gauge of global cognitive status. As expected, pathologic marker counts were higher in persons that experienced more severe antemortem cognitive impairment. Research subjects who had come to autopsy from the UK ADC cohort were the basis for the study, using the same methodology as described in detail in (244). The number of cases in each group of final MMSE scores: MMSE 26–30 (n = 47); MMSE 21–25 (n = 184); MMSE 16–20 (n = 63); MMSE 11–15 (n = 70); MMSE 6–10 (n = 53); and MMSE 0–5 (n = 186). Most of these individuals were last evaluated within 1 year of death. Information related to the UK-ADC cohort and neuropathology protocols, including lesion counting methods, were presented in detail elsewhere (2, 218, 244).
The amygdala also is the anatomical location where presumed secondary misfolding of α-SN and TDP-43 most often is observed in AD, particularly in advanced AD, when the amygdala appears to be a veritable “incubator” of protein misfolding. It has previously been shown that medial temporal lobe α-SN and TDP-43 pathologies with an epicenter in the amygdala are more likely to occur in advanced AD (Braak NFT stage VI) than in cases lacking, or with less severe, AD pathology (7, 65, 149, 150). Data from the UK-ADC autopsy series are shown in Figure 7. In this sample (n = 172), Braak NFT stage is associated with the combination of TDP-43 and LB pathologies ( p = 0.004; Fig. 7). Further examining the a priori hypothesis that subjects with both TDP-43 and LB are more likely to have advanced NFT pathology, we see that 61.1% of TDP-43-positive/LB-positive subjects were Braak NFT stage VI (61.1%) in comparison to 27.9% who were Braak NFT stage VI among those that lack either or both of TDP-43 and LB pathologies (p = 0.009). These data also underscore that a large percentage of brains harbor comorbid pathologies. Relatedly, both α-SN and TDP-43 pathologies occur in early-onset, autosomal dominant AD cases (see below), thereby providing added support for the hypothesis that these pathologies are downstream/secondary effects in brains with advanced AD pathology. However, LB pathology is also frequently seen in persons lacking advanced AD.
FIGURE 7.

In brains with Braak NFT Stage VI, there is a relatively high likelihood of having both TDP-43 and α-Synuclein (Lewy bodies, or LB) pathologies simultaneously in the medial temporal lobe (amygdala). We hypothesize that these data may indicate secondary (downstream of AD pathology) misfolding in advanced AD. Patients who had come to autopsy from UK ADC cohorts, and had hippocampal TDP-43 pathology and amygdala α-Synuclein neuropathologic assessment available (n = 172) were the basis for the study. For the statistical comparison of Braak NFT stage versus the combination of TDP-43 and LB pathologies, a 9 degree of freedom chi-square test with 10,000 Monte Carlo simulations conferred a p value of 0.004. In addition, a 2-sample test for proportions with a continuity correction was used to test the a priori hypothesis that the proportion of subjects with Braak stage VI NFTs was different between those with both TDP-43 and LB pathologies and individuals that lacked either or both TDP-43 and LB pathologies. The 2-sided test confers a p value of 0.009 suggesting a higher proportion of Stage VI in TDP-positive/LB-positive (61.1%) versus those that lacked TDP and/or LB pathology (27.9%). The number of cases in each group: TDP-negative/LB-negative (n = 93); TDP-negative/LB-positive (n = 17); TDP-positive/LB-negative (n = 44); TDP-positive/LB-positive (n = 18).
Prevalent LBDs include Parkinson disease (PD) and dementia with Lewy bodies (DLB). In a study of cases with a broad spectrum of PD severity, Braak et al reported that α-SN pathology develops in a predictable pattern with the dorsal motor nucleus of the vagus nerve affected first, followed by an expansion of the pathology through more brain regions in clinical PD (151, 152) (Table 1). Data from other high-quality autopsy series also support the hypothesis of a caudal-to-rostral directional spread of α-SN pathology in PD (153, 154). In the Braak PD staging scheme, the amygdala is affected in Stage 4 (out of 6) (137), and Braak et al also reported that the earliest areas affected in the amygdala are the central nucleus and the accessory cortical nucleus (155); Harding et al confirmed many early LBs in the cortical nucleus (156).
Yet there is appreciable interindividual variability in the anatomic distribution of α-SN pathology among LBD cases in comparison to AD cases (157–161). It may be that no single staging scheme could encompass all the persons who are affected by LBD while being predictive of the severity of each patient’s various clinical signs and symptoms. These findings partly indicate demographic differences in large autopsy cohorts. The reflections of Jellinger are also applicable: “It should be emphasized that interpretation of abnormal accumulations may be influenced by the conditions of staining, the antibodies used, of fixation” (153).
While cohort- and laboratory-specific technical factors are relevant, many clues implicate another potential source of variation in the anatomic distribution and clinical manifestations of α-SN pathology: pathologic synergies. For example, the presence of AD/Tau pathology seems to be associated with altered distribution of α-SN pathology. In a study of familial, early-onset AD brains, the amygdala showed α-SN pathology in >90% of patients with PSEN1 mutations and >70% of patients with PSEN2 mutations, whereas the dorsal motor nucleus of the vagus nerve usually was not affected (162). Further, studies from different centers have reported multiple brains where the amygdala was the area most strongly affected with α-SN pathology (160, 163–171), and still other cases where the olfactory bulb was the only brain area affected (172–174). A key observation is that α-SN aggregates tend to be present in diseases that also have Tau pathology (175). Further, misfolded Tau and α-SN were shown to colocalize in the same nerve cells, and those double-immunolabeled cells tended to be located in the limbic regions, particularly the amygdala (59, 176) and olfactory bulb (60). These findings were summarized by Schmidt et al, who studied Tau and α-SN inclusions in human brains: “…these 2 lesions frequently occurred together in the same neurons of the amygdala. These findings are in contrast in other sites that accumulate Lewy bodies and NFTs, but rarely both lesions in the same neuron” (176).
In some ways similar to α-SN pathology, TDP-43 pathology demonstrates tropism for the amygdala. TDP-43 pathology illustrates that the disease in which a pathological marker was discovered is not necessarily the most prevalent condition in which that misfolded protein may be exerting a deleterious impact on human brains. The phenomenon of pathologic misfolding of TDP-43 was discovered in diseases along the spectrum that include frontotemporal dementia (FTD), and its pathologic substrate frontotemporal lobar degeneration (FTLD), with or without motor neuron disease (177). However, it is now known that TDP-43 pathology is most likely to be present in individuals lacking either clinical FTD or motor neuron disease symptoms (37, 178, 179). Recent data indicate that the brains of 20%–50% of individuals in advanced old age contain TDP-43 pathology (34, 130, 179–185), in sharp contrast to FTLD, which has a lifetime incidence of ∼1:700 (186, 187). Thus, FTLD and motoneuron disease combined are more than a 100-fold less common than non-FTLD aging-associated TDP-43 pathology. This is important because the public health impact of TDP-43 pathology is far broader than was originally thought, with strong relevance to amygdalar pathology in elderly persons.
Analyses of data from large autopsy series have showed that the amygdala constitutes a primary anatomical location where TDP-43 pathology is observed in aged brains, with or without comorbid AD pathology (120, 149, 188–195). A significant proportion of subjects have been found to have TDP-43 pathology in the amygdala but essentially nowhere else in the brain: 18% (166/946) of the cohort in James et al (195), and 16% (31/193) in Josephs et al (188). As with α-SN pathology, TDP-43 pathology in the amygdala is a common, although not universal, feature of PSEN1 mutant early-onset AD cases (196).
There also is evidence for TDP-43 pathology, again like α-SN pathology, to be synergistic with Tau/NFT pathology. Non-AD tauopathy diseases, including argyrophilic grain disease (197), anti-IgLON5 tauopathy (198), dentate granule NFTs (66), corticobasal degeneration, and progressive supra-nuclear palsy (64, 199–202) have been reported to also have comorbid TDP-43 pathology, often both in the amygdala. Another intriguing [Tau + TDP-43] brain disease is chronic traumatic encephalopathy (CTE), which develops following repetitive trauma-induced brain injury (27, 71, 203). In the earliest stages of CTE, clusters of Tau and TDP-43 pathologic aggregates are observed in the depths of cerebral sulci and/or around blood vessels (27, 71, 204). Persons with this pathology may be normal functioning or they may manifest subtle symptoms that are relatively stable (205). An unknown, but possibly large, number of participants in contact sports have some version of this condition (206, 207). After years of clinical dormancy, even following decades during which traumatic injury had ceased, CTE may transform clinically into a progressive dementing disorder (208). In these cases, Tau and TDP-43 pathologies coexist in the amygdala and medial temporal lobe and spread through the cortex and brainstem (27, 67, 71, 208). This is yet another example where the amygdala region may be part of a pivotal transition wherein pathologic synergies contribute to what eventually becomes an auto-propagating, dementia-inducing neurodegenerative disease.
Although rare diseases (i.e. lifetime risk <1%) are not a focus of the current review, there are 2 themes that have emerged with direct relevance to this discussion. First, unusual diseases share with common ones the tendency for pathologic markers to be conspicuous in the amygdala. For example, in the parkinsonism-dementia complex of Guam, both Tau and α-SN aggregates are often colocalized in the amygdala (209–212). Second, there are many proteins other than Αβ, Tau, α-synuclein, or TDP-43 that misfold in the human brain, corresponding with rare neurodegenerative diseases: proteinaceous aggregates composed of misfolded fused in sarcoma (FUS), Rho-guanine nucleotide exchange factor (RGNEF), optineurin, and other proteins have been demonstrated within pathologic aggregates that correlate with neurological diseases (213–217). Thus, the amygdala appears to be a key nexus of neurodegenerative disease pathology, and proteins other than Αβ, Tau, α-SN, or TDP-43 may be prone to misfolding in the human brain.
SIX SELECTED CASES: Α β, TAU, α-SYNUCLEIN, AND TDP-43 in ADJACENT SECTIONS OF AMYGDALA
To convey how the common neurodegenerative disease-related pathologic markers are distributed in human amygdalae in a small sample, we show digitally rendered low-magnification photomicrographs from 6 human brains (Fig. 8). These individuals were followed longitudinally in a community-based cohort from normal clinical status at baseline examination until their eventual autopsy (by which time 4 individuals had documented cognitive impairment). The autopsies showed pathologic features commonly experienced in this autopsy series (2, 218) (Table 2). Immunohistochemical stains were used as described previously (66), along with digital pathologic methods that enable visualization of pathologic markers at low magnification (219). The consecutive adjacent sections of amygdala were all stained in the same order: Αβ (NAB228, gift from Dr. Eddie Lee; 1:10,000 dilution; positive staining in Table 2 indicates parenchymal plaque pathology), phospho-Tau (PHF-1, gift from Dr. Peter Davies; 1:500 dilution), α-synuclein (LB509, gift from Dr. Virginia Lee; 1:500 dilution), and phospho-TDP-43 (1D3 clone, EMD Millipore, Billerica, MA; 1:500 dilution). The next adjacent section was stained with the modified Klüver–Barrera technique to delineate the subfields of the amygdala and peri-amygdaloid regions. Labeling of the amygdalar anatomy was performed based on microscopic review of patient sections stained with a modified Klüver–Barrera protocol, incorporating 0.1% Luxol fast blue, Eosin Y, and a 0.5% aqueous solution of Cresyl violet. Anatomic references (220–226) were used in labeling the Klüver–Barrera-stained sections; the terminology of Crosby and Humphrey was applied for the amygdalar subnuclei (220). Note that each immunostain has distinct properties; for example, α-SN has the highest “background,” which is true normal staining.
FIGURE 8.

Histopathology of 6 amygdalae from the UK-ADC autopsy cohort. See Table 2 for demographic and pathologic information for these cases. The cases were selected to convey non-end-stage pathologies and relatively early clinical disease. Shown are 5 adjacent 8-µm-thick sections for each case. A Klüver–Barrera (modified) stained section (upper left) with labels is provided for anatomic orientation, followed by adjacent sections stained immunohistochemically for Aβ, phospho-Tau, α-Synuclein, and phospho-TDP-43. The immunopositive inclusions are highlighted using automated digital pathology (150, 219). Also shown in the lower right panel for each case is a “highlight” from one of the stained sections; the portion of the figure that is depicted as a highlight is indicated by a black arrow. Case 1 shows phospho-TDP-43 pathology, and phospho-Tau pathology that includes ARTAG (highlighted), particularly in the area medial to the lateral ventricle. Case 2 has large ectatic blood vessels between the lateral ventricle and the medial region of the amygdala, and near those blood vessels is scattered phospho-TDP-43 pathology (highlighted). Case 3 represents a relatively common clinical-pathologic scenario in the UK-ADC cohort: clinical dementia and a diagnosis of “Probable AD,” followed by an autopsy that revealed the presence of all 4 pathologic markers, including extensive phospho-TDP-43 pathology in the basal and lateral amygdalar subnuclei (highlighted). Case 4 also has both AD and α-synuclein pathologies. The highlighted region shows the pial surface with some scattered α-Synuclein aggregates (purple arrows). Case 5 is an interesting case of preclinical AD + LB, both within the amygdala and in the nearby cortex. The phospho-Tau antibody highlights abundant NFTs and neuritic plaques; the combination of dense plaques and tangles is rare in other brain areas. Case 6 is another case with preclinical or early disease (final MMSE score of 28), showing mild degree of LBD and phospho-TDP-43. The highlight depicts subpial Aβ deposits. Cases with comorbid Aβ, Tau, α-Synuclein, and phospho-TDP-43 pathologies are common among cognitively impaired subjects in this cohort, but are quite unusual among cognitively intact individuals (2) (Fig. 7). Abbreviations: ac – anterior commissure, ag – ambient gyrus, Acc. Bas.n. – Accessory basal nucleus, Bas.n. – basal nucleus, Bsl.Forbr. – basal forebrain, including magnocellular neurons (nucleus basalis), BV, blood vessel, Cent.Med.n.g. – Centromedian nuclear group, Claus – claustrum, Co.n. – Cortical nucleus, Co.A.Tr. – cortico-amygdaloid transition area, cs – collateral sulcus, Ectx - Entorhinal cortex, ers – endorhinal sulcus, fg – fusiform gyrus, Icm – intercalated cell mass, Ins – insula, Lat.n. – Lateral nucleus, lv – lateral ventricle, phg – parahippocampal gyrus, sas – semiannular sulcus, sg – semilunar gyrus, Sub – subiculum, Str. – striatum/ ventral extension of putamen, TEctx – transentorhinal cortex, TPC – temporopolar cortex (area TG, perirhinal cortex, Brodmann area 35-36).
TABLE 2.
Clinical and Pathological Data on 6 Cases From the University of Kentucky ADC Autopsy Cohort
| Case # | Age at Death (Years) | Final Clinical Diagnosis | Final Overall Diagnosis | Braak NFT Stage | Medial Temporal Lobe Pathology |
||
|---|---|---|---|---|---|---|---|
| Aβ+ | TDP-43 Rating (0–2) | α-Synuclein Severity Rating (0–4) | |||||
| 1 | 87 | MCI | CARTS + ARTAG + PART | III | N | 2 | 0 |
| 2 | 78 | AD | CARTS + PART | II | N | 2 | 0 |
| 3 | 88 | AD | AD + DLB + TDP-43 | IV | Y | 2 | 3 |
| 4 | 96 | AD + DLB | AD + DLB | V | Y | 0 | 4 |
| 5 | 87 | Normal | Preclinical AD (mixed) | V | Y | 1 | 2–3 |
| 6 | 96 | Normal | Preclinical AD (mixed) | V | Y | 1 | 1 |
AD, Alzheimer disease; ARTAG, aging-related tau astrogliopathy CARTS, cerebral age-related TDP-43 and sclerosis; DLB, dementia with Lewy bodies; N, no; MCI, mild cognitive impairment; PART, primary age-related tauopathy; Y, yes. Amygdalae were stained immunohistochemically for Aβ, Tau, TDP-43, and α-synuclein and are illustrated in Figure 8.
FIGURE 8.

Continued
FIGURE 8.

Continued
The stained brain sections showcase common patterns of pathologic markers seen in the UK-ADC cohort, and were selected to include pathologies in early and/or preclinical stages. From these photographs one can discern the following: (i) pathology often exists around the periphery of the amygdalae, near the meninges and/or lateral ventricle; (ii) peri-amygdaloid grey matter, including the entorhinal cortex, frequently shows pathologies; (iii) cortical and transitional regions also seem vulnerable to the accumulation of pathologic markers; and (iv) a constant in all cases among aged individuals is the presence of phospho-Tau pathology, which can be seen in gray or white matter, and within neurons (usually AD and/or PART) and/or astrocytes (usually ARTAG). As may be expected, there is not an easily deducible “one-to-one” overlap in the pathologies and the pathologic synergies (if they exist) may be specific for certain diseases and/or may be “seeded” within small subnuclei. A more systematic exploration of the anatomic associations between different pathologic markers requires far more cases and is beyond the scope of the current article.
ADDITIONAL CONSIDERATIONS AND CONCLUSIONS
In this review, we postulated that there is a synergistic relationship between misfolded protein species in some aged human brains and that these mechanisms may be extraordinarily strong within the amygdala. It is intuitively unrealistic to hypothesize that every neurodegenerative disease case is affected by pathologic synergies in the amygdala. And an alternative (arguably the null) hypothesis is that there is no pathologic synergy between misfolded proteins in neurodegenerative diseases, including in the amygdala. The underlying conditions in some aged brains may favor misfolding of multiple proteins, perhaps a “phenotype of neurodegeneration” (7). The misfolding of those proteins could be independent of each other. For example, the data in Figure 1C indicating more severe impairment for AD + DLB than pure AD may merely reflect the additive effect of separate underlying diseases. For there to be no pathologic synergies, many phenomena remain to be explained. For example, it is challenging to develop a hypothesis to explain the features of a neuritic amyloid plaque which does not include pathologic synergy (Fig. 2), and why, in the absence of pathologic synergy, there are α-SN and/or TDP-43 pathologies in middle-aged persons with APP mutations (227–229). However, it is useful to remind ourselves that biologic mechanisms in the brain may be neither all synergistic nor all independent of each other, but a combination of influences is quite possible.
Another concern is that much of the data gathered on pathologic synergies in neurodegeneration were from cross-sectional (postmortem) studies and therefore lack mechanistic insights. It remains unproven that any misfolded proteins in human brains are deleterious for the cells they are in or near. Whether or not a specific protein’s misfolding has a direct “toxic” impact, an assumption of this review is that the protein misfolding in the aged human brain heralds a deleterious process. This assumption is based on extensive prior studies from multiple institutions that indicate a strong correlation between the density and/or distribution of pathologic markers and clinical dysfunction (16, 17). If a pathologic marker were only a proxy for the presence of a “toxic oligomer,” or some other molecular adduct or agent(s), that fact could be important information for investigators seeking therapeutic strategies. However, if the correlation is reliable between a readily identified pathologic marker and something directly toxic that is less easily identified in situ, then the identifiable marker could still offer a robust indicator of the harmful process(es) that drive symptomatology.
We conclude by speculating that the amygdala may provide an anatomic setting for the pursuit of entirely new diagnostic and therapeutic targets. Given that the known pathologic protein species for common neurodegenerative diseases were characterized relatively recently—Αβ: 1984 (230), Tau: 1986–1987 (231–236), α-SN: 1997–1998 (237, 238), and TDP-43: 2006 (177, 239)—are we certain that there are no additional misfolded proteins that are directly relevant to prevalent brain diseases? And, if there are as yet unidentified proteins that could function as new pathologic markers and potential therapeutic targets, isn’t the amygdala the logical place to find them? Despite the great amount of scientific progress during the past century, the final sentences of Alois Alzheimer’s seminal case study, published in 1907, still ring true in translation: “There are without any doubt many more psychic illnesses than listed in our textbooks. In some of these instances a later histological examination will subsequently reveal peculiarities of the specific case. Then, we will gradually arrive at a stage, when we will be able to separate out individual disease from the large illness categories of our textbooks; to delineate them clinically more accurately.” (240).
Supplementary Material
ACKNOWLEDGMENTS
We are sincerely grateful for the research volunteers and colleagues at the University of Kentucky Alzheimer’s Disease Center. We thank Dr. Peter Davies, Dr. Eddie Lee, and Dr. Virginia Lee for antibodies and Dr. Frederick Schmitt for editorial comments.
REFERENCES
- 1. Rahimi J, Kovacs GG.. Prevalence of mixed pathologies in the aging brain. Alzheimers Res Ther 2014; 6:82.http://dx.doi.org/10.1186/s13195-014-0082-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nelson PT, Jicha GA, Schmitt FA, et al. Clinicopathologic correlations in a large Alzheimer disease center autopsy cohort: neuritic plaques and neurofibrillary tangles “do count” when staging disease severity. J Neuropathol Exp Neurol 2007; 66:1136–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schneider JA, Arvanitakis Z, Bang W, et al. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 2007; 69:2197–204http://dx.doi.org/10.1212/01.wnl.0000271090.28148.24 [DOI] [PubMed] [Google Scholar]
- 4. Milenkovic I, Petrov T, Kovacs GG.. Patterns of hippocampal tau pathology differentiate neurodegenerative dementias. Dement Geriatr Cogn Disord 2014; 38:375–88http://dx.doi.org/10.1159/000365548 [DOI] [PubMed] [Google Scholar]
- 5. Guo JL, Covell DJ, Daniels JP, et al. Distinct α-synuclein strains differentially promote tau inclusions in neurons. Cell 2013;154:103–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Clinton LK, Blurton-Jones M, Myczek K, et al. Synergistic Interactions between Aβ, tau, and α-synuclein: acceleration of neuropathology and cognitive decline. J Neurosci 2010;30:7281–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Spires-Jones TL, Attems J, Thal DR.. Interactions of pathological proteins in neurodegenerative diseases. Acta Neuropathol 2017; 134:187–205http://dx.doi.org/10.1007/s00401-017-1709-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lewis J, Dickson DW.. Propagation of tau pathology: hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol 2016; 131:27–48http://dx.doi.org/10.1007/s00401-015-1507-z [DOI] [PubMed] [Google Scholar]
- 9. Trojanowski JQ, Lee VM.. “Fatal attractions” of proteins. A comprehensive hypothetical mechanism underlying Alzheimer's disease and other neurodegenerative disorders. Ann N Y Acad Sci 2000; 924:62–7 [DOI] [PubMed] [Google Scholar]
- 10. Goedert M. NEURODEGENERATION. Alzheimer's and Parkinson's diseases: The prion concept in relation to assembled Aβ, tau, and α-synuclein. Science 2015;349:1255555. [DOI] [PubMed] [Google Scholar]
- 11. Jucker M, Walker LC.. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013; 501:45–51http://dx.doi.org/10.1038/nature12481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kapasi A, DeCarli C, Schneider JA.. Impact of multiple pathologies on the threshold for clinically overt dementia. Acta Neuropathol 2017; 134:171–86http://dx.doi.org/10.1007/s00401-017-1717-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nonaka T, Masuda-Suzukake M, Hasegawa M.. Molecular mechanisms of the co-deposition of multiple pathological proteins in neurodegenerative diseases. Neuropathology 2017. doi: 10.1111/neup.12427 [DOI] [PubMed] [Google Scholar]
- 14. Pellicciari C, Malatesta M.. Identifying pathological biomarkers: histochemistry still ranks high in the omics era. Eur J Histochem 2011; 55:e42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dugger BN, Dickson DW.. Pathology of neurodegenerative diseases. Cold Spring Harb Perspect Biol 2017; 9(7). pii: a028035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nelson PT, Alafuzoff I, Bigio EH, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. (Review). J Neuropathol Exp Neurol 2012; 71:362–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nelson PT, Braak H, Markesbery WR.. Neuropathology and cognitive impairment in Alzheimer disease: a complex but coherent relationship. J Neuropathol Exp Neurol 2009; 68:1–14http://dx.doi.org/10.1097/NEN.0b013e3181919a48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Paula-Barbosa MM, Brito R, Silva CA, et al. Neurofibrillary changes in the cerebral cortex of a patient with subacute sclerosing panencephalitis (SSPE). Acta Neuropathol 1979; 48:157–60http://dx.doi.org/10.1007/BF00691159 [DOI] [PubMed] [Google Scholar]
- 19. Ikeda K, Akiyama H, Kondo H, et al. Numerous glial fibrillary tangles in oligodendroglia in cases of subacute sclerosing panencephalitis with neurofibrillary tangles. Neurosci Lett 1995; 194:133–5http://dx.doi.org/10.1016/0304-3940(95)11713-7 [DOI] [PubMed] [Google Scholar]
- 20. Wong KT, Allen IV, McQuaid S, et al. An immunohistochemical study of neurofibrillary tangle formation in post-encephalitic Parkinsonism. Clin Neuropathol 1996; 15:22–5 [PubMed] [Google Scholar]
- 21. Brat DJ, Gearing M, Goldthwaite PT, et al. Tau-associated neuropathology in ganglion cell tumours increases with patient age but appears unrelated to ApoE genotype. Neuropathol Appl Neurobiol 2001; 27:197–205http://dx.doi.org/10.1046/j.1365-2990.2001.00311.x [DOI] [PubMed] [Google Scholar]
- 22. Batra A, Prayson RA.. Meningioangiomatosis associated with focal cortical dysplasia and neurofibrillary tangles. Clin Neuropathol 2013; 32:37–41http://dx.doi.org/10.5414/NP300501 [DOI] [PubMed] [Google Scholar]
- 23. Auer IA, Schmidt ML, Lee VM, et al. Paired helical filament tau (PHFtau) in Niemann-Pick type C disease is similar to PHFtau in Alzheimer's disease. Acta Neuropathol 1995; 90:547–51http://dx.doi.org/10.1007/BF00318566 [DOI] [PubMed] [Google Scholar]
- 24. Wisniewski K, Jervis GA, Moretz RC, et al. Alzheimer neurofibrillary tangles in diseases other than senile and presenile dementia. Ann Neurol 1979; 5:288–94http://dx.doi.org/10.1002/ana.410050311 [DOI] [PubMed] [Google Scholar]
- 25. Gelpi E, Hoftberger R, Graus F, et al. Neuropathological criteria of anti-IgLON5-related tauopathy. Acta Neuropathol 2016; 132:531–43http://dx.doi.org/10.1007/s00401-016-1591-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Niklowitz WJ, Mandybur TI.. Neurofibrillary changes following childhood lead encephalopathy. J Neuropathol Exp Neurol 1975; 34:445–55http://dx.doi.org/10.1097/00005072-197509000-00006 [DOI] [PubMed] [Google Scholar]
- 27. McKee AC, Cairns NJ, Dickson DW, et al. The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol 2016; 131:75–86http://dx.doi.org/10.1007/s00401-015-1515-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Williams DR. Tauopathies: classification and clinical update on neurodegenerative diseases associated with microtubule-associated protein tau. Intern Med J 2006; 36:652–60http://dx.doi.org/10.1111/j.1445-5994.2006.01153.x [DOI] [PubMed] [Google Scholar]
- 29. Goedert M, Spillantini MG.. Pathogenesis of the tauopathies. J Mol Neurosci 2011; 45:425–31http://dx.doi.org/10.1007/s12031-011-9593-4 [DOI] [PubMed] [Google Scholar]
- 30. Jicha GA, Abner EL, Schmitt FA, et al. Preclinical AD Workgroup staging: pathological correlates and potential challenges. Neurobiol Aging 2012; 33:622 e1–e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hardy J, Allsop D.. Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol Sci 1991; 12:383–8http://dx.doi.org/10.1016/0165-6147(91)90609-V [DOI] [PubMed] [Google Scholar]
- 32. Hardy J. Alzheimer's disease: the amyloid cascade hypothesis: an update and reappraisal. J Alzheimers Dis 2006; 9:151–3http://dx.doi.org/10.3233/JAD-2006-9S317 [DOI] [PubMed] [Google Scholar]
- 33. Irwin DJ, Lee VM, Trojanowski JQ.. Parkinson's disease dementia: convergence of α-synuclein, tau and amyloid-β pathologies. Nat Rev Neurosci 2013;14:626–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nelson PT, Trojanowski JQ, Abner EL, et al. “New Old Pathologies”: AD, PART, and cerebral age-related TDP-43 with sclerosis (CARTS). J Neuropathol Exp Neurol 2016; 75:482–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schneider JA, Arvanitakis Z, Yu L, et al. Cognitive impairment, decline and fluctuations in older community-dwelling subjects with Lewy bodies. Brain 2012; 135:3005–14http://dx.doi.org/10.1093/brain/aws234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nelson PT, Kryscio RJ, Abner EL, et al. Acetylcholinesterase inhibitor treatment is associated with relatively slow cognitive decline in patients with Alzheimer's disease and AD + DLB. J Alzheimers Dis 2009; 16:29–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nelson PT, Schmitt FA, Lin Y, et al. Hippocampal sclerosis in advanced age: clinical and pathological features. Brain 2011; 134:1506–18http://dx.doi.org/10.1093/brain/awr053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fischer O. Nekrosen mit drusigen Wucherungen der Neurofibrillen, eine regelmässige Veränderung der Hirnrinde bei seniler Demenz. Monatsschr Psychiat Neurol 1907; 22:361–72 [Google Scholar]
- 39. Merz PA, Wisniewski HM, Somerville RA, et al. Ultrastructural morphology of amyloid fibrils from neuritic and amyloid plaques. Acta Neuropathol 1983; 60:113–24http://dx.doi.org/10.1007/BF00685355 [DOI] [PubMed] [Google Scholar]
- 40. Yoshimura N. Evidence that paired helical filaments originate from neurofilaments–electron microscope observations of neurites in senile plaques in the brain in Alzheimer's disease. Clin Neuropathol 1984; 3:22–7 [PubMed] [Google Scholar]
- 41. Armstrong DM, Benzing WC, Evans J, et al. Substance P and somatostatin coexist within neuritic plaques: implications for the pathogenesis of Alzheimer's disease. Neuroscience 1989; 31:663–71http://dx.doi.org/10.1016/0306-4522(89)90431-4 [DOI] [PubMed] [Google Scholar]
- 42. Benzing WC, Mufson EJ, Armstrong DM.. Immunocytochemical distribution of peptidergic and cholinergic fibers in the human amygdala: their depletion in Alzheimer's disease and morphologic alteration in non-demented elderly with numerous senile plaques. Brain Res 1993; 625:125–38http://dx.doi.org/10.1016/0006-8993(93)90145-D [DOI] [PubMed] [Google Scholar]
- 43. Mandler M, Walker L, Santic R, et al. Pyroglutamylated amyloid-β is associated with hyperphosphorylated tau and severity of Alzheimer's disease. Acta Neuropathol 2014;128:67–79. [DOI] [PubMed] [Google Scholar]
- 44. Carter J, Lippa CF.. Beta-amyloid, neuronal death and Alzheimer's disease. Curr Mol Med 2001; 1:733–7http://dx.doi.org/10.2174/1566524013363177 [DOI] [PubMed] [Google Scholar]
- 45. Klein WL, Krafft GA, Finch CE.. Targeting small Aβ oligomers: the solution to an Alzheimer's disease conundrum?. Trends Neurosci 2001;24:219–24 [DOI] [PubMed] [Google Scholar]
- 46. Kayed R, Glabe CG.. Conformation-dependent anti-amyloid oligomer antibodies. Methods Enzymol 2006; 413:326–44 [DOI] [PubMed] [Google Scholar]
- 47. Butterfield DA, Reed T, Newman SF, et al. Roles of amyloid β-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer's disease and mild cognitive impairment. Free Radic Biol Med 2007;43:658–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Selkoe DJ. Soluble oligomers of the amyloid β-protein impair synaptic plasticity and behavior. Behav Brain Res 2008;192:106–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Koffie RM, Meyer-Luehmann M, Hashimoto T, et al. Oligomeric amyloid β associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A 2009;106:4012–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Darocha-Souto B, Scotton TC, Coma M, et al. Brain oligomeric β-amyloid but not total amyloid plaque burden correlates with neuronal loss and astrocyte inflammatory response in amyloid precursor protein/tau transgenic mice. J Neuropathol Exp Neurol 2011;70:360–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Glabe CG, Kayed R.. Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. Neurology 2006; 66:S74–8 [DOI] [PubMed] [Google Scholar]
- 52. Bolmont T, Clavaguera F, Meyer-Luehmann M, et al. Induction of tau pathology by intracerebral infusion of amyloid-β-containing brain extract and by amyloid-β deposition in APP × Tau transgenic mice. Am J Pathol 2007;171:2012–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. De Felice FG, Wu D, Lambert MP, et al. Alzheimer's disease-type neuronal tau hyperphosphorylation induced by A β oligomers. Neurobiol Aging 2008;29:1334–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jin M, Shepardson N, Yang T, et al. Soluble amyloid {β}-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci U S A 2011;108:5819–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Namba Y, Tomonaga M, Kawasaki H, et al. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer's disease and kuru plaque amyloid in Creutzfeldt–Jakob disease. Brain Res 1991; 541:163–6http://dx.doi.org/10.1016/0006-8993(91)91092-F [DOI] [PubMed] [Google Scholar]
- 56. Gozal YM, Cheng D, Duong DM, et al. Merger of laser capture microdissection and mass spectrometry: a window into the amyloid plaque proteome. Methods Enzymol 2006; 412:77–93 [DOI] [PubMed] [Google Scholar]
- 57. Sengupta U, Nilson AN, Kayed R.. The role of amyloid-β oligomers in toxicity, propagation, and immunotherapy. EBioMedicine 2016;6:42–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Barcikowska M, Wisniewski HM, Bancher C, et al. About the presence of paired helical filaments in dystrophic neurites participating in the plaque formation. Acta Neuropathol 1989; 78:225–31http://dx.doi.org/10.1007/BF00687751 [DOI] [PubMed] [Google Scholar]
- 59. Marui W, Iseki E, Ueda K, et al. Occurrence of human α-synuclein immunoreactive neurons with neurofibrillary tangle formation in the limbic areas of patients with Alzheimer's disease. J Neurol Sci 2000;174:81–4 [DOI] [PubMed] [Google Scholar]
- 60. Fujishiro H, Tsuboi Y, Lin WL, et al. Co-localization of tau and α-synuclein in the olfactory bulb in Alzheimer's disease with amygdala Lewy bodies. Acta Neuropathol 2008;116:17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ishizawa T, Mattila P, Davies P, et al. Colocalization of tau and α-synuclein epitopes in Lewy bodies. J Neuropathol Exp Neurol 2003;62:389–97. [DOI] [PubMed] [Google Scholar]
- 62. Yancopoulou D, Xuereb JH, Crowther RA, et al. Tau and α-synuclein inclusions in a case of familial frontotemporal dementia and progressive aphasia. J Neuropathol Exp Neurol 2005;64:245–53 [DOI] [PubMed] [Google Scholar]
- 63. Hishikawa N, Hashizume Y, Ujihira N, et al. Alpha-synuclein-positive structures in association with diffuse neurofibrillary tangles with calcification. Neuropathol Appl Neurobiol 2003; 29:280–7http://dx.doi.org/10.1046/j.1365-2990.2003.00470.x [DOI] [PubMed] [Google Scholar]
- 64. Yamashita S, Sakashita N, Yamashita T, et al. Concomitant accumulation of α-synuclein and TDP-43 in a patient with corticobasal degeneration. J Neurol 2014;261:2209–17. [DOI] [PubMed] [Google Scholar]
- 65. Higashi S, Iseki E, Yamamoto R, et al. Concurrence of TDP-43, tau and α-synuclein pathology in brains of Alzheimer's disease and dementia with Lewy bodies. Brain Res 2007;1184:284–94. [DOI] [PubMed] [Google Scholar]
- 66. Smith VD, Bachstetter AD, Ighodaro E, et al. Overlapping but distinct TDP-43 and tau pathologic patterns in aged hippocampi. Brain Pathol 2017. doi: 10.1111/bpa.12505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Saing T, Dick M, Nelson PT, et al. Frontal cortex neuropathology in dementia pugilistica. (Research Support, N.I.H., Extramural). J Neurotrauma 2012; 29:1054–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kokoulina P, Rohn TT.. Caspase-cleaved transactivation response DNA-binding protein 43 in Parkinson's disease and dementia with Lewy bodies. Neurodegener Dis 2010; 7:243–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hosokawa M, Kondo H, Serrano GE, et al. Accumulation of multiple neurodegenerative disease-related proteins in familial frontotemporal lobar degeneration associated with granulin mutation. Sci Rep 2017; 7:1513.http://dx.doi.org/10.1038/s41598-017-01587-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fujishiro H, Uchikado H, Arai T, et al. Accumulation of phosphorylated TDP-43 in brains of patients with argyrophilic grain disease. Acta Neuropathol 2009; 117:151–8http://dx.doi.org/10.1007/s00401-008-0463-2 [DOI] [PubMed] [Google Scholar]
- 71. McKee AC, Gavett BE, Stern RA, et al. TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol 2010; 69:918–29http://dx.doi.org/10.1097/NEN.0b013e3181ee7d85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Nakashima-Yasuda H, Uryu K, Robinson J, et al. Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol 2007; 114:221–9http://dx.doi.org/10.1007/s00401-007-0261-2 [DOI] [PubMed] [Google Scholar]
- 73. Castellani RJ, Nunomura A, Lee HG, et al. Phosphorylated tau: toxic, protective, or none of the above. J Alzheimers Dis 2008; 14:377–83http://dx.doi.org/10.3233/JAD-2008-14404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Castellani RJ, Lee HG, Siedlak SL, et al. Reexamining Alzheimer's disease: evidence for a protective role for amyloid-β protein precursor and amyloid-β. J Alzheimers Dis 2009;18:447–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Andreasson KI, Bachstetter AD, Colonna M, et al. Targeting innate immunity for neurodegenerative disorders of the central nervous system. J Neurochem 2016; 138:653–93http://dx.doi.org/10.1111/jnc.13667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Latta CH, Brothers HM, Wilcock DM.. Neuroinflammation in Alzheimer's disease; A source of heterogeneity and target for personalized therapy. Neuroscience 2015; 302:103–11http://dx.doi.org/10.1016/j.neuroscience.2014.09.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Goedert M. The ordered assembly of tau is the gain-of-toxic function that causes human tauopathies. Alzheimers Dement 2016; 12:1040–50http://dx.doi.org/10.1016/j.jalz.2016.09.001 [DOI] [PubMed] [Google Scholar]
- 78. Iguchi Y, Katsuno M, Takagi S, et al. Oxidative stress induced by glutathione depletion reproduces pathological modifications of TDP-43 linked to TDP-43 proteinopathies. Neurobiol Dis 2012; 45:862–70http://dx.doi.org/10.1016/j.nbd.2011.12.002 [DOI] [PubMed] [Google Scholar]
- 79. Kovacs GG. Molecular pathological classification of neurodegenerative diseases: turning towards precision medicine. Int J Mol Sci 2016;17:189.http://dx.doi.org/10.3390/ijms17020189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Frost B, Diamond MI.. Prion-like mechanisms in neurodegenerative diseases. Nat Rev Neurosci 2010; 11:155–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Schwarz AJ, Yu P, Miller BB, et al. Regional profiles of the candidate tau PET ligand 18F-AV-1451 recapitulate key features of Braak histopathological stages. Brain 2016; 139:1539–50http://dx.doi.org/10.1093/brain/aww023 [DOI] [PubMed] [Google Scholar]
- 82. Scholl M, Lockhart SN, Schonhaut DR, et al. PET imaging of Tau deposition in the aging human brain. Neuron 2016; 89:971–82http://dx.doi.org/10.1016/j.neuron.2016.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Dugger BN, Clark CM, Serrano G, et al. Neuropathologic heterogeneity does not impair florbetapir-positron emission tomography postmortem correlates. J Neuropathol Exp Neurol 2014; 73:72–80http://dx.doi.org/10.1097/NEN.0000000000000028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Murray ME, Lowe VJ, Graff-Radford NR, et al. Clinicopathologic and 11C-Pittsburgh compound B implications of Thal amyloid phase across the Alzheimer's disease spectrum. Brain 2015; 138:1370–81http://dx.doi.org/10.1093/brain/awv050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Braak H, Thal DR, Ghebremedhin E, et al. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol 2011; 70:960–9http://dx.doi.org/10.1097/NEN.0b013e318232a379 [DOI] [PubMed] [Google Scholar]
- 86. Haroutunian V, Purohit DP, Perl DP, et al. Neurofibrillary tangles in nondemented elderly subjects and mild Alzheimer disease. Arch Neurol 1999; 56:713–8http://dx.doi.org/10.1001/archneur.56.6.713 [DOI] [PubMed] [Google Scholar]
- 87. Crary JF, Trojanowski JQ, Schneider JA, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 2014; 128:755–66http://dx.doi.org/10.1007/s00401-014-1349-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Nelson PT, Abner EL, Schmitt FA, et al. Brains with medial temporal lobe neurofibrillary tangles but no neuritic amyloid plaques are a diagnostic dilemma but may have pathogenetic aspects distinct from Alzheimer disease. J Neuropathol Exp Neurol 2009; 68:774–84http://dx.doi.org/10.1097/NEN.0b013e3181aacbe9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kryscio RJ, Abner EL, Jicha GA, et al. Self-reported memory complaints: a comparison of demented and unimpaired outcomes. J Prev Alzheimer's Dis 2015; 3:13–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Abner EL, Kryscio RJ, Schmitt FA, et al. Outcomes after diagnosis of mild cognitive impairment in a large autopsy series. Ann Neurol 2017; 81:549–59http://dx.doi.org/10.1002/ana.24903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Mungas D, Tractenberg R, Schneider JA, et al. A 2-process model for neuropathology of Alzheimer's disease. Neurobiol Aging 2014; 35:301–8http://dx.doi.org/10.1016/j.neurobiolaging.2013.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. (Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't). Alzheimers Dement 2012; 8:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Swanson LW, Petrovich GD.. What is the amygdala?. Trends Neurosci 1998; 21:323–31http://dx.doi.org/10.1016/S0166-2236(98)01265-X [DOI] [PubMed] [Google Scholar]
- 94. De Olmos J, Amygdala In: Paxinos G, Mai JK, eds. The Human Nervous System. 2 ed.Boston, MA, USA: Academic Press; 2004: 739–868 [Google Scholar]
- 95. LeDoux J. The amygdala. Curr Biol 2007; 17:R868–74 [DOI] [PubMed] [Google Scholar]
- 96. Di Marino V, Etienne Y, Niddam M, The Amygdaloid Nuclear Complex: Anatomic Study of the Human Amygdala. New York: Springer, 2016 [Google Scholar]
- 97. Bickart KC, Dickerson BC, Barrett LF.. The amygdala as a hub in brain networks that support social life. Neuropsychologia 2014; 63:235–48http://dx.doi.org/10.1016/j.neuropsychologia.2014.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. O'Rahilly R, Muller F, The Embryonic Human Brain: An Atlas of Developmental Stages. 2 ed.New York, NY, USA: Wiley-Liss, 1999 [Google Scholar]
- 99. Butler AB, Hodos W, Comparative Vertebrate Neuroanatomy: Evolution and Adaptation. 2 ed.New York, NY, USA: Wiley-Liss, 2005 [Google Scholar]
- 100. Yilmazer-Hanke DM, Amygdala In: Mai JK, Paxinos G, eds. The Human Nervous System. 3 ed.Boston: Academic Press (Elsevier; ), 2012:759–835 [Google Scholar]
- 101. Baxter MG, Murray EA.. The amygdala and reward. Nat Rev Neurosci 2002; 3:563–73http://dx.doi.org/10.1038/nrn875 [DOI] [PubMed] [Google Scholar]
- 102. Pruessner JC, Li LM, Serles W, et al. Volumetry of hippocampus and amygdala with high-resolution MRI and three-dimensional analysis software: minimizing the discrepancies between laboratories. Cereb Cortex 2000; 10:433–42http://dx.doi.org/10.1093/cercor/10.4.433 [DOI] [PubMed] [Google Scholar]
- 103. Brabec J, Rulseh A, Hoyt B, et al. Volumetry of the human amygdala: an anatomical study. Psychiatry Res 2010; 182:67–72http://dx.doi.org/10.1016/j.pscychresns.2009.11.005 [DOI] [PubMed] [Google Scholar]
- 104. Smith CD, Andersen AH, Gold BT, et al. Structural brain alterations before mild cognitive impairment in ADNI: validation of volume loss in a predefined antero-temporal region. J Alzheimers Dis 2012; 31:S49–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Saygin ZM, Kliemann D, Iglesias JE, et al. High-resolution magnetic resonance imaging reveals nuclei of the human amygdala: manual segmentation to automatic atlas. Neuroimage 2017; 155:370–82http://dx.doi.org/10.1016/j.neuroimage.2017.04.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Barger N, Stefanacci L, Schumann CM, et al. Neuronal populations in the basolateral nuclei of the amygdala are differentially increased in humans compared with apes: a stereological study. J Comp Neurol 2012; 520:3035–54http://dx.doi.org/10.1002/cne.23118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Kuo JR, Kaloupek DG, Woodward SH.. Amygdala volume in combat-exposed veterans with and without posttraumatic stress disorder: a cross-sectional study. Arch Gen Psychiatry 2012; 69:1080–6http://dx.doi.org/10.1001/archgenpsychiatry.2012.73 [DOI] [PubMed] [Google Scholar]
- 108. Morey RA, Gold AL, LaBar KS, et al. Amygdala volume changes in posttraumatic stress disorder in a large case-controlled veterans group. Arch Gen Psychiatry 2012; 69:1169–78http://dx.doi.org/10.1001/archgenpsychiatry.2012.50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Brierley B, Shaw P, David AS.. The human amygdala: a systematic review and meta-analysis of volumetric magnetic resonance imaging. Brain Res Brain Res Rev 2002; 39:84–105http://dx.doi.org/10.1016/S0165-0173(02)00160-1 [DOI] [PubMed] [Google Scholar]
- 110. Watson C, Andermann F, Gloor P, et al. Anatomic basis of amygdaloid and hippocampal volume measurement by magnetic resonance imaging. Neurology 1992; 42:1743–50http://dx.doi.org/10.1212/WNL.42.9.1743 [DOI] [PubMed] [Google Scholar]
- 111. McDonald AJ. Is there an amygdala and how far does it extend? An anatomical perspective. Ann N Y Acad Sci 2003; 985:1–21http://dx.doi.org/10.1111/j.1749-6632.2003.tb07067.x [DOI] [PubMed] [Google Scholar]
- 112. Swanson LW. The amygdala and its place in the cerebral hemisphere. Ann N Y Acad Sci 2003; 985:174–84http://dx.doi.org/10.1111/j.1749-6632.2003.tb07081.x [DOI] [PubMed] [Google Scholar]
- 113. Alheid GF. Extended amygdala and basal forebrain. Ann N Y Acad Sci 2003; 985:185–205http://dx.doi.org/10.1111/j.1749-6632.2003.tb07082.x [DOI] [PubMed] [Google Scholar]
- 114. Sah P, Faber ES, Lopez De Armentia M, et al. The amygdaloid complex: anatomy and physiology. Physiol Rev 2003; 83:803–34http://dx.doi.org/10.1152/physrev.00002.2003 [DOI] [PubMed] [Google Scholar]
- 115. Price JL, Davis PB, Morris JC, et al. The distribution of tangles, plaques and related immunohistochemical markers in healthy aging and Alzheimer's disease. Neurobiol Aging 1991; 12:295–312http://dx.doi.org/10.1016/0197-4580(91)90006-6 [DOI] [PubMed] [Google Scholar]
- 116. Ighodaro ET, Jicha GA, Schmitt FA, et al. Hippocampal sclerosis of aging can be segmental: two cases and review of the literature. J Neuropathol Exp Neurol 2015; 74:642–52http://dx.doi.org/10.1097/NEN.0000000000000204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Insausti R, Amaral DG, Hippocampal Formation In: Mai J, Paxinos G, eds. The Human Nervous System. 3 ed.New York, NY, USA: Academic Press, 2012: 896–942 [Google Scholar]
- 118. Fudge JL, Haber SN.. Bed nucleus of the stria terminalis and extended amygdala inputs to dopamine subpopulations in primates. Neuroscience 2001; 104:807–27http://dx.doi.org/10.1016/S0306-4522(01)00112-9 [DOI] [PubMed] [Google Scholar]
- 119. Murphy GM Jr, Inger P, Mark K, et al. Volumetric asymmetry in the human amygdaloid complex. J Hirnforsch 1987;28:281–9 [PubMed] [Google Scholar]
- 120. Geser F, Robinson JL, Malunda JA, et al. Pathological 43-kDa transactivation response DNA-binding protein in older adults with and without severe mental illness. Arch Neurol 2010; 67:1238–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Kovacs GG, Ferrer I, Grinberg LT, et al. Aging-related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta Neuropathol 2016; 131:87–102http://dx.doi.org/10.1007/s00401-015-1509-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Snyder HM, Corriveau RA, Craft S, et al. Vascular contributions to cognitive impairment and dementia including Alzheimer's disease. Alzheimers Dement 2015; 11:710–7http://dx.doi.org/10.1016/j.jalz.2014.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Wilcock DM, Schmitt FA, Head E.. Cerebrovascular contributions to aging and Alzheimer's disease in Down syndrome. Biochim Biophys Acta 2016; 1862:909–14http://dx.doi.org/10.1016/j.bbadis.2015.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Nelson PT, Jicha GA, Wang WX, et al. ABCC9/SUR2 in the brain: Implications for hippocampal sclerosis of aging and a potential therapeutic target. Ageing Res Rev 2015; 24:111–25http://dx.doi.org/10.1016/j.arr.2015.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Huther G, Dorfl J, Van der Loos H, et al. Microanatomic and vascular aspects of the temporomesial region. Neurosurgery 1998; 43:1118–36http://dx.doi.org/10.1097/00006123-199811000-00065 [DOI] [PubMed] [Google Scholar]
- 126. Wen HT, Rhoton AL Jr, de Oliveira E, et al. Microsurgical anatomy of the temporal lobe: part 1: mesial temporal lobe anatomy and its vascular relationships as applied to amygdalohippocampectomy. Neurosurgery 1999;45:549–91 discussion 91-2 [DOI] [PubMed] [Google Scholar]
- 127. Herman LH, Fernando OU, Gurdjian ES.. The anterior choroidal artery: an anatomical study of its area of distribution. Anat Rec 1966; 154:95–101http://dx.doi.org/10.1002/ar.1091540109 [DOI] [PubMed] [Google Scholar]
- 128. Lin WL, Castanedes-Casey M, Dickson DW.. Transactivation response DNA-binding protein 43 microvasculopathy in frontotemporal degeneration and familial Lewy body disease. J Neuropathol Exp Neurol 2009; 68:1167–76http://dx.doi.org/10.1097/NEN.0b013e3181baacec [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Ighodaro ET, Abner EL, Fardo DW, et al. Risk factors and global cognitive status related to brain arteriolosclerosis in elderly individuals. J Cereb Blood Flow Metab 2017; 37:201–16http://dx.doi.org/10.1177/0271678X15621574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Neltner JH, Abner EL, Jicha GA, et al. Brain pathologies in extreme old age. Neurobiol Aging 2016; 37:1–11http://dx.doi.org/10.1016/j.neurobiolaging.2015.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Lim AS, Yu L, Schneider JA, et al. Sleep fragmentation, cerebral arteriolosclerosis, and brain infarct pathology in community-dwelling older people. Stroke 2016; 47:516–8http://dx.doi.org/10.1161/STROKEAHA.115.011608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Nelson PT, Head E, Schmitt FA, et al. Alzheimer's disease is not “brain aging”: neuropathological, genetic, and epidemiological human studies. Acta Neuropathol 2011; 121:571–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. McDonald AJ, Mott DD.. Functional neuroanatomy of amygdalohippocampal interconnections and their role in learning and memory. J Neurosci Res 2017; 95:797–820http://dx.doi.org/10.1002/jnr.23709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol 2012; 123:1–11http://dx.doi.org/10.1007/s00401-011-0910-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Thal DR, Capetillo-Zarate E, Del Tredici K, et al. The development of amyloid β protein deposits in the aged brain. Sci Aging Knowledge Environ 2006;2006:re1. [DOI] [PubMed] [Google Scholar]
- 136. Braak H, Braak E.. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991; 82:239–59http://dx.doi.org/10.1007/BF00308809 [DOI] [PubMed] [Google Scholar]
- 137. Braak H, Braak E, Yilmazer D, et al. Pattern of brain destruction in Parkinson's and Alzheimer's diseases. J Neural Transm (Vienna) 1996; 103:455–90http://dx.doi.org/10.1007/BF01276421 [DOI] [PubMed] [Google Scholar]
- 138. Scott SA, DeKosky ST, Scheff SW.. Volumetric atrophy of the amygdala in Alzheimer's disease: quantitative serial reconstruction. Neurology 1991; 41:351–6http://dx.doi.org/10.1212/WNL.41.3.351 [DOI] [PubMed] [Google Scholar]
- 139. Unger JW, Lapham LW, McNeill TH, et al. The amygdala in Alzheimer's disease: neuropathology and Alz 50 immunoreactivity. Neurobiol Aging 1991; 12:389–99http://dx.doi.org/10.1016/0197-4580(91)90063-P [DOI] [PubMed] [Google Scholar]
- 140. Tsuchiya K, Kosaka K.. Neuropathological study of the amygdala in presenile Alzheimer's disease. J Neurol Sci 1990; 100:165–73http://dx.doi.org/10.1016/0022-510X(90)90029-M [DOI] [PubMed] [Google Scholar]
- 141. Hopper MW, Vogel FS.. The limbic system in Alzheimer's disease. A neuropathologic investigation. Am J Pathol 1976; 85:1–20 [PMC free article] [PubMed] [Google Scholar]
- 142. Benzing WC, Ikonomovic MD, Brady DR, et al. Evidence that transmitter-containing dystrophic neurites precede paired helical filament and Alz-50 formation within senile plaques in the amygdala of nondemented elderly and patients with Alzheimer's disease. J Comp Neurol 1993; 334:176–91http://dx.doi.org/10.1002/cne.903340203 [DOI] [PubMed] [Google Scholar]
- 143. Kromer Vogt LJ, Hyman BT, Van Hoesen GW, et al. Pathological alterations in the amygdala in Alzheimer's disease. Neuroscience 1990; 37:377–85http://dx.doi.org/10.1016/0306-4522(90)90408-V [DOI] [PubMed] [Google Scholar]
- 144. Price JL, Morris JC.. Tangles and plaques in nondemented aging and “preclinical” Alzheimer's disease. Ann Neurol 1999; 45:358–68 [DOI] [PubMed] [Google Scholar]
- 145. Herzog AG, Kemper TL.. Amygdaloid changes in aging and dementia. Arch Neurol 1980; 37:625–9http://dx.doi.org/10.1001/archneur.1980.00500590049006 [DOI] [PubMed] [Google Scholar]
- 146. Sahin HA, Emre M, Ziabreva I, et al. The distribution pattern of pathology and cholinergic deficits in amygdaloid complex in Alzheimer's disease and dementia with Lewy bodies. Acta Neuropathol 2006; 111:115–25http://dx.doi.org/10.1007/s00401-005-0003-2 [DOI] [PubMed] [Google Scholar]
- 147. Tomlinson BE, Blessed G, Roth M.. Observations on the brains of demented old people. J Neurol Sci 1970; 11:205–42http://dx.doi.org/10.1016/0022-510X(70)90063-8 [DOI] [PubMed] [Google Scholar]
- 148. Scott SA, DeKosky ST, Sparks DL, et al. Amygdala cell loss and atrophy in Alzheimer's disease. Ann Neurol 1992; 32:555–63http://dx.doi.org/10.1002/ana.410320412 [DOI] [PubMed] [Google Scholar]
- 149. McAleese KE, Walker L, Erskine D, et al. TDP-43 pathology in Alzheimer's disease, dementia with Lewy bodies and ageing. Brain Pathol 2017; 27:472–9http://dx.doi.org/10.1111/bpa.12424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Attems J, Neltner JH, Nelson PT.. Quantitative neuropathological assessment to investigate cerebral multi-morbidity. Alzheimers Res Ther 2014; 6:85.http://dx.doi.org/10.1186/s13195-014-0085-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Braak H, Ghebremedhin E, Rub U, et al. Stages in the development of Parkinson's disease-related pathology. Cell Tissue Res 2004; 318:121–34http://dx.doi.org/10.1007/s00441-004-0956-9 [DOI] [PubMed] [Google Scholar]
- 152. Del Tredici K, Rub U, De Vos RA, et al. Where does parkinson disease pathology begin in the brain?. J Neuropathol Exp Neurol 2002; 61:413–26http://dx.doi.org/10.1093/jnen/61.5.413 [DOI] [PubMed] [Google Scholar]
- 153. Jellinger KA. Alpha-synuclein pathology in Parkinson's and Alzheimer's disease brain: incidence and topographic distribution: a pilot study. Acta Neuropathol 2003; 106:191–201http://dx.doi.org/10.1007/s00401-003-0725-y [DOI] [PubMed] [Google Scholar]
- 154. Dickson DW, Uchikado H, Fujishiro H, et al. Evidence in favor of Braak staging of Parkinson's disease. Mov Disord 2010; 25:S78–82 [DOI] [PubMed] [Google Scholar]
- 155. Braak H, Braak E, Yilmazer D, et al. Amygdala pathology in Parkinson's disease. Acta Neuropathol 1994; 88:493–500http://dx.doi.org/10.1007/BF00296485 [DOI] [PubMed] [Google Scholar]
- 156. Harding AJ, Stimson E, Henderson JM, et al. Clinical correlates of selective pathology in the amygdala of patients with Parkinson's disease. Brain 2002; 125:2431–45http://dx.doi.org/10.1093/brain/awf251 [DOI] [PubMed] [Google Scholar]
- 157. Burke RE, Dauer WT, Vonsattel JP.. A critical evaluation of the Braak staging scheme for Parkinson's disease. Ann Neurol 2008; 64:485–91http://dx.doi.org/10.1002/ana.21541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Jellinger KA. A critical evaluation of current staging of α-synuclein pathology in Lewy body disorders. Biochim Biophys Acta 2009;1792:730–40 [DOI] [PubMed] [Google Scholar]
- 159. McKeith IG, Boeve BF, Dickson DW, et al. Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology 2017; 89:88–100http://dx.doi.org/10.1212/WNL.0000000000004058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Toledo JB, Gopal P, Raible K, et al. Pathological α-synuclein distribution in subjects with coincident Alzheimer's and Lewy body pathology. Acta Neuropathol 2016;131:393–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Iseki E. Dementia with Lewy bodies: reclassification of pathological subtypes and boundary with Parkinson's disease or Alzheimer's disease. Neuropathology 2004; 24:72–8http://dx.doi.org/10.1111/j.1440-1789.2003.00530.x [DOI] [PubMed] [Google Scholar]
- 162. Leverenz JB, Fishel MA, Peskind ER, et al. Lewy body pathology in familial Alzheimer disease: evidence for disease- and mutation-specific pathologic phenotype. Arch Neurol 2006; 63:370–6http://dx.doi.org/10.1001/archneur.63.3.370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Zaccai J, Brayne C, McKeith I, et al. Patterns and stages of α-synucleinopathy: Relevance in a population-based cohort. Neurology 2008;70:1042–8 [DOI] [PubMed] [Google Scholar]
- 164. Hamilton RL. Lewy bodies in Alzheimer's disease: a neuropathological review of 145 cases using α-synuclein immunohistochemistry. Brain Pathol 2000;10:378–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Uchikado H, Lin WL, DeLucia MW, et al. Alzheimer disease with amygdala Lewy bodies: a distinct form of α-synucleinopathy. J Neuropathol Exp Neurol 2006;65:685–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Leverenz JB, Hamilton R, Tsuang DW, et al. Empiric refinement of the pathologic assessment of Lewy-related pathology in the dementia patient. Brain Pathol 2008; 18:220–4http://dx.doi.org/10.1111/j.1750-3639.2007.00117.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Alafuzoff I, Ince PG, Arzberger T, et al. Staging/typing of Lewy body related α-synuclein pathology: a study of the BrainNet Europe Consortium. Acta Neuropathol 2009;117:635–52. [DOI] [PubMed] [Google Scholar]
- 168. Lippa CF, Schmidt ML, Lee VM, et al. Antibodies to α-synuclein detect Lewy bodies in many Down's syndrome brains with Alzheimer's disease. Ann Neurol 1999;45:353–7. [DOI] [PubMed] [Google Scholar]
- 169. Parkkinen L, Soininen H, Alafuzoff I.. Regional distribution of α-synuclein pathology in unimpaired aging and Alzheimer disease. J Neuropathol Exp Neurol 2003;62:363–7 [DOI] [PubMed] [Google Scholar]
- 170. Rezaie P, Cairns NJ, Chadwick A, et al. Lewy bodies are located preferentially in limbic areas in diffuse Lewy body disease. Neurosci Lett 1996; 212:111–4http://dx.doi.org/10.1016/0304-3940(96)12775-0 [DOI] [PubMed] [Google Scholar]
- 171. Raunio A, Myllykangas L, Kero M, et al. Amygdala α-synuclein pathology in the population-based Vantaa 85+ study. J Alzheimers Dis 2017;58:669–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Beach TG, Adler CH, Lue L, et al. Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol 2009; 117:613–34http://dx.doi.org/10.1007/s00401-009-0538-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Markesbery WR, Jicha GA, Liu H, et al. Lewy body pathology in normal elderly subjects. J Neuropathol Exp Neurol 2009; 68:816–22http://dx.doi.org/10.1097/NEN.0b013e3181ac10a7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Sengoku R, Saito Y, Ikemura M, et al. Incidence and extent of Lewy body-related α-synucleinopathy in aging human olfactory bulb. J Neuropathol Exp Neurol 2008;67:1072–83. [DOI] [PubMed] [Google Scholar]
- 175. Popescu A, Lippa CF, Lee VM, et al. Lewy bodies in the amygdala: increase of α-synuclein aggregates in neurodegenerative diseases with tau-based inclusions. Arch Neurol 2004;61:1915–9 [DOI] [PubMed] [Google Scholar]
- 176. Schmidt ML, Martin JA, Lee VM, et al. Convergence of Lewy bodies and neurofibrillary tangles in amygdala neurons of Alzheimer's disease and Lewy body disorders. Acta Neuropathol 1996; 91:475–81http://dx.doi.org/10.1007/s004010050454 [DOI] [PubMed] [Google Scholar]
- 177. Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006; 314:130–3http://dx.doi.org/10.1126/science.1134108 [DOI] [PubMed] [Google Scholar]
- 178. Amador-Ortiz C, Ahmed Z, Zehr C, et al. Hippocampal sclerosis dementia differs from hippocampal sclerosis in frontal lobe degeneration. Acta Neuropathol (Berl) 2007; 113:245–52http://dx.doi.org/10.1007/s00401-006-0183-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Nag S, Yu L, Wilson RS, et al. TDP-43 pathology and memory impairment in elders without pathologic diagnoses of AD or FTLD. Neurology 2017; 88:653–60http://dx.doi.org/10.1212/WNL.0000000000003610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Arai T, Mackenzie IR, Hasegawa M, et al. Phosphorylated TDP-43 in Alzheimer's disease and dementia with Lewy bodies. Acta Neuropathol 2009; 117:125–36http://dx.doi.org/10.1007/s00401-008-0480-1 [DOI] [PubMed] [Google Scholar]
- 181. Nelson PT, Smith CD, Abner EL, et al. Hippocampal sclerosis of aging, a prevalent and high-morbidity brain disease. Acta Neuropathol 2013; 126:161–77http://dx.doi.org/10.1007/s00401-013-1154-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Murray ME, Cannon A, Graff-Radford NR, et al. Differential clinicopathologic and genetic features of late-onset amnestic dementias. Acta Neuropathol 2014; 128:411–21http://dx.doi.org/10.1007/s00401-014-1302-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Keage HA, Hunter S, Matthews FE, et al. TDP-43 pathology in the population: prevalence and associations with dementia and age. J Alzheimers Dis 2014; 42:641–50 [DOI] [PubMed] [Google Scholar]
- 184. Kadokura A, Yamazaki T, Lemere CA, et al. Regional distribution of TDP-43 inclusions in Alzheimer disease (AD) brains: their relation to AD common pathology. Neuropathology 2009; 29:566–73http://dx.doi.org/10.1111/j.1440-1789.2009.01017.x [DOI] [PubMed] [Google Scholar]
- 185. Nascimento C, Suemoto CK, Rodriguez RD, et al. Higher prevalence of TDP-43 proteinopathy in cognitively normal asians: a clinicopathological study on a multiethnic sample. Brain Pathol 2016; 26:177–85http://dx.doi.org/10.1111/bpa.12296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Coyle-Gilchrist IT, Dick KM, Patterson K, et al. Prevalence, characteristics, and survival of frontotemporal lobar degeneration syndromes. Neurology 2016; 86:1736–43http://dx.doi.org/10.1212/WNL.0000000000002638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Knopman DS, Roberts RO.. Estimating the number of persons with frontotemporal lobar degeneration in the US population. J Mol Neurosci 2011; 45:330–5http://dx.doi.org/10.1007/s12031-011-9538-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Josephs KA, Murray ME, Whitwell JL, et al. Updated TDP-43 in Alzheimer's disease staging scheme. Acta Neuropathol 2016; 131:571–85http://dx.doi.org/10.1007/s00401-016-1537-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Josephs KA, Murray ME, Whitwell JL, et al. Staging TDP-43 pathology in Alzheimer's disease. Acta Neuropathol 2014; 127:441–50http://dx.doi.org/10.1007/s00401-013-1211-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Hu WT, Josephs KA, Knopman DS, et al. Temporal lobar predominance of TDP-43 neuronal cytoplasmic inclusions in Alzheimer disease. Acta Neuropathol 2008; 116:215–20http://dx.doi.org/10.1007/s00401-008-0400-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Josephs KA, Whitwell JL, Knopman DS, et al. Abnormal TDP-43 immunoreactivity in AD modifies clinicopathologic and radiologic phenotype. Neurology 2008; 70:1850–7http://dx.doi.org/10.1212/01.wnl.0000304041.09418.b1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Nag S, Yu L, Capuano AW, et al. Hippocampal sclerosis and TDP-43 pathology in aging and Alzheimer disease. Ann Neurol 2015; 77:942–52http://dx.doi.org/10.1002/ana.24388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Uchino A, Takao M, Hatsuta H, et al. Incidence and extent of TDP-43 accumulation in aging human brain. Acta Neuropathol Commun 2015; 3:35.http://dx.doi.org/10.1186/s40478-015-0215-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. King A, Sweeney F, Bodi I, et al. Abnormal TDP-43 expression is identified in the neocortex in cases of dementia pugilistica, but is mainly confined to the limbic system when identified in high and moderate stages of Alzheimer's disease. Neuropathology 2010; 30:408–19http://dx.doi.org/10.1111/j.1440-1789.2009.01085.x [DOI] [PubMed] [Google Scholar]
- 195. James BD, Wilson RS, Boyle PA, et al. TDP-43 stage, mixed pathologies, and clinical Alzheimer's-type dementia. Brain 2016; pii: aww224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Lippa CF, Rosso AL, Stutzbach LD, et al. Transactive response DNA-binding protein 43 burden in familial Alzheimer disease and Down syndrome. Arch Neurol 2009; 66:1483–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Arnold SJ, Dugger BN, Beach TG.. TDP-43 deposition in prospectively followed, cognitively normal elderly individuals: correlation with argyrophilic grains but not other concomitant pathologies. Acta Neuropathol 2013; 126:51–7http://dx.doi.org/10.1007/s00401-013-1110-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Cagnin A, Mariotto S, Fiorini M, et al. Microglial and neuronal TDP-43 pathology in anti-IgLON5-related tauopathy. J Alzheimers Dis 2017; 59:13–20http://dx.doi.org/10.3233/JAD-170189 [DOI] [PubMed] [Google Scholar]
- 199. Yokota O, Davidson Y, Bigio EH, et al. Phosphorylated TDP-43 pathology and hippocampal sclerosis in progressive supranuclear palsy. Acta Neuropathol 2010; 120:55–66http://dx.doi.org/10.1007/s00401-010-0702-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Koga S, Sanchez-Contreras M, Josephs KA, et al. Distribution and characteristics of transactive response DNA binding protein 43 kDa pathology in progressive supranuclear palsy. Mov Disord 2017; 32:246–55http://dx.doi.org/10.1002/mds.26809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Kertesz A, Finger E, Murrell J, et al. Progressive supranuclear palsy in a family with TDP-43 pathology. Neurocase 2015; 21:178–84http://dx.doi.org/10.1080/13554794.2013.878729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Kouri N, Oshima K, Takahashi M, et al. Corticobasal degeneration with olivopontocerebellar atrophy and TDP-43 pathology: an unusual clinicopathologic variant of CBD. Acta Neuropathol 2013; 125:741–52http://dx.doi.org/10.1007/s00401-013-1087-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Omalu BI, DeKosky ST, Minster RL, et al. Chronic traumatic encephalopathy in a National Football League player. Neurosurgery 2005; 57:128–34http://dx.doi.org/10.1227/01.NEU.0000163407.92769.ED [DOI] [PubMed] [Google Scholar]
- 204. Armstrong RA, McKee AC, Alvarez VE, et al. Clustering of tau-immunoreactive pathology in chronic traumatic encephalopathy. J Neural Transm (Vienna) 2017; 124:185–92http://dx.doi.org/10.1007/s00702-016-1635-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. McKee AC, Stern RA, Nowinski CJ, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain 2013; 136:43–64http://dx.doi.org/10.1093/brain/aws307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Dickstein DL, Pullman MY, Fernandez C, et al. Cerebral [18 F]T807/AV1451 retention pattern in clinically probable CTE resembles pathognomonic distribution of CTE tauopathy. Transl Psychiatry 2016; 6:e900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Sundman M, Doraiswamy PM, Morey RA.. Neuroimaging assessment of early and late neurobiological sequelae of traumatic brain injury: implications for CTE. Front Neurosci 2015; 9:334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Kriegel J, Papadopoulos Z, McKee AC.. Chronic traumatic encephalopathy: is latency in symptom onset explained by Tau propagation? Cold Spring Harb Perspect Med 2017. doi: 10.1101/cshperspect.a024059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Forman MS, Schmidt ML, Kasturi S, et al. Tau and α-synuclein pathology in amygdala of Parkinsonism-dementia complex patients of Guam. Am J Pathol 2002;160:1725–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Yamazaki M, Arai Y, Baba M, et al. Alpha-synuclein inclusions in amygdala in the brains of patients with the parkinsonism-dementia complex of Guam. J Neuropathol Exp Neurol 2000; 59:585–91http://dx.doi.org/10.1093/jnen/59.7.585 [DOI] [PubMed] [Google Scholar]
- 211. Miklossy J, Steele JC, Yu S, et al. Enduring involvement of tau, β-amyloid, α-synuclein, ubiquitin and TDP-43 pathology in the amyotrophic lateral sclerosis/parkinsonism-dementia complex of Guam (ALS/PDC). Acta Neuropathol 2008;116:625–37. [DOI] [PubMed] [Google Scholar]
- 212. Takeda T, Seilhean D, Le Ber I, et al. Amygdala TDP-43 pathology in frontotemporal lobar degeneration and motor neuron disease. J Neuropathol Exp Neurol 2017; 76:800–12http://dx.doi.org/10.1093/jnen/nlx063 [DOI] [PubMed] [Google Scholar]
- 213. Kwiatkowski TJ Jr., Bosco DA, Leclerc AL, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009;323:1205–8http://dx.doi.org/10.1126/science.1166066 [DOI] [PubMed] [Google Scholar]
- 214. Keller BA, Volkening K, Droppelmann CA, et al. Co-aggregation of RNA binding proteins in ALS spinal motor neurons: evidence of a common pathogenic mechanism. Acta Neuropathol 2012; 124:733–47http://dx.doi.org/10.1007/s00401-012-1035-z [DOI] [PubMed] [Google Scholar]
- 215. Neumann M, Roeber S, Kretzschmar HA, et al. Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol 2009; 118:605–16http://dx.doi.org/10.1007/s00401-009-0581-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Vance C, Rogelj B, Hortobagyi T, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009; 323:1208–11http://dx.doi.org/10.1126/science.1165942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Maruyama H, Morino H, Ito H, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010; 465:223–6http://dx.doi.org/10.1038/nature08971 [DOI] [PubMed] [Google Scholar]
- 218. Schmitt FA, Nelson PT, Abner E, et al. University of Kentucky Sanders-Brown healthy brain aging volunteers: donor characteristics, procedures, and neuropathology. Curr Alzheimer Res 2012; 9:724–33http://dx.doi.org/10.2174/156720512801322591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Neltner JH, Abner EL, Schmitt FA, et al. Digital pathology and image analysis for robust high-throughput quantitative assessment of Alzheimer disease neuropathologic changes. J Neuropathol Exp Neurol 2012; 71:1075–85http://dx.doi.org/10.1097/NEN.0b013e3182768de4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Crosby EC, Humphrey T.. Studies of the vertebrate telencephalon. II. The nuclear pattern of the anterior olfactory nucleus, tuberculum olfactorium and the amygdaloid complex in adult man. J Comp Neurol 1941; 74:309–52 [Google Scholar]
- 221. Carpenter MB, Core Text of Neuroanatomy. 3rd ed Baltimore: Williams and Wilkins, 1985 [Google Scholar]
- 222. Economo C, Triarhou LC, Cellular Structure of the Human Cerebral Cortex. Basel; New York: Karger, 2009 [Google Scholar]
- 223. Braak H, Architectonics of the Human Telencephalic Cortex. Berlin; New York: Springer-Verlag, 1980 [Google Scholar]
- 224. Amunts K, Kedo O, Kindler M, et al. Cytoarchitectonic mapping of the human amygdala, hippocampal region and entorhinal cortex: intersubject variability and probability maps. Anat Embryol (Berl) 2005; 210:343–52http://dx.doi.org/10.1007/s00429-005-0025-5 [DOI] [PubMed] [Google Scholar]
- 225. Yilmazer-Hanke DM, Amygdala In: Mai JK, Paxinos G, eds. The Human Nervous System. 3rd ed Amsterdam; Boston: Elsevier Academic Press, 2012:759–835 [Google Scholar]
- 226. Insausti R, Amaral DG, Hippocampal Formation In: Mai JK, Paxinos G, eds. The Human Nervous System. 3rd ed Amsterdam; Boston: Elsevier Academic Press; 2012:896–942 [Google Scholar]
- 227. Lippa CF, Schmidt ML, Lee VM, et al. Alpha-synuclein in familial Alzheimer disease: epitope mapping parallels dementia with Lewy bodies and Parkinson disease. Arch Neurol 2001; 58:1817–20http://dx.doi.org/10.1001/archneur.58.11.1817 [DOI] [PubMed] [Google Scholar]
- 228. Rosenberg CK, Pericak-Vance MA, Saunders AM, et al. Lewy body and Alzheimer pathology in a family with the amyloid-β precursor protein APP717 gene mutation. Acta Neuropathol 2000;100:145–52 [DOI] [PubMed] [Google Scholar]
- 229. Lippa CF, Fujiwara H, Mann DM, et al. Lewy bodies contain altered α-synuclein in brains of many familial Alzheimer's disease patients with mutations in presenilin and amyloid precursor protein genes. Am J Pathol 1998;153:1365–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Glenner GG, Wong CW.. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 1984; 120:885–90http://dx.doi.org/10.1016/S0006-291X(84)80190-4 [DOI] [PubMed] [Google Scholar]
- 231. Grundke-Iqbal I, Iqbal K, Tung YC, et al. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A 1986; 83:4913–7http://dx.doi.org/10.1073/pnas.83.13.4913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Kosik KS, Joachim CL, Selkoe DJ.. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci U S A 1986; 83:4044–8http://dx.doi.org/10.1073/pnas.83.11.4044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Wood JG, Mirra SS, Pollock NJ, et al. Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule-associated protein tau (tau). Proc Natl Acad Sci U S A 1986; 83:4040–3http://dx.doi.org/10.1073/pnas.83.11.4040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Wolozin BL, Pruchnicki A, Dickson DW, et al. A neuronal antigen in the brains of Alzheimer patients. Science 1986; 232:648–50http://dx.doi.org/10.1126/science.3083509 [DOI] [PubMed] [Google Scholar]
- 235. Delacourte A, Defossez A.. Alzheimer's disease: Tau proteins, the promoting factors of microtubule assembly, are major components of paired helical filaments. J Neurol Sci 1986; 76:173–86http://dx.doi.org/10.1016/0022-510X(86)90167-X [DOI] [PubMed] [Google Scholar]
- 236. Ihara Y, Nukina N, Miura R, et al. Phosphorylated tau protein is integrated into paired helical filaments in Alzheimer's disease. J Biochem 1986; 99:1807–10http://dx.doi.org/10.1093/oxfordjournals.jbchem.a135662 [DOI] [PubMed] [Google Scholar]
- 237. Spillantini MG, Schmidt ML, Lee VM, et al. Alpha-synuclein in Lewy bodies. Nature 1997; 388:839–40http://dx.doi.org/10.1038/42166 [DOI] [PubMed] [Google Scholar]
- 238. Wakabayashi K, Matsumoto K, Takayama K, et al. NACP, a presynaptic protein, immunoreactivity in Lewy bodies in Parkinson's disease. Neurosci Lett 1997; 239:45–8http://dx.doi.org/10.1016/S0304-3940(97)00891-4 [DOI] [PubMed] [Google Scholar]
- 239. Arai T, Hasegawa M, Akiyama H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 2006; 351:602–11http://dx.doi.org/10.1016/j.bbrc.2006.10.093 [DOI] [PubMed] [Google Scholar]
- 240. Alzheimer A, Stelzmann RA, Schnitzlein HN, et al. An English translation of Alzheimer's 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde.” Clin Anat 1995; 8:429–31 [DOI] [PubMed] [Google Scholar]
- 241. Thal DR, Rub U, Orantes M, et al. Phases of A β-deposition in the human brain and its relevance for the development of AD. Neurology 2002;58:1791–800. [DOI] [PubMed] [Google Scholar]
- 242. Saito Y, Ruberu NN, Sawabe M, et al. Staging of argyrophilic grains: an age-associated tauopathy. J Neuropathol Exp Neurol 2004; 63:911–8http://dx.doi.org/10.1093/jnen/63.9.911 [DOI] [PubMed] [Google Scholar]
- 243. Martins CA, Oulhaj A, de Jager CA, et al. APOE alleles predict the rate of cognitive decline in Alzheimer disease: a nonlinear model. Neurology 2005; 65:1888–93http://dx.doi.org/10.1212/01.wnl.0000188871.74093.12 [DOI] [PubMed] [Google Scholar]
- 244. Nelson PT, Abner EL, Scheff SW, et al. Alzheimer's-type neuropathology in the precuneus is not increased relative to other areas of neocortex across a range of cognitive impairment. Neurosci Lett 2009; 450:336–9http://dx.doi.org/10.1016/j.neulet.2008.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.