

Abstract
Among flowering plants, the synthesis of choline (Cho) from ethanolamine (EA) can potentially occur via three parallel, interconnected pathways involving methylation of free bases, phospho-bases, or phosphatidyl-bases. We investigated which pathways operate in tobacco (Nicotiana tabacum L.) because previous work has shown that the endogenous Cho supply limits accumulation of glycine betaine in transgenic tobacco plants engineered to convert Cho to glycine betaine. The kinetics of metabolite labeling were monitored in leaf discs supplied with [33P]phospho-EA, [33P]phospho-monomethylethanolamine, or [14C]formate, and the data were subjected to computer modeling. Because partial hydrolysis of phospho-bases occurred in the apoplast, modeling of phospho-base metabolism required consideration of the re-entry of [33P]phosphate into the network. Modeling of [14C]formate metabolism required consideration of the labeling of the EA and methyl moieties of Cho. Results supported the following conclusions: (a) The first methylation step occurs solely at the phospho-base level; (b) the second and third methylations occur mainly (83%–92% and 65%–85%, respectively) at the phospho-base level, with the remainder occurring at the phosphatidyl-base level; and (c) free Cho originates predominantly from phosphatidylcholine rather than from phospho-Cho. This study illustrates how computer modeling of radiotracer data, in conjunction with information on chemical pool sizes, can provide a coherent, quantitative picture of fluxes within a complex metabolic network.
All plants synthesize modest amounts of choline (Cho) for incorporation into the membrane phospholipid phosphatidylcholine (Ptd-Cho); the Cho requirement for Ptd-Cho synthesis is approximately 1 to 2 μmol g−1 fresh weight (Rhodes and Hanson, 1993). In certain plants, Cho has an additional fate—oxidation to the osmoprotectant glycine betaine (GlyBet) (Gorham, 1995). Such plants may accumulate GlyBet to levels exceeding 20 μmol g−1 fresh weight, and so must produce far more Cho than those that lack GlyBet (Rhodes and Hanson, 1993). This difference between GlyBet accumulators and non-accumulators is highlighted by the results of expressing bacterial or plant genes for GlyBet synthesis in non-accumulators such as tobacco (Nicotiana tabacum L.) and Arabidopsis. The engineered plants produce relatively little GlyBet (<0.2–2 μmol g−1 fresh weight) unless they are given exogenous Cho, whereupon GlyBet levels increase by >20-fold (Hayashi et al., 1997; Nuccio et al., 1998; Huang et al., 2000, and refs. therein). The evidence that the endogenous Cho supply in tobacco cannot support high levels of GlyBet synthesis focused our attention on how Cho is made in this species, and on how metabolic flux to Cho is regulated.
Cho biogenesis has not been investigated in tobacco or other Solanaceae, but radiotracer and enzymatic evidence for diverse plants (both GlyBet accumulators and nonaccumulators) indicates that it can proceed via three parallel, interconnected paths involving sequential methylations of an ethanolamine (EA) moiety at the free base, phospho-base (P-base), or phosphatidyl-base (Ptd-base) levels (for review, see Rhodes and Hanson, 1993). Figure 1 shows the full network of these reactions. The situation may be simpler in leaves, where there is evidence only for P-base and Ptd-base pathways, the free base route having been found only in endosperm (Prud'homme and Moore, 1992a, 1992b). A further simplifying hypothesis is that the first methylation in leaves occurs only at the P-base level (Datko and Mudd, 1988a, 1988b). Results for leaves or cultured cells of species representing five families are all consistent with there being a common phospho-EA (P-EA) → phospho-monomethylethanolamine (P-MME) step, followed by methylations at the P-base level, the Ptd-base level, or both, depending on the species (Rhodes and Hanson, 1993). There is evidence from both GlyBet accumulators and non-accumulators that the enzyme mediating the P-EA → P-MME step, P-EA N-methyltransferase, exerts substantial control over flux through the entire pathway. The activity of this enzyme is down-regulated when Cho is supplied to Lemna, carrot, or soybean, and up-regulated in response to salt stress and light in spinach (Mudd and Datko, 1989a, 1989b; Summers and Weretilnyk, 1993; Weretilnyk et al., 1995).
Figure 1.

The network of reactions that potentially participate in Cho synthesis in flowering plants. Note that P-bases are converted to Ptd-bases via cytidyldiphospho-base intermediates, which have been omitted for simplicity. Enzymes mediating the free base and P-base methylations are cytosolic (Prud'homme and Moore, 1992b; Weretilnyk et al., 1995); those mediating Ptd-base methylations are microsomal (Datko and Mudd, 1988b). The two-step chloroplastic pathway leading to GlyBet is also shown; this step is only present in certain species (e.g. Chenopodiaceae) and is absent from wild-type tobacco. Bet ald, Betaine aldehyde.
In this study, we set out to establish the route(s) by which tobacco makes Cho by combining the power of in vivo radiotracer labeling with that of computer modeling. In moderately complex metabolic networks such as that shown in Figure 1, computer modeling is indispensable for coherent and quantitative calculation of fluxes (Bailey, 1998; Nuccio et al., 1999). The modeling work aimed to identify which of the pathways in Figure 1 carry most of the biosynthetic flux. This knowledge is essential to understanding the metabolic constraints on Cho production and, therefore, to designing engineering strategies to overcome them.
RESULTS
Measurements of Growth and Pool Sizes
Our labeling experiments were carried out on discs from half-expanded leaves because these appear to be the most active in Cho synthesis (Nuccio et al., 1998). As growth must be factored into metabolic models, we measured the expansion rate of such leaves, and the results indicated increases in leaf area and mass of approximately 10% d−1. As we could find no published values for Ptd-base levels in expanding tobacco leaf tissue, these were also measured. Values (nmol g−1 fresh weight; mean ± se; n = 3) were: Ptd-Cho, 1,820 ± 490; Phosphatidyl-ethanolamine (Ptd-EA), 752 ± 96; phosphatidyl-MME (Ptd-MME), <10. The phospho-Cho (P-Cho) (95 ± 17), Cho (58 ± 8), P-EA (131 ± 5), and EA (95 ± 4) pool sizes used to guide model development were as reported by Nuccio et al. (1998).
Partial Hydrolysis of P-Bases in the Apoplast
P-Cho has been shown to be hydrolyzed in the apoplast of cultured sycamore cells (Gout et al., 1990), and there is indirect evidence that this also occurs in tobacco plants (Nuccio et al., 1998). To determine whether 33P-MME and 33P-EA are also attacked by apoplastic phosphatase, we supplied them to leaf discs and measured the levels of the supplied 33P-base and of 33Pi in the extracellular (apoplastic) and intracellular compartments. Table I summarizes results for 33P-MME. Label uptake was rapid (approximately 90% within 30 min), and at 30 min the majority of the absorbed label was in 33P-MME. Of the label remaining in the apoplast at 30 min, 30% was in the form of 33Pi, indicating partial hydrolysis of 33P-MME before uptake. More extensive hydrolysis of 33P-EA was observed: after 30 min, 61% of the unabsorbed label was in the form of 33Pi (not shown).
Table I.
Partial apoplastic hydrolysis of 33P-MME during uptake by tobacco leaf discs
| Time | 33P Absorption | Intracellular
|
Extracellular
|
||
|---|---|---|---|---|---|
| P-MME | Pi | P-MME | Pi | ||
| min | nCi/3 discs | ||||
| 30 | 2,738 (91.3)a | 1,919 | 431 | 188 | 78 |
| 120 | 2,970 (99.0) | 1,072 | 507 | NDb | ND |
| 240 | 2,991 (99.7) | 369 | 620 | ND | ND |
Discs were each given 2 μL of a solution containing 1 nmol (1,000 nCi) of 33P-MME and 10 nmol of Pi. Batches of three discs (approximately 50 mg fresh wt total) were incubated for the times indicated, then washed for 15 min as described in “Materials and Methods,” and extracted. The label that was washed out was taken as being extracellular (apoplastic) and the rest as being intracellular.
Values in parentheses are 33P absorption expressed as a percentage of the 3,000 nCi supplied.
ND, Not determined.
In experiments with 33P-MME or 33P-EA, we therefore minimized potential complications arising from apoplastic hydrolysis by supplying them together with a 10-fold excess of unlabeled Pi. We reasoned that a large isotopic dilution of 33Pi upon its release in the apoplast, coupled with further dilution by the intracellular Pi pool (also large; Bligny et al., 1990), would make indirect labeling of Cho synthesis intermediates from 33Pi less important relative to their direct labeling from 33P-bases. To check this, we compared 33P incorporation into Ptd-Cho, the end product of the Cho synthesis pathway, using batches of three discs given 3 μCi (3 nmol) of 33Pi or a similar amount of 33P-MME plus 30 nmol of unlabeled Pi. At 360 min, [33P]-Ptd-Cho formation from 33Pi was approximately 8% of that from 33P-MME. As the 33Pi was fed without a 10-fold excess of unlabeled Pi, the contribution to Ptd-Cho synthesis of 33Pi released from 33P-MME can be conservatively estimated to be less than 8%. The model developed to interpret the labeling patterns from supplied 33P-MME and 33P-EA accounted for apoplastic hydrolysis and for re-incorporation of a small amount of 33Pi of low specific activity via the synthesis of P-Cho, P-EA, and P-MME from the respective bases. Nuccio et al. (1998) showed that tobacco plants readily phosphorylate Cho, EA, and MME.
Metabolism of 33P-MME and 33P-EA
Figure 2 is a schematic representation of the model developed for 33P-MME and 33P-EA labeling experiments. The fluxes in this scheme are designated K1 through K16; diagonal arrows designate transport into and out of metabolically inactive storage pools (these storage pools, of P-EA, P-MME, and Cho, are omitted from Fig. 2 for clarity). The fluxes and pool sizes generated by modeling the data from the various labeling experiments are presented in Tables II and III, respectively (the pool size values in Table III that were based primarily on measurements are italicized to distinguish them from those that were model derived). Figure 3 shows the fit between the model-generated curves and experimental data points.
Figure 2.

Scheme of the model developed from the 33P-labeling experiments. K1 through K16 adjacent to arrows refer to flux rates given in Table II; metabolite pool sizes are presented in Table III. All flux rates are assumed to remain constant except for rates A, A′, B, B′, C, and C′, which determine uptake and apoplastic hydrolysis of supplied P-MME and P-EA. Exogenously supplied 33P-MME (33P-MMEex) (specific activity 1,057 nCi nmol−1) or 33P-EA (33P-EAex) (specific activity 1,000 nCi nmol−1) is taken up into the apoplast at a rate proportional to the exogenous pool size (A = 33P-EAex × 0.25 min−1; A′ = 33P-MMEex × 0.3 min−1). The exogenous pool is taken up to an apoplastic pool, of initial pool size 0 nmol g−1 fresh weight (specific activity 0 nCi nmol−1). Rapid influx of the exogenous pool into the apoplastic pool (33P-MMEap and 33P-EAap) causes expansion of the apoplastic pool during the first 10 min. The apoplastic pool has two fates: hydrolysis to base and 33Pi at a variable rate (B = 33P-EAap × 0.05 min−1; B′ = 33P-MMEap × 0.014 min−1), or uptake into a metabolic symplastic pool at a variable rate (C = 33P-EAap × 0.0033 min−1; C′ = 33P-MMEap × 0.015 min−1). The 33Pi released from hydrolysis is diluted by a large endogenous unlabeled Pi pool (10,000 nmol g−1 fresh weight). Diagonal arrows designate transport to and from separate storage pools; rates are given in the text. The asterisk denotes a flux (40 pmol min−1 g−1 fresh weight) from an EA source to Ptd-EA, apparent only in [14C]formate labeling experiments. In the 33P-EA and formate models, rate K10 refers to the flux EA + Pi → P-EA, whereas in the 33P-MME model, rate K10 refers to the flux MME + Pi → P-MME (Table III). PA, Phosphatidic acid.
Table II.
Model-generated metabolic fluxes through the Cho synthesis pathway for 33P-EA, 33P-MME, and [14C]formate labeling experiments
| Flux |
33P-MME
|
33P-EA
|
[14C]Formate
|
Meana (se) | ||
|---|---|---|---|---|---|---|
| Expt. 1 | Expt. 2 | Expt. 1 | Expt. 1 | Expt. 2 | ||
| pmol min−1 g−1 fresh wt | ||||||
| K1 | – | – | 90 | 104 | 104 | 99 (4) |
| K2 | 240 | 200 | 75 | 92 | 92 | 140 (33) |
| K3 | 220 | 150 | 67 | 80 | 80 | 119 (29) |
| K4 | 820 | 820 | 800 | 780 | 780 | 800 (8) |
| K5 | 400 | 400 | 490 | 380 | 380 | 410 (18) |
| K6 | 350 | 350 | 220 | 320 | 320 | 312 (21) |
| K7 | 350 | 350 | 220 | – | – | 307 (35) |
| K8 | 370 | 370 | 240 | 340 | 340 | 332 (21) |
| K9 | 20 | 20 | 20 | 20 | 20 | 20 (0) |
| K10 | 260 | 240 | 240 | 150 | 150 | 208 (24) |
| K11 | 20 | 30 | 8 | 12 | 12 | 16 (3) |
| K12 | 20 | 30 | 8 | 12 | 12 | 16 (3) |
| K13 | 20 | 30 | 8 | 12 | 12 | 16 (3) |
| K14 | 40 | 60 | 16 | 24 | 24 | 33 (7) |
| K15 | – | – | 50 | 110 | 110 | 90 (13) |
| K16 | – | – | 0 | 80 | 80 | 53 (21) |
Each numbered K value represents a metabolic flux from Figure 2. Fluxes in and out of storage pools are given in the text.
Mean of model output values from all the labeling experiments.
Table III.
Pool sizes of the intermediates in Cho synthesis used to model 33P-EA, 33P-MME, and [14C]formate labeling experiments
| Metabolitea |
33P-MME
|
33P-EA
|
[14C]Formate
|
Meana (se) | ||
|---|---|---|---|---|---|---|
| Expt. 1 | Expt. 2 | Expt. 1 | Expt. 1 | Expt. 2 | ||
| nmol g−1 fresh wt | ||||||
| P-EA | – | – | 15 | 6 | 6 | 9 (3) |
| P-EA (storage) | – | – | 100 | 100 | 100 | 100 (0) |
| P-MME | 10 | 10 | 2 | 3 | 1.5 | 5.3 (1.9) |
| P-MME (storage) | 5 | 5 | 10 | 5 | 5 | 6 (1) |
| P-DME | 12 | 5 | 0.3 | 1 | 1 | 3.9 (2.2) |
| P-Cho | 100b | 40 | 30 | 30 | 40 | 48 (13) |
| Ptd-EA | – | – | 750 | 750 | 750 | 750 (0) |
| Ptd-MME | 4 | 6 | 2.5 | 3 | 5 | 4.1 (0.6) |
| Ptd-DME | 1 | 3 | 2 | 2 | 3 | 2.2 (0.4) |
| Ptd-Cho | 1,600 | 1,800 | 1,500 | 1,500 | 1,500 | 1,580 (58) |
| Cho | – | – | – | 2 | 2 | 2 (0) |
| Cho (storage) | – | – | – | 70 | 70 | 70 (0) |
| Phosphatidic acid | 100 | 100 | 100 | – | – | 100 (0) |
| Pi | 10,000 | 10,000 | 10,000 | – | – | 10,000 (0) |
Mean of model output values from all the labeling experiments.
Values in italics are based directly on the range of measured values; nonitalicized values are model-derived estimates.
Figure 3.

Observed and simulated radiolabeling of metabolites in the Cho synthesis pathway in 33P-MME experiment one (A–C), 33P-EA experiment one (D–F), and [14C]formate experiment one (G–I). Lines are model-generated curves, and symbols are observed radioactivity values. ○, Pi; ●, P-MME; □, P-DME; ▪, P-Cho; ▴, Ptd-MME; ▿, Ptd-DME; Δ, Ptd-Cho; ♦, P-EA; ⋄, Ptd-EA; ▾, Cho; ★, sum of (Ptd-EA plus phosphatidyl-DME). The P-MME and P-EA data points and curves refer to the sum of all the pools (apoplastic, metabolic, and storage) of these intermediates; Cho data points and curves are the sum of the metabolic and storage pools. In C and F, Ptd-MME and Ptd-DME data have been multiplied by 5 for graphical display on a scale comparable to Ptd-Cho. In H, Ptd-MME and Ptd-EA plus Ptd-DME data have been multiplied by 2.
The flux rates A, A′, B, B′, C, and C′ in the model (Fig. 2) are proportional to pool size. These rates determine the kinetics of label entry into Pi and label recovery in the total endogenous P-EA and P-MME pools (i.e. the sum of apoplastic and symplastic pools; Fig. 3, A and D). The 33Pi released from hydrolysis of the apoplastic 33P-EA and 33P-MME is assumed to be diluted by a large endogenous pool of Pi (10,000 nmol g−1 fresh weight) that includes the equivalent of 600 nmol g−1 fresh weight of unlabeled Pi supplied (as potassium phosphate) with the 33P-bases (Table I). All other fluxes in the model are held constant. This simplification was adopted instead of making all fluxes dependent on pool size, because the values generated from more complex models with pool-size-dependent fluxes were similar to those obtained using invariant fluxes (data not shown).
33P-MME
When 33P-MME was supplied, both P-DME and P-Cho acquired 33P rapidly (Fig. 3B). P-DME lost label after 120 min as label continued to accumulate in P-Cho (Fig. 3B). The Ptd-bases Ptd-MME and Ptd-DME acquired much less label than the P-bases (note that radioactivity recovered in Ptd-MME and Ptd-DME is plotted on a 5-fold-expanded scale in Fig. 3C). As expected for the end product of the pathway, Ptd-Cho continued to accumulate 33P throughout the experiment (Fig. 3C). In qualitative terms, these labeling patterns indicate that the methylations in the Cho synthesis pathway are probably taking place mainly at the P-base level. Modeling permitted quantitative and specific conclusions: (a) the P-MME → Ptd-DME step (K2) carries 12 times more flux than the Ptd-MME → Ptd-DME step (K13); (b) the P-DME → P-Cho step (K3) carries 5.5 times more flux than the Ptd-DME → Ptd-Cho step (K14); (c) the labeling patterns of P-Cho and Ptd-Cho are consistent with substantial recycling of Cho moieties; this includes catabolism of Ptd-Cho to both P-Cho (K5) and free Cho (K6) at estimated rates of 400 and 350 pmol min−1 g−1 fresh weight, respectively. Note that the catabolism of Ptd-Cho to Cho is assumed to liberate phosphatidic acid that is further assumed to be hydrolyzed to Pi; (d) it is necessary to invoke a small, metabolically inactive (storage) pool of P-MME in slow exchange (10 pmol min−1 g−1 fresh weight) with a metabolically active pool; (e) the net synthesis rate of Ptd-Cho [(K4 + K14)−(K5 + K6)], 110 pmol min−1 g−1 fresh weight, is adequate to meet growth requirements, i.e. to maintain a pool of Ptd-Cho of 1600 nmol g−1 fresh weight when the leaf expansion rate is approximately 10% per day (160 nmol day−1 g−1 fresh weight = 111 pmol min−1 g−1 fresh weight); (f) free Cho is formed mainly from Ptd-Cho rather than from P-Cho.
In a second experiment, the supplied 33P-MME was more extensively hydrolyzed to 33Pi. This was accommodated in the model by diverting a greater proportion of the apoplastic P-MME to hydrolysis. Other minor differences in model parameters from the first 33P-MME labeling experiment were: (a) the P-Cho pool size was 2.5-fold lower, and (b) the methylation fluxes via the Ptd-bases (K13, K14) were 1.5-fold higher, and those via P-base (K2, K3) correspondingly lower. With these modest exceptions, the model predictions (Tables II and III) were essentially the same as for the first experiment.
33P-EA
The dose of 33P-EA substrate given was similar to that in the 33P-MME experiments, but the 33P recovered in Ptd-Cho was about 10-fold less, and label incorporation into the other metabolites monitored was similarly lower (Fig. 3, D–F). This was attributable to faster hydrolysis of P-EA in the apoplast and to a somewhat lower endogenous flux in the pathway, presumably due to variation between batches of leaf discs. Quantitative explanation of the labeling kinetics of P-EA specifically required that there be a small metabolic pool of P-EA (≈15 nmol g−1 fresh weight) and that the flux from this pool to a large storage pool of P-EA (160 pmol min−1 g−1 fresh weight) be twice that of the P-MME pool. In considering the labeling kinetics of intermediates derived from 33P-EA, the labeling pattern of P-MME was also best accommodated by assuming separate metabolically active and storage pools (Table III) in slow exchange with one another (fluxes of 9 and 4 pmol min−1 g−1 fresh weight into and out of the storage pool, respectively).
Although Ptd-EA acquired significant 33P label within the first 60 min (Fig. 3F), modeling indicated that it was not further metabolized to Ptd-MME or degraded to P-EA at a detectable rate. Net synthesis of Ptd-EA (K15−K16) was estimated to be 50 pmol min−1 g−1 fresh weight, a rate that is adequate to maintain the Ptd-EA pool of 750 nmol g−1 fresh weight, assuming a leaf expansion rate of approximately 10% per day.
Metabolism of [14C]Formate
Plant tissues rapidly incorporate formate carbon into C1-substituted folate pools, and then, via Met and S-adenosyl L-Met (AdoMet), into the methyl groups of Cho (Hitz et al., 1981; Hanson and Rhodes; 1983; Cossins and Chen, 1997). This allowed us to test the model developed for 33P-EA and 33P-MME experiments by applying it to [14C]formate labeling data. These experiments also enabled investigation of a possible free base pathway to Cho, which cannot be detected by 33P labeling. No changes were made to the model network except those specified below. Two points were considered when designing and interpreting [14C]formate-labeling experiments. First, formate dehydrogenase activity in leaves can oxidize formate to CO2 (Hanson and Nelsen, 1978; Hourton-Cabassa et al., 1998). Relatively large [14C]formate doses (41 or 127 nmol per three discs) were therefore used to compensate for 14CO2 losses (>95% within 1 h), and incubation was in darkness to avoid refixation by photosynthesis of the 14CO2 released.
Second, label from formate can enter the β-carbon of Ser (via C1-folate pools and Ser hydroxymethyltransferase) and then EA, so that 14C in methylated EA derivatives may reside in the C2 moiety as well as the methyl groups (Cossins and Chen, 1997). In our experiments, relatively little 14C entered the C2 moiety, as judged from the low amount of label in P-EA (≤0.2 nCi g−1 fresh weight; data not shown) and from the low initial rate of 14C incorporation into Ptd-EA plus Ptd-DME (≤6% of that into Ptd-Cho; Fig. 3H). In barley leaves, labeling in the C2 moiety was also minimal (≤6% of the total labeling in GlyBet; Hanson and Nelsen 1978). In modeling [14C]formate data, we therefore assumed that supplying [14C]formate resulted in the immediate labeling of AdoMet and EA pools to different specific activities, initially 2.6 nCi nmol−1 for AdoMet and 0.11 nCi nmol−1 for EA. Thereafter, the specific activities of AdoMet and EA were further assumed to decline exponentially:
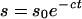 |
where s0 is the initial specific activity, s is specific activity at time t, and c is a time constant. The time constants for AdoMet and EA were 0.02 min−1 and 0.003 min−1, respectively.
When [14C]formate (41 nmol per three discs) was supplied, no labeling was detected in MME and DME (≤0.4 nCi g−1 fresh weight; data not shown); a free base route for Cho synthesis is therefore unlikely and so was not incorporated into the model (Fig. 2). Tables II and III summarize the fluxes and pool sizes required in the model to account for the observed labeling patterns. These values agree closely (generally within ±30%) with those required for the 33P-base experiments, which attests to the model's robustness. Cho labeled only slightly and progressively with time (Fig. 3G), consistent with it originating from the turnover of Ptd-Cho, rather than from dephosphorylation of P-Cho or direct N-methylation of EA (compare with Fig. 1). In the model, Cho was partitioned into a metabolic pool (2 nmol g−1 fresh weight) and a storage pool (70 nmol g−1 fresh weight) with equal steady-state fluxes between the two pools (20 pmol min−1 g−1 fresh weight). As observed for 33P-labeling experiments, 14C labeling of all the P-bases reached a maximum at 60 to 120 min and thereafter declined markedly (Fig. 3G). Also as observed in 33P experiments, Ptd-MME labeling peaked early and then fell slowly, while label continued to accumulate in Ptd-Cho throughout the experiment (Fig. 3H).
Ptd-EA and Ptd-DME were not separated in this experiment, and therefore the sum of label recovered in these two compounds is shown in Figure 3H, superimposed on the simulated time course for the sum of these two metabolites. However, the model permitted estimates to be made of the relative label contributions of Ptd-EA and Ptd-DME to the total radioactivity in the Ptd-EA plus Pdt-DME fraction (Fig. 3I). This analysis indicated that the failure of label to chase from the total Ptd-EA plus Pdt-DME fraction (Fig. 3H) is due to progressive accumulation of label in the EA moiety of Ptd-EA (Fig. 3I). The Ptd-EA pool contained greater than 4 nCi g−1 fresh weight by 600 min (Fig. 3I), while its precursor, P-EA, acquired very little label (<0.2 nCi g−1 fresh weight; Fig. 3I). To account for this labeling pattern and also to maintain the size of the Ptd-EA pool as growth occurs, it was necessary to invoke an additional flux of EA moieties to Ptd-EA, aside from that through P-EA (see “Discussion”). This additional flux would not have been detected in the preceding 33P-EA-labeling experiment as it concerns only the EA moiety of Ptd-EA. The initial specific activity of the source of this flux was set to a low value (0.11 nCi nmol−1), with a time constant (c) of 0.003 min−1 and a flux into the Ptd-EA pool of 40 pmol min−1 g−1 fresh weight.
A second labeling experiment was conducted with a higher dose of [14C]formate (127 nmol per three discs). As before, no label was detected in MME or DME. The experimental data for the second [14C]formate experiment were effectively modeled using assumptions very similar to those used for the first, except that the initial specific activities of AdoMet and EA were 2-fold higher (Tables II and III).
DISCUSSION
The radiotracer and computer modeling evidence presented here indicate that the main route of Cho synthesis in tobacco is at the P-base level, with a minor contribution at the Ptd-base level. The first N-methylation appears to occur exclusively at the P-base level (P-EA → P-MME). Computer-assisted analyses of the experimental data also show that choline synthesis occurs mainly through Ptd-Cho turnover rather than through the dephosphorylation of P-Cho. These points were not evident by graphical interpretation of the radiotracer data alone. This illustrates the power of metabolic models, in conjunction with information on chemical pool sizes, in providing a self-consistent, quantitative account of fluxes within a complex metabolic network. These conclusions are discussed in detail below, together with further insights provided by modeling.
Cho Synthesis
Modeling indicated that regardless of whether 33P-EA, 33P-MME, or [14C]formate was the source of label and whether the experiments were in light or darkness, there was always at least 2.5-fold more flux via the P-base N-methylation pathway (K2, K3) than via the Ptd-base pathway (K13, K14). This finding appears robust, since models that assumed a greater flux via the Ptd-base pathway than via the P-base pathway invariably failed to fit the experimental data (not shown). Furthermore, the [14C]formate labeling data indicate that Cho synthesis does not occur via a free base pathway in tobacco leaves, which is consistent with the findings for leaves of all other species so far tested (Rhodes and Hanson, 1993).
The labeling kinetics of Ptd-MME in 33P-EA labeling experiments were accounted for by allowing the first N-methylation to occur solely at the P-base level. Moreover, modeling of the [14C]formate data showed that allowing significant conversion of Ptd-EA to Ptd-MME would have resulted in much more 14C accumulation in Ptd-MME than was observed. This follows, because at early time points it was necessary to assign a specific activity to AdoMet that was approximately 20-fold greater than that of EA to account for labeling patterns of network intermediates. Conversion of Ptd-EA to Ptd-MME would therefore be accompanied by incorporation of a heavily labeled methyl group into Ptd-MME. Thus, no support for a significant flux from Ptd-EA → Ptd-MME was found, and the labeling pattern of Ptd-MME in [14C]formate experiments was accounted for by a modest flux from P-MME of the same magnitude as that needed to account for the 33P data. Our finding that the first N-methylation in the network appears to occur exclusively at the P-base level supports the hypothesis of Datko and Mudd (1988a, 1988b).
Modeling of Cho synthesis required partitioning of P-EA and P-MME between metabolically active and inactive (storage) pools. The inactive pools may be sequestered in a compartment such as the vacuole. However, kinetically distinct metabolically active and inactive pools might also arise if there is metabolic channeling of the flux P-EA → P-MME → P-DME → P-Cho via an enzyme complex. It is therefore noteworthy that P-EA N-methyltransferase has been proposed to catalyze not only the conversion of P-EA to P-MME but also the two subsequent methylation steps (Smith et al., 2000).
The Origin of EA
There is good evidence that EA moieties are derived from the decarboxylation of Ser, but it is not clear whether the reaction takes place at the level of free Ser, P-Ser, or Ptd-Ser (Rhodes and Hanson, 1993). It is therefore interesting that modeling of the [14C]formate data indicated that the EA moiety of Ptd-EA came from a P-EA pool of low specific activity and from another source, also of low specific activity. This other source could be Ptd-Ser decarboxylation or a base exchange reaction between free EA and phospholipids (Kinney and Moore, 1987; Mudd and Datko, 1989c). Consistent with these possibilities, preliminary analyses showed that 14C was present in products with the chromatographic properties of Ptd-Ser and free EA. While these data do not show how EA moieties are derived from Ser, they do suggest that the pathway(s) include phospholipid and soluble intermediates, and, because their specific activities are low, that some of the intermediate pools are large.
Ptd-Cho Catabolism
Modeling indicated that free Cho is generated in tobacco leaves mainly through Ptd-Cho catabolism. This resembles the situation in barley (Hitz et al., 1981) but differs from that in spinach, where the main source of free Cho is dephosphorylation of P-Cho (Rhodes and Hanson, 1993). Modeling further suggested that tobacco leaves catabolize Ptd-Cho to P-Cho and to free Cho, i.e. via both phospholipase C and phospholipase D reactions. A high flux from Ptd-Cho to P-Cho has also been observed in Suc-starved sycamore cells undergoing autophagy (Aubert et al., 1996). Phospholipase C-mediated phospholipid degradation may therefore be important both in normal phospholipid turnover and during net phospholipid breakdown.
Metabolism of Cytoplasmic Phosphate
Modeling of the 33P-base labeling data indicated that considerably more dilution of Pi label was occurring than could be accounted for by a cytoplasmic free Pi pool of normal size (≤1,000 nmol g−1 fresh weight), and it was necessary to invoke a pool of 10,000 nmol g−1 fresh weight. In other plant systems, the cytoplasmic Pi pool is in rapid exchange with the large pool of phosphoesters that include sugar phosphates, UDP-Glc, and adenylates (Roscher et al., 1998). This suggests an explanation for the large Pi pools seen in our model: that cytoplasmic free Pi equilibrated so rapidly with organic phosphates that the cytoplasmic Pi pool in effect included the phosphate esters.
Implications for Metabolic Engineering
The results described here have obvious implications for engineering Cho synthesis to meet the demand for GlyBet production (Nuccio et al., 1998). Because the first N-methylation occurs at the P-base level, a rational target for increasing flux to Cho moieties is overexpression of P-EA N-methyltransferase. Our models do not address the regulatory architecture of the network (Stephanopoulos and Vallino, 1991). A key question to be resolved is whether P-Cho exerts feedback regulation on P-EA N-methyltransferase in tobacco—in vivo tracer studies in sugar beet (Hanson and Rhodes, 1983) and in vitro studies in Lemna suggest this possibility (Mudd and Datko, 1989a). Perhaps equally important to future attempts at engineering Cho supply by manipulating P-EA N-methyltransferase activity and/or its regulatory properties will be whether the potential supply of P-EA is adequate to support greatly increased flux from P-EA to P-Cho. Our modeling data suggest that the metabolically active P-EA pool is small and sequestered from the bulk P-EA of the tissue.
MATERIALS AND METHODS
Plant Material
Tobacco (N. tabacum L. cv Wisconsin 38) plants were grown in a light soil mix (one plant per 16-cm pot) in a greenhouse in natural daylight; the minimum temperature was 18°C. Irrigation was with 0.5× Hoagland nutrient solution. Half-expanded leaves (blade length about 15 cm) for radiotracer experiments were taken between February and May 1998 from mature plants that had not yet started to flower. The leaf expansion rate was estimated by planimetry.
Radiochemicals
[14C]Formic acid (Na salt in 70% [v/v] ethanol; 48.5 μCi μmol−1) and [γ-33P]ATP (2 Ci μmol−1) were from NEN Life Science Products (Boston). Before use, the [14C]formate was freed of ethanol by evaporation under reduced pressure, and redissolved in water. 33P-EA and 33P-MME (approximately 1 μCi nmol−1) were synthesized from EA and MME, respectively, using yeast Cho kinase and [γ-33P]ATP. Reaction mixtures (100 μL) contained 0.1 m Tris-HCl, pH 8.5, 1 mm MgCl2, 1 mm ATP, approximately 100 μCi of [γ-33P]ATP, 2 mm EA-HCl or MME-HCl, and 0.1 unit of Cho kinase. After incubation for 18 h at 25°C, residual [33P]ATP was hydrolyzed by adding 0.5 mL of 1.2 m HCl and heating at 100°C for 1 h (P-EA and P-MME are not hydrolyzed by these conditions), and the reaction mixture was lyophilized to remove HCl. The dried mixture was redissolved in 2 mL of water and applied to a 2-mL column of BioRex-70 (H+) followed by 1-mL columns of AG-50 (H+) and AG-1 (OH−), arranged in series. After washing the column series with 35 mL of water, 33P-EA and 33P-MME (plus 33Pi from unreacted [33P]ATP) were eluted from the AG-1 column with 5 mL of 2.5 n HCl, and the eluate was lyophilized. The 33P-bases were separated from Pi by thin-layer electrophoresis on cellulose plates in 1.5 n formic acid (1.8 kV for 20 min at 3°C). 33P-EA was further purified by thin-layer chromatography (TLC) on silica gel G plates developed with methanol:acetone:conc. HCl (90:10:4, v/v; TLC system A); this removed a minor contaminant that appeared to be a phosphoester of Tris. Radiochemical yields were 56% to 58%, and radiochemical purities were >95%. For feeding to leaf discs, the purified 33P-bases were dissolved at a concentration of 0.5 μCi μL−1 (approximately 0.5 nmol μL−1) in 5 mm potassium phosphate buffer, final pH approximately 6.0, giving a Pi/P-base ratio of 10. 33Pi (1 μCi nmol−1) was prepared by hydrolysis of [γ-33P]ATP followed by ion-exchange purification as above.
Leaf Disc Experiments
Discs (11 mm in diameter) were cut from the mid-blade region of a single, half-expanded leaf for each experiment. Eight shallow radial incisions were made on the abaxial surface of each disc using a sharp scalpel and thereby minimizing tissue damage. Samples were batches of three discs (approximately 50 mg fresh weight). Labeled solutions were applied (2 μL per disc) to the center of the abaxial surface (at the junction of the cuts), and the discs were then incubated, abaxial surface uppermost, on moist filter paper circles in Petri dishes. Incubation was at 25°C ± 2°C in darkness for [14C]formate experiments and in fluorescent light (photon flux density, 150 μE m−2 s−1) for 33P-base experiments. Discs that had been fed 33P-bases were in some cases washed after incubation to analyze label remaining in the apoplast. Washing was for 15 min, with rotary shaking, in 5 mL of 5 mm potassium phosphate (pH 7.5) containing 0.1 mm P-EA as carrier.
Analysis of Labeled Metabolites
Procedures were essentially as described previously (Hitz et al., 1981; Hanson and Rhodes, 1983; Nuccio et al., 1998). Discs were extracted by a methanol-chloroform-water procedure after boiling in isopropanol to denature phospholipases. Lipids were separated by TLC on silica gel 60 plates developed first with acetone:petroleum ether (3:1; v/v), then with chloroform:methanol:acetic acid:water (85:15:10:3.5, v/v). Because Ptd-EA and Ptd-DME comigrate in this system, when necessary, they were resolved by rechromatography on silica gel 60 using chloroform:methanol:concentrated NH4OH (65:25:5, v/v); the Ptd-MME zone was rechromatographed likewise for some samples. Phospholipid zones were located by autoradiography and iodine staining, and identified by reference to standards. The identity of Ptd-Cho was confirmed by conversion to phosphatidic acid upon treatment with phospholipase D (Kates, 1972). Radioactivity was quantified by scintillation counting of TLC zones. Water-soluble compounds were fractionated by ion-exchange followed by TLC. In 33P experiments, the samples were applied to a 1-mL column of AG-1 (OH−); 8 mL of 1 n formic acid was used to elute P-bases, followed by 5 mL of 2.5 n HCl to elute Pi. After lyophilization, P-bases were chromatographed twice in TLC system A. In 14C experiments, 1-mL columns of AG-1 (OH−) and BioRex-70 (H+) were arranged in series; free bases were eluted from BioRex-70 with 5 mL of 1 n HCl, and P-bases (plus [14C]formate) from AG-1 with 5 mL of 2.5 n HCl. The eluates were lyophilized; 14C loss during lyophilization of AG-1 eluates was used as a measure of their [14C]formate content. Phospho bases were hydrolyzed to free bases; both this hydrolyzate and the free bases eluted from BioRex-70 were analyzed using TLC system A. Data were corrected for recovery using samples spiked with 33P-EA, 33P-MME, [14C]P-Cho, or [14C]Cho. Recoveries of the P-bases were similar, and averages of these values were applied to 33P-DME data; the value for [14C]Cho was applied to data for all free bases. When necessary, data were corrected for spillover of radioactivity from adjacent TLC zones. 33P-labeling data were corrected for radioactive decay using a half-life of 25.4 d.
Measurement of Ptd-Base Pool Sizes
The organic phase from methanol-chloroform-water extracts of 0.5 g fresh weight of leaf tissue was dried in a N2 stream and treated with 0.5 mL of 4 n HCl at l00°C for l5 to 18 h to hydrolyze Ptd-bases. Cho was then determined by the method of Nie et al. (1993) as modified by Nuccio et al. (1998), and EA was determined by the TLC assay method given by Nuccio et al. (1998). Data were corrected for the recovery of Pdt-base standards added to samples before extraction.
Computer Modeling of 33P- and 14C-Labeling Data
The computer model used was similar to the metabolic flux analysis model described by Kocsis et al. (1998). Programs were written in Microsoft Visual Basic. Key model parameters are the initial pool sizes and their specific activities, and the flux rates connecting the various pools.
For all metabolites except the apoplastic and metabolic pools of the supplied precursors P-EA and P-MME and the Pi pool, the rate of change of the concentration of metabolite M (nmol g−1 fresh weight) is taken as:
 |
where Ki (pmol min−1 g−1 fresh weight) is the fixed rate of production of M from its various precursor pools and the Kj (pmol min−1 g−1 fresh weight) is the fixed rate of conversion of M to its various fates (see “Results” for the justification for using fixed rates for most fluxes in the model).
The variable rates A, B, C, and A′, B′, C′ in Figure 2 describe the uptake and apoplastic hydrolysis of exogenously added 33P-EA and 33P-MME, respectively. Exogenous precursor is taken up into the apoplast at a variable rate (A or A′) that is proportional to the precursor pool size, so that as the pool of precursor declines, so does uptake rate. The precursor is taken up into an apoplastic pool of initial size 0 nmol g−1 fresh weight (and initial specific activity 0 nCi nmol−1). The apoplastic pool has two fates—hydrolysis to free base and 33Pi at a variable rate (B or B′), or uptake into a metabolic symplastic pool at a variable rate (C or C′). Rates B, B′, C, and C′ are proportional to the apoplastic pool size.
During short time intervals (0.4- or 0.6-min iterations), material of the current specific activity is drawn from one pool to another at specified rates, new specific activities and pool sizes are computed, and total radioactivity in each pool is plotted (superimposed on observed data) as a function of time. Flux rates and pool sizes are progressively adjusted (within limits determined by experimentally observed or literature values) until a close match between observed and simulated radioactivity is obtained, as judged graphically or by computing absolute deviations between observed and simulated values. Further details on the development of the type of metabolic models used here are given at http://www.hort.purdue.edu/cfpesp/models/models.htm.
Footnotes
This work was supported in part by the U.S. Department of Agriculture National Research Initiative Competitive Grants Program (grant no. 98–35100–6149 to A.D.H.), by the National Science Foundation (grant no. IBN–9813999 to A.D.H.), by the Department of Energy (grant no. DE–FG02–99ER20344 to D.R.), by a National Institute of Science and Technology grant (to Y.S.-H.), by an endowment from the C.V. Griffin, Sr. Foundation, and by the Florida Agricultural Experiment Station. This paper is journal series no. R–07256.
LITERATURE CITED
- Aubert S, Gout E, Bligny R, Marty-Mazars D, Barrieu F, Alabouvette J, Marty F, Douce R. Ultrastructural and biochemical characterization of autophagy in higher plant cells subjected to carbon deprivation: control by the supply of mitochondria with respiratory substrates. J Cell Biol. 1996;133:1251–1263. doi: 10.1083/jcb.133.6.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JE. Mathematical modeling and analysis in biochemical engineering: past accomplishments and future opportunities. Biotechnol Prog. 1998;14:8–20. doi: 10.1021/bp9701269. [DOI] [PubMed] [Google Scholar]
- Bligny R, Gardestrom P, Roby C, Douce R. 31P-NMR studies of spinach leaves and their chloroplasts. J Biol Chem. 1990;265:1319–1326. [PubMed] [Google Scholar]
- Cossins EA, Chen L. Folates and one-carbon metabolism in plants and fungi. Phytochemistry. 1997;45:437–452. doi: 10.1016/s0031-9422(96)00833-3. [DOI] [PubMed] [Google Scholar]
- Datko AH, Mudd SH. Phosphatidylcholine synthesis: differing patterns in soybean and carrot. Plant Physiol. 1988a;88:854–861. doi: 10.1104/pp.88.3.854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datko AH, Mudd SH. Enzymes of phosphatidylcholine synthesis in Lemna, soybean, and carrot. Plant Physiol. 1988b;88:1338–1348. doi: 10.1104/pp.88.4.1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorham J. Betaines in higher plants: biosynthesis and role in stress metabolism. In: Wallsgrove RM, editor. Amino Acids and Their Derivatives in Higher Plants. Cambridge: Cambridge University Press; 1995. pp. 173–203. [Google Scholar]
- Gout E, Bligny R, Roby C, Douce R. Transport of phosphocholine in higher plant cells: 31P nuclear magnetic resonance studies. Proc Natl Acad Sci USA. 1990;87:4280–4283. doi: 10.1073/pnas.87.11.4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson AD, Nelsen CE. Betaine accumulation and [14C]formate metabolism in water-stressed barley leaves. Plant Physiol. 1978;62:305–312. doi: 10.1104/pp.62.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson AD, Rhodes D. 14C-Tracer evidence for synthesis of Cho and betaine via phosphoryl base intermediates in salinized sugarbeet leaves. Plant Physiol. 1983;71:692–700. doi: 10.1104/pp.71.3.692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi H, Alia, Mustardy L, Deshnium P, Ida M, Murata N. Transformation of Arabidopsis thaliana with the codA gene for Cho oxidase: accumulation of glycinebetaine and enhanced tolerance to salt and cold stress. Plant J. 1997;12:133–142. doi: 10.1046/j.1365-313x.1997.12010133.x. [DOI] [PubMed] [Google Scholar]
- Hitz WD, Rhodes D, Hanson AD. Radiotracer evidence implicating phosphoryl and phosphatidyl bases as intermediates in betaine synthesis by water-stressed barley leaves. Plant Physiol. 1981;68:814–812. doi: 10.1104/pp.68.4.814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hourton-Cabassa C, Ambard-Bretteville F, Moreau F, Davy de Virville J, Remy R, Colas des Francs-Small C. Stress induction of mitochondrial formate dehydrogenase in potato leaves. Plant Physiol. 1998;116:627–635. doi: 10.1104/pp.116.2.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Hirji R, Adam L, Rozwadowski KI, Hammerlindl J, Keller WA, Selvaraj G. Genetic engineering of glycinebetaine production toward enhancing stress tolerance in plants: metabolic limitations. Plant Physiol. 2000;122:747–756. doi: 10.1104/pp.122.3.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kates M. Techniques of Lipidology. Amsterdam: North Holland Publishing; 1972. [Google Scholar]
- Kinney AJ, Moore TS. Phosphatidylcholine synthesis in castor bean endosperm: I. Metabolism of l-serine. Plant Physiol. 1987;84:78–81. doi: 10.1104/pp.84.1.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocsis MG, Nolte KD, Rhodes D, Shen T-L, Gage DA, Hanson AD. Dimethylsulfoniopropionate biosynthesis in Spartina alterniflora. Plant Physiol. 1998;117:273–281. doi: 10.1104/pp.117.1.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudd SH, Datko AH. Synthesis of methylated EA moieties: regulation by Cho in Lemna. Plant Physiol. 1989a;90:296–305. doi: 10.1104/pp.90.1.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudd SH, Datko AH. Synthesis of methylated EA moieties: regulation by Cho in soybean and carrot. Plant Physiol. 1989b;90:306–310. doi: 10.1104/pp.90.1.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudd SH, Datko AH. Synthesis of EA and its regulation in Lemna paucicostata. Plant Physiol. 1989c;91:587–597. doi: 10.1104/pp.91.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Y, He JL, Hsia SL. A micro enzymatic method for determination of Cho-containing phospholipids in serum and high density lipoproteins. Lipids. 1993;28:949–951. doi: 10.1007/BF02537506. [DOI] [PubMed] [Google Scholar]
- Nuccio ML, Rhodes D, McNeil SD, Hanson AD. Metabolic engineering of plants for osmotic stress resistance. Curr Opin Plant Biol. 1999;2:128–134. doi: 10.1016/s1369-5266(99)80026-0. [DOI] [PubMed] [Google Scholar]
- Nuccio ML, Russell BL, Nolte KD, Rathinasabapathi B, Gage DA, Hanson AD. The endogenous Cho supply limits glycine betaine synthesis in transgenic tobacco expressing Cho monooxygenase. Plant J. 1998;16:487–496. doi: 10.1046/j.1365-313x.1998.00316.x. [DOI] [PubMed] [Google Scholar]
- Prud'homme M-P, Moore TS. Phosphatidylcholine synthesis in castor bean endosperm: free bases as intermediates. Plant Physiol. 1992a;100:1527–1535. doi: 10.1104/pp.100.3.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prud'homme M-P, Moore TS. Phosphatidylcholine synthesis in castor bean endosperm: the occurrence of an S-adenosyl-l-methionine:ethanolamine N-methyltransferase. Plant Physiol. 1992b;100:1536–1540. doi: 10.1104/pp.100.3.1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes D, Hanson AD. Quaternary ammonium and tertiary sulfonium compounds in higher plants. Annu Rev Plant Physiol Plant Mol Biol. 1993;44:357–384. [Google Scholar]
- Roscher A, Emsley L, Raymond P, Roby C. Unidirectional steady state rates of central metabolism enzymes measured simultaneously in a living plant tissue. J Biol Chem. 1998;273:25053–25061. doi: 10.1074/jbc.273.39.25053. [DOI] [PubMed] [Google Scholar]
- Smith DD, Summers PS, Weretilnyk EA. Phosphocholine synthesis in spinach: characterization of phosphoethanolamine N-methyltransferase. Physiol Plant. 2000;108:286–294. [Google Scholar]
- Stephanopoulos G, Vallino JJ. Network rigidity and metabolic engineering in metabolite overproduction. Science. 1991;252:1675–1681. doi: 10.1126/science.1904627. [DOI] [PubMed] [Google Scholar]
- Summers PS, Weretilnyk EA. Choline synthesis in spinach in relation to salt stress. Plant Physiol. 1993;103:1269–1276. doi: 10.1104/pp.103.4.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weretilnyk EA, Smith DD, Wilch GA, Summers PS. Enzymes of Cho synthesis in spinach: response of P-base N-methytransferase activities to light and salinity. Plant Physiol. 1995;109:1085–1091. doi: 10.1104/pp.109.3.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]