

Abstract
In the infarcted heart, the damage‐associated molecular pattern proteins released by necrotic cells trigger both myocardial and systemic inflammatory responses. Induction of chemokines and cytokines and up‐regulation of endothelial adhesion molecules mediate leukocyte recruitment in the infarcted myocardium. Inflammatory cells clear the infarct of dead cells and matrix debris and activate repair by myofibroblasts and vascular cells, but may also contribute to adverse fibrotic remodelling of viable segments, accentuate cardiomyocyte apoptosis and exert arrhythmogenic actions. Excessive, prolonged and dysregulated inflammation has been implicated in the pathogenesis of complications and may be involved in the development of heart failure following infarction. Studies in animal models of myocardial infarction (MI) have suggested the effectiveness of pharmacological interventions targeting the inflammatory response. This article provides a brief overview of the cell biology of the post‐infarction inflammatory response and discusses the use of pharmacological interventions targeting inflammation following infarction. Therapy with broad anti‐inflammatory and immunomodulatory agents may also inhibit important repair pathways, thus exerting detrimental actions in patients with MI. Extensive experimental evidence suggests that targeting specific inflammatory signals, such as the complement cascade, chemokines, cytokines, proteases, selectins and leukocyte integrins, may hold promise. However, clinical translation has proved challenging. Targeting IL‐1 may benefit patients with exaggerated post‐MI inflammatory responses following infarction, not only by attenuating adverse remodelling but also by stabilizing the atherosclerotic plaque and by inhibiting arrhythmia generation. Identification of the therapeutic window for specific interventions and pathophysiological stratification of MI patients using inflammatory biomarkers and imaging strategies are critical for optimal therapeutic design.
Abbreviations
- 11β‐HSD
11‐β hydroxysteroid dehydrogenase
- ACS
acute coronary syndrome
- CRP
C‐reactive protein
- ECM
extracellular matrix
- GR
glucocorticoid receptor
- iNKT
invariant natural killer T cells
- IRAK
IL‐1 receptor associated kinase
- MI
myocardial infarction
- MR
mineralocorticoid receptor
- NSAIDs
nonsteroidal anti‐inflammatory drugs
- PCI
percutaneous coronary intervention
- SDF
stromal cell‐derived factor
- STEMI
ST elevation myocardial infarction
- Tregs
regulatory T cells
- α‐SMA
α‐smooth muscle actin
Introduction
Myocardial infarction (MI) is a major cause of morbidity and mortality worldwide. Implementation of reperfusion strategies in patients presenting with ST elevation MI (STEMI) has significantly reduced acute mortality. However, this remarkable therapeutic success resulted in an expansion of the pool of patients who, while surviving the acute event, remain at risk for development of heart failure. The pathogenesis of heart failure following MI is intricately linked with repair and remodelling of the infarcted heart. The term ‘post‐infarction ventricular remodelling’ describes a constellation of cellular, molecular and proteomic changes in both infarcted and non‐infarcted myocardium that ultimately result in chamber dilation, hypertrophy of viable segments and progressive myocardial dysfunction. In human patients, dilative remodelling of the ventricle is associated with higher mortality and increased incidence of ventricular arrhythmias. The severity of adverse post‐infarction remodelling is dependent on the size of the infarct but is also affected by the qualitative characteristics of cardiac repair and by the profile of cellular and molecular alterations in the viable myocardium.
Extensive experimental evidence suggests that MI is intricately associated with activation of an inflammatory reaction (Frangogiannis, 2014a). Inflammatory mediators are directly involved in the pathogenesis of the vulnerable plaque, leading to occlusion of the coronary vessel and subsequent necrosis of the myocardial territory served by the vessel. Cardiomyocyte necrosis triggers both a systemic inflammatory response, mobilizing bone marrow‐derived immune cells, and a local reaction, leading to recruitment of circulating inflammatory cells that serve to clear the infarct from dead cells and matrix debris. Although leukocyte subsets play an important role in repair of the infarcted heart, prolonged activation of inflammatory pathways is involved in chronic adverse remodelling of the ventricle. Despite an impressive growth in our understanding of the role of inflammation in the pathogenesis of coronary occlusion and in the pathophysiology of cardiac repair remodelling and fibrosis, development of therapeutic strategies targeting inflammatory signals in patients with MI poses major challenges. This review provides a brief overview of the role of inflammatory cascades in injury, repair and remodelling of the infarcted heart, describes the long history of failed attempts to attenuate post‐ischaemic dysfunction and to reduce adverse remodelling by targeting inflammation, and discusses promising new therapeutic approaches and the challenges of clinical implementation.
The role of inflammation in plaque rupture
In patients, ruptured atherosclerotic plaques are responsible for the majority of cases of fatal MI (Davies and Thomas, 1984). Both systemic inflammation and local activation of macrophage‐driven inflammatory signalling in the micro‐environment of the plaque have been implicated in the pathogenesis of plaque rupture (Crea and Libby, 2017). Induction of chemokines, such as http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=771 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=856, mediate recruitment of macrophages in atherosclerotic plaques (Gu et al., 1998; Lesnik et al., 2003). The diverse phenotypic profiles of macrophages critically regulate progression, evolution and even regression of the atherosclerotic process (Mantovani et al., 2009; Rahman et al., 2017). http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2504 (Schieffer et al., 2000), oxidized LDL (Xu et al., 1999), http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1874 signalling (Mach et al., 1997) and pro‐inflammatory cytokines stimulate macrophage‐derived expression of proteases (including http://www.guidetopharmacology.org/GRAC/FamilyIntroductionForward?familyId=738 and cathepsins), degrading the extracellular matrix (ECM) of the fibrous cap (Shah et al., 1995) and promoting plaque fissuring. Moreover, local release of cholesterol crystals activates the inflammasome, generating active http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4974 and triggering pro‐inflammatory signalling (Freigang et al., 2011). In addition to the direct actions of pro‐inflammatory mediators on macrophage phenotype, mast cell degranulation, dysregulation of T cell subsets, B‐cell‐derived cytokine synthesis and stimulation of vascular cells in the plaque environment have also been implicated in activation of inflammatory macrophages in atherosclerotic plaques (Kaartinen et al., 1996; Mazzolai et al., 2004; Tay et al., 2016; Sage and Mallat, 2017; Tabas and Lichtman, 2017).
Activation of the post‐infarction inflammatory response
Prolonged coronary occlusion leads to death of the cardiomyocytes in the tissues served by the vessel, triggering activation of an intense inflammatory reaction. The post‐infarction inflammatory response can be divided in three phases: the alarm phase characterized by release of damage‐associated molecular pattern (DAMP) proteins that stimulate innate immune pathways; the leukocyte mobilization phase, marked by recruitment of neutrophils, monocytes and lymphocytes in the infarcted area; and the resolution phase, associated with suppression of pro‐inflammatory signalling and clearance of the leukocyte infiltrate (Figure 1).
Figure 1.
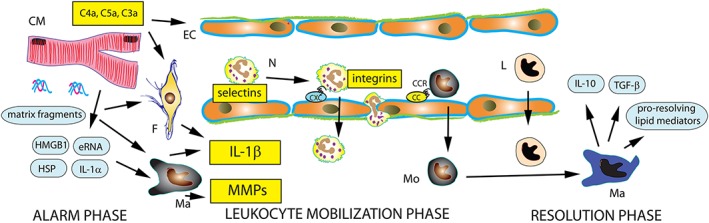
The inflammatory response following MI can be divided into three phases: the alarm phase, the leukocyte mobilization phase and the resolution phase. Necrotic cardiomyocytes (CM) release alarmins (heat shock proteins [HSP], high mobility group box 1 [HMGB1], extracellular RNA/eRNA, IL‐1α and other danger signals) that activate innate immune signalling pathways. ECM fragments also trigger inflammatory signalling. Induction of pro‐inflammatory cytokines, such as IL‐1, and chemokines mediates recruitment of neutrophils (N) and pro‐inflammatory monocytes (Mo) through interactions with endothelial cells (EC) that involve selectins and integrins. Clearance of dead cells and matrix debris from the infarct triggers transition to the resolution phase. Anti‐inflammatory lymphocyte (L) and macrophage (Ma) subsets release mediators that suppress pro‐inflammatory signalling, such as IL‐10, TGF‐β and pro‐resolving lipid mediators. Experimental studies suggest that inhibition of the complement cascade, IL‐1β antagonism, CCL2 inhibition, selectin and leukocyte integrin neutralization may be promising therapeutic strategies for patients with MI. F, fibroblast.
During the alarm phase, necrotic cardiomyocytes release danger signals (such as high mobility group box‐1, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4953, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2844, extracellular RNA, and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4973) that stimulate innate immune signalling (Andrassy et al., 2008; Chen et al., 2014; Lugrin et al., 2015). Generation of ECM fragments also contributes to the intense inflammatory reaction in the infarcted area (Huebener et al., 2008). Stimulation of innate immune responses following infarction involves effects of alarmins on http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=316 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2843‐dependent pathways in leukocytes, vascular cells and fibroblasts triggering transcription of pro‐inflammatory cytokines and chemokines (Arslan et al., 2011a; Zhang et al., 2015). Activation of the complement cascade also contributes to the post‐infarction inflammatory response (Hill and Ward, 1971; De Hoog et al., 2014). Post‐infarction inflammation not only serves to clear dead cells and matrix debris from the infarcted tissue but also sets the stage for repair of the infarcted area. In addition to activation of a local myocardial inflammatory response, infarction also triggers systemic inflammation, stimulating release of bone marrow‐derived leukocytes. In mouse models of MI, the spleen has also been suggested as an important contributor of inflammatory leukocytes (Swirski et al., 2009). Although the relative role of the cardio‐splenic axis in human MI remains unclear, clinical investigations have suggested that patients with acute coronary syndromes (ACS) have increased splenic metabolic activity and that activation of the spleen independently predicts cardiovascular events (Emami et al., 2015).
The cytokines and chemokines
Induction of pro‐inflammatory cytokines is a hallmark of the post‐infarction inflammatory response. Early release of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5074 (Frangogiannis et al., 1998) triggers a cytokine cascade that mediates recruitment of leukocytes in the infarcted myocardium. Activation of the inflammasome platform in fibroblasts, cardiomyocytes and immune cells (Kawaguchi et al., 2011; Mezzaroma et al., 2011) stimulates release of active IL‐1β, a critical mediator in regulation of cardiac inflammation and repair. IL‐1 signalling stimulates chemokine synthesis and promotes leukocyte infiltration in the infarcted myocardium (Bujak et al., 2008). Cardiac fibroblasts also respond to IL‐1, by acquiring a pro‐inflammatory and matrix‐degrading phenotype and by secreting cytokines, chemokines and MMPs. Moreover, IL‐1 delays myofibroblast conversion, suppressing synthesis of α‐smooth muscle actin (α‐SMA) (Saxena et al., 2013). The effects of IL‐1 on cardiac fibroblasts may serve to prevent premature acquisition of a matrix‐synthetic phenotype, until the infarct is cleared of dead cells and matrix debris.
Chemokines are also markedly up‐regulated in the infarcted heart and have been demonstrated to mediate leukocyte recruitment. Induction of both CXC and CC chemokines has been consistently demonstrated in experimental models of MI. CXC chemokines containing the ELR motif (Glu‐Leu‐Arg), such as http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=821, have been implicated in neutrophil recruitment (Ivey et al., 1995). On the other hand, members of the CC chemokine subfamily, such as CCL2 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=759, mediate recruitment of pro‐inflammatory monocytes (Dewald et al., 2005; Nahrendorf et al., 2007; Zouggari et al., 2013). Some members of the chemokine family may have effects on non‐haematopoietic cells, such as cardiomyocytes, fibroblasts and vascular cells. The CXC chemokine http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4465 may recruit progenitor cells with angiogenic potential (Liehn et al., 2011), contributing to neovascularization of the scar, and may stimulate pro‐survival cascades in ischaemic cardiomyocytes (Aiuti et al., 1997; Askari et al., 2003). The CXC chemokine http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=835 is markedly up‐regulated in experimental models of MI and may suppress fibrosis by inhibiting growth factor‐mediated fibroblast migration (Bujak et al., 2009; Saxena et al., 2014a).
Recruitment of leukocytes
Chemokines and cytokines play a critical role in recruitment of inflammatory leukocytes in the infarcted myocardium. Cytokine‐mediated induction of adhesion molecules in endothelial cells and integrin activation in leukocytes trigger adhesive interactions, ultimately leading to neutrophil, monocyte and lymphocyte extravasation in the infarcted area (Yamazaki et al., 1993; Frangogiannis, 2014a). Leukocyte subpopulations have been suggested to play important roles in both injurious and repair processes following MI. Early studies suggested that infiltrating neutrophils may extend ischaemic injury by exerting cytotoxic effects on viable cardiomyocytes in the infarct border zone (Entman et al., 1992). On the other hand, neutrophils have been suggested to orchestrate repair of the infarcted heart by modulating macrophage phenotype (Horckmans et al., 2017).
The macrophages
Monocytes recruited to the infarct region differentiate into macrophages and phagocytose dead cells and matrix debris, while secreting cytokines and growth factors that orchestrate repair. Clearance of apoptotic cells by professional phagocytes, a process known as efferocytosis (Wan et al., 2013), triggers cascades that suppress inflammation and promote activation of reparative mesenchymal cells. Ingestion of apoptotic cells is associated with release of anti‐inflammatory cytokines, such as http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4975 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5060 (Huynh et al., 2002), suppressing inflammation and activating a fibrogenic and matrix‐preserving programme. Several lines of evidence suggest crucial protective actions of macrophages in cardiac repair. First, macrophage depletion increased adverse remodelling in infarcted mice (van Amerongen et al., 2007). Second, in experimental models, macrophages played a crucial role in preventing mural thrombus formation following MI (Ben‐Mordechai et al., 2013; Frantz et al., 2013). Third, generation of alternatively activated macrophages exhibiting an M2‐like phenotype is critical to protect the infarcted heart from cardiac rupture (Shiraishi et al., 2016). Transition of macrophages into an anti‐inflammatory phenotype may also require activation of intracellular inhibitory cascades that restrain the immune response, such as expression of http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=579, an inhibitory member of the IRAK family that suppresses innate immune signalling (Chen et al., 2012). It has been suggested that therapeutic activation of the reparative properties of macrophages through administration of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4996 may exert protective actions in acute MI (Shintani et al., 2017). However, such therapeutic approaches need to be cautiously implemented, considering the known pro‐fibrotic actions of IL‐4 in the remodelling heart (Peng et al., 2015).
The lymphocytes
Early infiltration of the infarcted heart with lymphocyte subsets has been extensively documented in experimental models of MI (Frangogiannis et al., 2000a; Yan et al., 2013). Moreover, clinical studies have suggested that effector T cells may be trapped in the coronary microcirculation following reperfusion of the infarcted area and may contribute to the pathogenesis of microvascular obstruction, extending ischaemic cardiomyocyte injury (Boag et al., 2015). Early recruitment of lymphocyte subpopulations to the infarcted myocardium has been suggested to stimulate neutrophil and monocyte infiltration. B cells have been demonstrated to promote mobilization of pro‐inflammatory monocytes, thus playing a central role in activation of the inflammatory cascade (Zouggari et al., 2013). CD4−γδT‐cells have been implicated in neutrophil and macrophage infiltration and may promote adverse remodelling following MI (Yan et al., 2012). It should be emphasized that other lymphocyte subsets, such as regulatory T cells (Tregs), CD4+ helper T cells and invariant natural killer T (iNKT) cells, may have important repair functions following MI, negatively regulating inflammation, and activating mesenchymal and angiogenic cell populations to limit adverse remodelling (Dobaczewski et al., 2010; Hofmann et al., 2012; Sobirin et al., 2012; Weirather et al., 2014; Saxena et al., 2014b).
Negative regulation of the post‐infarction inflammatory response
Although macrophages are key effector cells in suppression of the post‐infarction inflammatory response, several other cell types may contribute to downmodulation of pro‐inflammatory signalling. Anti‐inflammatory lymphocyte subsets, such as Tregs (Dobaczewski et al., 2010; Weirather et al., 2014; Saxena et al., 2014b), iNKT cells (Sobirin et al., 2012) and dendritic cells (Anzai et al., 2012) have been identified as important sources of anti‐inflammatory cytokines in the healing infarct. Surviving cardiomyocytes in the infarct border zone may also limit and restrain inflammation by secreting mediators that recruit and activate regulatory and reparative macrophages (Lorchner et al., 2015). Acquisition of an anti‐inflammatory phenotype by vascular cells may also contribute to negative regulation of post‐infarction inflammation. Members of the TGF‐β family may inhibit adhesion molecule expression by endothelial cells and leukocytes, inhibiting leukocyte‐endothelial cell interactions and preventing uncontrolled leukocyte recruitment (Kempf et al., 2011). Recruitment of mural cells by infarct neovessels may serve to suppress endothelial pro‐inflammatory activation (Zymek et al., 2006). Thus, timely suppression and spatial containment of the inflammatory response following infarction is dependent on activation of a wide range of molecular signals with actions on several different cell types. In animal models, defects in these regulatory mechanisms result in unrestrained, prolonged or expanded inflammation, leading to accentuated cardiac remodelling and worse dysfunction following infarction. In patients, defective negative regulation of the post‐infarction inflammatory response may be involved in the pathogenesis of adverse remodelling and heart failure in patients surviving an acute MI (Frangogiannis, 2014a).
Myofibroblast activation
Because the adult mammalian heart has negligible regenerative capacity, repair of the infarcted myocardium is dependent on fibroblast activation and subsequent formation of a collagen‐based scar. Perturbations in fibroblast activation and in the profile of ECM proteins in the infarcted and remodelling myocardium can be associated with increased dysfunction and adverse remodelling (Frangogiannis et al., 2005; Kong et al., 2017). During the proliferative phase of infarct healing, the cardiac fibroblast population markedly expands (Frangogiannis et al., 2000b). Activated myofibroblasts form organized arrays in the infarct border zone (Blankesteijn et al., 1997). These cells incorporate into their cytoskeleton contractile proteins (such as α‐SMA and the embryonal isoform of smooth muscle myosin) (Willems et al., 1994; Frangogiannis et al., 2000b; Shinde et al., 2017) but do not express markers of mature vascular smooth muscle cells, such as the SM1 and SM2 isoforms of smooth muscle myosin heavy chain (Frangogiannis et al., 2000b). Infarct myofibroblasts are predominantly derived from epicardium‐derived fibroblast populations (Ruiz‐Villalba et al., 2015; Kanisicak et al., 2016). During the proliferative phase of cardiac repair, suppression of pro‐inflammatory signals, activation of TGF‐β cascades and deposition of specialized matrix proteins, such as ED‐A http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6754 (Arslan et al., 2011b) and matricellular proteins (Frangogiannis, 2017a), trigger conversion of interstitial fibroblasts into myofibroblasts.
Activated myofibroblasts have been identified as the main source of ECM proteins in the healing infarct (Cleutjens et al., 1995). In addition to their matrix synthetic capacity, activated infarct fibroblasts may also contribute to phagocytosis of dead cells (Nakaya et al., 2017) and may secrete mediators that modulate cardiomyocyte survival (Abrial et al., 2014) or mediate activation of immune cells (Anzai et al., 2017). Whether distinct subpopulations are responsible for the functional pleiotropy of infarct myofibroblasts remains unknown. Excessive fibroblast activation may lead to expansion of the fibrotic area, increasing myocardial stiffness and promoting diastolic dysfunction. The potential involvement of negative regulatory mechanisms that restrain fibrogenic signals, in the prevention of uncontrolled fibrosis following MI, has not been investigated.
Inflammation in the remodelling myocardium
In the presence of a large infarction, massive loss of contractile myocardium is associated with activation of an inflammatory response in remote remodelling myocardial segments, accompanied by progressive interstitial fibrosis (Sager et al., 2016b). Several mechanisms may contribute to inflammatory activation in the remodelling myocardium. First, volume and pressure loads, related to dilation of the chamber following infarction and to the elevation of filling pressures. Mechanical stress in the remodelling myocardium may locally activate macrophages stimulating their proliferation and promoting a fibrogenic environment (Sager et al., 2016b). Second, defective suppression or impaired spatial containment of the inflammatory response in the infarct border zone may lead to prolonged activation of inflammatory pathways or expansion of the inflammatory infiltrate to viable segments (Frangogiannis et al., 2005). Third, an immune‐mediated response triggered by poorly defined antigens may mediate chronic inflammation in the remodelling myocardium (Ismahil et al., 2014). In human ischaemic heart failure, subpopulations of patients may exhibit dysregulated inflammatory responses that may contribute to the pathogenesis of the cardiomyopathy.
The rationale for targeting inflammation after MI
The critical role of inflammation in all aspects of the myocardial response to injury suggests that targeting inflammatory signals may hold promise to reduce mortality and prevent heart failure in patients surviving an acute MI. The cell biological basis supporting therapeutic interventions that modulate the post‐MI inflammatory cascade may involve several distinct beneficial actions. First, attenuation of inflammation during the early post‐ischaemic inflammatory phase may prevent leukocyte‐mediated cardiomyocyte injury in surviving cardiomyocytes of the border zone. Second, inhibition of inappropriate late activation of pro‐inflammatory signalling may protect cardiomyocytes in the remodelling area from chronic apoptosis. Third, attenuation of inflammation may restrain protease activation, increasing the tensile strength of the healing scar and preventing adverse remodelling. Fourth, suppression of inflammation‐driven fibrogenic signalling may protect the heart from dysregulated fibrotic remodelling. Fifth, selective activation of chemokine‐dependent recruitment of progenitor cells into the area of infarction may promote angiogenesis and even contribute to the ultimate goal of myocardial regeneration. Sixth, pro‐inflammatory signalling has been linked to ventricular arrhythmias; thus, attenuation of inflammation may have direct anti‐arrhythmic actions. Finally, anti‐inflammatory approaches may prevent plaque rupture reducing the incidence of recurrent coronary events.
It should be emphasized that some of the protective effects of certain established therapeutic approaches in patients with MI, such as angiotensin converting enzyme inhibition (Leuschner et al., 2010), angiotensin receptor blockade (Kohno et al., 2008), mineralocorticoid receptor (MR) inhibition (Fraccarollo et al., 2008), β‐adrenoceptor blockade (Garcia‐Prieto et al., 2017) and administration of statins (Zhang et al., 2005), may involve direct modulation of inflammation. For example, leukocyte‐specific http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=29 signalling has been reported to mediate leukocyte recruitment in the infarcted heart (Grisanti et al., 2016), and a recent study suggested that the infarct‐limiting effects of β‐blockade with metoprolol in a mouse model of MI were lost following neutrophil depletion or through genetic knockdown of genes associated with platelet:neutrophil interactions (Garcia‐Prieto et al., 2017). However, considering the broad effects of neurohumoral pathways on both cardiomyocyte and non‐cardiomyocyte populations, the relative contribution of inflammatory cell modulation remains unclear.
Early attempts to inhibit inflammation were primarily focused on the use of broad anti‐inflammatory strategies, such as glucocorticoids. These approaches were often associated with adverse consequences. Over the last 30 years, progress in fundamental immunology and better understanding of cardiac pathophysiology led to implementation of new therapeutic strategies targeting specific inflammatory pathways. Despite the increasing sophistication of the approaches, therapeutic implementation of inflammatory targets in patients with MI remains challenging, in part due to the remarkable pathophysiological heterogeneity of the human condition.
Broad anti‐inflammatory interventions
Glucocorticoids
Due to the ubiquitous expression of the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=625) in all nucleated cells, glucocorticoids have a wide range of effects on many different cell types and are potent regulators of the inflammatory response (Cain and Cidlowski, 2017). During the alarm phase, glucocorticoids attenuate responses to danger signals, suppressing production of inflammatory mediators. Glucocorticoids also markedly reduce leukocyte infiltration into the tissues, by decreasing chemokine expression and by suppressing adhesive interactions between leukocytes and endothelial cells. During the resolution phase, glucocorticoids exert a wide range of actions on both inflammatory and reparative cells. Glucocorticoids are known to promote clearance of apoptotic cells (Liu et al., 1999) and direct macrophages towards an anti‐inflammatory phenotype (Snyder and Unanue, 1982). In addition to their effects on immune cells, glucocorticoids also inhibit repair, by reducing fibroblast‐derived collagen synthesis and by inhibiting the angiogenic response. It should be emphasized that the effects of glucocorticoids are not limited to activation of GR signalling. Glucocorticoids also bind with high affinity to the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=626s (Arriza et al., 1987) and may also act by activating transcription‐independent non‐classical pathways, involving cell surface receptors (Samarasinghe et al., 2012). Stimulation of MR signalling mediates pro‐inflammatory macrophage activation in vitro and in vivo (Usher et al., 2010), whereas activation of endogenous myeloid cell‐specific GR signalling has been suggested to mediate reparative pathways (Galuppo et al., 2017). The relative role of GR and MR activation in the response to glucocorticoid treatment is likely to depend on the specific agent used, the dose and on the cell biological context.
Numerous experimental studies have examined the effects of glucocorticoids in experimental models of MI (Table 1). Unfortunately, most of the studies were performed in models of non‐reperfused MI, limiting the value of the conclusions in the current era of myocardial reperfusion. Although the effects are dependent on the agent used, the dose and duration of treatment, several studies have suggested that glucocorticoids may protect the infarcted myocardium, reducing cardiomyocyte necrosis and apoptosis (Libby et al., 1973; Xu et al., 2011). The basis for these protective actions is unclear, especially considering the use of permanent coronary occlusion models that would be expected to cause death of most cardiomyocytes in the area at risk. Other in vivo studies suggested that high‐dose corticosteroid therapy impairs clearance of dead cells from the infarct and may disrupt fibroblast function (Kloner et al., 1978), leading to formation of thinner scars. These effects would be expected to be detrimental in repair of the infarcted heart and may precipitate adverse remodelling. Clinical studies on the use of corticosteroids in patients with MI have produced conflicting results (Table 2). Although some studies reported protective effects, in other studies, significant concerns were raised regarding the safety of the approach (Roberts et al., 1976; Giugliano et al., 2003). Considering their broad actions on all cell types involved in cardiac injury and repair and their effects on several molecular cascades, glucocorticoids cause a wide range of adverse effects and are unattractive therapeutic options for patients with MI. However, understanding the cell‐specific actions of GR activation may suggest more targeted approaches with therapeutic potential. Moreover, recent insights into the pathways involved in tissue‐specific intracellular metabolism of glucocorticoids have suggested novel therapeutic directions (Gray et al., 2017). The enzyme http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2763) catalyses intracellular regeneration of glucocorticoids from inert metabolites. In a mouse model, global deletion of 11‐β‐HSD1, the more widely distributed isoform of the enzyme, promoted angiogenesis and attenuated infarct expansion following MI (White et al., 2016), suggesting that endogenous glucocorticoid regeneration may inhibit repair and exacerbate remodelling following MI. Thus, approaches inhibiting 11‐β‐HSD1 may be of therapeutic value to prevent development of heart failure following MI.
Table 1.
Effects of glucocorticoids in experimental models of MI
| Model | Animal | Agent, dose and duration of treatment | Major findings | Ref. |
|---|---|---|---|---|
| Non‐reperfused MI | Cat | http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7088 (MP, 30 mg·kg−1, i.v.) 30 min prior or 60 min following occlusion | Both pretreatment and post‐occlusion administration reduced myocardial injury, assessed through reduction of ST segment elevation. | (Spath Jr et al., 1974) |
| Non‐reperfused MI | Dog |
Group1: http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2868 (HC, 50 mg·kg−1, i.v.) 30 min after occlusion, followed by supplementary dose of 25 mg·kg−1 12 h after occlusion; Group2: HC (50 mg·kg−1, i.v.) 6 h after occlusion, followed by supplementary dose of 25 mg·kg−1 12 h after occlusion |
HC reduced infarct size and attenuated cardiomyocyte necrosis, even when administered 6 h after occlusion. | (Libby et al., 1973) |
| Non‐reperfused MI | Rat | MP (50 mg·kg−1 i.v.) 5 min after occlusion, followed by (50 mg·kg−1 i.m.) 3, 6 and 24 h after occlusion | MP treatment was associated with reduced collagen deposition, delayed inflammation and repair, and persistent presence of ‘mummified’ cardiomyocytes (cells with preservation of striations and sarcolemmal membrane that exhibited nuclear degeneration). | (Kloner et al., 1978) |
| Non‐reperfused MI | Rat |
1. HC: 50 mg·kg−1 i.v. 5 min after occlusion; 2. Single‐dose MP: 50 mg·kg−1 i.v. 5 min after occlusion; 3. Multiple dose MP: 50 mg·kg−1 i.v. 5 min after occlusion, followed by 50 mg·kg−1 i.m. at 3, 6 and 24 h |
1. Based on histological analysis and measurements of creatine kinase (CK) activity, glucocorticoids salvaged injured myocardium (by 15% in the HC group, 21% in MP single dose group, and 21% in the MP multiple dose group. 2. Multiple dose MP caused infarct thinning |
(Maclean et al., 1978) |
| Non‐reperfused MI | Cat | http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2768 (Dx), 8 mg·kg−1 i.v., 30 min prior to or 60 min following occlusion |
1. Dx pre‐ or post‐ administration significantly attenuated the increase in plasma CPK activity. 2. Dx had no effects on the haemodynamic response within the first 5 h following MI. |
(Spath and Lefer, 1975) |
| Non‐reperfused MI | Dog |
Group1: high dose MP (50 mg·kg−1 i.v). 15 min and 3, 24 and 48 h after occlusion; Group2: low dose MP (30 mg·kg−1 i.v). 15 min after occlusion |
1. Low dose MP reduced the size of the infarct. 2. The high dose MP protocol (but not low dose MP) caused infarct thinning and worsened systolic dysfunction without affecting collagen content. |
(Hammerman et al., 1983a) |
| Non‐reperfused MI | Dog | MP, 7.5 mg·kg−1, i.v. twice daily for 7 days after occlusion | MP reduced infarct size and attenuated compensatory hypertrophy without affecting haemodynamics. | (Slutsky and Murray, 1985) |
| Non‐reperfused MI | Rat | MP (50 mg·kg−1, i.v.) immediately after occlusion, followed by 50 mg·kg−1, i.p., q6 h for 3 days | MP decreased collagen content in the infarcted heart. | (Vivaldi et al., 1987) |
| Non‐reperfused MI | Mouse | Dx (20 mg·kg−1, i.p.) 20 h prior to occlusion | Dx treatment reduced infarct size, attenuating cardiomyocyte apoptosis. | (Xu et al., 2011) |
| Non‐reperfused MI | Rat | MP (5 mg·kg−1, i.p.) starting 7 days post‐MI and continued to 21 day after occlusion | MP did not affect mortality or infarct size, but attenuated hypertrophic remodelling and significantly increased capillary density. | (Van Kerckhoven et al., 2004) |
| Reperfused MI | Dog | MP single dose (50 mg·kg−1, i.v.) after occlusion | MP did not affect infarct size and haemodynamic variables. | (Genth et al., 1982) |
| Reperfused MI | Dog | MP (30 mg·kg−1, i.v.) after occlusion; | MP increased myocardial blood flow during ischaemia, but had no effect on blood flow after reperfusion. | (da‐Luz et al., 1982) |
| Ischaemia and reperfusion in vivo | Rabbit | low dose http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2866 (5 mg·kg−1/24 h i.m.) or high dose prednisolone (10 mg·kg−1/24 h i.m.) protocols | Infarct healing was significantly delayed in both low and high dose groups. However, infarct thinning was not affected. | (Shizukuda et al., 1991) |
Table 2.
Effects of glucocorticoid treatment in patients with MI
| Type of study | Number of patients | Agent, dose and duration | Major findings | Ref. |
|---|---|---|---|---|
| Double blind clinical trial | 132 | oral http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7096 (starting dose: 30 mg·day−1), for 12 days |
No difference in acute mortality. No difference in rhythm and conduction disturbance, and cardiac rupture. |
(Sievers et al., 1964) |
| Prospective cohort study | 446 | Hydrocortisone (HC), 500 mg i.v. for the first 4 days |
HC significantly reduced mortality. There were no differences in the incidence of acute complications (acute heart failure, shock, cardiac arrhythmia, infections). |
(Barzilai et al., 1972) |
| Prospective cohort study | 39 | Methylprednisolone (MP): 3 g, 7–12 h following rise of serum CPK | MP treatment reduced infarct size. | (Morrison et al., 1975) |
| Prospective cohort study | 66 |
MP (i.v.): 1. Single 2.0 g dose 7–25 h following initial rise of CPK; 2. Two 2.0 g doses, 3–12 h apart |
Reduction of infarct size and mortality in both MP treatment groups. | (Morrison et al., 1976) |
| Prospective cohort study | 44 |
1. Single dose: MP, 30 mg·kg−1, i.v., 7 h from first CPK elevation; 2. Multiple dose: MP, 30 mg·kg−1, i.v. starting after 7 h from first CPK elevation, every 6 h, for 48 h |
Neither single dose nor multiple dose affected haemodynamics. Multiple dose MP (but not single dose) extended infarct size, increased ventricular dysrhythmias, and caused hyperglycaemia. | (Roberts et al., 1976) |
| Prospective cohort study | 29 | MP 30 mg·kg−1, i.v. 7 h and 10 h from onset of symptoms | High doses of MP given early in the course of MI have neither deleterious nor beneficial effects. | (Peters et al., 1978) |
| Prospective cohort study | 10 | MP, 2.0 g i.v. single dose, average of 13 h from onset of chest pain | MP administration had no short‐term protective effects and worsened haemodynamics. | (Heikkila and Nieminen, 1978) |
| Prospective cohort study | 45 | Methylprednisolone, single dose 25 mg·kg−1, i.v. within 4 h after onset of chest pain | MP delayed cardiomyocyte injury and may stabilize lysosomal membranes during acute myocardial ischaemia. | (Welman et al., 1979) |
| Prospective cohort study | 42 | MP, 30 mg·kg−1, i.v four doses, q6 h. | Early short‐term high‐dose MP had no effects on infarct size, dysrhythmias, complications, or left ventricular function 2 weeks after infarction. | (Bush et al., 1980) |
| Prospective cohort study | 28 | MP (30 mg·kg−1), i.v. two doses, 2.5 h apart | MP administration in AMI patients had no effect on survival, infarct size and metabolic parameters, but increased cardiac output and reduced systemic vascular resistance. | (Henning et al., 1981) |
| Retrospective cohort study | 1746 | N/A | Previous or inpatient corticosteroid use did not affect the incidence of cardiac rupture, or non‐rupture related mortality in MI patients. | (Dellborg et al., 1985) |
| Retrospective study | 41 | N/A | This uncontrolled retrospective study suggested that use of anti‐inflammatory agents (glucocorticoids and NSAIDs) before and after MI may be associated with a high incidence of cardiac rupture. | (Silverman and Pfeifer, 1987) |
| Double blind, randomized trial (RCT) | 1118 |
Group 1: early MP: 30 mg·kg−1, i.v. within 6 h of chest pain; repeated administration 3 h later; Group 2: late MP (30 mg·kg−1, i.v.) 6–12 h from onset of chest pain; repeated administration 3 h later |
Late treatment (6–12 h) with MP reduced mortality without affecting cardiac rupture, early malignant ventricular arrhythmias or other adverse cardiac events. Late MP treatment reduced 28 day and 6 month mortality in patients with inferior/posterior infarction, but not in anterior MI. Early treatment had no effects. | (Metz et al., 1986) (The Solu‐Medrol Sterile Powder AMI Study Group, 1986) |
| RCT | 40 | MP, 2.0 g, i.v. within 6 h, repeated same dose 3 h later | MP infusion had no effects on mortality, death from cardiac rupture, peak cardiac injury enzymes, arrhythmias, haemodynamics and 6 months hospitalization rates. | (Madias and Hood Jr, 1982) |
Nonsteroidal anti‐inflammatory drugs (NSAIDs)
http://www.guidetopharmacology.org/GRAC/DatabaseSearchForward?page=1&searchString=Nonsteroidal%20anti-inflammatory%20drugs&searchCategories=all&order=rank (including aspirin) have broad anti‐inflammatory actions as their effects are mediated through inhibition of http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=269, the rate limiting enzyme in prostaglandin synthesis. There are two major isoforms of COX, the constitutively expressed COX‐1 and COX‐2, which is not found in normal tissues but is induced by inflammation, ischaemia and stress. Non‐selective NSAIDs, introduced in the 1950s, inhibit both COX isoforms. Inhibition of COX‐1‐induced prostaglandins in the gastric mucosa by traditional NSAIDs is often associated with gastrointestinal toxicity, including peptic ulcer disease. Thus, selective COX‐2 inhibitors were introduced in the late 1990s in an attempt to develop anti‐inflammatory strategies without the risk of gastrointestinal side effects (Boulakh and Gislason, 2016).
Aspirin acts through non‐competitive, irreversible acetylation of COX. In nucleated cells, the ability of the cells to synthesize COX‐1 and COX‐2 de novo allows recovery of prostaglandin synthesis despite inhibition by aspirin. In contrast, in platelets, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4482 production is dependent on preformed COX‐1. Thus, irreversible binding of aspirin to platelet COX‐1 results in inhibition of platelet aggregation for the entire life of the platelet and mediates aspirin's potent cardioprotective actions by reducing the incidence of new cardiovascular events (ISIS‐2 Collaborative Group, 1988). In contrast, because the NSAIDs, apart from aspirin, competitively and reversibly inhibit COX, they do not cause sustained inhibition of platelet aggregation and do not provide long‐term protection from atherothrombotic cardiovascular events (Vonkeman and van de Laar, 2010).
Although some early studies in animal models suggested that both non‐selective non‐aspirin NSAIDs and selective COX‐2 inhibitors may have protective effects following MI (Lefer and Polansky, 1979), by attenuating adverse remodelling, by reducing cardiomyocyte apoptosis and by increasing arteriolar density (Abbate et al., 2006; Straino et al., 2007), other experimental studies demonstrated detrimental effects on infarct healing, resulting in scar thinning (Brown Jr et al., 1983; Hammerman et al., 1984a; Hammerman et al., 1983b) and accentuated systolic dysfunction (Table 3) (Timmers et al., 2007). Moreover, genetic loss‐of‐function studies demonstrated protective effects for endogenous COX‐2 in myocardial ischaemia/reperfusion models (Camitta et al., 2001; Bolli et al., 2002).
Table 3.
Effects of NSAIDs in experimental models of MI
| Model | Species | Agents dose duration delivery method | Major findings | Ref. |
|---|---|---|---|---|
| Reperfused MI | Dog |
Group1: BW755C (inhibitor of both lipoxygenase and COX, 10 mg·kg−1, i.v.) 30 min after reperfusion Group2: http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1909 (5 mg·kg−1, i.v.) 10 min before reperfusion |
BW755C treatment reduced infarct size and decreased the incidence of arrhythmias, attenuating leukocyte infiltration. Treatment with indomethacin did not affect infarct size and leukocyte migration into the ischaemic myocardium. | (Mullane et al., 1984) |
| Non reperfused MI | Dog | Indomethacin (10 mg·kg−1, i.v.) at 15 min and 3 h after occlusion | Indomethacin caused infarct expansion and scar thinning. | (Hammerman et al., 1984b) |
| Non reperfused MI | Cat | http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2713 (12.5 mg·kg−1, i.v.) 0 and 2.5 h after occlusion | Ibuprofen attenuated myocardial injury without affecting haemodynamics during the early stage of infarction. | (Lefer and Polansky, 1979) |
| Non reperfused MI | Rat | http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4139 (25 mg·kg−1·day−1, i.p.) 2 days before occlusion until the end of the experiment | Aspirin significantly attenuated interstitial and perivascular fibrosis in the spared myocardium, without affecting wound healing, compensatory hypertrophy and LV dysfunction. | (Kalkman et al., 1995) |
| Non reperfused MI | Mouse | Aspirin (120 mg·kg−1·day−1, subcutaneously) after occlusion for 4 weeks | High dose aspirin did not affect post‐infarct cardiac remodelling and cardiac dysfunction, but attenuated pro‐inflammatory cytokine levels. | (Adamek et al., 2007) |
| Non‐reperfused MI | Pig | http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2892 (COX‐2i; 400 mg p.o. bid) after occlusion until end of protocol | Celecoxib increased mortality, promoted infarct thinning, LV dilatation, and accentuated systolic dysfunction. | (Timmers et al., 2007) |
| Non‐reperfused MI | Rat | http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2895 (COX‐2i, 0.75 mg·kg−1 i.p.) 24 h after occlusion, daily for 5 days | Parecoxib ameliorated remodelling, reducing peri‐infarct apoptosis and preserving arteriolar density. Parecoxib did not affect mortality, infarct size and plasma inflammatory cytokines. | (Straino et al., 2007) |
| Non‐reperfused MI | Mouse | Parecoxib (COX‐2i, 0.75 mg·kg−1, i.p.) 24 h after surgery, daily for 5 days |
Parecoxib treatment significantly reduced apoptosis in the peri‐infarct region; No difference of mortality on day 7. |
(Abbate et al., 2006) |
| Reperfused and non‐reperfused MI | Mouse | Parecoxib (COX2i, 0.75 mg·kg−1·day−1, i.p.) for 7 days | Parecoxib treatment reduced mortality and attenuated apoptosis in non‐reperfused infarcts, but had no effects in reperfused infarction. | (Salloum et al., 2009) |
| Non‐reperfused MI | Rat |
Group 1: DFU (COX2i, 5 mg·kg−1·day−1, p.o.); Group 2: low dose aspirin (1 mg·kg−1·day−1, p.o.); Group 3: high dose aspirin (25 mg·kg−1·day−1, p.o.) 30 min prior to occlusion for 3 months |
DFU treatment significantly reduced left ventricular end‐diastolic pressure, reduced infarct size and improved cardiac contractility without affecting mortality. Aspirin had no effects on cardiac function. |
(Saito et al., 2004) |
| Non‐reperfused MI | Mouse |
NS‐398 (COX‐2i, 3 mg·kg−1·day−1, p.o.) 48 h after occlusion for 2 weeks; Rofecoxib (COX2i, 15 mg·kg−1·day−1, p.o.), 48 h after occlusion for 2 weeks |
NS‐398 attenuated adverse remodelling and dysfunction following MI without affecting infarct size. Both COX2i reduced cardiac hypertrophy, collagen production, and TGF‐β expression in the infarcted heart. | (LaPointe et al., 2004) |
| Non‐reperfused MI | Dog | http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7273,(1 mg·kg−1 i.v.) 15 min, 3 h after occlusion | Piroxicam caused moderate scar thinning without perturbing regional function. | (Hammerman et al., 1984a) |
In the clinical setting, a large body of evidence suggests that exposure to NSAIDs increases risk of cardiovascular events in patients with known cardiovascular disease. In post‐MI patients, administration of NSAIDs is associated with a high risk of death and reinfarction and an increased risk of hospitalization due to heart failure (Gislason et al., 2006; Brophy et al., 2007). Several mechanisms may explain the detrimental effects of NSAIDs in patients with MI. First, COX‐2 inhibition may promote pro‐thrombotic events by inhibiting generation of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1915. Second, loss of renal actions of COX‐2 may elevate blood pressure and promote heart failure decompensation. Third, COX‐2 inhibition may block atheroprotective actions, thus accelerating atherosclerosis (Egan et al., 2004). Fourth, COX‐2 targeting may inhibit important cardioprotective actions and exert pro‐arrhythmic effects. Fifth, COX inhibition may disrupt important reparative functions, leading to formation of a defective scar (Hammerman et al., 1984b). Considering the abundant clinical evidence suggesting detrimental effects, all non‐aspirin NSAIDs should be avoided in patients with established cardiovascular disease (Schmidt et al., 2016).
Immunomodulatory strategies
Few studies have tested the effects of non‐specific immunomodulation and immunosuppression in MI. In animal models, early administration of low‐dose immunosuppressive agents has been reported to exert beneficial actions. However, clinical studies have provided disappointing results. In a rat model of reperfused MI, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7154 administration attenuated leukocyte infiltration and reduced ventricular dysfunction (Zhu et al., 2008). http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4815 treatment was also found to exert protective effects on the ischaemic and reperfused myocardium in both large animal (Asanuma et al., 2004) and rodent models (Maranhao et al., 2017). However, in a small clinical trial, methotrexate administration to patients with STEMI did not affect acute infarct size and worsened systolic dysfunction 3 months after the acute event (Moreira et al., 2017). Immune modulation with intravenous immunoglobulin also failed to reduce infarct size and attenuate adverse remodelling in STEMI patients (Gullestad et al., 2013).
Cyclosporine, another potent immunosuppressive agent, has attracted interest as a therapeutic agent for patients with MI, because of its effects as a cyclophilin B inhibitor. It has been proposed that cyclophilin B inhibition may inhibit opening of the mitochondrial permeability transition pore, thus protecting ischaemic cardiomyocytes from death. In addition to its protective actions on cardiomyocytes, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1024 may also reduce inflammation in the infarcted heart (Squadrito et al., 1999). Despite promising early results in pilot studies, suggesting reduced infarct size in STEMI patients treated with cyclosporine (Piot et al., 2008), a large randomized double‐blind controlled trial showed no beneficial effects of a bolus dose of cyclosporine in STEMI patients undergoing percutaneous coronary intervention (PCI) (Cung et al., 2015).
The failures of broad anti‐inflammatory inhibition with corticosteroids and NSAIDs and advances in understanding the biology of the inflammatory response led to the development of specific inflammatory targets following MI.
Targeted anti‐inflammatory interventions
Experimental studies have identified crucial molecular signals mediating the inflammatory response following MI. Therapeutic interventions in animal models suggested that targeted inhibition of specific inflammatory signals may protect the infarcted heart from acute injury and prevent adverse remodelling following MI. Despite promising results in animal models, therapeutic implementation of inflammatory targets in patients with MI has been challenging (Table 4).
Table 4.
Targeted anti‐inflammatory and immunomodulatory therapies in patients with MI
| Type of study | Number of patients | Agent, dose and duration | Major findings | Ref. |
|---|---|---|---|---|
| Anti‐integrin | ||||
| RCT: FESTIVAL | 88 |
http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9598 (anti‐CD11/18, also known as LeukArrest or Hu23F2G) Low dose: (0.3 mg·kg−1) High dose: (1.0 mg·kg−1) i.v. after coronary angiography |
Hu23F2G was well tolerated, with no increase in adverse events, including infections. Single‐photon emission computed tomographic (SPECT) imaging showed no significant effects of anti‐CD11/CD18 treatment on myocardial infarct size in STEMI patients. |
(Rusnak et al., 2001) |
| RCT: HALT‐MI | 420 |
Rovelizumab (anti‐CD11/18) Low dose: (0.3 mg·kg−1) High dose: (1.0 mg·kg−1) i.v. before coronary angioplasty |
Treatment with anti‐CD11/CD18 did not affect infarct size in STEMI patients who underwent primary angioplasty. | (Faxon et al., 2002) |
| RCT: LIMIT‐AMI | 394 | Anti‐CD18 (i.v. bolus of 0.5 or 2.0 mg·kg−1, before commencing recombinant tissue plasminogen activator (rtPA) | No significant effects on coronary blood flow, infarct size, or the rate of ECG ST‐segment elevation resolution STEMI patients treated with rtPA. | (Baran et al., 2001) |
| Anti‐Selectin | ||||
| RCT: SELECT‐ACS | 544 | Inclacumab (anti‐P‐Selectin), single infusion 5 or 20 mg·kg−1, 1–12 h before PCI | Inclacumab at a dose of 20 mg·kg−1 appeared to reduce myocardial damage after PCI in non‐STEMI patients, without significant difference in adverse events. | (Tardif et al., 2013) |
| IL‐1 inhibition | ||||
| Pilot Study: VCU‐ART | 10 | Anakinra (IL‐1RN) 100 mg·day−1 subcutaneously for 14 days | IL‐1 blockade with anakinra was safe and ameliorated LV remodelling in STEMI patients. | (Abbate et al., 2010) |
| Pilot Study: VCU‐ART2 | 30 | Anakinra (IL‐1RN) 100 mg·day−1 subcutaneously for 14 days | Anakinra blunted the acute inflammatory response in STEMI patients, without showing benefits of LV remodelling or function. | (Abbate et al., 2013) |
| RCT: MRC‐ILA Heart Study | 182 | Anakinra (IL‐1RN) 100 mg·day−1 subcutaneously for 14 days |
Following 14 day treatment of IL‐1RN, inflammatory markers were reduced; In patients with NSTE‐ACS, IL‐1RN treatment significantly increased major adverse cardiac events at 1 year, but not at day 30 or 3 months. |
(Morton et al., 2015) |
| RCT: CANTOS | 10 061 | Canakinumab (anti‐IL‐1β) at three different doses: 50, 150 and 300 mg subcutaneously, once every 3 months |
In patients with previous MI and an hs‐CRP level ≥ 2 mg·L−1, canakinumab (150 mg) significantly reduced hs‐CRP, but increased the incidence of fatal infection and sepsis. The 300 mg dose had similar effects, but the multiple statistical comparisons yielded non‐significant differences in comparison to the placebo group. The reduced rate of recurrent cardiovascular events in treated patients was independent of lipid level lowering. |
(Ridker et al., 2017) |
| Anti‐complement | ||||
| Pilot study | 31 | Complement 1 inhibitor (loading dose 50 or 100 U·kg−1 6 h after MI, followed by continuous infusion 1.25 or 2 U·kg−1·h−1 for 48 h) | In MI patients who received early thrombolytic therapy, C1 inhibitor treatment reduced troponin T and creatine kinase‐MB levels, without causing adverse effects. | (de Zwaan et al., 2002) |
| RCT: COMPLY | 943 | Pexelizumab (anti‐C5) bolus (2 mg·kg−1), or pexelizumab bolus (2 mg·kg−1) followed by pexelizumab infusion (0.05 mg·kg−1·h−1) for 20 h | In STEMI patients receiving fibrinolysis, adjunct treatment with pexelizumab neither reduced infarct size nor improved clinical outcomes. | (Mahaffey et al., 2003) |
| RCT: COMMA | 960 | Pexelizumab bolus (2 mg·kg−1), or pexelizumab bolus (2 mg·kg−1) followed by pexelizumab infusion (0.05 mg·kg−1·h−1) for 20 h | In STEMI patients undergoing PCI, adjunct treatment with pexelizumab had no measurable effect on infarct size or on the composite of 90 day death, new or worsening heart failure, shock and stroke. 90 day mortality was significantly reduced in pexelizumab bolus plus infusion. | (Granger et al., 2003) |
| RCT: APEX‐AMI | 5745 | Pexelizumab prior to PCI, i.v. bolus 2 mg·kg−1, followed by 0.05 mg·kg−1·h−1 infusion over the subsequent 24 h | In patients treated with primary PCI for STEMI, adjunct pexelizumab treatment showed no significant effect on mortality or the composite endpoint of death, cardiogenic shock, and heart failure at day 30 or day 90. | (Armstrong et al., 2007) |
| http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4998 antagonism | ||||
| RCT | 117 | http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6881 (IL‐6Ra, single dose 280 mg, i.v.) prior to coronary angiography |
Tocilizumab attenuated the inflammatory response and PCI‐related troponin T (TnT) release in NSTEMI patients. Tocilizumab did not affect coronary flow reserve during hospitalization or after 6 months. |
PMID: (Kleveland et al.,
2016) (Holte et al., 2017) |
| TNF‐α antagonism | ||||
| RCT | 26 | http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6789 (TNF‐α antagonist) at a single intravenous dose of 10 mg | Following acute MI, etanercept reduced systemic inflammation but increased platelet activation without affecting peripheral vasomotor or fibrinolytic function. | (Padfield et al., 2013) |
| Proteinase inhibition | ||||
| RCT: TIPTOP | 429 | Doxycycline (non‐selective MMP inhibitor, 100 mg p.o.) immediately after primary PCI and then twice daily for 7 days | In patients with a first STEMI and LV dysfunction treated with primary PCI, a timely short‐term treatment with doxycycline significantly reduced adverse LV remodelling and decreased infarct size assessed through SPECT. | (Cerisano et al., 2014) |
| RCT: PREMIER | 253 | PG‐116800 (MMP inhibitor), 200 mg oral dose taken twice daily for 90 days | MMPs inhibition with PG‐116800 after MI failed to reduce LV remodelling or improve clinical outcomes in patients with STEMI. | (Hudson et al., 2006) |
| Pilot clinical trial | 10 | http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7487, human plasma‐derived http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7487), single infusion of 60 mg·kg−1, within 12 h of revascularization | A single administration of Prolastin C in patients with STEMI is well tolerated and is associated with a blunted acute inflammatory response. | (Abbate et al., 2015b) |
Targeting the complement cascade
Activation of the complement cascade is a critical part of the innate immune response following MI and has been suggested to extend ischaemic injury (Yasojima et al., 1998). In experimental studies, complement inhibition strategies have consistently reduced the size of the infarct and improved function in both rodent and large animal models of reperfused MI (Vakeva et al., 1998; Pischke et al., 2017). Unfortunately, clinical studies have been disappointing. Approaches targeting the complement system, an upstream activator of the innate immune response, were equally disappointing. In the Assessment of Pexelizumab in Acute Myocardial infarction clinical trial, treatment of STEMI patients with the anti‐http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=8712 antibody pexelizumab did not affect 30 day mortality and the composite endpoint of death, cardiogenic shock and congestive heart failure (Armstrong et al., 2007).
Targeting C‐reactive protein (CRP)
Inflammatory injury is associated with release of http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=906 (such as CRP), prototypical acute phase proteins involved in host defense (Deban et al., 2011). In an experimental model of non‐reperfused infarction, CRP injection accentuated cardiomyocyte injury by activating the complement cascade, whereas (somewhat predictably) administration of low MW inhibitors of CRP blocked the deleterious effects of CRP (Pepys et al., 2006). Despite these early promising findings, enthusiasm regarding the potential value of CRP inhibition in patients with MI is dampened by conflicting findings on the effects of CRP in atherosclerosis and thrombosis. Although, in apolipoprotein‐E null mice, treatment with human native CRP accelerated atherosclerotic disease (Schwedler et al., 2005), in other studies, transgenic overexpression of human CRP had no effects on atherosclerosis, thrombosis and inflammation (Hirschfield et al., 2005; Tennent et al., 2008) or even delayed plaque formation (Kovacs et al., 2007). The conflicting in vivo effects of CRP may be explained by the contextually regulated formation of CRP isoforms with distinct functional properties. CRP is known to undergo dissociation from a native pentameric form (pCRP) to potently pro‐inflammatory monomeric subunits (mCRP) that may serve to localize the inflammatory response (Eisenhardt et al., 2009). Inhibition of CRP dissociation has been suggested as a promising anti‐inflammatory strategy in MI.
Integrins and selectins
Selectins and leukocyte http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=760 are critically implicated in leukocyte extravasation in the infarcted myocardium (Figure 1). Numerous experimental studies demonstrated that neutralizing antibodies targeting members of the integrin and selectin families reduced the size of the infarct in myocardial ischaemia/reperfusion models (Simpson et al., 1988; Ma et al., 1991; Aversano et al., 1995; Arai et al., 1996; Christia and Frangogiannis, 2013). More recently, a nanoparticle‐based strategy silencing five key adhesion molecules was reported to preserve function in the infarcted myocardium (Sager et al., 2016a). Protection of the myocardium was presumably due to attenuation of leukocyte‐mediated cardiomyocyte injury. Unfortunately, clinical trials did not confirm the impressive protective effects of anti‐adhesion molecule approaches observed in experimental studies. Anti‐http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2451/http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2456 and anti‐CD18 integrin targeting failed to reduce infarct size in STEMI patients (Baran et al., 2001; Rusnak et al., 2001; Faxon et al., 2002) (Table 4). On the other hand, administration of the P‐selectin inhibitor inclacumab in patients with ACS reduced cardiomyocyte injury but did not affect clinical outcome (Tardif et al., 2013; Seropian et al., 2014).
Chemokines as therapeutic targets
Approaches targeting chemokines involved in recruitment of pro‐inflammatory leukocytes have shown promising results in experimental animal models. Anti‐CCL2 therapy reduced mortality, attenuated chamber dilation and improved systolic function in a model of non‐reperfused infarction (Hayashidani et al., 2003). Inhibition of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=758 also exerted protective effects, improving cardiac function and attenuating fibrotic remodelling (Montecucco et al., 2012). Silencing the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=59, the main receptor for CCL2, that mediates recruitment of pro‐inflammatory monocytes in sites of inflammation, was reported to have beneficial effects not only in the infarcted heart but also in the composition of atherosclerotic plaques and in metabolic dysfunction (Leuschner et al., 2011). However, it should be emphasized that broad targeting of the effects of CC chemokines may also have detrimental actions. Chemokine‐mediated signalling is important for recruitment of leukocyte subsets with anti‐inflammatory properties and may be involved in activation of a programme of repair. In a mouse model of reperfused MI, genetic disruption of http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=62 was associated with accentuated dilative remodelling, presumably due to impaired recruitment of anti‐inflammatory monocyte subsets and of Tregs (Dobaczewski et al., 2010). Thus, recruitment of specific leukocyte subsets through chemokine‐chemokine receptor interactions may be critical for repression and resolution of post‐infarction inflammation.
Administration of the chemokines that recruit reparative cells, such as CXCL12, may also hold therapeutic promise. Therapy with CXCL12 reduced infarct size and accentuated angiogenesis in experimental models of MI, attenuating systolic dysfunction and improving left ventricular mechanics (Hu et al., 2007; Saxena et al., 2008; Macarthur Jr et al., 2014; MacArthur Jr et al., 2013). Although these experimental findings seem promising, clinical translation is challenging. Loss‐of‐function studies have suggested important pro‐inflammatory actions of CXCL12 in the infarcted heart (Proulx et al., 2007; Jujo et al., 2010) raising concerns that administration of this chemokine may also have injurious effects, accentuating or prolonging inflammatory cascades. Clinical evidence remains extremely limited, as the effects of CXCL12 therapy in MI has not been studied. However, in a Phase II clinical trial in patients with high‐risk ischaemic cardiomyopathy, CXCL12 gene therapy was safe but did not meet the primary endpoint for functional improvement (Chung et al., 2015).
Targeting the cytokines
Although a growing body of experimental evidence suggests that pro‐inflammatory cytokines may be promising therapeutic targets in patients with MI, their pleiotropic and multifunctional effects, and their involvement in both injurious and reparative responses pose major therapeutic challenges. Recent experimental and clinical studies have suggested that the IL‐1 system may represent a promising therapeutic target in patients with MI (Saxena et al., 2016). Safe and effective strategies for IL‐1 inhibition are extensively used treatment of patients with inflammatory arthritides or auto‐inflammatory syndromes. Anakinra is a non‐glycosylated recombinant form of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5878 that binds to the type I IL‐1 receptor but does not activate a signalling response, thus functioning as a competitive IL‐1α/IL‐1β inhibitor. On the other hand, anti‐IL‐1β antibodies (such as http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6773) selectively target IL‐β‐mediated responses. In most experimental MI studies, inhibition of the IL‐1 system with anakinra or anti‐IL‐1β antibodies exerted protective effects, reducing chamber dilation and improving dysfunction (Abbate et al., 2008; Toldo et al., 2013). Early evidence from clinical studies has also produced promising results. Pilot studies demonstrated that http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6972 can be safely administered as a 2 week course in STEMI patients and may attenuate adverse remodelling, while protecting from the development of post‐infarction heart failure (Abbate et al., 2010; Abbate et al., 2013; Abbate et al., 2015a). In the recently reported Canakinumab Antiinflammatory Thrombosis Outcome Study trial (Ridker et al., 2017), IL‐1β inhibition in high‐risk patients with atherosclerotic disease attenuated inflammation and reduced cardiovascular events. In patients treated with 150 mg of canakinumab, the primary endpoint (the composite of MI, nonfatal stroke and cardiovascular death) was significantly lower. The beneficial effects of canakinumab were modest: to avoid one primary endpoint event, 156 patients had to be treated for 1 year. Moreover, the canakinumab group exhibited a very low, but significantly higher than placebo, death rate due to infection. Despite these issues and the concerns regarding the very high cost of the antibody, this landmark clinical trial supports the case for targeted anti‐cytokine therapy in selected patients with MI. An emerging body of evidence suggesting that IL‐1‐mediated inflammation accentuates adverse remodelling post‐MI (Bujak et al., 2008) and may promote arrhythmia generation (Monnerat et al., 2016; De Jesus et al., 2017) further strengthens the rationale for targeting IL‐1 in high‐risk subpopulations of MI patients, suggesting that protection may not be limited to reduction of new atherothrombotic events.
The TGF‐β system
Despite the critical involvement of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5060 signalling in cardiac injury and repair (Frangogiannis, 2017b), targeting TGF‐β following MI poses several major challenges. First, TGF‐βs are known to modulate phenotype and function of all cell types involved in cardiac injury and repair. Thus, TGF‐β inhibition would be expected to affect both injurious and protective actions. The effects of TGF‐β inhibition may be dependent on the timing of the intervention. Thus, early neutralization of TGF‐β may block anti‐inflammatory signalling in macrophages, accentuating inflammation and increasing the incidence of cardiac rupture, whereas late suppression may attenuate pro‐fibrotic signals, improving diastolic function (Ikeuchi et al., 2004). Second, because TGF‐β is involved in preservation of cardiac and vascular homeostasis, TGF‐β inhibition following MI may carry significant risks, promoting aneurysmal rupture in vulnerable patients (Engebretsen et al., 2014; Frangogiannis, 2014b; Biernacka et al., 2015). Third, the complex biology of TGF‐β signalling further complicates design of therapeutic strategies. TGF‐β signals through intracellular effectors, the Smads and through activation of non‐Smad pathways (such as http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=519, http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=514, and http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=518). Design of effective therapeutic strategies requires understanding of the relative role of Smad‐dependent and Smad‐independent signalling in vivo (Bujak et al., 2007; Rainer et al., 2014). In the infarcted heart, experimental studies have suggested distinct effects of Smad‐dependent signalling in cardiomyocytes and fibroblasts (Kong et al., 2017). Moreover, non‐Smad pathways may also contribute to activation of interstitial cells towards a fibrogenic phenotype (Molkentin et al., 2017). Dissection of cell‐specific responses to TGF‐β and understanding of the temporal sequence of its cellular actions in the infarcted heart are needed to design safe and effective therapeutic approaches.
Targeting the MMP system
MMPs are involved in post‐MI repair and remodelling, not only by critically regulating ECM metabolism but also by processing inflammatory mediators, such as chemokines and cytokines (Fingleton, 2017; Frangogiannis, 2017a). Genetic deletion of MMP2 and MMP9 has been shown to attenuate post‐MI ventricular dilation and to protect from cardiac rupture in mouse models of non‐reperfused MI (Ducharme et al., 2000; Matsumura et al., 2005). However, pharmacological inhibition of MMPs in animal models of MI has produced conflicting results, depending on the timing of the intervention, the inhibition profile of the agent used and the experimental model. Early treatment with http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6464, a non‐selective MMP inhibitor, attenuated adverse remodelling in a rat model of non‐reperfused infarction (Villarreal et al., 2003). In contrast, early inhibition of MMP9 delayed resolution of inflammation and worsened dysfunction in a mouse model of permanent coronary occlusion (Iyer et al., 2016). In clinical studies, no consistent beneficial effects of MMP inhibition have been reported. In the TIPTOP trial, administration of doxycycline (100 mg p.o. bid for 7 days) in patients with STEMI and left ventricular dysfunction reduced the size of the infarct and attenuated cardiac remodelling (Cerisano et al., 2014). In contrast, in the PREMIER trial, administration of an MMP inhibitor with high affinity for MMP2, MMP3, MMP8, MMP9, MMP13 and MMP14 did not improve clinical outcomes and left ventricular remodelling in STEMI patients (Hudson et al., 2006).
Challenges in targeting inflammation following MI
The diverse roles of inflammatory cascades in injury and repair
The critical involvement of inflammation in both injury and repair of the infarcted heart complicates attempts to target inflammatory signals in patients with MI (Saxena et al., 2016). Inflammatory pathways have been implicated in extending cardiomyocyte death and in triggering matrix degradation but also play a critical role in clearance of dead cells from the infarct and in formation of a scar that preserves the structural integrity of the ventricle. Moreover, inflammatory mediators have been implicated in recruitment of progenitor cells involved in infarct angiogenesis (Taghavi and George, 2013). Thus, inhibition of an inflammatory signal involved in early injury may also inhibit a crucial repair response. Design of therapeutic strategies targeting inflammation in patients with MI needs to take into account important temporal and spatial considerations. There is ample evidence to suggest that prolonged or expanded pro‐inflammatory signalling may accentuate adverse remodelling by activating proteases that degrade the cardiac ECM, by transducing pro‐apoptotic responses in cardiomyocytes and by promoting fibrogenic signalling in the viable non‐infarcted myocardium (Chen et al., 2012; Frangogiannis et al., 2005). It is likely that following MI, there is a therapeutic window of opportunity for safe and effective targeting of specific inflammatory signals. Understanding the time course of the cellular actions of specific inflammatory signals is critical for optimal design of therapeutic strategies.
The pathophysiological heterogeneity of human post‐infarction remodelling
The remarkable pathophysiological heterogeneity in human patients surviving MI further complicates therapeutic implementation of promising targets. The extent of adverse post‐MI remodelling is only partly dependent on the size of the infarct. Differences in susceptibility to adverse remodelling between patients may be explained by age and gender, genetic substrate, the presence or absence of concomitant conditions, the pattern of atherosclerotic disease, administration of medications and other poorly understood factors. Certain subpopulations of patients may have defective mechanisms for negative regulation of inflammation, thus exhibiting prolonged or expanded inflammatory responses. Others may have accentuated fibrotic reactions. Pathophysiological stratification of the patients on the basis of their biochemical profile, clinical characteristics and functional responses may identify patients with overactive post‐infarction inflammatory responses that may benefit from targeted anti‐inflammatory strategies (Frangogiannis, 2014a). Clinical and experimental studies suggest that certain patient subpopulations, such as diabetics, may exhibit dysregulated inflammatory reactions following MI that may be responsible for accentuated remodelling and worse dysfunction. Patients with diabetes have a high incidence of diastolic dysfunction following MI, despite a smaller infarct size and comparable systolic dysfunction (Stone et al., 1989). In experimental models, diabetes and obesity are associated with cardiomyocyte hypertrophy and interstitial fibrosis. These changes may reflect exaggerated angiotensin‐mediated responses and increased TGF‐β/Smad signalling (Biernacka et al., 2015). Targeting fibrogenic mediators may be a promising therapeutic strategy in these patients. On the other hand, other patients may exhibit prolonged activation of pro‐inflammatory signals. These patients may benefit from strategies targeting critical inflammatory cascades, such as IL‐1. Biomarkers (Seropian et al., 2016) and imaging approaches (Wollenweber et al., 2014; Nahrendorf et al., 2015) may be used to assess inflammatory activation in these patients, in order to design personalized therapeutic approaches.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b,c,d,e).
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
Dr Frangogiannis' laboratory is supported by NIH grants R01 HL76246 and R01 HL85440, and by U.S. Department of Defense grants PR151134 and PR151029. Dr Huang is supported by China Scholarship Council grant 201603170222.
Huang, S. , and Frangogiannis, N. G. (2018) Anti‐inflammatory therapies in myocardial infarction: failures, hopes and challenges. British Journal of Pharmacology, 175: 1377–1400. doi: 10.1111/bph.14155.
References
- Abbate A, Kontos MC, Abouzaki NA, Melchior RD, Thomas C, Van Tassell BW et al (2015a). Comparative safety of interleukin‐1 blockade with anakinra in patients with ST‐segment elevation acute myocardial infarction (from the VCU‐ART and VCU‐ART2 pilot studies). Am J Cardiol 115: 288–292. [DOI] [PubMed] [Google Scholar]
- Abbate A, Kontos MC, Grizzard JD, Biondi‐Zoccai GG, Van Tassell BW, Robati R et al (2010). Interleukin‐1 blockade with anakinra to prevent adverse cardiac remodeling after acute myocardial infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU‐ART] pilot study). Am J Cardiol 105: 1371, e1371–1377. [DOI] [PubMed] [Google Scholar]
- Abbate A, Limana F, Capogrossi MC, Santini D, Biondi‐Zoccai GG, Scarpa S et al (2006). Cyclo‐oxygenase‐2 (COX‐2) inhibition reduces apoptosis in acute myocardial infarction. Apoptosis 11: 1061–1063. [DOI] [PubMed] [Google Scholar]
- Abbate A, Salloum FN, Vecile E, Das A, Hoke NN, Straino S et al (2008). Anakinra, a recombinant human interleukin‐1 receptor antagonist, inhibits apoptosis in experimental acute myocardial infarction. Circulation 117: 2670–2683. [DOI] [PubMed] [Google Scholar]
- Abbate A, Van Tassell BW, Biondi‐Zoccai G, Kontos MC, Grizzard JD, Spillman DW et al (2013). Effects of interleukin‐1 blockade with anakinra on adverse cardiac remodeling and heart failure after acute myocardial infarction [from the Virginia Commonwealth University‐Anakinra Remodeling Trial (2) (VCU‐ART2) pilot study]. Am J Cardiol 111: 1394–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbate A, Van Tassell BW, Christopher S, Abouzaki NA, Sonnino C, Oddi C et al (2015b). Effects of prolastin C (plasma‐derived alpha‐1 antitrypsin) on the acute inflammatory response in patients with ST‐segment elevation myocardial infarction (from the VCU‐alpha 1‐RT pilot study). Am J Cardiol 115: 8–12. [DOI] [PubMed] [Google Scholar]
- Abrial M, Da Silva CC, Pillot B, Augeul L, Ivanes F, Teixeira G et al (2014). Cardiac fibroblasts protect cardiomyocytes against lethal ischemia‐reperfusion injury. J Mol Cell Cardiol 68: 56–65. [DOI] [PubMed] [Google Scholar]
- Adamek A, Hu K, Bayer B, Wagner H, Ertl G, Bauersachs J et al (2007). High dose aspirin and left ventricular remodeling after myocardial infarction: aspirin and myocardial infarction. Basic Res Cardiol 102: 334–340. [DOI] [PubMed] [Google Scholar]
- Aiuti A, Webb IJ, Bleul C, Springer T, Gutierrez‐Ramos JC (1997). The chemokine SDF‐1 is a chemoattractant for human CD34+ hematopoietic progenitor cells and provides a new mechanism to explain the mobilization of CD34+ progenitors to peripheral blood. J Exp Med 185: 111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Cidlowski JA, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Nuclear hormone receptors. Br J Pharmacol 174: S208–S224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. Br J Pharmacol 174: S225–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017d). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017e). The Concise Guide to PHARMACOLOGY 2017/18: Other proteins. Br J Pharmacol 174: S1–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrassy M, Volz HC, Igwe JC, Funke B, Eichberger SN, Kaya Z et al (2008). High‐mobility group box‐1 in ischemia‐reperfusion injury of the heart. Circulation 117: 3216–3226. [DOI] [PubMed] [Google Scholar]
- Anzai A, Anzai T, Nagai S, Maekawa Y, Naito K, Kaneko H et al (2012). Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling. Circulation 125: 1234–1245. [DOI] [PubMed] [Google Scholar]
- Anzai A, Choi JL, He S, Fenn AM, Nairz M, Rattik S et al (2017). The infarcted myocardium solicits GM‐CSF for the detrimental oversupply of inflammatory leukocytes. J Exp Med 214: 3293–3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai M, Lefer DJ, So T, DiPaula A, Aversano T, Becker LC (1996). An anti‐CD18 antibody limits infarct size and preserves left ventricular function in dogs with ischemia and 48‐hour reperfusion. J Am Coll Cardiol 27: 1278–1285. [DOI] [PubMed] [Google Scholar]
- Armstrong PW, Granger CB, Adams PX, Hamm C, Holmes D Jr, O'Neill WW et al (2007). Pexelizumab for acute ST‐elevation myocardial infarction in patients undergoing primary percutaneous coronary intervention: a randomized controlled trial. JAMA 297: 43–51. [DOI] [PubMed] [Google Scholar]
- Arriza JL, Weinberger C, Cerelli G, Glaser TM, Handelin BL, Housman DE et al (1987). Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. Science 237: 268–275. [DOI] [PubMed] [Google Scholar]
- Arslan F, de Kleijn DP, Pasterkamp G (2011a). Innate immune signaling in cardiac ischemia. Nat Rev Cardiol 8: 292–300. [DOI] [PubMed] [Google Scholar]
- Arslan F, Smeets MB, Riem Vis PW, Karper JC, Quax PH, Bongartz LG et al (2011b). Lack of fibronectin‐EDA promotes survival and prevents adverse remodeling and heart function deterioration after myocardial infarction. Circ Res 108: 582–592. [DOI] [PubMed] [Google Scholar]
- Asanuma H, Sanada S, Ogai A, Minamino T, Takashima S, Asakura M et al (2004). Methotrexate and MX‐68, a new derivative of methotrexate, limit infarct size via adenosine‐dependent mechanisms in canine hearts. J Cardiovasc Pharmacol 43: 574–579. [DOI] [PubMed] [Google Scholar]
- Askari AT, Unzek S, Popovic ZB, Goldman CK, Forudi F, Kiedrowski M et al (2003). Effect of stromal‐cell‐derived factor 1 on stem‐cell homing and tissue regeneration in ischaemic cardiomyopathy. Lancet 362: 697–703. [DOI] [PubMed] [Google Scholar]
- Aversano T, Zhou W, Nedelman M, Nakada M, Weisman H (1995). A chimeric IgG4 monoclonal antibody directed against CD18 reduces infarct size in a primate model of myocardial ischemia and reperfusion. J Am Coll Cardiol 25: 781–788. [DOI] [PubMed] [Google Scholar]
- Baran KW, Nguyen M, McKendall GR, Lambrew CT, Dykstra G, Palmeri ST et al (2001). Double‐blind, randomized trial of an anti‐CD18 antibody in conjunction with recombinant tissue plasminogen activator for acute myocardial infarction: limitation of myocardial infarction following thrombolysis in acute myocardial infarction (LIMIT AMI) study. Circulation 104: 2778–2783. [DOI] [PubMed] [Google Scholar]
- Barzilai D, Plavnick J, Hazani A, Einath R, Kleinhaus N, Kanter Y (1972). Use of hydrocortisone in the treatment of acute myocardial infarction. Summary of a clinical trial in 446 patients. Chest 61: 488–491. [DOI] [PubMed] [Google Scholar]
- Ben‐Mordechai T, Holbova R, Landa‐Rouben N, Harel‐Adar T, Feinberg MS, Abd Elrahman I et al (2013). Macrophage subpopulations are essential for infarct repair with and without stem cell therapy. J Am Coll Cardiol 62: 1890–1901. [DOI] [PubMed] [Google Scholar]
- Biernacka A, Cavalera M, Wang J, Russo I, Shinde A, Kong P et al (2015). Smad3 signaling promotes fibrosis while preserving cardiac and aortic geometry in obese diabetic mice. Circ Heart Fail 8: 788–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankesteijn WM, Essers‐Janssen YP, Verluyten MJ, Daemen MJ, Smits JF (1997). A homologue of Drosophila tissue polarity gene frizzled is expressed in migrating myofibroblasts in the infarcted rat heart. Nat Med 3: 541–544. [DOI] [PubMed] [Google Scholar]
- Boag SE, Das R, Shmeleva EV, Bagnall A, Egred M, Howard N et al (2015). T lymphocytes and fractalkine contribute to myocardial ischemia/reperfusion injury in patients. J Clin Invest 125: 3063–3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolli R, Shinmura K, Tang XL, Kodani E, Xuan YT, Guo Y et al (2002). Discovery of a new function of cyclooxygenase (COX)‐2: COX‐2 is a cardioprotective protein that alleviates ischemia/reperfusion injury and mediates the late phase of preconditioning. Cardiovasc Res 55: 506–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulakh L, Gislason GH (2016). Treatment with non‐steroidal anti‐inflammatory drugs in patients after myocardial infarction – a systematic review. Expert Opin Pharmacother 17: 1387–1394. [DOI] [PubMed] [Google Scholar]
- Brophy JM, Levesque LE, Zhang B (2007). The coronary risk of cyclo‐oxygenase‐2 inhibitors in patients with a previous myocardial infarction. Heart 93: 189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EJ Jr, Kloner RA, Schoen FJ, Hammerman H, Hale S, Braunwald E (1983). Scar thinning due to ibuprofen administration after experimental myocardial infarction. Am J Cardiol 51: 877–883. [DOI] [PubMed] [Google Scholar]
- Bujak M, Dobaczewski M, Chatila K, Mendoza LH, Li N, Reddy A et al (2008). Interleukin‐1 receptor type I signaling critically regulates infarct healing and cardiac remodeling. Am J Pathol 173: 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bujak M, Dobaczewski M, Gonzalez‐Quesada C, Xia Y, Leucker T, Zymek P et al (2009). Induction of the CXC chemokine interferon‐gamma‐inducible protein 10 regulates the reparative response following myocardial infarction. Circ Res 105: 973–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bujak M, Ren G, Kweon HJ, Dobaczewski M, Reddy A, Taffet G et al (2007). Essential role of Smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation 116: 2127–2138. [DOI] [PubMed] [Google Scholar]
- Bush CA, Renner W, Boudoulas H (1980). Corticosteroids in acute myocardial infarction. Angiology 31: 710–714. [DOI] [PubMed] [Google Scholar]
- Cain DW, Cidlowski JA (2017). Immune regulation by glucocorticoids. Nat Rev Immunol 17: 233–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camitta MG, Gabel SA, Chulada P, Bradbury JA, Langenbach R, Zeldin DC et al (2001). Cyclooxygenase‐1 and ‐2 knockout mice demonstrate increased cardiac ischemia/reperfusion injury but are protected by acute preconditioning. Circulation 104: 2453–2458. [DOI] [PubMed] [Google Scholar]
- Cerisano G, Buonamici P, Valenti R, Sciagra R, Raspanti S, Santini A et al (2014). Early short‐term doxycycline therapy in patients with acute myocardial infarction and left ventricular dysfunction to prevent the ominous progression to adverse remodelling: the TIPTOP trial. Eur Heart J 35: 184–191. [DOI] [PubMed] [Google Scholar]
- Chen C, Feng Y, Zou L, Wang L, Chen HH, Cai JY et al (2014). Role of extracellular RNA and TLR3‐Trif signaling in myocardial ischemia‐reperfusion injury. J Am Heart Assoc 3: e000683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Saxena A, Li N, Sun J, Gupta A, Lee DW et al (2012). Endogenous IRAK‐M attenuates postinfarction remodeling through effects on macrophages and fibroblasts. Arterioscler Thromb Vasc Biol 32: 2598–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christia P, Frangogiannis NG (2013). Targeting inflammatory pathways in myocardial infarction. Eur J Clin Invest 43: 986–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung ES, Miller L, Patel AN, Anderson RD, Mendelsohn FO, Traverse J et al (2015). Changes in ventricular remodelling and clinical status during the year following a single administration of stromal cell‐derived factor‐1 non‐viral gene therapy in chronic ischaemic heart failure patients: the STOP‐HF randomized Phase II trial. Eur Heart J 36: 2228–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleutjens JP, Verluyten MJ, Smiths JF, Daemen MJ (1995). Collagen remodeling after myocardial infarction in the rat heart. Am J Pathol 147: 325–338. [PMC free article] [PubMed] [Google Scholar]
- Crea F, Libby P (2017). Acute coronary syndromes: the way forward from mechanisms to precision treatment. Circulation 136: 1155–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cung TT, Morel O, Cayla G, Rioufol G, Garcia‐Dorado D, Angoulvant D et al (2015). Cyclosporine before PCI in patients with acute myocardial infarction. N Engl J Med 373: 1021–1031. [DOI] [PubMed] [Google Scholar]
- da‐Luz PL, Leite JJ, Barros LF, Dias‐Neto A, Zanarco EL, Pileggi FJ (1982). Experimental myocardial infarction: effect of methylprednisolone on myocardial blood flow after reperfusion. Braz J Med Biol Res 15: 355–360. [PubMed] [Google Scholar]
- Davies MJ, Thomas A (1984). Thrombosis and acute coronary‐artery lesions in sudden cardiac ischemic death. N Engl J Med 310: 1137–1140. [DOI] [PubMed] [Google Scholar]
- De Hoog VC, Timmers L, Van Duijvenvoorde A, De Jager SC, Van Middelaar BJ, Smeets MB et al (2014). Leucocyte expression of complement C5a receptors exacerbates infarct size after myocardial reperfusion injury. Cardiovasc Res 103: 521–529. [DOI] [PubMed] [Google Scholar]
- De Jesus NM, Wang L, Lai J, Rigor RR, Francis Stuart SD, Bers DM et al (2017). Antiarrhythmic effects of interleukin 1 inhibition after myocardial infarction. Heart Rhythm 14: 727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Zwaan C, Kleine AH, Diris JH, Glatz JF, Wellens HJ, Strengers PF et al (2002). Continuous 48‐h C1‐inhibitor treatment, following reperfusion therapy, in patients with acute myocardial infarction. Eur Heart J 23: 1670–1677. [DOI] [PubMed] [Google Scholar]
- Deban L, Jaillon S, Garlanda C, Bottazzi B, Mantovani A (2011). Pentraxins in innate immunity: lessons from PTX3. Cell Tissue Res 343: 237–249. [DOI] [PubMed] [Google Scholar]
- Dellborg M, Held P, Swedberg K, Vedin A (1985). Rupture of the myocardium. Occurrence and risk factors. Br Heart J 54: 11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, Abou‐Khamis T et al (2005). CCL2/monocyte chemoattractant protein‐1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res 96: 881–889. [DOI] [PubMed] [Google Scholar]
- Dobaczewski M, Xia Y, Bujak M, Gonzalez‐Quesada C, Frangogiannis NG (2010). CCR5 signaling suppresses inflammation and reduces adverse remodeling of the infarcted heart, mediating recruitment of regulatory T cells. Am J Pathol 176: 2177–2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducharme A, Frantz S, Aikawa M, Rabkin E, Lindsey M, Rohde LE et al (2000). Targeted deletion of matrix metalloproteinase‐9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J Clin Invest 106: 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan KM, Lawson JA, Fries S, Koller B, Rader DJ, Smyth EM et al (2004). COX‐2‐derived prostacyclin confers atheroprotection on female mice. Science 306: 1954–1957. [DOI] [PubMed] [Google Scholar]
- Eisenhardt SU, Habersberger J, Peter K (2009). Monomeric C‐reactive protein generation on activated platelets: the missing link between inflammation and atherothrombotic risk. Trends Cardiovasc Med 19: 232–237. [DOI] [PubMed] [Google Scholar]
- Emami H, Singh P, MacNabb M, Vucic E, Lavender Z, Rudd JH et al (2015). Splenic metabolic activity predicts risk of future cardiovascular events: demonstration of a cardiosplenic axis in humans. JACC Cardiovasc Imaging 8: 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engebretsen KV, Skardal K, Bjornstad S, Marstein HS, Skrbic B, Sjaastad I et al (2014). Attenuated development of cardiac fibrosis in left ventricular pressure overload by SM16, an orally active inhibitor of ALK5. J Mol Cell Cardiol 76: 148–157. [DOI] [PubMed] [Google Scholar]
- Entman ML, Youker K, Shoji T, Kukielka G, Shappell SB, Taylor AA et al (1992). Neutrophil induced oxidative injury of cardiac myocytes. A compartmented system requiring CD11b/CD18‐ICAM‐1 adherence. J Clin Invest 90: 1335–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faxon DP, Gibbons RJ, Chronos NA, Gurbel PA, Sheehan F (2002). The effect of blockade of the CD11/CD18 integrin receptor on infarct size in patients with acute myocardial infarction treated with direct angioplasty: the results of the HALT‐MI study. J Am Coll Cardiol 40: 1199–1204. [DOI] [PubMed] [Google Scholar]
- Fingleton B (2017). Matrix metalloproteinases as regulators of inflammatory processes. Biochim Biophys Acta 1864: 2036–2042. [DOI] [PubMed] [Google Scholar]
- Fraccarollo D, Galuppo P, Schraut S, Kneitz S, van Rooijen N, Ertl G et al (2008). Immediate mineralocorticoid receptor blockade improves myocardial infarct healing by modulation of the inflammatory response. Hypertension 51: 905–914. [DOI] [PubMed] [Google Scholar]
- Frangogiannis NG (2014a). The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol 11: 255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangogiannis NG (2014b). Targeting the transforming growth factor (TGF)‐beta cascade in the remodeling heart: benefits and perils. J Mol Cell Cardiol 76: 169–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangogiannis NG (2017a). The extracellular matrix in myocardial injury, repair, and remodeling. J Clin Invest 127: 1600–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangogiannis NG (2017b). The role of transforming growth factor (TGF)‐beta in the infarcted myocardium. J Thorac Dis 9: S52–S63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangogiannis NG, Lindsey ML, Michael LH, Youker KA, Bressler RB, Mendoza LH et al (1998). Resident cardiac mast cells degranulate and release preformed TNF‐alpha, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusion. Circulation 98: 699–710. [DOI] [PubMed] [Google Scholar]
- Frangogiannis NG, Mendoza LH, Lindsey ML, Ballantyne CM, Michael LH, Smith CW et al (2000a). IL‐10 is induced in the reperfused myocardium and may modulate the reaction to injury. J Immunol 165: 2798–2808. [DOI] [PubMed] [Google Scholar]
- Frangogiannis NG, Michael LH, Entman ML (2000b). Myofibroblasts in reperfused myocardial infarcts express the embryonic form of smooth muscle myosin heavy chain (SMemb). Cardiovasc Res 48: 89–100. [DOI] [PubMed] [Google Scholar]
- Frangogiannis NG, Ren G, Dewald O, Zymek P, Haudek S, Koerting A et al (2005). The critical role of endogenous thrombospondin (TSP)‐1 in preventing expansion of healing myocardial infarcts. Circulation 111: 2935–2942. [DOI] [PubMed] [Google Scholar]
- Frantz S, Hofmann U, Fraccarollo D, Schafer A, Kranepuhl S, Hagedorn I et al (2013). Monocytes/macrophages prevent healing defects and left ventricular thrombus formation after myocardial infarction. FASEB J 27: 871–881. [DOI] [PubMed] [Google Scholar]
- Freigang S, Ampenberger F, Spohn G, Heer S, Shamshiev AT, Kisielow J et al (2011). Nrf2 is essential for cholesterol crystal‐induced inflammasome activation and exacerbation of atherosclerosis. Eur J Immunol 41: 2040–2051. [DOI] [PubMed] [Google Scholar]
- Galuppo P, Vettorazzi S, Hovelmann J, Scholz CJ, Tuckermann JP, Bauersachs J et al (2017). The glucocorticoid receptor in monocyte‐derived macrophages is critical for cardiac infarct repair and remodeling. FASEB J 31: 5122–5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Prieto J, Villena‐Gutierrez R, Gomez M, Bernardo E, Pun‐Garcia A, Garcia‐Lunar I et al (2017). Neutrophil stunning by metoprolol reduces infarct size. Nat Commun 8: 14780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genth K, Hofmann M, Schaper W (1982). Ineffectiveness of methylprednisolone to reduce infarct size in experimental coronary occlusion. Basic Res Cardiol 77: 182–187. [DOI] [PubMed] [Google Scholar]
- Gislason GH, Jacobsen S, Rasmussen JN, Rasmussen S, Buch P, Friberg J et al (2006). Risk of death or reinfarction associated with the use of selective cyclooxygenase‐2 inhibitors and nonselective nonsteroidal antiinflammatory drugs after acute myocardial infarction. Circulation 113: 2906–2913. [DOI] [PubMed] [Google Scholar]
- Giugliano GR, Giugliano RP, Gibson CM, Kuntz RE (2003). Meta‐analysis of corticosteroid treatment in acute myocardial infarction. Am J Cardiol 91: 1055–1059. [DOI] [PubMed] [Google Scholar]
- Granger CB, Mahaffey KW, Weaver WD, Theroux P, Hochman JS, Filloon TG et al (2003). Pexelizumab, an anti‐C5 complement antibody, as adjunctive therapy to primary percutaneous coronary intervention in acute myocardial infarction: the COMplement inhibition in Myocardial infarction treated with Angioplasty (COMMA) trial. Circulation 108: 1184–1190. [DOI] [PubMed] [Google Scholar]
- Gray GA, White CI, Castellan RF, McSweeney SJ, Chapman KE (2017). Getting to the heart of intracellular glucocorticoid regeneration: 11beta‐HSD1 in the myocardium. J Mol Endocrinol 58: R1–R13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grisanti LA, Traynham CJ, Repas AA, Gao E, Koch WJ, Tilley DG (2016). beta2‐Adrenergic receptor‐dependent chemokine receptor 2 expression regulates leukocyte recruitment to the heart following acute injury. Proc Natl Acad Sci U S A 113: 15126–15131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu L, Okada Y, Clinton SK, Gerard C, Sukhova GK, Libby P et al (1998). Absence of monocyte chemoattractant protein‐1 reduces atherosclerosis in low density lipoprotein receptor‐deficient mice. Mol Cell 2: 275–281. [DOI] [PubMed] [Google Scholar]
- Gullestad L, Orn S, Dickstein K, Eek C, Edvardsen T, Aakhus S et al (2013). Intravenous immunoglobulin does not reduce left ventricular remodeling in patients with myocardial dysfunction during hospitalization after acute myocardial infarction. Int J Cardiol 168: 212–218. [DOI] [PubMed] [Google Scholar]
- Hammerman H, Alker KJ, Schoen FJ, Kloner RA (1984a). Morphologic and functional effects of piroxicam on myocardial scar formation after coronary occlusion in dogs. Am J Cardiol 53: 604–607. [DOI] [PubMed] [Google Scholar]
- Hammerman H, Kloner RA, Hale S, Schoen FJ, Braunwald E (1983a). Dose‐dependent effects of short‐term methylprednisolone on myocardial infarct extent, scar formation, and ventricular function. Circulation 68: 446–452. [DOI] [PubMed] [Google Scholar]
- Hammerman H, Kloner RA, Schoen FJ, Brown EJ Jr, Hale S, Braunwald E (1983b). Indomethacin‐induced scar thinning after experimental myocardial infarction. Circulation 67: 1290–1295. [DOI] [PubMed] [Google Scholar]
- Hammerman H, Schoen FJ, Braunwald E, Kloner RA (1984b). Drug‐induced expansion of infarct: morphologic and functional correlations. Circulation 69: 611–617. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashidani S, Tsutsui H, Shiomi T, Ikeuchi M, Matsusaka H, Suematsu N et al (2003). Anti‐monocyte chemoattractant protein‐1 gene therapy attenuates left ventricular remodeling and failure after experimental myocardial infarction. Circulation 108: 2134–2140. [DOI] [PubMed] [Google Scholar]
- Heikkila J, Nieminen MS (1978). Failure of methylprednisolone to protect acutely ischemic myocardium: a contrast with subsequent beta‐adrenergic blockade in man. Chest 73: 577–582. [DOI] [PubMed] [Google Scholar]
- Henning RJ, Becker H, Vincent JL, Thijs L, Kalter E, Weil MH (1981). Use of methylprednisolone in patients following acute myocardial infarction. Hemodynamic and metabolic effects. Chest 79: 186–194. [DOI] [PubMed] [Google Scholar]
- Hill JH, Ward PA (1971). The phlogistic role of C3 leukotactic fragments in myocardial infarcts of rats. J Exp Med 133: 885–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschfield GM, Gallimore JR, Kahan MC, Hutchinson WL, Sabin CA, Benson GM et al (2005). Transgenic human C‐reactive protein is not proatherogenic in apolipoprotein E‐deficient mice. Proc Natl Acad Sci U S A 102: 8309–8314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann U, Beyersdorf N, Weirather J, Podolskaya A, Bauersachs J, Ertl G et al (2012). Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation 125: 1652–1663. [DOI] [PubMed] [Google Scholar]
- Holte E, Kleveland O, Ueland T, Kunszt G, Bratlie M, Broch K et al (2017). Effect of interleukin‐6 inhibition on coronary microvascular and endothelial function in myocardial infarction. Heart 103: 1521–1527. [DOI] [PubMed] [Google Scholar]
- Horckmans M, Ring L, Duchene J, Santovito D, Schloss MJ, Drechsler M et al (2017). Neutrophils orchestrate post‐myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur Heart J 38: 187–197. [DOI] [PubMed] [Google Scholar]
- Hu X, Dai S, Wu WJ, Tan W, Zhu X, Mu J et al (2007). Stromal cell derived factor‐1 alpha confers protection against myocardial ischemia/reperfusion injury: role of the cardiac stromal cell derived factor‐1 alpha CXCR4 axis. Circulation 116: 654–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson MP, Armstrong PW, Ruzyllo W, Brum J, Cusmano L, Krzeski P et al (2006). Effects of selective matrix metalloproteinase inhibitor (PG‐116800) to prevent ventricular remodeling after myocardial infarction: results of the PREMIER (Prevention of Myocardial Infarction Early Remodeling) trial. J Am Coll Cardiol 48: 15–20. [DOI] [PubMed] [Google Scholar]
- Huebener P, Abou‐Khamis T, Zymek P, Bujak M, Ying X, Chatila K et al (2008). CD44 is critically involved in infarct healing by regulating the inflammatory and fibrotic response. J Immunol 180: 2625–2633. [DOI] [PubMed] [Google Scholar]
- Huynh ML, Fadok VA, Henson PM (2002). Phosphatidylserine‐dependent ingestion of apoptotic cells promotes TGF‐beta1 secretion and the resolution of inflammation. J Clin Invest 109: 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeuchi M, Tsutsui H, Shiomi T, Matsusaka H, Matsushima S, Wen J et al (2004). Inhibition of TGF‐beta signaling exacerbates early cardiac dysfunction but prevents late remodeling after infarction. Cardiovasc Res 64: 526–535. [DOI] [PubMed] [Google Scholar]
- ISIS‐2 (Second International Study of Infarct Survival) Collaborative Group (1988). Randomised trial of intravenous streptokinase, oral aspirin, both, or neither among 17,187 cases of suspected acute myocardial infarction: ISIS‐2. Lancet 2: 349–360. [PubMed] [Google Scholar]
- Ismahil MA, Hamid T, Bansal SS, Patel B, Kingery JR, Prabhu SD (2014). Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: critical importance of the cardiosplenic axis. Circ Res 114: 266–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivey CL, Williams FM, Collins PD, Jose PJ, Williams TJ (1995). Neutrophil chemoattractants generated in two phases during reperfusion of ischemic myocardium in the rabbit. Evidence for a role for C5a and interleukin‐8. J Clin Invest 95: 2720–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer RP, de Castro Bras LE, Patterson NL, Bhowmick M, Flynn ER, Asher M et al (2016). Early matrix metalloproteinase‐9 inhibition post‐myocardial infarction worsens cardiac dysfunction by delaying inflammation resolution. J Mol Cell Cardiol 100: 109–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jujo K, Hamada H, Iwakura A, Thorne T, Sekiguchi H, Clarke T et al (2010). CXCR4 blockade augments bone marrow progenitor cell recruitment to the neovasculature and reduces mortality after myocardial infarction. Proc Natl Acad Sci U S A 107: 11008–11013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaartinen M, Penttila A, Kovanen PT (1996). Mast cells in rupture‐prone areas of human coronary atheromas produce and store TNF‐alpha. Circulation 94: 2787–2792. [DOI] [PubMed] [Google Scholar]
- Kalkman EA, van Suylen RJ, van Dijk JP, Saxena PR, Schoemaker RG (1995). Chronic aspirin treatment affects collagen deposition in non‐infarcted myocardium during remodeling after coronary artery ligation in the rat. J Mol Cell Cardiol 27: 2483–2494. [DOI] [PubMed] [Google Scholar]
- Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN et al (2016). Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun 7: 12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H et al (2011). Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 123: 594–604. [DOI] [PubMed] [Google Scholar]
- Kempf T, Zarbock A, Widera C, Butz S, Stadtmann A, Rossaint J et al (2011). GDF‐15 is an inhibitor of leukocyte integrin activation required for survival after myocardial infarction in mice. Nat Med 17: 581–588. [DOI] [PubMed] [Google Scholar]
- Kleveland O, Kunszt G, Bratlie M, Ueland T, Broch K, Holte E et al (2016). Effect of a single dose of the interleukin‐6 receptor antagonist tocilizumab on inflammation and troponin T release in patients with non‐ST‐elevation myocardial infarction: a double‐blind, randomized, placebo‐controlled phase 2 trial. Eur Heart J 37: 2406–2413. [DOI] [PubMed] [Google Scholar]
- Kloner RA, Fishbein MC, Lew H, Maroko PR, Braunwald E (1978). Mummification of the infarcted myocardium by high dose corticosteroids. Circulation 57: 56–63. [DOI] [PubMed] [Google Scholar]
- Kohno T, Anzai T, Naito K, Sugano Y, Maekawa Y, Takahashi T et al (2008). Angiotensin‐receptor blockade reduces border zone myocardial monocyte chemoattractant protein‐1 expression and macrophage infiltration in post‐infarction ventricular remodeling. Circ J 72: 1685–1692. [DOI] [PubMed] [Google Scholar]
- Kong P, Shinde AV, Su Y, Russo I, Chen B, Saxena A et al (2017). Opposing actions of fibroblast and cardiomyocyte smad3 signaling in the infarcted myocardium. Circulation: 2017; 2010.1161/CIRCULATIONAHA.2117.029622 (epub ahead of print). [DOI] [PMC free article] [PubMed]
- Kovacs A, Tornvall P, Nilsson R, Tegner J, Hamsten A, Bjorkegren J (2007). Human C‐reactive protein slows atherosclerosis development in a mouse model with human‐like hypercholesterolemia. Proc Natl Acad Sci U S A 104: 13768–13773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaPointe MC, Mendez M, Leung A, Tao Z, Yang XP (2004). Inhibition of cyclooxygenase‐2 improves cardiac function after myocardial infarction in the mouse. Am J Physiol Heart Circ Physiol 286: H1416–H1424. [DOI] [PubMed] [Google Scholar]
- Lefer AM, Polansky EW (1979). Beneficial effects of ibuprofen in acute myocardial ischemia. Cardiology 64: 265–279. [DOI] [PubMed] [Google Scholar]
- Lesnik P, Haskell CA, Charo IF (2003). Decreased atherosclerosis in CX3CR1−/− mice reveals a role for fractalkine in atherogenesis. J Clin Invest 111: 333–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuschner F, Dutta P, Gorbatov R, Novobrantseva TI, Donahoe JS, Courties G et al (2011). Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat Biotechnol 29: 1005–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuschner F, Panizzi P, Chico‐Calero I, Lee WW, Ueno T, Cortez‐Retamozo V et al (2010). Angiotensin‐converting enzyme inhibition prevents the release of monocytes from their splenic reservoir in mice with myocardial infarction. Circ Res 107: 1364–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P, Maroko PR, Bloor CM, Sobel BE, Braunwald E (1973). Reduction of experimental myocardial infarct size by corticosteroid administration. J Clin Invest 52: 599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liehn EA, Tuchscheerer N, Kanzler I, Drechsler M, Fraemohs L, Schuh A et al (2011). Double‐edged role of the CXCL12/CXCR4 axis in experimental myocardial infarction. J Am Coll Cardiol 58: 2415–2423. [DOI] [PubMed] [Google Scholar]
- Liu Y, Cousin JM, Hughes J, Van Damme J, Seckl JR, Haslett C et al (1999). Glucocorticoids promote nonphlogistic phagocytosis of apoptotic leukocytes. J Immunol 162: 3639–3646. [PubMed] [Google Scholar]
- Lorchner H, Poling J, Gajawada P, Hou Y, Polyakova V, Kostin S et al (2015). Myocardial healing requires Reg3beta‐dependent accumulation of macrophages in the ischemic heart. Nat Med 21: 353–362. [DOI] [PubMed] [Google Scholar]
- Lugrin J, Parapanov R, Rosenblatt‐Velin N, Rignault‐Clerc S, Feihl F, Waeber B et al (2015). Cutting edge: IL‐1alpha is a crucial danger signal triggering acute myocardial inflammation during myocardial infarction. J Immunol 194: 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XL, Tsao PS, Lefer AM (1991). Antibody to CD‐18 exerts endothelial and cardiac protective effects in myocardial ischemia and reperfusion. J Clin Invest 88: 1237–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macarthur JW Jr, Cohen JE, McGarvey JR, Shudo Y, Patel JB, Trubelja A et al (2014). Preclinical evaluation of the engineered stem cell chemokine stromal cell‐derived factor 1alpha analog in a translational ovine myocardial infarction model. Circ Res 114: 650–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur JW Jr, Purcell BP, Shudo Y, Cohen JE, Fairman A, Trubelja A et al (2013). Sustained release of engineered stromal cell‐derived factor 1‐alpha from injectable hydrogels effectively recruits endothelial progenitor cells and preserves ventricular function after myocardial infarction. Circulation 128: S79–S86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mach F, Schonbeck U, Bonnefoy JY, Pober JS, Libby P (1997). Activation of monocyte/macrophage functions related to acute atheroma complication by ligation of CD40: induction of collagenase, stromelysin, and tissue factor. Circulation 96: 396–399. [DOI] [PubMed] [Google Scholar]
- Maclean D, Fishbein MC, Braunwald E, Maroko PR (1978). Long‐term preservation of ischemic myocardium after experimental coronary artery occlusion. J Clin Invest 61: 541–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madias JE, Hood WB Jr (1982). Effects of methylprednisolone on the ischemic damage in patients with acute myocardial infarction. Circulation 65: 1106–1113. [DOI] [PubMed] [Google Scholar]
- Mahaffey KW, Granger CB, Nicolau JC, Ruzyllo W, Weaver WD, Theroux P et al (2003). Effect of pexelizumab, an anti‐C5 complement antibody, as adjunctive therapy to fibrinolysis in acute myocardial infarction: the COMPlement inhibition in myocardial infarction treated with thromboLYtics (COMPLY) trial. Circulation 108: 1176–1183. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Garlanda C, Locati M (2009). Macrophage diversity and polarization in atherosclerosis: a question of balance. Arterioscler Thromb Vasc Biol 29: 1419–1423. [DOI] [PubMed] [Google Scholar]
- Maranhao RC, Guido MC, de Lima AD, Tavares ER, Marques AF, Tavares de Melo MD et al (2017). Methotrexate carried in lipid core nanoparticles reduces myocardial infarction size and improves cardiac function in rats. Int J Nanomedicine 12: 3767–3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura S, Iwanaga S, Mochizuki S, Okamoto H, Ogawa S, Okada Y (2005). Targeted deletion or pharmacological inhibition of MMP‐2 prevents cardiac rupture after myocardial infarction in mice. J Clin Invest 115: 599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzolai L, Duchosal MA, Korber M, Bouzourene K, Aubert JF, Hao H et al (2004). Endogenous angiotensin II induces atherosclerotic plaque vulnerability and elicits a Th1 response in ApoE−/− mice. Hypertension 44: 277–282. [DOI] [PubMed] [Google Scholar]
- Metz CA, Stubbs DF, Hearron MS (1986). Significance of infarct site and methylprednisolone on survival following acute myocardial infarction. J Int Med Res 14 (Suppl 1): 11–14. [DOI] [PubMed] [Google Scholar]
- Mezzaroma E, Toldo S, Farkas D, Seropian IM, Van Tassell BW, Salloum FN et al (2011). The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc Natl Acad Sci U S A 108: 19725–19730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molkentin JD, Bugg D, Ghearing N, Dorn LE, Kim P, Sargent MA et al (2017). Fibroblast‐specific genetic manipulation of p38 mitogen‐activated protein kinase in vivo reveals its central regulatory role in fibrosis. Circulation 136: 549–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monnerat G, Alarcon ML, Vasconcellos LR, Hochman‐Mendez C, Brasil G, Bassani RA et al (2016). Macrophage‐dependent IL‐1beta production induces cardiac arrhythmias in diabetic mice. Nat Commun 7: 13344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montecucco F, Braunersreuther V, Lenglet S, Delattre BM, Pelli G, Buatois V et al (2012). CC chemokine CCL5 plays a central role impacting infarct size and post‐infarction heart failure in mice. Eur Heart J 33: 1964–1974. [DOI] [PubMed] [Google Scholar]
- Moreira DM, Lueneberg ME, da Silva RL, Fattah T, Gottschall CAM (2017). Methotrexate therapy in st‐segment elevation myocardial infarctions: a randomized double‐blind, placebo‐controlled trial (TETHYS Trial). J Cardiovasc Pharmacol Ther 22: 538–545. [DOI] [PubMed] [Google Scholar]
- Morrison J, Maley T, Reduto L, Victa C, Pyros I, Brandon J et al (1975). Effect of methylprednisolone on predicted myocardial infarction size in man. Crit Care Med 3: 94–102. [DOI] [PubMed] [Google Scholar]
- Morrison J, Reduto L, Pizzarello R, Geller K, Maley T, Gulotta S (1976). Modification of myocardial injury in man by corticosteroid administration. Circulation 53: I200–I204. [PubMed] [Google Scholar]
- Morton AC, Rothman AM, Greenwood JP, Gunn J, Chase A, Clarke B et al (2015). The effect of interleukin‐1 receptor antagonist therapy on markers of inflammation in non‐ST elevation acute coronary syndromes: the MRC‐ILA heart study. Eur Heart J 36: 377–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullane KM, Read N, Salmon JA, Moncada S (1984). Role of leukocytes in acute myocardial infarction in anesthetized dogs: relationship to myocardial salvage by anti‐inflammatory drugs. J Pharmacol Exp Ther 228: 510–522. [PubMed] [Google Scholar]
- Nahrendorf M, Frantz S, Swirski FK, Mulder WJ, Randolph G, Ertl G et al (2015). Imaging systemic inflammatory networks in ischemic heart disease. J Am Coll Cardiol 65: 1583–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL et al (2007). The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med 204: 3037–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakaya M, Watari K, Tajima M, Nakaya T, Matsuda S, Ohara H et al (2017). Cardiac myofibroblast engulfment of dead cells facilitates recovery after myocardial infarction. J Clin Invest 127: 383–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padfield GJ, Din JN, Koushiappi E, Mills NL, Robinson SD, Cruden Nle M et al (2013). Cardiovascular effects of tumour necrosis factor alpha antagonism in patients with acute myocardial infarction: a first in human study. Heart 99: 1330–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng H, Sarwar Z, Yang XP, Peterson EL, Xu J, Janic B et al (2015). Profibrotic role for interleukin‐4 in cardiac remodeling and dysfunction. Hypertension 66: 582–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepys MB, Hirschfield GM, Tennent GA, Gallimore JR, Kahan MC, Bellotti V et al (2006). Targeting C‐reactive protein for the treatment of cardiovascular disease. Nature 440: 1217–1221. [DOI] [PubMed] [Google Scholar]
- Peters RW, Norman A, Parmley WW, Emilson BB, Scheinman MM, Cheitlin M (1978). Effect of therapy with methylprednisolone on the size of myocardial infarcts in man. Chest 73: 483–488. [DOI] [PubMed] [Google Scholar]
- Piot C, Croisille P, Staat P, Thibault H, Rioufol G, Mewton N et al (2008). Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med 359: 473–481. [DOI] [PubMed] [Google Scholar]
- Pischke SE, Gustavsen A, Orrem HL, Egge KH, Courivaud F, Fontenelle H et al (2017). Complement factor 5 blockade reduces porcine myocardial infarction size and improves immediate cardiac function. Basic Res Cardiol 112: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proulx C, El‐Helou V, Gosselin H, Clement R, Gillis MA, Villeneuve L et al (2007). Antagonism of stromal cell‐derived factor‐1alpha reduces infarct size and improves ventricular function after myocardial infarction. Pflugers Arch 455: 241–250. [DOI] [PubMed] [Google Scholar]
- Rahman K, Vengrenyuk Y, Ramsey SA, Vila NR, Girgis NM, Liu J et al (2017). Inflammatory Ly6Chi monocytes and their conversion to M2 macrophages drive atherosclerosis regression. J Clin Invest 127: 2904–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainer PP, Hao S, Vanhoutte D, Lee DI, Koitabashi N, Molkentin JD et al (2014). Cardiomyocyte‐specific transforming growth factor beta suppression blocks neutrophil infiltration, augments multiple cytoprotective cascades, and reduces early mortality after myocardial infarction. Circ Res 114: 1246–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C et al (2017). Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 377: 1119–1131. [DOI] [PubMed] [Google Scholar]
- Roberts R, DeMello V, Sobel BE (1976). Deleterious effects of methylprednisolone in patients with myocardial infarction. Circulation 53: I204–I206. [PubMed] [Google Scholar]
- Ruiz‐Villalba A, Simon AM, Pogontke C, Castillo MI, Abizanda G, Pelacho B et al (2015). Interacting resident epicardium‐derived fibroblasts and recruited bone marrow cells form myocardial infarction scar. J Am Coll Cardiol 65: 2057–2066. [DOI] [PubMed] [Google Scholar]
- Rusnak JM, Kopecky SL, Clements IP, Gibbons RJ, Holland AE, Peterman HS et al (2001). An anti‐CD11/CD18 monoclonal antibody in patients with acute myocardial infarction having percutaneous transluminal coronary angioplasty (the FESTIVAL study). Am J Cardiol 88: 482–487. [DOI] [PubMed] [Google Scholar]
- Sage AP, Mallat Z (2017). Readapting the adaptive immune response – therapeutic strategies for atherosclerosis. Br J Pharmacol 174: 3926–3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sager HB, Dutta P, Dahlman JE, Hulsmans M, Courties G, Sun Y et al (2016a). RNAi targeting multiple cell adhesion molecules reduces immune cell recruitment and vascular inflammation after myocardial infarction. Sci Transl Med 8: 342ra380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sager HB, Hulsmans M, Lavine KJ, Moreira MB, Heidt T, Courties G et al (2016b). Proliferation and recruitment contribute to myocardial macrophage expansion in chronic heart failure. Circ Res 119: 853–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Rodger IW, Hu F, Robinson R, Huynh T, Giaid A (2004). Inhibition of COX pathway in experimental myocardial infarction. J Mol Cell Cardiol 37: 71–77. [DOI] [PubMed] [Google Scholar]
- Salloum FN, Hoke NN, Seropian IM, Varma A, Ownby ED, Houser JE et al (2009). Parecoxib inhibits apoptosis in acute myocardial infarction due to permanent coronary ligation but not due to ischemia‐reperfusion. J Cardiovasc Pharmacol 53: 495–498. [DOI] [PubMed] [Google Scholar]
- Samarasinghe RA, Witchell SF, DeFranco DB (2012). Cooperativity and complementarity: synergies in non‐classical and classical glucocorticoid signaling. Cell Cycle 11: 2819–2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena A, Bujak M, Frunza O, Dobaczewski M, Gonzalez‐Quesada C, Lu B et al (2014a). CXCR3‐independent actions of the CXC chemokine CXCL10 in the infarcted myocardium and in isolated cardiac fibroblasts are mediated through proteoglycans. Cardiovasc Res 103: 217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena A, Chen W, Su Y, Rai V, Uche OU, Li N et al (2013). IL‐1 induces proinflammatory leukocyte infiltration and regulates fibroblast phenotype in the infarcted myocardium. J Immunol 191: 4838–4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena A, Dobaczewski M, Rai V, Haque Z, Chen W, Li N et al (2014b). Regulatory T cells are recruited in the infarcted mouse myocardium and may modulate fibroblast phenotype and function. Am J Physiol Heart Circ Physiol 307: H1233–H1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena A, Fish JE, White MD, Yu S, Smyth JW, Shaw RM et al (2008). Stromal cell‐derived factor‐1alpha is cardioprotective after myocardial infarction. Circulation 117: 2224–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena A, Russo I, Frangogiannis NG (2016). Inflammation as a therapeutic target in myocardial infarction: learning from past failures to meet future challenges. Transl Res 167: 152–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schieffer B, Schieffer E, Hilfiker‐Kleiner D, Hilfiker A, Kovanen PT, Kaartinen M et al (2000). Expression of angiotensin II and interleukin 6 in human coronary atherosclerotic plaques: potential implications for inflammation and plaque instability. Circulation 101: 1372–1378. [DOI] [PubMed] [Google Scholar]
- Schmidt M, Lamberts M, Olsen AM, Fosboll E, Niessner A, Tamargo J et al (2016). Cardiovascular safety of non‐aspirin non‐steroidal anti‐inflammatory drugs: review and position paper by the working group for Cardiovascular Pharmacotherapy of the European Society of Cardiology. Eur Heart J 37: 1015–1023. [DOI] [PubMed] [Google Scholar]
- Schwedler SB, Amann K, Wernicke K, Krebs A, Nauck M, Wanner C et al (2005). Native C‐reactive protein increases whereas modified C‐reactive protein reduces atherosclerosis in apolipoprotein E‐knockout mice. Circulation 112: 1016–1023. [DOI] [PubMed] [Google Scholar]
- Seropian IM, Sonnino C, Van Tassell BW, Biasucci LM, Abbate A (2016). Inflammatory markers in ST‐elevation acute myocardial infarction. Eur Heart J Acute Cardiovasc Care 5: 382–395. [DOI] [PubMed] [Google Scholar]
- Seropian IM, Toldo S, Van Tassell BW, Abbate A (2014). Anti‐inflammatory strategies for ventricular remodeling following ST‐segment elevation acute myocardial infarction. J Am Coll Cardiol 63: 1593–1603. [DOI] [PubMed] [Google Scholar]
- Shah PK, Falk E, Badimon JJ, Fernandez‐Ortiz A, Mailhac A, Villareal‐Levy G et al (1995). Human monocyte‐derived macrophages induce collagen breakdown in fibrous caps of atherosclerotic plaques. Potential role of matrix‐degrading metalloproteinases and implications for plaque rupture. Circulation 92: 1565–1569. [PubMed] [Google Scholar]
- Shinde AV, Humeres C, Frangogiannis NG (2017). The role of alpha‐smooth muscle actin in fibroblast‐mediated matrix contraction and remodeling. Biochim Biophys Acta 1863: 298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani Y, Ito T, Fields L, Shiraishi M, Ichihara Y, Sato N et al (2017). IL‐4 as a repurposed biological drug for myocardial infarction through augmentation of reparative cardiac macrophages: proof‐of‐concept data in mice. Sci Rep 7: 6877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraishi M, Shintani Y, Shintani Y, Ishida H, Saba R, Yamaguchi A et al (2016). Alternatively activated macrophages determine repair of the infarcted adult murine heart. J Clin Invest 126: 2151–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shizukuda Y, Miura T, Ishimoto R, Itoya M, Iimura O (1991). Effect of prednisolone on myocardial infarct healing: characteristics and comparison with indomethacin. Can J Cardiol 7: 447–454. [PubMed] [Google Scholar]
- Sievers J, Johansson BW, Nilsson SE (1964). The corticosteroid treatment of acute myocardial infarction. Cardiologia 45: 65–76. [DOI] [PubMed] [Google Scholar]
- Silverman HS, Pfeifer MP (1987). Relation between use of anti‐inflammatory agents and left ventricular free wall rupture during acute myocardial infarction. Am J Cardiol 59: 363–364. [DOI] [PubMed] [Google Scholar]
- Simpson PJ, Todd RF 3rd, Fantone JC, Mickelson JK, Griffin JD, Lucchesi BR (1988). Reduction of experimental canine myocardial reperfusion injury by a monoclonal antibody (anti‐Mo1, anti‐CD11b) that inhibits leukocyte adhesion. J Clin Invest 81: 624–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slutsky RA, Murray M (1985). Computed tomographic analysis of the effects of hyperosmolar mannitol and methylprednisolone on myocardial infarct size. J Am Coll Cardiol 5: 273–279. [DOI] [PubMed] [Google Scholar]
- Snyder DS, Unanue ER (1982). Corticosteroids inhibit murine macrophage Ia expression and interleukin 1 production. J Immunol 129: 1803–1805. [PubMed] [Google Scholar]
- Sobirin MA, Kinugawa S, Takahashi M, Fukushima A, Homma T, Ono T et al (2012). Activation of natural killer T cells ameliorates postinfarct cardiac remodeling and failure in mice. Circ Res 111: 1037–1047. [DOI] [PubMed] [Google Scholar]
- Spath JA Jr, Lane DL, Lefer AM (1974). Protective action of methylprednisolone on the myocardium during experimental myocardial ischemia in the cat. Circ Res 35: 44–51. [DOI] [PubMed] [Google Scholar]
- Spath JA, Lefer AM (1975). Effects of dexamethasone on myocardial cells in the early phase of acute myocardial infarction. Am Heart J 90: 50–55. [DOI] [PubMed] [Google Scholar]
- Squadrito F, Altavilla D, Squadrito G, Saitta A, Campo GM, Arlotta M et al (1999). Cyclosporin‐A reduces leukocyte accumulation and protects against myocardial ischaemia reperfusion injury in rats. Eur J Pharmacol 364: 159–168. [DOI] [PubMed] [Google Scholar]
- Stone PH, Muller JE, Hartwell T, York BJ, Rutherford JD, Parker CB et al (1989). The effect of diabetes mellitus on prognosis and serial left ventricular function after acute myocardial infarction: contribution of both coronary disease and diastolic left ventricular dysfunction to the adverse prognosis. The MILIS Study Group. J Am Coll Cardiol 14: 49–57. [DOI] [PubMed] [Google Scholar]
- Straino S, Salloum FN, Baldi A, Ockaili RA, Piro M, Das A et al (2007). Protective effects of parecoxib, a cyclo‐oxygenase‐2 inhibitor, in postinfarction remodeling in the rat. J Cardiovasc Pharmacol 50: 571–577. [DOI] [PubMed] [Google Scholar]
- Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez‐Retamozo V, Panizzi P et al (2009). Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 325: 612–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I, Lichtman AH (2017). Monocyte‐macrophages and T cells in atherosclerosis. Immunity 47: 621–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taghavi S, George JC (2013). Homing of stem cells to ischemic myocardium. Am J Transl Res 5: 404–411. [PMC free article] [PubMed] [Google Scholar]
- Tardif JC, Tanguay JF, Wright SS, Duchatelle V, Petroni T, Gregoire JC et al (2013). Effects of the P‐selectin antagonist inclacumab on myocardial damage after percutaneous coronary intervention for non‐ST‐segment elevation myocardial infarction: results of the SELECT‐ACS trial. J Am Coll Cardiol 61: 2048–2055. [DOI] [PubMed] [Google Scholar]
- Tay C, Liu YH, Hosseini H, Kanellakis P, Cao A, Peter K et al (2016). B‐cell‐specific depletion of tumour necrosis factor alpha inhibits atherosclerosis development and plaque vulnerability to rupture by reducing cell death and inflammation. Cardiovasc Res 111: 385–397. [DOI] [PubMed] [Google Scholar]
- Tennent GA, Hutchinson WL, Kahan MC, Hirschfield GM, Gallimore JR, Lewin J et al (2008). Transgenic human CRP is not pro‐atherogenic, pro‐atherothrombotic or pro‐inflammatory in apoE−/− mice. Atherosclerosis 196: 248–255. [DOI] [PubMed] [Google Scholar]
- The Solu‐Medrol Sterile Powder AMI Studies Group (1986). Methylprednisolone as an intervention following myocardial infarction. J Int Med Res 14 (Suppl 1): 1–10. [PubMed] [Google Scholar]
- Timmers L, Sluijter JP, Verlaan CW, Steendijk P, Cramer MJ, Emons M et al (2007). Cyclooxygenase‐2 inhibition increases mortality, enhances left ventricular remodeling, and impairs systolic function after myocardial infarction in the pig. Circulation 115: 326–332. [DOI] [PubMed] [Google Scholar]
- Toldo S, Mezzaroma E, Van Tassell BW, Farkas D, Marchetti C, Voelkel NF et al (2013). Interleukin‐1beta blockade improves cardiac remodelling after myocardial infarction without interrupting the inflammasome in the mouse. Exp Physiol 98: 734–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usher MG, Duan SZ, Ivaschenko CY, Frieler RA, Berger S, Schutz G et al (2010). Myeloid mineralocorticoid receptor controls macrophage polarization and cardiovascular hypertrophy and remodeling in mice. J Clin Invest 120: 3350–3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakeva AP, Agah A, Rollins SA, Matis LA, Li L, Stahl GL (1998). Myocardial infarction and apoptosis after myocardial ischemia and reperfusion: role of the terminal complement components and inhibition by anti‐C5 therapy. Circulation 97: 2259–2267. [DOI] [PubMed] [Google Scholar]
- van Amerongen MJ, Harmsen MC, van Rooijen N, Petersen AH, van Luyn MJ (2007). Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am J Pathol 170: 818–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Kerckhoven R, van Veghel R, Saxena PR, Schoemaker RG (2004). Pharmacological therapy can increase capillary density in post‐infarction remodeled rat hearts. Cardiovasc Res 61: 620–629. [DOI] [PubMed] [Google Scholar]
- Villarreal FJ, Griffin M, Omens J, Dillmann W, Nguyen J, Covell J (2003). Early short‐term treatment with doxycycline modulates postinfarction left ventricular remodeling. Circulation 108: 1487–1492. [DOI] [PubMed] [Google Scholar]
- Vivaldi MT, Eyre DR, Kloner RA, Schoen FJ (1987). Effects of methylprednisolone on collagen biosynthesis in healing acute myocardial infarction. Am J Cardiol 60: 424–425. [DOI] [PubMed] [Google Scholar]
- Vonkeman HE, van de Laar MA (2010). Nonsteroidal anti‐inflammatory drugs: adverse effects and their prevention. Semin Arthritis Rheum 39: 294–312. [DOI] [PubMed] [Google Scholar]
- Wan E, Yeap XY, Dehn S, Terry R, Novak M, Zhang S et al (2013). Enhanced efferocytosis of apoptotic cardiomyocytes through myeloid‐epithelial‐reproductive tyrosine kinase links acute inflammation resolution to cardiac repair after infarction. Circ Res 113: 1004–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirather J, Hofmann UD, Beyersdorf N, Ramos GC, Vogel B, Frey A et al (2014). Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ Res 115: 55–67. [DOI] [PubMed] [Google Scholar]
- Welman E, Selwyn AP, Fox KM (1979). Lysosomal and cytosolic enzyme release in acute myocardial infarction: effects of methylprednisolone. Circulation 59: 730–733. [DOI] [PubMed] [Google Scholar]
- White CI, Jansen MA, McGregor K, Mylonas KJ, Richardson RV, Thomson A et al (2016). Cardiomyocyte and vascular smooth muscle‐independent 11beta‐hydroxysteroid dehydrogenase 1 amplifies infarct expansion, hypertrophy, and the development of heart failure after myocardial infarction in male mice. Endocrinology 157: 346–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems IE, Havenith MG, De Mey JG, Daemen MJ (1994). The alpha‐smooth muscle actin‐positive cells in healing human myocardial scars. Am J Pathol 145: 868–875. [PMC free article] [PubMed] [Google Scholar]
- Wollenweber T, Roentgen P, Schafer A, Schatka I, Zwadlo C, Brunkhorst T et al (2014). Characterizing the inflammatory tissue response to acute myocardial infarction by clinical multimodality noninvasive imaging. Circ Cardiovasc Imaging 7: 811–818. [DOI] [PubMed] [Google Scholar]
- Xu B, Strom J, Chen QM (2011). Dexamethasone induces transcriptional activation of Bcl‐xL gene and inhibits cardiac injury by myocardial ischemia. Eur J Pharmacol 668: 194–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XP, Meisel SR, Ong JM, Kaul S, Cercek B, Rajavashisth TB et al (1999). Oxidized low‐density lipoprotein regulates matrix metalloproteinase‐9 and its tissue inhibitor in human monocyte‐derived macrophages. Circulation 99: 993–998. [DOI] [PubMed] [Google Scholar]
- Yamazaki T, Seko Y, Tamatani T, Miyasaka M, Yagita H, Okumura K et al (1993). Expression of intercellular adhesion molecule‐1 in rat heart with ischemia/reperfusion and limitation of infarct size by treatment with antibodies against cell adhesion molecules. Am J Pathol 143: 410–418. [PMC free article] [PubMed] [Google Scholar]
- Yan X, Anzai A, Katsumata Y, Matsuhashi T, Ito K, Endo J et al (2013). Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J Mol Cell Cardiol 62: 24–35. [DOI] [PubMed] [Google Scholar]
- Yan X, Shichita T, Katsumata Y, Matsuhashi T, Ito H, Ito K et al (2012). Deleterious effect of the IL‐23/IL‐17A axis and gammadeltaT cells on left ventricular remodeling after myocardial infarction. J Am Heart Assoc 1: e004408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasojima K, Schwab C, McGeer EG, McGeer PL (1998). Human heart generates complement proteins that are upregulated and activated after myocardial infarction. Circ Res 83: 860–869. [DOI] [PubMed] [Google Scholar]
- Zhang J, Cheng X, Liao YH, Lu B, Yang Y, Li B et al (2005). Simvastatin regulates myocardial cytokine expression and improves ventricular remodeling in rats after acute myocardial infarction. Cardiovasc Drugs Ther 19: 13–21. [DOI] [PubMed] [Google Scholar]
- Zhang W, Lavine KJ, Epelman S, Evans SA, Weinheimer CJ, Barger PM et al (2015). Necrotic myocardial cells release damage‐associated molecular patterns that provoke fibroblast activation in vitro and trigger myocardial inflammation and fibrosis in vivo. J Am Heart Assoc 4: e001993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Qiu Y, Wang Q, Zhu Y, Hu S, Zheng L et al (2008). Low dose cyclophosphamide rescues myocardial function from ischemia‐reperfusion in rats. Eur J Cardiothorac Surg 34: 661–666. [DOI] [PubMed] [Google Scholar]
- Zouggari Y, Ait‐Oufella H, Bonnin P, Simon T, Sage AP, Guerin C et al (2013). B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat Med 19: 1273–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zymek P, Bujak M, Chatila K, Cieslak A, Thakker G, Entman ML et al (2006). The role of platelet‐derived growth factor signaling in healing myocardial infarcts. J Am Coll Cardiol 48: 2315–2323. [DOI] [PubMed] [Google Scholar]