

Abstract
Preeclampsia (PE), a hypertensive disorder of pregnancy, is a leading cause of maternal and fetal morbidity and mortality. Although the etiology is unknown, PE is thought to be caused by defective implantation and decidualization in pregnancy. Pregnant blood pressure high (BPH)/5 mice spontaneously develop placentopathies and maternal features of human PE. We hypothesized that BPH/5 implantation sites have transcriptomic alterations. Next-generation RNA sequencing of implantation sites at peak decidualization, embryonic day (E)7.5, revealed complement gene up-regulation in BPH/5 vs. controls. In BPH/5, expression of complement factor 3 was increased around the decidual vasculature of E7.5 implantation sites and in the trophoblast giant cell layer of E10.5 placentae. Altered expression of VEGF pathway genes in E5.5 BPH/5 implantation sites preceded complement dysregulation, which correlated with abnormal vasculature and increased placental growth factor mRNA and VEGF164 expression at E7.5. By E10.5, proangiogenic genes were down-regulated, whereas antiangiogenic sFlt-1 was up-regulated in BPH/5 placentae. We found that early local misexpression of VEGF genes and abnormal decidual vasculature preceded sFlt-1 overexpression and increased complement deposition in BPH/5 placentae. Our findings suggest that abnormal decidual angiogenesis precedes complement activation, which in turn contributes to the aberrant trophoblast invasion and poor placentation that underlie PE.—Sones, J. L., Merriam, A. A., Seffens, A., Brown-Grant, D.-A., Butler, S. D., Zhao, A. M., Xu, X., Shawber, C. J., Grenier, J. K., Douglas, N. C. Angiogenic factor imbalance precedes complement deposition in placentae of the BPH/5 model of preeclampsia.
Keywords: decidual vasculature, VEGF, sFlt-1, RGC32, implantation, trophoblast
Preeclampsia (PE) is a common manifestation of placental dysfunction that affects up to 8% of pregnancies and is one of the leading causes of maternal and neonatal morbidity and mortality worldwide (1, 2). PE is defined as hypertension developing after 20 wk of gestation in combination with one of the following: renal insufficiency, liver function abnormalities, neurologic symptoms, or thrombocytopenia (2, 3). Complications of PE include fetal growth restriction, neonatal morbidities associated with preterm birth, and long-standing maternal hypertension and cardiovascular disease (1, 4). PE arises in the placenta and progresses in two distinct stages. The first stage involves shallow trophoblast invasion into the maternal spiral arteries in the uterus with incomplete vascular remodeling leading to decreased blood flow and placental ischemia (5–7). The second stage is the maternal syndrome of PE, which is characterized by hypertension and systemic endothelial dysfunction. It has been proposed that transition from the first to the second stage of PE is mediated by placental release of antiangiogenic factors into the maternal circulation (5, 8). Although the defects that lead to PE are proposed to occur early in pregnancy, PE is typically diagnosed only in the third trimester of pregnancy when maternal sequelae of PE become clinically evident. Thus, we are still unable to reliably predict who will develop PE, and delivery is the only cure.
The complement system, a key component of the innate immune response, is necessary to coordinate inflammatory responses and communicate to the adaptive immune system. Inappropriate complement activation increases the systemic inflammatory response, contributing to maternal features similar to those observed in the second stage of PE, demonstrating that complement pathways are important in normal human pregnancy for development of the placenta (9). The major complement pathways—classic, alternative and lectin—converge to activate complement component C3, leading to release of activation fragments C3a and C3b. Biologic functions of the complement system are mediated through the activation fragments, including C1q, C3a, C3b, and Bb, and through formation of the terminal C5b-9 membrane attack complex, the final effector common to all complement pathways (9). In humans, excess complement activation has been found at sites of placental injury (10, 11), and abnormal expression of activation fragments has been associated with PE (11–15). Increased serum levels of complement Bb, C3a, and C5a before 20 wk of gestation have been found to be predictive of future risk of PE (12–14), and inhibition of complement C5 has been associated with improvement in maternal PE parameters (16). These findings implicate inappropriate complement system activation in the manifestation of PE.
The relationship between abnormal activation of the complement system and angiogenic factor imbalance in human PE is an area of active investigation. Angiogenesis in the pregnant mammalian uterus and developing placenta is mediated by VEGF and placental growth factor (PlGF), key angiogenic regulators acting through VEGF receptor-1 (VEGFR1) and -2 (VEGFR2) (17–20). Antiangiogenic soluble VEGFR1 (sFlt-1), antagonizes the proangiogenic ligands, VEGF and PlGF. In PE, the placenta is thought to abnormally release antiangiogenic factors, with a notable early-to-midgestation imbalance of circulating proangiogenic and antiangiogenic VEGF proteins in women in whom PE subsequently develops (8, 13, 21–27). Increased circulating levels of sFlt-1 and decreased VEGF and PlGF have been consistently observed in PE and an elevated sFlt-1:PlGF ratio can predict PE before the appearance of clinical symptoms (8, 25). Abnormal circulating sFlt-1:PlGF ratios have also been described in other placenta-based diseases (28, 29), suggesting that aberrant VEGF family signaling is integral to the development of placentopathy.
As in human PE, abnormal activation of the complement system and excessive placental production of antiangiogenic factors have been associated with development of a PE-like syndrome in rodent pregnancies (30–36). Pregnant mice lacking C1q have maternal features of human PE, including hypertension, proteinuria, and elevated sFlt-1 levels (37). Further, misexpression of placental C1qa, a polypeptide chain of complement subcomponent C1q, has been associated with murine fetal growth restriction (38). Administration of a C3 inhibitor during the first trimester of DBA/2×CBA/J pregnancies prevented development of the PE-like syndrome (36). Dysregulated VEGF family angiogenic factors also contribute to adverse pregnancy outcomes in rodents. Response gene to complement (RGC)-32, a gene induced by complement activation, is a mediator of VEGFR2 and PlGF (39, 40). Loss of RGC32 during pregnancy impaired placental angiogenesis, resulting in fetal growth restriction (39). Exogenous administration of the antiangiogenic sFlt-1 protein in the second trimester of mouse pregnancy resulted in maternal features of PE (30, 31). These rodent models of placental dysfunction and the PE-like phenotype show that altered activation of complement C1q and C3 and dysregulated VEGF family angiogenic factors result in abnormal placental development and function, supporting conclusions from human studies.
Blood pressure high (BPH)/5 mice are an inbred strain with borderline hypertension that spontaneously develops both maternal and fetal hallmarks of PE (32, 41–43). BPH/5 placental pathologies include shallow trophoblast invasion and inadequate spiral artery remodeling within the decidua at midgestation (32, 43). Studies in BPH/5 mice showed alterations in angiogenic factors in the maternal circulation and in the placenta in early pregnancy (32). Recent work showed abnormal spacing of implantation sites (embryo clustering) and defective decidualization with increased immune activation and inflammation at the maternal–fetal interface before placenta formation in BPH/5 pregnancies (33, 35, 44, 45). Prior studies in the BPH/5 model support a role for both abnormal angiogenesis and immune system activation in development of the PE-like BPH/5 phenotype. In this study, we characterized the onset of angiogenic factor imbalance and abnormal immune activation in early BPH/5 pregnancy with respect to decidualization and placenta formation.
MATERIALS AND METHODS
Animals
Virgin BPH/5 and control C57Bl/6 (C57) mice (8–12 wk of age) were obtained from in-house colonies. C57 mice have been used as control mice in previous BPH/5 studies, because they were used in the original 8-way cross to derive the BPH/5 strain (41, 46). Noon on the day a mating plug was observed was designated as E0.5. Uterine implantation sites from pregnant females at E5.5 and E7.5 and placentae from pregnant females at E10.5 were collected, snap frozen, and stored at −80°C for analyses. The Cornell University Animal Care and Use Committee approved all animal procedures. Care of the mice met the standards set forth by the Guide for the Care and Use of Laboratory Animals (National Institutes of Health, Bethesda, MD, USA), the U.S. Department of Agriculture (USDA, Washington, DC, USA) regulations, and the American Veterinary Medical Association (AVMA, Schaumburg, IL, USA) Panel on Euthanasia.
Next-generation sequencing
RNA sequencing (RNA-Seq) was performed on RNA isolated from E7.5 BPH/5 and C57 implantation sites (n = 4). Total RNA was purified with Trizol (Thermo Fisher Scientific, Waltham, MA, USA) according to the commercial protocol. The RapidOUT DNA Removal kit (Thermo Fisher Scientific) was then used to remove any copurified genomic DNA. The RNA samples were subjected to spectrophotometry (Nanodrop; Thermo Fisher Scientific) to determine concentration and chemical purity (A260:230 and A260:280 ratios). The Fragment Analyzer (Advanced Analytical, Ankeny, IA, USA) was used to determine RNA integrity. RNA samples with an RNA integrity number >7 were used for RNA-Seq. PolyA+TruSeq-barcoded RNAseq libraries were generated with the NEBNext Ultra Directional RNA Library Prep Kit (New England Biolabs, Ipswich, MA, USA). Each library was quantified with a Qubit 2.0 (dsDNA HS kit; Thermo Fisher Scientific) and the size distribution was determined with the Fragment Analyzer (Advanced Analytical) before pooling. Libraries were sequenced on a NextSeq500 instrument (Illumina, San Diego, CA, USA) and at least 20 M single-end 75 bp reads were generated per library. The reads were trimmed for low quality and adaptor sequences with Cutadapt v1.8 (47). Trimmed reads were mapped to the mm10 reference genome with Tophat v. 2.1 (48) for analysis of annotated genes [University of California Santa Cruz (UCSC) mm10 transcriptome; UCSC, Santa Cruz, CA, USA]. Cufflinks v2.2 was used to generate normalized gene expression values [units: fragments per kilobase of transcript per million mapped reads, (FPKM)] for annotated genes and for statistical analysis of differential gene expression (49). Additional criteria used to filter for stringent differential expression included minimum 2-fold change and average FPKM ≥5 in at least 1 condition. DAVID 6.7 was used for pathway analysis and JMP v. 11 (SAS Institute, Cary, NC, USA) was used for hierarchical clustering heat maps. The RNA-Seq data have been uploaded to the Genome Expression Omnibus (GEO, accession number GSE103670; National Center for Biotechnology Information, Bethesda, MD, USA).
Quantitative reverse transcription-PCR
Total RNA was extracted from implantation sites and placentae, cDNA was synthesized, and gene expression was determined by quantitative RT-PCR (qRT-PCR) by using SYBR Green (Qiagen, Frederick, MD, USA) (50). All primer sequences are listed in Table 1. Each qRT-PCR was performed in triplicate with 25 ng cDNA. The relative expression levels of the target genes were calculated and expressed as fold change, 2−ΔΔCt (51).
TABLE 1.
Sequences of forward and reverse primers used in qRT-PCR
| Gene | Primer, 5′-3′ | GenBank accession number | Reference | |
|---|---|---|---|---|
| Forward | Reverse | |||
| 18s | CCGGGCTTCTATTTTGTTGGT | TAGCGGCGCAATACGAATG | 35 | |
| C1qa | CAGTTTGATCGGACCACGGA | CAAGCGTCATTGGGTTCTGC | NM 007572.2 | |
| C3 | CACCGCCAAGAATCGCTAC | GATCAGGTGTTTCAGCCGC | 66 | |
| Ccl8 | TCTACGCAGTGCTTCTTTGCC | AAGGGGGATCTTCAGCTTTAGTA | NM 021443.3 | |
| Ccl21a | GTGATGGAGGGGGTCAGGA | GGGATGGGACAGCCTAAACT | NM 011124.4 | |
| CfB | GAAACCCTGTCACTGTCATTC | CCCCAAACACATACACATCC | 67 | |
| Il1f6 | GCAGCATCACCTTCGCTTAGA | CAGATATTGGCATGGGAGCAAG | NM 019450.3 | |
| PlGF | AGTGGAAGTGGTGCCTTTCAA | GTGAGACACCTCATCAGGGTA | 39 | |
| Prl3d1 | CCAGAGAATCGAGAGGAAGTCC | ACCAGGTGTTTCAGAGGTTCTT | 68 | |
| RGC32 | CCGATCTGGACAGGACCTTA | TTCAGCACTCTCCGAACTGC | 39 | |
| sVEGFR-1 (sFlt-1) | GGGAAGACATCCTTCGGAAGA | TCCGAGAGAAAATGGCCTTTT | 69 | |
| VEGFa | CATCTTCAAGCCGTCCTGTGT | CTCCAGGGCTTCATCGTTACA | 70 | |
| VEGFR-1 | TTCGGAAGACAGAAGTTCTCGTT | GACCTCGTAGTCACTGAGGTTTTG | 69 | |
| VEGFR-2 | AGAACACCAAAAGAGAGAGGAACG | GCACACAGGCAGAAACCAGTAG | 71 | |
GenBank, National Center for Biotechnology Information, Bethesda, MD, USA (https://www.ncbi.nlm.nih.gov/genbank)
Immunofluorescence and immunohistochemistry
Implantation sites and placenta specimens for histologic examination were fixed in 4% paraformaldehyde at 4°C, infiltrated with 30% sucrose in PBS, embedded in Tissue-Tek OCT Compound (Sakura Fine Technical, Torrance, CA, USA), and cryosectioned at 7 μm. Implantation sites showing interembryonic regions and central parts of the decidua were confirmed by hematoxylin and eosin (H&E) staining of every fifth section. Orientation of placenta sections was verified by H&E staining of every fifth section.
IF staining was performed 3 times on 5–6 different BPH/5 and C57 implantation sites (52, 53). Primary antibodies used for immunofluorescence (IF) were goat anti-mouse C3 (55463; MP Cappel, Santa Ana, CA, USA) and rat anti-mouse CD31 (553370; BD Biosciences, San Jose, CA, USA). Secondary antibodies used for IF were Alexa Fluor594 (A11058) and Alexa Fluor488 (A21208; both from Thermo Fisher Scientific).
Immunohistochemical (IHC) staining was performed 3 times on 4–6 different BPH/5 and C57 placentae or implantation sites from 3 pregnancies per group (54). Primary antibodies used for IHC were goat anti-mouse C3, rabbit anti-mouse C9 (PAB823Mu01; Cloud-Clone, Katy, TX, USA), rat anti-mouse CD31 (553370; BD Biosciences) and goat anti-mouse VEGFR2 (AF644; R&D Systems, Inc., Minneapolis, MN, USA). Secondary antibodies used for IHC were biotin rabbit anti-goat IgGn (BA-5000; Vector, Torrance, CA, USA), biotin goat anti-rabbit IgG (BA-1000; Vector) and biotin rabbit anti-rat IgG (BA-4001; Vector). Sections were counterstained with hematoxylin or methylene green. Slides stained with secondary antibody alone served as negative controls for IF and IHC staining.
Histologic analyses
Fluorescent images were captured with a Nikon A1 scanning confocal microscope on an Eclipse Ti microscope stand (Nikon Instruments, Melville, NY, USA). Standard lasers and filters were used. Maximum intensity projections are shown. Confocal images of E7.5 implantation sites were examined with ImageJ software (NIH, Bethesda, MD, USA) to measure cell fluorescence. Quantification of C3 deposition in implantation sites was determined by calculating the corrected total cell fluorescence (CTCF). CTCF from 5 different BPH/5 and C57 implantation sites was determined and median CTCF was compared.
IHC staining was examined with a Nikon Microphot-FXA microscope and images were captured using NIS-Elements D3.10 software (Melville, NY, USA). For placentae at E10.5, areas of positive staining were determined and normalized to the total area of the trophoblast giant cell (TGC) layer in representative sections. Three independent observers quantified the images. Ratios (sum of positive staining:total area of the TGC layer) from the 3 observers were averaged and compared for BPH/5 vs. C57 control placentae (n = 4 per group). For implantation sites at E7.5, vascular space was defined as lumen area surrounded by VEGFR2+ endothelial cells. Vascular space was measured with ImageJ software (NIH) and normalized to area of decidua analyzed. Mean vascular space/decidua (pixels/inch) was compared for BPH/5 vs. C57 controls (n = 4 per group).
Western Blot Analysis
Protein was measured in E7.5 BPH/5 and C57 implantation sites (n = 7) according to previously published methods (32). Band intensity was measured by densitometric analysis with ImageJ software (NIH) and expressed relative to actin.
Statistics
Statistical significance was determined for RNA-Seq data by the q-value (multiple-hypothesis–corrected false discovery rate ≤0.05) generated by Cuffdiff2 (49). Statistical analyses for all other data were performed with Prism, v6.0f (GraphPad, La Jolla, CA, USA). Medians were compared with Mann-Whitney U tests and data are expressed as medians with interquartile range (IQR). For normally distributed data, means were compared by using Student’s t test. Statistical significance was defined as P < 0.05.
RESULTS
BPH/5 implantation sites have dysregulated complement gene expression
Early pregnancy events, including implantation and decidualization, are key to a successful pregnancy (55). Recently, it was reported that pregnant BPH/5 mice have several genes misexpressed in the implantation site during peak decidualization (E7.5) that may play a causal role in fetal and maternal features associated with PE (35). To further explore the molecular signature of BPH/5 implantation sites at E7.5, next-generation sequencing was performed. Expression of 14,368 genes was detected by RNA-Seq expression profiling, and of those, 484 differed significantly (q ≤ 0.05) between the BPH/5 and C57 E7.5 implantation sites and met stringent criteria for minimum expression and minimum 2-fold change (Fig. 1). Pathway analysis was performed to categorize the genes into biologically relevant groups. Because of our recent finding that BPH/5 mice have a robust increase in inflammation, specifically cyclooxygenase-2 (Cox2) up-regulation, at the maternal–fetal interface at E7.5 (35), we focused on pathways associated with inflammation.
Figure 1.
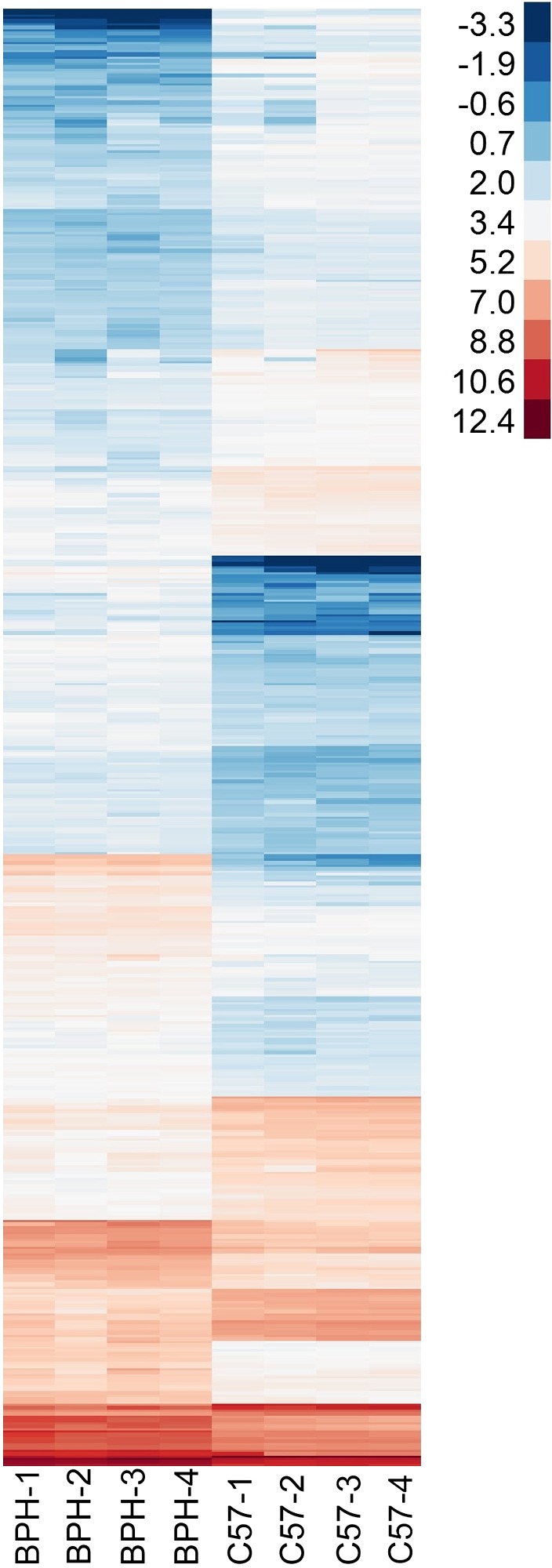
Gene expression profile measured by RNA-Seq at E7.5. Heat map showing log2-FPKM values for 484 genes found to have differential expression between BPH/5 and C57 (q ≤ 0.05, minimum FPKM = 5 in at least 1 group, minimum 2-fold change between groups).
RNA-Seq analysis identified 22 inflammatory response genes that were differentially expressed between BPH/5 and C57 (Table 2). We validated gene expression changes with qRT-PCR for 3 chemokines. IL-36α (Il1f6), C-C motif chemokine 8 (Ccl8), and C-C motif chemokine 21a (Ccl21a) were up-regulated 6.8-, 5.0- and 3.7-fold, respectively, in BPH/5 pregnancies (Supplemental Fig. S1). Expression of prolactin-3D1 isoform-2 precursor (Prl3d1), a critical factor in placental development, was down-regulated 10.3-fold in BPH/5 pregnancies. These data confirm both up-regulated and down-regulated gene expression changes that were detected by RNA-Seq at E7.5.
TABLE 2.
Differentially expressed inflammatory response genes in BPH/5 and C57 implantation sites at E7.5
| Gene | Description | BPH/5 (avgFPKM) | C57 (avgFPKM) | Fold change (log2)a |
|---|---|---|---|---|
| C1rb | Complement C1r-B subcomponent | 7.0 | 0.1 | 6.0 |
| Cxcl5 | C-X-C motif chemokine 5 | 7.1 | 0.8 | 3.1 |
| Il1f6 | IL-36 α | 8.5 | 1.2 | 2.8 |
| Kng1 | Kininogen-1 isoform 2 | 52.9 | 10.2 | 2.4 |
| Ccl8 | C-C motif chemokine 8 | 9.8 | 1.9 | 2.3 |
| Ccl27a | C-C motif chemokine 27 | 82.6 | 16.8 | 2.3 |
| Hfe | Hereditary hemochromatosis protein homolog | 20.1 | 4.5 | 2.2 |
| Pla2g7 | Platelet-activating factor acetylhydrolase | 6.5 | 1.6 | 2.0 |
| Ccl21a | C-C motif chemokine 21a | 12.1 | 3.3 | 1.9 |
| Ctsg | Cathepsin G | 15.6 | 4.7 | 1.7 |
| C3 | Complement C3 | 103.3 | 31.2 | 1.7 |
| CfB | Complement factor B | 15.0 | 4.8 | 1.7 |
| Enpp1 | Ectonucleotide pyrophosphatase/phosphodiesterase Family member 1 | 6.2 | 2.0 | 1.6 |
| Ada | Adenosine deaminase | 58.0 | 20.1 | 1.5 |
| Wfdc15b | WAP four-disulfide core domain protein 15B | 7.4 | 2.6 | 1.5 |
| Tpsb2 | Tryptase β-2 | 21.8 | 8.8 | 1.3 |
| S1pr3 | Sphingosine 1-phosphate receptor 3 | 7.5 | 3.4 | 1.1 |
| Rarres2 | Retinoic acid receptor responder protein 2 | 231.5 | 107.3 | 1.1 |
| Spon2 | Spondin-2 | 13.0 | 6.2 | 1.1 |
| Entpd2 | Ectonucleoside triphosphate diphosphohydrolase 2 | 6.7 | 3.2 | 1.1 |
| Cd14 | Monocyte differentiation antigen CD14 | 9.7 | 4.7 | 1.0 |
| Lyz2 | Lysozyme C-2 | 101.7 | 49.8 | 1.0 |
All q = 0.00023, except Ccl21a = 0.00043.
Among differentially expressed inflammatory response genes were members of the complement family, complement factor 1r-B subcomponent (C1rb), complement factor 3 (C3), and complement factor B (CfB), representing both the classic and alternative arms of the complement pathway. C1rb expression was increased 65.8-fold in BPH/5 pregnancies, a significant finding because C1q, C1r, and C1s comprise the C1 complex that initiates activation of the classic arm of the complement system. CfB, a member of the alternative arm, and C3, a factor downstream of both classic and alternative arms of the complement pathway, were increased 3.1- and 3.3-fold, respectively, in BPH/5 pregnancies.
Complement expression is increased in BPH/5 decidual vasculature at the peak of decidualization
Because placental complement dysregulation is proposed to play a role in the pathogenesis of PE and several complement genes were shown to be altered in the BPH/5 implantation site before placenta formation, we sought to further characterize their temporospatial expression in early BPH/5 pregnancy. We determined expression, by qRT-PCR and immunostaining, of specific complement components at E7.5, the peak of decidualization, in BPH/5 and C57 pregnancies. Based on our RNA-Seq data and PE studies in humans and rodents, we chose to determine mRNA expression for C1qa, C3, and CfB. mRNA expression of C1qa was comparable between BPH/5 and C57 implantation sites at E7.5 (Fig. 2A). mRNA expression of C3 and CfB was significantly increased in BPH/5 implantation sites as compared to C57 controls (Fig. 2B, C). Thus, there is an increase in components that function in both the classic and alternative arms of the complement pathway at the beginning of placentation, supporting the RNA-Seq findings (Table 2).
Figure 2.

Increased decidual vascular complement deposition before placentation in BPH/5 pregnancies. A–C) qRT-PCR analysis of C1qa, C3, and CfB mRNA expression in C57 and BPH/5 implantation sites at E7.5. A) C1qa mRNA expression was unchanged. B) C3 mRNA expression was increased in BPH/5 pregnancies. **P = 0.004. C) CfB mRNA expression was increased in BPH/5 pregnancies. *P = 0.05. D–F) IF staining was performed on tissue sections of implantation sites at E7.5. Nuclei were stained with DAPI (blue). White boxes: areas magnified in the insets. Scale bars, 500 µm. C3 deposition was detected in the ectoplacental cone (area between the dashed yellow line and embryo) and around the decidual vasculature (arrowheads) of the C57 (D) and BPH/5 (E) implantation sites. Anti-goat secondary antibody alone (F) was used for C3 controls. G–J) Double staining IF was performed for CD31 and C3. Representative areas of the decidual vasculature are shown. Scale bars, 100 µm. CD31 identifies decidual vessels (G, I) C3 deposition around the vasculature was increased in BPH/5 pregnancies (J) as compared to C57 controls (H). To quantitatively assess the relative amount of C3 deposition in BPH/5 and C57 implantation sites, we calculated and compared CTCF. K) CTCF for C3 was significantly higher in BPH/5 pregnancies as compared to C57 controls. **P = 0.002. Results represent the median and IQR (n = 5–12 implantation sites/group). am, antimesometrial; e, embryo; m, mesometrial; v, vessels.
To localize C3 expression, we costained tissue sections of E7.5 implantation sites for C3 and the endothelial cell marker, CD31. Relative to C57 decidua, C3 expression was strongly increased in the mesometrial pole of BPH/5 implantation sites (Fig. 2D, E). C3 deposition was detected in the ectoplacental cone (highlighted by dashed yellow lines). C3 colocalized with CD31 (Fig. 2I, J), demonstrating increased C3 deposition around the decidual vasculature of BPH/5 pregnancies. At E7.5, C3 deposition was significantly increased around decidual vessels in BPH/5 pregnancies as compared to C57 controls, as determined by CTCF (Fig. 2K). Our data demonstrate an increase in C3 deposition around the vasculature in a murine PE-like mouse model.
Complement expression is increased in the TGC layer of midgestation placentae
Given our findings of excessive C3 deposition before placentation (Fig. 2), we hypothesized that aberrant complement expression also occurred in BPH/5 placentae. We used qRT-PCR and immunostaining of tissue sections to characterize complement expression in midgestation placentae at E10.5. Whereas BPH/5 placentae had comparable expression of C1qa and CfB mRNA to C57 controls, expression of C3 mRNA was increased in BPH/5 (Fig. 3A–C). We detected increased C3 and C9 deposition in BPH/5 placentae as compared to C57 controls, with staining localized to the TGC layer (Fig. 3D–I). Deposition of both C3 and C9 in the TGC layer of BPH/5 placentae was significantly increased as compared to C57 controls (Fig. 3J, K). Excessive deposition of C9, a component of the membrane attack (C5b-9) terminal complex, suggests activation of the final step of the complement cascade.
Figure 3.

Midgestational BPH/5 placentae have excessive complement expression and deposition. A–C) qRT-PCR analysis of C1qa, C3, and CfB mRNA expression in C57 and BPH/5 placentae at E10.5. A) C1qa mRNA expression was unchanged. B) C3 mRNA expression was increased in BPH/5 placentae. **P = 0.001. C) CfB mRNA expression was unchanged. D–I) IHC was performed to detect C3 and C9 deposition in placental sections at E10.5. Red asterisks: TGCs; yellow arrows: positive staining, indicative of complement deposition. Scale bar, 500 µm. Representative sections of the TGC layer show C3 deposition (D, E) and C9 deposition (G, H). Anti-goat secondary antibody (F) or anti-rabbit secondary antibody (I) alone was used for C3 and C9 controls, respectively. To quantitatively assess the amount of C3 and C9 deposition in the TGC layer of BPH/5 and C57 placentae, we measured the total amount of complement deposition and normalized that to the total TGC area in a placenta section. J) Median C3 deposition in the TGC layer of BPH/5 placentae was significantly increased as compared to C57 controls. **P = 0.001. K) Median C9 deposition in the TGC layer of BPH/5 placentae was significantly increased as compared to C57 controls. **P = 0.009. Results represent the median and IQR (n = 4–12 placentas/group).
BPH/5 implantation sites have angiogenic imbalances before placenta formation
To determine the relationship between aberrant complement expression and angiogenic factor expression in BPH/5 pregnancy, we characterized the temporal expression pattern of complement and angiogenic factors during the peri-implantation period (E5.5; Fig. 4) and at the peak of decidualization (E7.5; Fig. 5). During the peri-implantation period and onset of decidualization (E5.5), we found that C1qa, C3, and CfB mRNA expression was comparable in BPH/5 and C57 pregnancies (Fig. 4A–C). Although sFlt-1 mRNA expression was similar in BPH/5 and C57 implantation sites at E5.5 (Fig. 4G), VEGFa mRNA was significantly up-regulated, and VEGFR1 and VEGFR2 mRNA was down-regulated in BPH/5 pregnancies as compared to C57 controls (Fig. 4D–F). At the peak of decidualization (E7.5), VEGFa mRNA expression trended upward in BPH/5 implantation sites (Fig. 5A). VEGF164 is the most biologically relevant VEGFa isoform found to be the expressed in the peri-implantation mouse uterus (56). Western blot showed significantly increased VEGF164 protein expression in BPH/5 implantation sites as compared to C57 controls (Fig. 5B, C). Whereas, mRNA expression of proangiogenic PlGF was significantly up-regulated, mRNA expression of RGC32, a positive mediator of PlGF and VEGFR2, was down-regulated in BPH/5 implantation sites, as compared to C57 controls at E7.5 (Fig. 5D, E). mRNA expression of sFlt-1 was similar in BPH/5 and C57 controls at E7.5 (Fig. 5F). Taken together, these data show aberrant VEGF family gene expression before abnormal complement deposition early in BPH/5 pregnancies.
Figure 4.

Angiogenic gene expression is altered in BPH/5 implantation sites during decidualization. mRNA expression of complement and angiogenic genes in C57 control and BPH/5 implantation sites at E5.5 was quantified by qRT-PCR. mRNA expression of C1qa (A), C3 (B), CfB (C), and sFlt-1(G) was similar in C57 control and BPH/5 implantation sites. Expression of VEGFa (D) was significantly increased in BPH/5 pregnancies. *P = 0.05. mRNA expression of both VEGFR1 (E) *P = 0.03 and VEGFR2 (F) *P = 0.02 was decreased in BPH/5 pregnancies. Results represent the median and IQR; n = 5–9 implantation sites/group.
Figure 5.

Angiogenic gene expression remains altered in BPH/5 implantation sites before placentation. A) VEGFa mRNA expression in C57 and BPH/5 implantation sites at E7.5 was quantified using qRT-PCR. mRNA expression of VEGFa was not significantly increased in BPH/5 pregnancies. P = 0.06. B) Representative Western blots of VEGF164 and actin in BPH/5 and C57 implantation sites at E7.5. C) Quantification of VEGF164 protein expression relative to actin by Western blot analysis. **P = 0.004. D–F) Quantification by qRT-PCR of PlGF, RGC32, and sFlt-1 transcripts in implantation sites at E7.5. D) mRNA expression of PlGF was increased in BPH/5 pregnancies. **P = 0.01. E) mRNA expression of RGC32 was decreased in BPH/5 pregnancies. **P = 0.01. F) mRNA expression of sFlt-1 showed no significant difference between BPH/5 and C57 pregnancies. Results represents the median and IQR; n = 6–10 implantation sites/group.
BPH/5 implantation sites have abnormal decidual vasculature
Because altered expression of VEGF family members was observed in BPH/5 pregnancies at E5.5 and E7.5, we determined whether there were changes in the decidual vasculature (Fig. 6). Uterine vascular density peaks between E6.5 and E7.5, and nearly all newly formed decidual vasculature expresses VEGFR2 (17). Using VEGFR2 expression as a marker of decidual vessels, we assessed development of decidual vasculature in BPH/5 and C57 control implantation sites at E7.5 (Fig. 6A–D). Measurement of decidual capillary blood space area was compared to the total area of decidua analyzed. Vascular space area:decidua ratio was significantly decreased in BPH/5 pregnancies, when compared to C57 controls (Fig. 6E). These findings suggest that postimplantation decidual angiogenesis is compromised in BPH/5 pregnancies, resulting in decreased decidual vascular blood space at E7.5.
Figure 6.

Development of the decidual vasculature was reduced in BPH/5 implantation sites. A, C) H&E staining was performed on tissue sections of implantation sites at E7.5. B, D) IHC was performed to detect VEGFR2 expression in the decidual vasculature. Nuclei were stained with methylene green. Scale bars, 500 µm. To quantitatively assess development of the decidual vasculature in BPH/5 and C57 pregnancies at E7.5, we measured the total amount of vascular space [red arrows in capillary lumens in (B, D)] and normalized that to the area of decidua measured in each section. E) Representative images used to quantify vascular space in ImageJ are shown. Mean vascular space/area of decidua in BPH/5 implantation sites was significantly decreased as compared to C57 controls. P = 0.03. Results represent the mean and sem (n = 4 implantation sites/group). am, antimesometrial; e, embryo; m, mesometrial.
Midgestation BPH/5 placentae have angiogenic imbalance
It has been reported that BPH/5 pregnant mice have altered placental and circulating levels of VEGFa and PlGF angiogenic factors; however neither those nor sFlt-1 was specifically examined in E10.5 placentae (32). Analysis of angiogenic factor gene expression was performed in E10.5 BPH/5 placentae. mRNA expression of proangiogenic VEGFa, PlGF, and RGC32 was decreased in BPH/5 placentae, compared with C57 controls (Fig. 7A–C), whereas mRNA expression of antiangiogenic factor sFlt-1 mRNA was up-regulated (Fig. 7D). Thus, we found an inverse relationship in mRNA expression of placenta-derived PlGF and sFlt-1, as is seen in the circulation of pregnant women in whom PE eventually develops.
Figure 7.

Midgestational BPH/5 placentae have abnormal angiogenic gene expression. mRNA expression of angiogenic genes in C57 and BPH/5 placentae at E10.5 was quantified with qRT-PCR. mRNA expression of proangiogenic was significantly decreased in BPH/5 placentae: VEGFa (A) *P = 0.02; PlGF (B) **P = 0.002; and RGC32 (C), **P = 0.004. D) mRNA expression of antiangiogenic sFlt-1 was significantly increased in BPH/5 placentae. *P = 0.04. Results represent the median and IQR (n = 6–12 placentae/group).
DISCUSSION
Previous reports showed increased complement expression in the ectoplacental cone and lower circulating and placental levels of VEGF and P1GF, starting during placentation at E9.5, in BPH/5 pregnancies (32, 33). Further, inhibition of complement led to an increase in VEGF and exogenous VEGF addition rescued the PE-like syndrome (32, 33). Herein, we demonstrate temporospatial changes in these pathways during the critical postimplantation period and at the beginning of placentation in BPH/5 mice. Complement gene expression was significantly up-regulated in BPH/5 pregnancies at E7.5, the peak of decidualization, and in midgestation BPH/5 placentae at E10.5, whereas angiogenic factor imbalance was initially detected at the start of decidualization (E5.5), with up-regulation of antiangiogenic sFlt-1 in midgestation BPH/5 placentae (E10.5). Thus, we found that angiogenic imbalance preceded an increase in complement pathway components at the maternal–fetal interface, suggesting that these alterations occur before placentation and may contribute to development of the maternal PE syndrome in BPH/5 pregnancy (Fig. 8).
Figure 8.

Summary of dynamic complement and angiogenic factor expression in BPH/5 pregnancy at the maternal–fetal interface as compared controls.
At E7.5, RNA-Seq demonstrated an up-regulation of complement factors C1rb, C3, and CfB, suggesting activation of both the classic and alternative arms of the complement cascade at the peak of decidualization in BPH/5 pregnancies (Table 2). We found a significant increase in the levels of both C3 and CfB in implantation sites at the peak of decidualization in BPH/5. Increased C3 and CfB expression in BPH/5 is consistent with studies abnormal C3 and CfB activation in adverse pregnancy outcomes (14, 33, 34, 36, 57, 58). Gelber et al. (33) showed increased C3 expression in the ectoplacental cone of BPH/5 pregnancies at E6.5 and E8.5. Thus, our observation of excessive C3 deposition in the ectoplacental cone at E7.5 was expected. We demonstrate excessive C3 deposition around decidual vasculature at E7.5 and in the TGC layer of BPH/5 placentae at midgestation. Levels of C9, a component of the final complement effector complex common to all complement pathways, were also increased in the TGC layer of midgestation BPH/5 placentae. Although pregnant mice lacking C1q developed features of PE (37), we found similar C1qa levels in BPH/5 and control implantation sites and placentae, suggesting that C1q does not contribute to the PE phenotype in BPH/5. As complement inhibition early in BPH/5 pregnancy (E5.5) improved fetoplacental outcomes, including trophoblast invasion (33), we hypothesize that the complement inhibition early in BPH/5 pregnancies reduces aberrant complement deposition around decidual vasculature and the ectoplacental cone before placenta formation and within the TGC layer of placentae, preventing the onset of PE-associated adverse outcomes. These findings suggest that local activation of complement pathways early in BPH/5 pregnancy is integral to developing PE-like features.
We sought to gain insight into the temporal relationship between excess complement deposition and misexpression of VEGF family genes in BPH/5 pregnancies. In the present study, we found dysregulated VEGF pathway genes at the start of decidualization, before abnormal complement expression in BPH/5. VEGF family ligands and receptors, including VEGF, PlGF, VEGFR1, VEGFR2, and sFlt-1, are implicated in decidual angiogenesis, placentation, and PE (17–20, 30, 31). In mice, VEGF is expressed in the endometrium and postimplantation decidua (56, 59). Activation of the VEGF receptors VEGFR1 and -2 promotes decidual angiogenesis (17, 18). Our data suggest that decreased VEGFR1 and -2 expression in BPH/5 pregnancies at E5.5 contributes to an antiangiogenic environment that inhibits development of decidual vasculature. At E7.5, we detected reduced vascular spaces and increased expression of proangiogenic ligands, VEGFa and PlGF in response to the antiangiogenic state. The latter may be related to a feedback loop in which reduced vessel density leads to hypoxia-induced expression of VEGFa (60, 61). This abnormal development of decidual vasculature and early local misexpression of VEGF family genes precedes both sFlt-1 overexpression and aberrant complement deposition in BPH/5 placentae (Fig. 8). Elucidating the exact mechanism whereby early angiogenic imbalances may contribute the PE phenotype is an exciting area of ongoing research in BPH/5.
We are the first to determine that RGC32 is misexpressed in a PE-like mouse model. RGC32, a coactivator that mediates cellular proliferation and differentiation, was originally identified because of differential expression of RGC32 in response to complement activation (40). Recent studies showed that expression of RGC32 was significantly lower in human PE placentae (62). We found that expression of RGC32 was down-regulated in BPH/5 implantation sites (E7.5) and midgestation placentae (E10.5), concurrent with onset of complement overexpression. Regulation of placental angiogenesis by RGC32-mediated changes in VEGF family members, PlGF and VEGFR2, was recently described. Specifically, loss of murine RGC32 decreased PlGF expression in trophoblasts and VEGFR2 expression in endothelial cells, leading to impaired placentation, fetal growth restriction, and increased fetal loss (39). We found that expression of proangiogenic PlGF was dynamic in BPH/5 pregnancies, up-regulated at E7.5 and down-regulated at E10.5. The lower placental PlGF in BPH/5 may reflect a decrease in trophoblast-specific PlGF expression as a result of decreased RGC32 expression. Mechanistic studies are needed to understand the role of RGC32 in BPH/5 and other PE mouse models.
We showed that BPH/5 placentae have decreased PlGF, VEGF, and RGC32, with increased sFlt-1, suggesting an imbalance of proangiogenic and antiangiogenic factors, as seen in human PE. Further, we found abnormal decidual vasculature with excess complement deposition around the vasculature in BPH/5 pregnancies. Based on our findings, we propose that aberrant decidual angiogenesis triggers complement activation before placentation. This early, postimplantation decidual aberration initiates adverse ripple effects that then contribute to shallow trophoblast invasion into the maternal spiral arteries, the first stage of PE. It is important to determine the cause of these early angiogenic defects during implantation in BPH/5 and whether amelioration prevents overactivation of complement at the maternal–fetal interface.
Because BPH/5 mice spontaneously develop a PE-like phenotype, we can investigate early pregnancy and even preconception risk factors that may lead to the heightened state of inflammation seen in this model. Our RNA-Seq analysis showed significant chemokine up-regulation. Decidual NK cells are dysregulated in BPH/5 pregnancies, and there is a reduction in the number of macrophages in BPH/5 decidua with a concomitant increase in activated T cells (44, 45). Ongoing investigations seek to identify the cell types contributing to aberrant gene expression in the BPH/5 decidua. Further, recently published data from the Sones laboratory demonstrated that BPH/5 female mice have an adverse metabolic phenotype from early adulthood, which includes hyperphagia and obesity with increased white adipose tissue (WAT), proinflammatory reproductive WAT, and leptin resistance (63). Obesity before and at the time of conception has been linked to adverse pregnancy outcomes (64, 65). Studies are ongoing to determine whether preconception WAT reduction in BPH/5 mice can decrease placental complement expression and normalize the decidual inflammation that plays an important role in the pathogenesis of PE in this model.
Supplementary Material
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
ACKNOWLEDGMENTS
The authors thank Robin L. Davisson for the generous gift of BPH/5 mice and additional support; Michelle A. Hoch for assistance with data analysis; Jan K. Kitajewski and Virginia E. Papaioannou for in-depth discussions and assistance with manuscript editing; and Theresa Swayne and Emilia Laura Munteanu for technical assistance with confocal microscopy. This work was supported by U.S. National Institutes of Health (NIH) National Heart Lung and Blood Institute Grant 1R01HL127013-01A1 (to N.C.D) and NIH Eunice Kennedy Shriver National Institute of Child Health and Human Development Grant P50-HD076210 (to J.L.S. and J.K.G.). Immunofluorescence images were collected in the Confocal and Specialized Microscopy Shared Resource of the Herbert Irving Comprehensive Cancer Center at Columbia University (New York, NY, USA), supported by NIH National Cancer Institute Grant P30 CA013696. The confocal microscope was purchased with NIH National Center for Research Resources Grant S10 RR025686. The authors declare no conflicts of interest.
Glossary
- BPH
blood pressure high
- Cox2
cyclooxygenase 2
- CTCF
corrected total cell fluorescence
- FPKM
fragments per kilobase per million
- H&E
hematoxylin and eosin
- IF
immunofluorescence
- IHC
immunohistochemistry
- IQR
interquartile range
- PE
preeclampsia
- PlGF
placental growth factor
- qRT-PCR
quantitative RT-PCR
- RGC
response gene to complement
- sFlt
antiangiogenic soluble VEGF receptor 1
- TGC
trophoblast giant cell
- VEGFR
vascular endothelial growth factor receptor
- WAT
white adipose tissue
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
AUTHOR CONTRIBUTIONS
J. L. Sones, A. A. Merriam, and N. C. Douglas designed and performed the research, analyzed data, and wrote the paper; A. Seffens and J. K. Grenier designed and performed the research and analyzed the data; D.-A. Brown-Grant, S. D. Butler, and X. Xu performed the research; A. M. Zhao analyzed the data; and C. J. Shawber wrote the paper.
REFERENCES
- 1.Ghulmiyyah L., Sibai B. (2012) Maternal mortality from preeclampsia/eclampsia. Semin. Perinatol. 36, 56–59 10.1053/j.semperi.2011.09.011 [DOI] [PubMed] [Google Scholar]
- 2.American College of Obstetricians and Gynecologists ; Task Force on Hypertension in Pregnancy . (2013) Hypertension in pregnancy: report of the American College of Obstetricians and gynecologists’ task force on hypertension in pregnancy. Obstet. Gynecol. 122, 1122–1131 10.1097/01.AOG.0000437382.03963.88 [DOI] [PubMed] [Google Scholar]
- 3.Tranquilli A. L., Dekker G., Magee L., Roberts J., Sibai B. M., Steyn W., Zeeman G. G., Brown M. A. (2014) The classification, diagnosis and management of the hypertensive disorders of pregnancy: a revised statement from the ISSHP. Pregnancy Hypertens. 4, 97–104 10.1016/j.preghy.2014.02.001 [DOI] [PubMed] [Google Scholar]
- 4.Chen C. W., Jaffe I. Z., Karumanchi S. A. (2014) Pre-eclampsia and cardiovascular disease. Cardiovasc. Res. 101, 579–586 10.1093/cvr/cvu018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huppertz B. (2008) Placental origins of preeclampsia: challenging the current hypothesis. Hypertension 51, 970–975 10.1161/HYPERTENSIONAHA.107.107607 [DOI] [PubMed] [Google Scholar]
- 6.Redman C. W., Staff A. C. (2015) Preeclampsia, biomarkers, syncytiotrophoblast stress, and placental capacity. Am. J. Obstet. Gynecol. 213, S9.e1, S9–S11. [DOI] [PubMed] [Google Scholar]
- 7.Tomimatsu T., Mimura K., Endo M., Kumasawa K., Kimura T. (2017) Pathophysiology of preeclampsia: an angiogenic imbalance and long-lasting systemic vascular dysfunction. Hypertens. Res. 40, 305–310 10.1038/hr.2016.152 [DOI] [PubMed] [Google Scholar]
- 8.Triunfo S., Crovetto F., Crispi F., Rodriguez-Sureda V., Dominguez C., Nadal A., Peguero A., Gratacos E., Figueras F. (2016) Association of first-trimester angiogenic factors with placental histological findings in late-onset preeclampsia. Placenta 42, 44–50 10.1016/j.placenta.2016.04.005 [DOI] [PubMed] [Google Scholar]
- 9.Regal J. F., Gilbert J. S., Burwick R. M. (2015) The complement system and adverse pregnancy outcomes. Mol. Immunol. 67, 56–70 10.1016/j.molimm.2015.02.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rampersad R., Barton A., Sadovsky Y., Nelson D. M. (2008) The C5b-9 membrane attack complex of complement activation localizes to villous trophoblast injury in vivo and modulates human trophoblast function in vitro. Placenta 29, 855–861 10.1016/j.placenta.2008.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lokki A. I., Heikkinen-Eloranta J., Jarva H., Saisto T., Lokki M. L., Laivuori H., Meri S. (2014) Complement activation and regulation in preeclamptic placenta. Front. Immunol. 5, 312 10.3389/fimmu.2014.00312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lynch A. M., Murphy J. R., Byers T., Gibbs R. S., Neville M. C., Giclas P. C., Salmon J. E., Holers V. M. (2008) Alternative complement pathway activation fragment Bb in early pregnancy as a predictor of preeclampsia. Am. J. Obstet. Gynecol. 198, 385.e1–385.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lynch A. M., Gibbs R. S., Murphy J. R., Giclas P. C., Salmon J. E., Holers V. M. (2011) Early elevations of the complement activation fragment C3a and adverse pregnancy outcomes. Obstet. Gynecol. 117, 75–83 10.1097/AOG.0b013e3181fc3afa [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lynch A. M., Eckel R. H., Murphy J. R., Gibbs R. S., West N. A., Giclas P. C., Salmon J. E., Holers V. M. (2012) Prepregnancy obesity and complement system activation in early pregnancy and the subsequent development of preeclampsia. Am. J. Obstet. Gynecol. 206, 428.e1–428.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burwick R. M., Fichorova R. N., Dawood H. Y., Yamamoto H. S., Feinberg B. B. (2013) Urinary excretion of C5b-9 in severe preeclampsia: tipping the balance of complement activation in pregnancy. Hypertension 62, 1040–1045 10.1161/HYPERTENSIONAHA.113.01420 [DOI] [PubMed] [Google Scholar]
- 16.Burwick R. M., Feinberg B. B. (2013) Eculizumab for the treatment of preeclampsia/HELLP syndrome. Placenta 34, 201–203 10.1016/j.placenta.2012.11.014 [DOI] [PubMed] [Google Scholar]
- 17.Douglas N. C., Tang H., Gomez R., Pytowski B., Hicklin D. J., Sauer C. M., Kitajewski J., Sauer M. V., Zimmermann R. C. (2009) Vascular endothelial growth factor receptor 2 (VEGFR-2) functions to promote uterine decidual angiogenesis during early pregnancy in the mouse. Endocrinology 150, 3845–3854 10.1210/en.2008-1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Douglas N. C., Zimmermann R. C., Tan Q. K., Sullivan-Pyke C. S., Sauer M. V., Kitajewski J. K., Shawber C. J. (2014) VEGFR-1 blockade disrupts peri-implantation decidual angiogenesis and macrophage recruitment. Vasc. Cell 6, 16 10.1186/2045-824X-6-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim M., Park H. J., Seol J. W., Jang J. Y., Cho Y. S., Kim K. R., Choi Y., Lydon J. P., Demayo F. J., Shibuya M., Ferrara N., Sung H. K., Nagy A., Alitalo K., Koh G. Y. (2013) VEGF-A regulated by progesterone governs uterine angiogenesis and vascular remodelling during pregnancy. EMBO Mol. Med. 5, 1415–1430 10.1002/emmm.201302618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vrachnis N., Kalampokas E., Sifakis S., Vitoratos N., Kalampokas T., Botsis D., Iliodromiti Z. (2013) Placental growth factor (PlGF): a key to optimizing fetal growth. J. Matern. Fetal Neonatal Med. 26, 995–1002 10.3109/14767058.2013.766694 [DOI] [PubMed] [Google Scholar]
- 21.Levine R. J., Maynard S. E., Qian C., Lim K. H., England L. J., Yu K. F., Schisterman E. F., Thadhani R., Sachs B. P., Epstein F. H., Sibai B. M., Sukhatme V. P., Karumanchi S. A. (2004) Circulating angiogenic factors and the risk of preeclampsia. N. Engl. J. Med. 350, 672–683 10.1056/NEJMoa031884 [DOI] [PubMed] [Google Scholar]
- 22.Thadhani R., Mutter W. P., Wolf M., Levine R. J., Taylor R. N., Sukhatme V. P., Ecker J., Karumanchi S. A. (2004) First trimester placental growth factor and soluble fms-like tyrosine kinase 1 and risk for preeclampsia. J. Clin. Endocrinol. Metab. 89, 770–775 10.1210/jc.2003-031244 [DOI] [PubMed] [Google Scholar]
- 23.Taylor R. N., Grimwood J., Taylor R. S., McMaster M. T., Fisher S. J., North R. A. (2003) Longitudinal serum concentrations of placental growth factor: evidence for abnormal placental angiogenesis in pathologic pregnancies. Am. J. Obstet. Gynecol. 188, 177–182 10.1067/mob.2003.111 [DOI] [PubMed] [Google Scholar]
- 24.Tidwell S. C., Ho H. N., Chiu W. H., Torry R. J., Torry D. S. (2001) Low maternal serum levels of placenta growth factor as an antecedent of clinical preeclampsia. Am. J. Obstet. Gynecol. 184, 1267–1272 10.1067/mob.2001.113129 [DOI] [PubMed] [Google Scholar]
- 25.Sovio U., Gaccioli F., Cook E., Hund M., Charnock-Jones D. S., Smith G. C. (2017) Prediction of preeclampsia using the soluble fms-like tyrosine kinase 1 to placental growth factor ratio: a prospective cohort study of unselected nulliparous women. Hypertension 69, 731–738 10.1161/HYPERTENSIONAHA.116.08620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He Y., Xu B., Song D., Yu F., Chen Q., Zhao M. (2016) Correlations between complement system’s activation factors and anti-angiogenesis factors in plasma of patients with early/late-onset severe preeclampsia. Hypertens. Pregnancy 35, 499–509 10.1080/10641955.2016.1190845 [DOI] [PubMed] [Google Scholar]
- 27.He Y., Xu B., Song D., Yu F., Chen Q., Zhao M. (2016) Expression of the complement system’s activation factors in plasma of patients with early/late-onset severe pre-eclampsia. Am. J. Reprod. Immunol. 76, 205–211 10.1111/aji.12541 [DOI] [PubMed] [Google Scholar]
- 28.Chang Y. S., Chen C. N., Jeng S. F., Su Y. N., Chen C. Y., Chou H. C., Tsao P. N., Hsieh W. S. (2017) The sFlt-1/PlGF ratio as a predictor for poor pregnancy and neonatal outcomes. Pediatr. Neonatol. 58, 529–533 [DOI] [PubMed] [Google Scholar]
- 29.Dröge L. A., Höller A., Ehrlich L., Verlohren S., Henrich W., Perschel F. H. (2017) Diagnosis of preeclampsia and fetal growth restriction with the sFlt-1/PlGF ratio: diagnostic accuracy of the automated immunoassay Kryptor®. Pregnancy Hypertens. 8, 31–36 10.1016/j.preghy.2017.02.005 [DOI] [PubMed] [Google Scholar]
- 30.Maynard S. E., Min J. Y., Merchan J., Lim K. H., Li J., Mondal S., Libermann T. A., Morgan J. P., Sellke F. W., Stillman I. E., Epstein F. H., Sukhatme V. P., Karumanchi S. A. (2003) Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J. Clin. Invest. 111, 649–658 10.1172/JCI17189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Szalai G., Xu Y., Romero R., Chaiworapongsa T., Xu Z., Chiang P. J., Ahn H., Sundell B., Plazyo O., Jiang Y., Olive M., Wang B., Jacques S. M., Qureshi F., Tarca A. L., Erez O., Dong Z., Papp Z., Hassan S. S., Hernandez-Andrade E., Than N. G. (2014) In vivo experiments reveal the good, the bad and the ugly faces of sFlt-1 in pregnancy. PLoS One 9, e110867 10.1371/journal.pone.0110867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Woods A. K., Hoffmann D. S., Weydert C. J., Butler S. D., Zhou Y., Sharma R. V., Davisson R. L. (2011) Adenoviral delivery of VEGF121 early in pregnancy prevents spontaneous development of preeclampsia in BPH/5 mice. Hypertension 57, 94–102 10.1161/HYPERTENSIONAHA.110.160242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gelber S. E., Brent E., Redecha P., Perino G., Tomlinson S., Davisson R. L., Salmon J. E. (2015) Prevention of defective placentation and pregnancy loss by blocking innate immune pathways in a syngeneic model of placental insufficiency. J. Immunol. 195, 1129–1138 10.4049/jimmunol.1402220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang W., Irani R. A., Zhang Y., Ramin S. M., Blackwell S. C., Tao L., Kellems R. E., Xia Y. (2012) Autoantibody-mediated complement C3a receptor activation contributes to the pathogenesis of preeclampsia. Hypertension 60, 712–721 10.1161/HYPERTENSIONAHA.112.191817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sones J. L., Cha J., Woods A. K., Bartos A., Heyward C. Y., Lob H. E., Isroff C. E., Butler S. D., Shapiro S. E., Dey S. K., Davisson R. L. (2016) Decidual Cox2 inhibition improves fetal and maternal outcomes in a preeclampsia-like mouse model. JCI Insight 1, e75351 10.1172/jci.insight.75351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qing X., Redecha P. B., Burmeister M. A., Tomlinson S., D’Agati V. D., Davisson R. L., Salmon J. E. (2011) Targeted inhibition of complement activation prevents features of preeclampsia in mice. Kidney Int. 79, 331–339 10.1038/ki.2010.393 [DOI] [PubMed] [Google Scholar]
- 37.Singh J., Ahmed A., Girardi G. (2011) Role of complement component C1q in the onset of preeclampsia in mice. Hypertension 58, 716–724 10.1161/HYPERTENSIONAHA.111.175919 [DOI] [PubMed] [Google Scholar]
- 38.Chu A., Thamotharan S., Ganguly A., Wadehra M., Pellegrini M., Devaskar S. U. (2016) Gestational food restriction decreases placental interleukin-10 expression and markers of autophagy and endoplasmic reticulum stress in murine intrauterine growth restriction. Nutr. Res. 36, 1055–1067 10.1016/j.nutres.2016.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cui X. B., Guo X., Chen S. Y. (2013) Response gene to complement 32 deficiency causes impaired placental angiogenesis in mice. Cardiovasc. Res. 99, 632–639 10.1093/cvr/cvt121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vlaicu S. I., Cudrici C., Ito T., Fosbrink M., Tegla C. A., Rus V., Mircea P. A., Rus H. (2008) Role of response gene to complement 32 in diseases. Arch. Immunol. Ther. Exp. (Warsz.) 56, 115–122 10.1007/s00005-008-0016-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davisson R. L., Hoffmann D. S., Butz G. M., Aldape G., Schlager G., Merrill D. C., Sethi S., Weiss R. M., Bates J. N. (2002) Discovery of a spontaneous genetic mouse model of preeclampsia. Hypertension 39, 337–342 10.1161/hy02t2.102904 [DOI] [PubMed] [Google Scholar]
- 42.Hoffmann D. S., Weydert C. J., Lazartigues E., Kutschke W. J., Kienzle M. F., Leach J. E., Sharma J. A., Sharma R. V., Davisson R. L. (2008) Chronic tempol prevents hypertension, proteinuria, and poor feto-placental outcomes in BPH/5 mouse model of preeclampsia. Hypertension 51, 1058–1065 10.1161/HYPERTENSIONAHA.107.107219 [DOI] [PubMed] [Google Scholar]
- 43.Dokras A., Hoffmann D. S., Eastvold J. S., Kienzle M. F., Gruman L. M., Kirby P. A., Weiss R. M., Davisson R. L. (2006) Severe feto-placental abnormalities precede the onset of hypertension and proteinuria in a mouse model of preeclampsia. Biol. Reprod. 75, 899–907 10.1095/biolreprod.106.053603 [DOI] [PubMed] [Google Scholar]
- 44.Sones J. L., Lob H. E., Isroff C. E., Davisson R. L. (2014) Role of decidual natural killer cells, interleukin-15, and interferon-γ in placental development and preeclampsia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 307, R490–R492 10.1152/ajpregu.00176.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heyward C. Y., Sones J. L., Lob H. E., Yuen L. C., Abbott K. E., Huang W., Begun Z. R., Butler S. D., August A., Leifer C. A., Davisson R. L. (2017) The decidua of preeclamptic-like BPH/5 mice exhibits an exaggerated inflammatory response during early pregnancy. J. Reprod. Immunol. 120, 27–33 10.1016/j.jri.2017.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schlager G. (1974) Selection for blood pressure levels in mice. Genetics 76, 537–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martin M. (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12 [Google Scholar]
- 48.Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S. L. (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36 10.1186/gb-2013-14-4-r36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trapnell C., Hendrickson D. G., Sauvageau M., Goff L., Rinn J. L., Pachter L. (2013) Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 31, 46–53 10.1038/nbt.2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Young C. N., Cao X., Guruju M. R., Pierce J. P., Morgan D. A., Wang G., Iadecola C., Mark A. L., Davisson R. L. (2012) ER stress in the brain subfornical organ mediates angiotensin-dependent hypertension. J. Clin. Invest. 122, 3960–3964 10.1172/JCI64583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lacko L. A., Massimiani M., Sones J. L., Hurtado R., Salvi S., Ferrazzani S., Davisson R. L., Campagnolo L., Stuhlmann H. (2014) Novel expression of EGFL7 in placental trophoblast and endothelial cells and its implication in preeclampsia. Mech. Dev. 133, 163–176 10.1016/j.mod.2014.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vorontchikhina M. A., Zimmermann R. C., Shawber C. J., Tang H., Kitajewski J. (2005) Unique patterns of Notch1, Notch4 and Jagged1 expression in ovarian vessels during folliculogenesis and corpus luteum formation. Gene Expr. Patterns 5, 701–709 10.1016/j.modgep.2005.02.001 [DOI] [PubMed] [Google Scholar]
- 53.Levin H. I., Sullivan-Pyke C. S., Papaioannou V. E., Wapner R. J., Kitajewski J. K., Shawber C. J., Douglas N. C. (2017) Dynamic maternal and fetal Notch activity and expression in placentation. Placenta 55, 5–12 10.1016/j.placenta.2017.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Douglas N. C., Wang J. G., Yu B., Gaddipati S., Guarnaccia M. M., Sauer M. V. (2009) A systematic, multidisciplinary approach to address the reproductive needs of HIV-seropositive women. Reprod. Biomed. Online 19, 257–263 10.1016/S1472-6483(10)60082-X [DOI] [PubMed] [Google Scholar]
- 55.Cha J., Sun X., Dey S. K. (2012) Mechanisms of implantation: strategies for successful pregnancy. Nat. Med. 18, 1754–1767 10.1038/nm.3012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Halder J. B., Zhao X., Soker S., Paria B. C., Klagsbrun M., Das S. K., Dey S. K. (2000) Differential expression of VEGF isoforms and VEGF(164)-specific receptor neuropilin-1 in the mouse uterus suggests a role for VEGF(164) in vascular permeability and angiogenesis during implantation. Genesis 26, 213–224 [DOI] [PubMed] [Google Scholar]
- 57.Thurman J. M., Kraus D. M., Girardi G., Hourcade D., Kang H. J., Royer P. A., Mitchell L. M., Giclas P. C., Salmon J., Gilkeson G., Holers V. M. (2005) A novel inhibitor of the alternative complement pathway prevents antiphospholipid antibody-induced pregnancy loss in mice. Mol. Immunol. 42, 87–97 10.1016/j.molimm.2004.07.043 [DOI] [PubMed] [Google Scholar]
- 58.Lynch A. M., Gibbs R. S., Murphy J. R., Byers T., Neville M. C., Giclas P. C., Salmon J. E., Van Hecke T. M., Holers V. M. (2008) Complement activation fragment Bb in early pregnancy and spontaneous preterm birth. Am. J. Obstet. Gynecol. 199, 354.e1–354.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fan X., Rai A., Kambham N., Sung J. F., Singh N., Petitt M., Dhal S., Agrawal R., Sutton R. E., Druzin M. L., Gambhir S. S., Ambati B. K., Cross J. C., Nayak N. R. (2014) Endometrial VEGF induces placental sFLT1 and leads to pregnancy complications. J. Clin. Invest. 124, 4941–4952 10.1172/JCI76864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dey S. K., Lim H., Das S. K., Reese J., Paria B. C., Daikoku T., Wang H. (2004) Molecular cues to implantation. Endocr. Rev. 25, 341–373 10.1210/er.2003-0020 [DOI] [PubMed] [Google Scholar]
- 61.Daikoku T., Matsumoto H., Gupta R. A., Das S. K., Gassmann M., DuBois R. N., Dey S. K. (2003) Expression of hypoxia-inducible factors in the peri-implantation mouse uterus is regulated in a cell-specific and ovarian steroid hormone-dependent manner: evidence for differential function of HIFs during early pregnancy. J. Biol. Chem. 278, 7683–7691 10.1074/jbc.M211390200 [DOI] [PubMed] [Google Scholar]
- 62.Wang Q. J., Song B. F., Zhang Y. H., Ma Y. Y., Shao Q. Q., Liu J., Qu X. (2015) Expression of RGC32 in human normal and preeclamptic placentas and its role in trophoblast cell invasion and migration. Placenta 36, 350–356 10.1016/j.placenta.2014.12.012 [DOI] [PubMed] [Google Scholar]
- 63.Sutton E. F., Lob H. E., Song J., Xia Y., Butler S., Liu C. C., Redman L. M., Sones J. L. (2017) Adverse metabolic phenotype of female offspring exposed to preeclampsia in utero: a characterization of the BPH/5 mouse in postnatal life. Am. J. Physiol. Regul. Integr. Comp. Physiol. 312, R485–R491 10.1152/ajpregu.00512.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Spradley F. T., Palei A. C., Granger J. P. (2015) Immune mechanisms linking obesity and preeclampsia. Biomolecules 5, 3142–3176 10.3390/biom5043142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Spradley F. T., Palei A. C., Granger J. P. (2015) Increased risk for the development of preeclampsia in obese pregnancies: weighing in on the mechanisms. Am. J. Physiol. Regul. Integr. Comp. Physiol. 309, R1326–R1343 10.1152/ajpregu.00178.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rupprecht T. A., Angele B., Klein M., Heesemann J., Pfister H. W., Botto M., Koedel U. (2007) Complement C1q and C3 are critical for the innate immune response to Streptococcus pneumoniae in the central nervous system. J. Immunol. 178, 1861–1869 [DOI] [PubMed] [Google Scholar]
- 67.Zou L., Feng Y., Xu G., Jian W., Chao W. (2016) Splenic RNA and microRNA mimics promote complement factor B production and alternative pathway activation via innate immune signaling. J. Immunol. 196, 2788–2798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang Y., Arenas-Hernandez M., Gomez-Lopez N., Dai J., Parker G. C., Puscheck E. E., Rappolee D. A. (2016) Hypoxic stress forces irreversible differentiation of a majority of mouse trophoblast stem cells despite FGF4. Biol. Reprod. 95, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huckle W. R., Roche R. I. (2004) Post-transcriptional control of expression of sFlt-1, an endogenous inhibitor of vascular endothelial growth factor. J. Cell. Biochem. 93, 120–132 [DOI] [PubMed] [Google Scholar]
- 70.Yang W. J., Yang D. D., Na S., Sandusky G. E., Zhang Q., Zhao G. (2005) Dicer is required for embryonic angiogenesis during mouse development. J. Biol. Chem. 280, 9330–9335 [DOI] [PubMed] [Google Scholar]
- 71.Hazarika S., Dokun A. O., Li Y., Popel A. S., Kontos C. D., Annex B. H. (2007) Impaired angiogenesis after hindlimb ischemia in type 2 diabetes mellitus: differential regulation of vascular endothelial growth factor receptor 1 and soluble vascular endothelial growth factor receptor 1. Circ. Res. 101, 948–956 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.