

Abstract
Disrupting a gene to determine its effect on an organism's phenotype is an indispensable tool in molecular biology. Such techniques are critical for understanding how a gene product contributes to the development and cellular identity of organisms. The explosion of genomic sequencing technologies combined with recent advances in genome‐editing techniques has elevated the possibilities of genetic manipulations in numerous organisms in which these experiments were previously not readily accessible or possible. Introducing the next generation of molecular biologists to these emerging techniques is key in the modern biology classroom. This comprehensive review introduces undergraduates to CRISPR/Cas9 editing and its uses in genetic studies. The goals of this review are to explain how CRISPR functions as a prokaryotic immune system, describe how researchers generate mutations with CRISPR/Cas9, highlight how Cas9 has been adapted for new functions, and discuss ethical considerations of genome editing. Additionally, anticipatory guides and questions for discussion are posed throughout the review to encourage active exploration of these topics in the classroom. Finally, the supplement includes a study guide and practical suggestions to incorporate CRISPR/Cas9 experiments into lab courses at the undergraduate level. © 2018 The Authors Biochemistry and Molecular Biology Education published by Wiley Periodicals, Inc. on behalf of International Union of Biochemistry and Molecular Biology, 46(2):195–205, 2018.
Keywords: CRISPR, Cas9, dCas9
Introduction
Scientists can probe the function of a gene, open reading frame, or other genomic feature by mutating or deleting a locus of interest and observing the resulting phenotype. However, even though these experiments are highly informative, these techniques could not be adapted in most organisms. In the past decade, researchers hypothesized that by exploiting endogenous, cellular DNA repair pathways, one could create mutations or precise edits at a desired location in the genome, termed genome editing. Double‐strand breaks are toxic to cells, thus organisms evolved mechanisms to repair these lesions. Scientists proposed that by generating a targeted, double‐strand break at a site of interest, then during the repair process errors may occur, resulting in a mutation at a desired site. Additionally, endogenous double‐strand break repair pathways could also stimulate the incorporation of exogenous DNA, creating very specific researcher‐designed edits. Thus, researchers started to identify ways to direct enzymes called nucleases that generate double‐strand breaks at specific regions of the genome. An RNA‐directed nuclease from a bacterial immune system called Cas9 has proven to be an easily programmed enzyme that when introduced into a cell or organism can create double‐strand breaks across eukaryotes 1, 2, 3.
Here we introduce novice biologists to the expanding world of genome editing. Specifically, we focus on CRISPR/Cas9 technology including how it evolved as a bacterial and archeal immune system and how and why the technology is adapted and useful for genome editing in different eukaryotes. We include practical knowledge about how genome edits are achieved and highlight how the Cas9 enzyme has been modified to expand the range of possible experiments. Finally, we explore ethical issues that have arisen around this technology. Each section begins with anticipatory guides to confront possible misconceptions common in molecular biology and facilitate discussions for a better understanding of the upcoming text. To assess and ensure that desired learning outcomes have been met, each section ends with questions for discussion to solidify and encourage further exploration of the material. Through reading this primer and discussion in class with peers, students should achieve the following learning outcomes:
Explain how CRISPR results in bacterial immunity
Define the different components necessary for genome editing by CRISPR
Describe how screening and selection are used to identify mutations
Design a CRISPR experiment to mutate a gene of interest
Evaluate potential ethical concerns raised by genome editing technologies
CRISPR: A Bacterial and Archeal Immune System Adapted for Eukaryotic Gene Editing
Microbiologists identified a unique pathway that bacteria and archea use to defend themselves from cellular invaders. Years later, molecular biologists recognized the potential of this basic scientific discovery to cut genomic DNA at precise sites in eukaryotic cells. We will explore how the CRISPR/Cas system works in prokaryotes and the key discoveries that adapted this defense mechanism for genetic engineering.
Anticipatory guides:
What do bacteria and archaea need to defend themselves against?
What are the hallmarks of an adaptive immune system?
Why would making a double‐strand break in the genome at a specific locus be useful?
Adaptive Immunity in Bacteria and Archea
Prokaryotes have defense mechanisms against viral and plasmid cellular invaders, just as multicellular organisms. One of these defense mechanisms is an adaptive immune system found in many bacteria and most archea called Clustered Regulatory Interspaced Short Palindromic Repeats or CRISPR, along with the CRISPR‐associated Proteins or Cas proteins. By integrating DNA sequences that are identical to past invaders into their genome, bacteria and archea generate a cellular memory of past invaders. These acquired sequences allow the bacteria or archea to recognize viral or plasmid invaders as non‐self, resulting in the degradation of the invading sequence 4 and functioning as an adaptive immune system for prokaryotes.
CRISPR immunity is characterized by distinct phases. First, during the adaptation phase bacteria or archaea gain a cellular memory of the invading virus or plasmid. Short sequences of the viral or plasmid genomes are integrated into the CRISPR locus of the bacterial or archaeal genome (Fig. 1A). These CRISPR loci were first identified by scientists working in the fermentation industry, where prokaryotes are essential to the production of fermented products. Through comparative genomic analysis of different S. thermophilus strains (a microbe used in producing yogurt), scientists identified a highly variable locus in the genome of these bacteria 5. This highly variable region had two distinct features: many non‐contiguous repeats that are separated by variable sequences, termed spacers. Upon closer inspection, researchers found that the spacer sequences matched those found in phage (viruses that infect bacteria) genomes 6. Interestingly, when researchers compared phage resistant and phage sensitive S. thermophilus, the phage resistant bacteria had spacer sequences that matched regions of that phage's genome 7. Thus, spacer content correlated with phage resistance leading to the model that short regions of the invader's genome are integrated into the CRISPR loci as a spacer, separated by repeat sequences, resulting in a cellular memory of previous infections (Fig. 1A).
Figure 1.
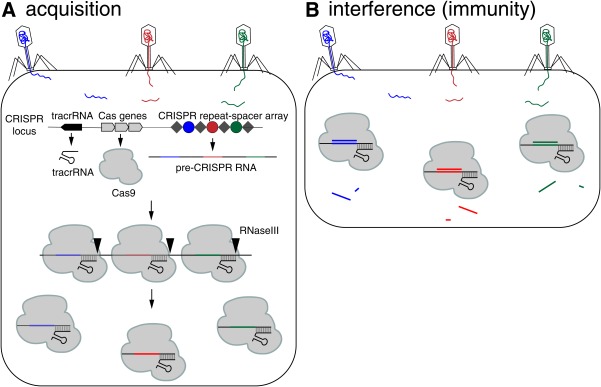
CRISPR/Cas9 mediated acquired immunity in prokaryotes. During the acquisition phase (A), cellular invaders such as phage virus inject nucleic acid sequences into the host cell. After infection, novel DNA sequences from the cellular invaders are incorporated into the host CRIPSPR locus as spacers (colored circles) flanked by repeat sequences (gray diamonds). As a result, when the CRISPR locus is transcribed, the pre‐CRISPR RNAs (crRNAs) encode the newly acquired protospacer sequences. The pre‐crRNA is cleaved to produce individual crRNAs that will associate with Cas proteins. The Cas protein utilizes the crRNAs as guides to silence foreign DNA that matches the crRNA sequence (B, interference phase). As a result, the second time a bacteria encounters the same foreign DNA, the crRNA/Cas9 complex is able to identify and silence the DNA.
After the acquisition of spacers, RNA, termed the CRISPR RNA (crRNA), is generated from spacers at the CRISPR locus and loaded onto a Cas protein. crRNA directs the Cas protein to recognize invading sequences and cleave the incoming phage or plasmid DNA (Fig. 1B). Three different types of CRISPR–Cas systems have been identified in bacteria and archea: Type I, Type II, and Type III. Each system utilizes a different mechanism to generate crRNA and Cas proteins that catalyze the nucleic acid cleavage 4. Here we will focus on the Type II CRISPR system, which has been most commonly adapted for genome editing due to its simplicity requiring just one Cas protein, Cas9, and two RNA components. To generate the crRNA, the CRISPR locus is transcribed, generating a long RNA molecule with sequences homologous to past invaders. This RNA molecule is termed the pre‐crRNA (Fig. 1A). A second RNA from a genomic locus upstream of the CRISPR locus is also transcribed. This RNA is called the trans‐activating CRISPR RNA (tracrRNA) 8 (Fig. 1A). The tracrRNA has a region that is complementary to the repeat region of the CRISPR locus, and binds to the newly transcribed pre‐crRNA creating a double‐stranded RNA which gets cleaved by RNaseIII (an enzyme that recognizes and cuts double‐stranded RNA) resulting in a crRNA:tracrRNA complex containing just one spacer sequence (Fig. 1B). This RNA complex then associates with a single Cas9 protein, creating an active ribonucleoprotein (RNP) complex (Fig. 1A).
Once the crRNA:tracrRNA is Cas9 bound, Cas9 is activated and can cleave invading nucleic acid sequences (interference) (Fig. 1B). Cas9 is termed an RNA‐guided endonuclease: it cleaves DNA at sequences that bind to the crRNA of the Cas9 RNP. Searching the invading DNA for sequences complementary to the crRNA occurs through Cas9 binding to sequences in the invading viral or plasmid genome termed Proto‐spacer Adjacent Motifs or PAMs 9, 10. Different Cas9 proteins from different species of bacteria or archea recognize different PAM sites. To date, S. pyogenes Cas9 (SpCas9) which recognizes a 5′‐NGG‐3′ PAM is the most commonly used for genome editing (Fig. 2A). Two critical arginine residues in SpCas9, Arg1333 and Arg1335, interact with the guanine nucleobases of the PAM on the noncomplementary strand 11. This interaction between the guanines of the PAM and the arginines in SpCas9, positions the phosphate of the DNA backbone 5’ to the PAM to interact with a phosphate‐lock loop in Cas9 and facilitate DNA strand unwinding 11. If the DNA is complementary to the guide RNA, an RNA:DNA hybrid forms, called an R loop, and cleavage follows. DNA cleavage results from the action of two different Cas9 nuclease domains: the HNH domain nicks the DNA strand that is complementary to the crRNA and the RuvC‐like domain nicks the strand that is not complementary to the crRNA 10, 12 (Fig. 3A). Cas9 cleaves the DNA 3 base pairs upstream of the PAM, resulting in a blunt‐end cleavage of DNA. Cleaving the DNA is deleterious to the invading plasmid or virus, resulting in degradation and protection against these invaders.
Figure 2.
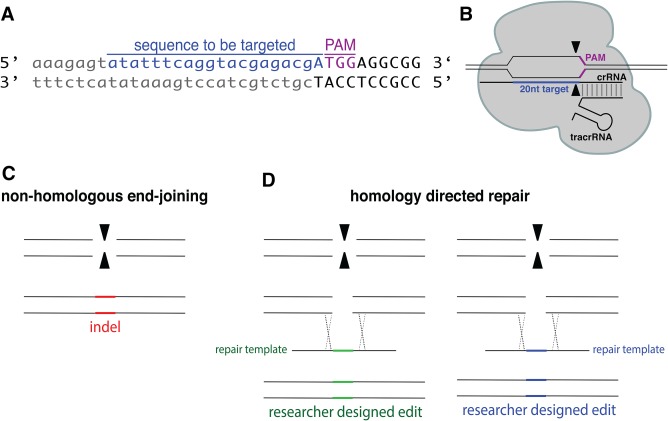
Cas9 induced double‐strand breaks can be repaired by both nonhomologous end‐joining (NHEJ) or homology‐directed repair (HDR). (A) sequence of a targeted genomic locus in relation to the PAM (5′‐NGG‐3′) site. (B) Cartoon representation of crRNA, tracrRNA, and Cas9 protein assembly. (C) NHEJ results in random insertions, deletions, and indels. (D) HDR results in precise researcher‐designed edits. To achieve HDR, the researcher also introduces a repair template that contains the desired edit in which the HDR repair machinery of the cell uses to repair the induced double strand break.
Figure 3.
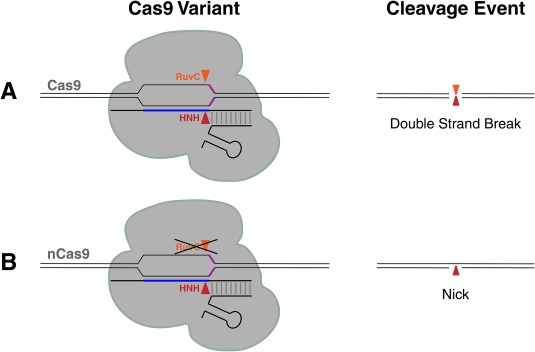
Cas9 has two nuclease domains each cutting a different strand of DNA. (A) Wildtype Cas9 contains two nuclease domains, RuvC and HNH which each cut a different strand of the DNA. When the RuvC nuclease domain is mutated, Cas9 will act as a nickase and produce a nicked DNA product (B).
The Power of Making Programmed Double‐Strand Breaks for Genome Editing in Eukaryotes
After initial characterization of the CRISPR/Cas9 microbial immune system, molecular biologists recognized how it could be exploited for precise genome editing in eukaryotes. In response to Cas9 induced double‐strand breaks, cells employ one of two DNA repair pathways to repair the damage: either through non‐homologous end joining (NHEJ) or homology‐directed repair (HDR) (Fig. 2) 13. NHEJ can occur through canonical NHEJ (C‐NHEJ), which ligates or essentially “glues” the broken ends back together. Additionally, there is an alternative end joining pathway (alt‐NHEJ), in which one strand of the DNA on either side of the break is resected to repair the lesion 14. Both of these repair methods are error‐prone, meaning that the lesion is repaired imperfectly, resulting in insertions or deletions (Fig. 2C). Alternatively, if there is a nearby DNA molecule with homology to the region around the double‐strand break, then the homologous DNA can be used as a template to repair the break through the homology‐directed repair (HDR) pathway. Ordinarily, this repair mechanism happens after DNA replication, but before cell division, so the break can be repaired off the newly replicated sister chromatid without any mutations 15. However, this form of repair can be exploited to introduce precise edits or large insertions or deletions by introducing a donor template for repair (Fig. 2D). Thus, by making a cut at a specific locus and taking advantage of the cellular DNA repair pathways, there is the potential to generate targeted mutations and insert sequences of interest. However, creating double‐strand breaks at precise genomic locations has been challenging due to the difficulty of directing DNA nucleases to specific sequences. Cas9 can easily be targeted by a unique crRNA to cut at any desired site. Since a PAM site is required for Cas9 binding, the target must be upstream of a 5′‐NGG‐3′ site (in the case of SpCas9) (Fig. 2A). Thus, as long as the sequence of your target gene is known, Cas9 can be targeted to almost any site given the presence of a nearby PAM (5′‐NGG‐3′).
To adapt CRISPR for genome editing in eukaryotes, first researchers characterized Cas9 and the role of the crRNA:tracrRNA complex. Through in vitro studies utilizing purified Cas9 to cut a DNA template and either adding or omitting the tracrRNA, researchers found that the tracrRNA is required for cleavage by Cas9 12. Additionally researchers found that the crRNA and tracrRNA could be combined into a single guide RNA or sgRNA 3, 12, 16, limiting the number of components needed to introduce into the cell. Next, three different studies showed that SpCas9 expression with a sgRNA precisely targets Cas9 resulting in a cut at a researcher specified location in the mouse or human genome 1, 2, 3, demonstrating the feasibility of CRISPR/Cas9 as a eukaryotic genome editing tool.
Questions for discussion:
What differences between prokaryotic and eukaryotic cells are important to consider when adapting Cas9 for eukaryotic gene editing?
What components need to be introduced to make a Cas9 induced break in eukaryotic cells?
Are Cas9 proteins found in humans? If so, what is their role? If not, why not?
How Do Researchers Exploit CRISPR for Genome Editing?
CRISPR mediated genome editing combined with the ease of whole genome sequencing has revolutionized genetics. Below we discuss the steps required to generate a desired CRISPR/Cas9 mutation, including (1) target selection, (2) generation and delivery of CRISPR/Cas9 components, and (3) identification of the desired mutation.
Anticipatory guides:
What are the limitations of other reverse genetic techniques?
How can we use CRISPR/Cas9 to create mutations?
What elements are necessary to express heterologous genes (like Cas9) in an organism?
Target and Guide Selection
The first step to generating a desired mutation is guide RNA design. There are many guidelines to consider when creating a guide RNA. Most importantly, the 20‐nucleotide target region of the guide RNA must be adjacent to a PAM site, 5′‐NGG‐3′ in the case of SpCas9. Therefore, one must identify the genomic region where a desired mutation is to be generated and select a 20‐nucleotide target in that region that is adjacent to a PAM site (Fig. 2A). For best results, a PAM site should be as close to the location of the desired mutation as possible. In the worm C. elegans, edits have been reported up to 50 bp from the PAM site, however efficiency for inducing a desired mutation or edit is inversely correlated to the number of base pairs from a PAM site 17. To facilitate guide RNA design, CRISPR design tools, such as http://crispor.tefor.net/18, scan the specificity of a target sequence to minimize off‐target effects. If target sequences are not specific enough, Cas9 can bind and cut in a different place than intended and result in background mutations that could confound experimental results. Additionally, guide RNA sequences can have very different efficiencies. Although it is not completely understood what affects guide RNA efficiency, and this is an active area of research in CRISPR biology, numerous studies have helped to establish characteristics of effective guide sequences 19, 20, 21, 22, including the presence of a purine (G or A) at the 3′ end of the 20‐nucleotide target 23, 24, 25.
Generation and Delivery of Components
Once optimal guide RNAs have been designed, Cas9 and sgRNAs can be introduced using three different strategies: The sgRNA or crRNA and tracrRNA and Cas9 can be expressed as DNA, RNA, or RNA/protein complexes (Fig. 4). The nucleic acid and/or protein can be introduced using microinjection (worms, fruit flies, and zebrafish) or electroporation or transfection (mammalian cell culture). For all methods described below, a single‐stranded or double‐stranded DNA can be included as a HDR template to generate a researcher‐designed edit (Fig. 2D).
Figure 4.
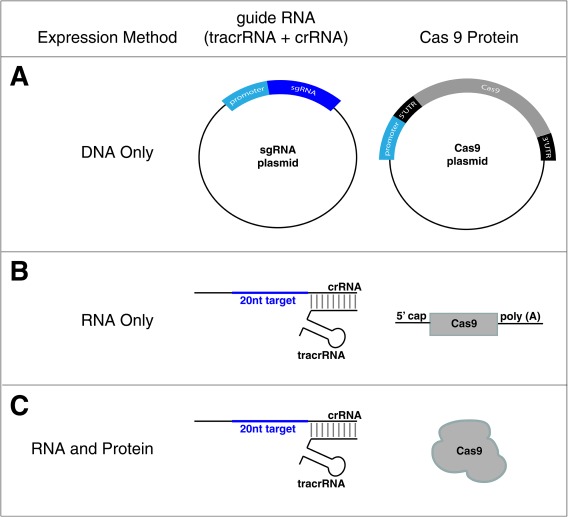
Different methods to introduce CRISPR/Cas9 components. CRISPR guides and the Cas9 protein required for genome editing can be introduced into organisms or cells both as DNA plasmids (A), both as RNA molecules (B), or RNA and Protein complexes (RNPs) (C).
DNA
To express from DNA, two plasmids are introduced: one encoding the sgRNA and one encoding the Cas9 protein 26, 27, 28 (Fig. 4A). The sgRNA (tracrRNA:crRNA hybrid), is used for simplicity so that two plasmids (tracrRNA:crRNA hybrid + Cas9), rather than three (tracrRNA + crRNA + Cas9), are required. Cas9 and sgRNA plasmids must be designed to ensure proper expression in the targeted eukaryotic cell. First, the Cas9 coding sequence is codon optimized for efficient expression in different organisms 26, 28, 29. Thus, by changing the native sequence of Cas9 (how it is normally encoded in the S. pyogenes genome) to one that uses codons most commonly used in that organism being edited, translation can be greatly improved. Additionally, appropriate 5′ and 3′ untranslated regions (UTR) must be included for efficient translation of Cas9 mRNA to protein. Lastly, when generating the DNA construct, a promoter, which is a sequence directly upstream of the transcription start site of the gene and recruits RNA polymerase to initiate transcription, needs to also be included to transcribe the encoded protein. The sgRNA must also be expressed from an appropriate promoter. Unlike Cas9, the sgRNA gene's ultimate product is RNA, not protein. Thus, the sgRNA is transcribed by RNA polymerase III, which is a polymerase specialized for the expression of ribosomal RNAs and tRNAs.
RNA and RNA/Protein (RNP) Complexes
Given that the introduction of DNA constructs requires the transcription of sgRNA and transcription and translation of Cas9 within the organism, researchers can lessen the in vivo burden by introducing mRNAs encoding Cas9 and sgRNAs (Fig. 4B) 30. To ensure appropriate expression of Cas9, in vitro‐synthesized mRNA needs to be post‐transcriptionally modified with a 5′ cap and 3′ Poly‐A tail (Fig. 4B). Alternatively, the Cas9 protein can be expressed and purified then complexed with the crRNA and tracrRNA, to be introduced into the cell as an RNA/protein (RNP) complex (Fig. 4C) 31, 32, 33. This method may be most ideal as it eliminates the need to optimize species‐specific promoters and UTRs. The main drawback of introduction by RNPs is that purifying the Cas9 protein and ordering RNA is costlier. Thus, depending on the desired edit and the reagents present on hand, injecting different DNA, RNA, or RNP protein complexes can be the most efficient and cost‐effective way to generate a desired edit.
Identification of Desired Mutation
Lastly, a researcher must identify a successful genome edit. Genome editing, including CRISPR/Cas9, is nowhere near 100% efficient creating a potential “needle in the haystack” problem. How does a researcher identify the individual or cell that harbors a successful edit from the many progeny of the injected animal or the many electroporated cells? Two different genetic strategies can be used to identify desired mutants: screens and selections. Screens effectively make the “needle” easier to identify in the haystack and selections reduce the size of the haystack to make “needle” identification easier. Both methods ease the burden of successful genome‐edit identification, a significant bottleneck in the CRISPR workflow.
A genetic screen examines many individuals to identify a successful edit. If there is a known phenotype associated with the desired mutation made, the progeny or cells can be screened directly for the associated phenotype. For example, when GFP is inserted utilizing HDR to tag a protein of interest, organisms or cells can be screened directly for fluorescent expression 34. Alternatively, rather than screening for a desired phenotype, one can screen for the molecular lesion generated by the genome edit. These methods are especially useful when NHEJ results in a small insertion or deletion. One such molecular assay for detecting molecular lesions is a restriction fragment length polymorphisms (RFLP) analysis, which takes advantage of differences in the presence of restriction sites between the wild‐type DNA and the mutation generated through CRISPR/Cas9 editing 35. The region of interest that was targeted for a mutation is PCR amplified and then digested with an enzyme called a restriction endonuclease, which cuts at a specific DNA sequence. If the enzyme is able to cut, then no mutation was generated and the wild‐type sequence is still present, which can be visualized by gel electrophoresis. However, if the PCR product is not cut, then a successful edit was generated. This assay also works in the opposite direction: mutations generated by CRISPR can create a restriction site. Alternatively, different endonuclease‐based assays such as CEL1 36 and bacterial T7 endonuclease 37 recognize and cleave single base mismatches, insertions, or deletions. These endonucleases are similar to RFLP analysis, but have the advantage that they do not rely on the creation or destruction of a restriction site. Unfortunately, both of these molecular screening approaches may require screening hundreds of candidates, a time‐consuming process.
Alternatively, a genetic selection could reduce the screening burden. Selection strategies utilize lethal mutations so that only the individuals of interest survive a particular condition. One selection strategy utilizes a specialized template for repair, which introduces a gene for drug resistance into the animal or cell 26. This drug resistance reporter gene provides the cell or organism resistance to that drug. The organisms or cells are then exposed to the drug after introduction of Cas9, the sgRNA, and the HDR template, which is normally toxic and kills the cells or organism, and only the individuals that successfully integrated the drug‐resistance marker gene at the desired locus through HDR of the Cas9 induced break survive and no molecular screening is required.
Generation of CRISPR/Cas9 Mutation in C. elegans: A Case Study
Above, we describe generally how CRISPR and Cas9 can be introduced and expressed to generate a mutation in the genome. Here we give an example of how CRISPR/Cas9 has been adapted in the model organism nematode worm, C. elegans to generate heritable genomic edits. CRISPR/Cas9 components are introduced into C. elegans through microinjection into the gonads of young adult hermaphrodites. Microinjection is an effective way to introduce exogenous DNA, RNA, and/or protein into C. elegans due to the large ratio of gonads as compared to C. elegans body size. Briefly, C. elegans are immobilized on a slide under a microscope and using a very thin and sharp glass capillary, small amounts of liquid can be injected into the worm's gonad by applying a burst of pressure through the needle. For further reading and visualization of the injection process see 38 and 39. When injecting DNA, special consideration must be taken to ensure that Cas9 is expressed as the correct time and tissue in C. elegans. To effectively generate mutations in the germ cells that can be passed onto the next generation (heritable mutations), Cas9 must be expressed in the gonad while the germ cells are developing. For transcripts to be translated at the correct time in development, C. elegans relies heavily on the 5′ and 3′ untranslated regions (UTR) of an mRNA for efficient translation in the appropriate tissue. Thus, 5′ and 3′ UTRs that are permissive to translation in the germ line were added to the Cas9 DNA expression 26, 27, 28. Different research groups have expressed Cas9 from different promoters in C. elegans to ensure expression in the correct tissue or in response to different stimuli. In C. elegans, some constructs utilize a promoter that expresses in tissues throughout the organism, the eft‐3 promoter 26, 28. Alternatively, other constructs expressed Cas9 from the C. elegans heat shock promoter 27. The heat shock promoter is an inducible promoter: under normal conditions the gene is not transcribed, however in response to elevated temperature the promoter is turned on and the gene is expressed. Thus, this inducible promoter allows the added advantage of only expressing Cas9 when the worms are transferred to elevated temperature. Reducing the duration that Cas9 is active reduces the potential for embryonic lethality that has been observed with the eft‐3 promoter, potentially due to off‐target cleavage by the continual expression of Cas9. The sgRNA must also be expressed from an appropriate promoter in its DNA expression construct. Like in other organisms, the sgRNA is transcribed by RNA polymerase III, which is a polymerase specialized for the expression of ribosomal RNAs and tRNAs (genes whose ultimate product are RNAs in the cell). The sgRNA is expressed from the U6 small RNA promoter, which is transcribed by RNA polymerase III 26, 27, 28.
Questions for discussion:
How would you determine and what strategies could you use to ensure that no off‐target background mutations occurred when editing with Cas9?
What advantages and disadvantages are there to each of the different ways of introducing Cas9 and sgRNA?
What type of desired mutations require a donor template to also be injected in addition to the sgRNA and Cas9?
Variations on a Theme: Alternatives to Cas9 and Novel Applications
Not only can CRISPR be used to make double‐strand breaks near a 5′‐NGG‐3′ PAM, other studies have identified different uses for Cas9 highlighted below.
Anticipatory guides:
Are there possible limitations of the CRISPR/Cas9 as it has been described so far?
What other uses could there be for targeting a protein to a specific region of the genome?
What are mechanisms that regulate gene expression at the different levels of the Central Dogma?
One reservation researchers have regarding Cas9 is the enzyme's specificity. To be an effective therapeutic, Cas9 must make precise edits without making unintended cuts. One strategy to increase Cas9 specificity is to pair two different Cas9 enzymes such that each enzyme cuts just one strand of DNA. This can be easily achieved since Cas9 has two distinct catalytic domains to cut each strand of DNA (Fig. 3A). By mutating the RuvC catalytic domain with a single amino acid mutation that inactivates this domain, termed D10A, then only the HNH catalytic domain cuts just the one strand of the DNA that is complementary to the sgRNA 12. This variant is called nickase Cas9 or nCas9 (Fig. 3B). A double‐strand break can be generated by targeting two nCas9s to adjacent PAMs at a desired locus with two different sgRNAs. Thus, the double‐strand break only occurs if both Cas9 nicking events happen, greatly improving specificity 40, 41, 42.
Additional Cas nucleases from bacteria and archaea that recognize alternative PAMs have been explored 43. One such promising enzyme is Cpf1 44. Cpf1 recognizes a T‐rich PAM, making it useful in regions of desired edits that are AT‐rich. Cpf1 also only requires a crRNA, and does not require a tracrRNA, for binding and cleavage, possibly simplifying editing. Unlike Cas9 that cuts both strands of DNA at the same location, Cpf1 generates a cut with a 5′ overhang, creating “sticky” ends that could be exploited to insert a sequence of interest through complementation and ligation. The continued investigation and characterization of microbial immune system promises to elucidate other potential editing enzymes.
Two key features are pivotal to Cas9's utility: it can generate double‐stranded breaks and the double‐stranded break location is determined by guide RNAs. This second feature has not gone unnoticed in the molecular biology community. The ability to specifically target a protein to a region of interest in the genome is a very valuable asset. Mutating each of the catalytic domains of Cas9 results in a protein (dCas9) that can be targeted to a desired site in the genome by the sgRNA without cutting the DNA 12 (Fig. 5). By tethering other proteins to dCas9, researchers can target specific molecular activities to genomic regions of interest. For example, by targeting dCas9 fused to a protein domain that represses transcription to the open reading frame or promoter of a gene through the sgRNA, this chimeric protein effectively silences targeted genes (Fig. 5A) 45. Alternatively, by fusing a transcriptional activator, a protein that recruits RNA polymerase, to dCas9 and targeting this construct to the promoter of a desired gene through the sgRNA, transcription could be activated (Fig. 5B) 45. Utilizing these same principles of dCas9 fused to a protein, dCas9 has also been used to direct enzymatic activity to a specific genomic location (Fig. 5C). Epigenetic modifiers can be targeted to a genomic site of interest by attaching proteins, such as DNA methylating or histone modifying enzymes 46, 47, 48, 49, modulating transcription at a desired genomic location. dCas9 can also be used to visualize where a specific genomic sequence is in the nucleus. By attaching a fluorescent protein to dCas9, such as the Green Fluorescent Protein (GFP) from jellyfish 50, the location in the nucleus of a genomic sequence complementary to the expressed sgRNA can be identified in live cells (Fig. 5D) 51. By recruiting proteins and enzymes to desired locations, dCas9 has provided new opportunities to interrogate the molecular biology of the nucleus in living cells.
Figure 5.
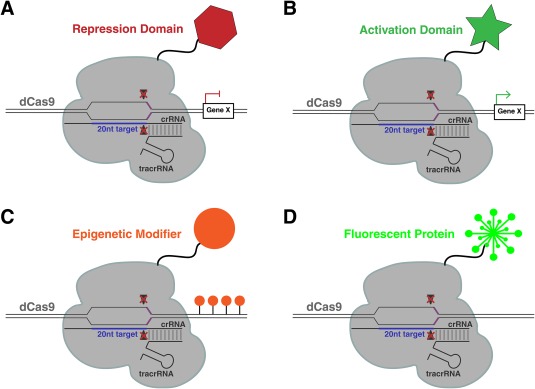
dCas9 can bring different enzymatic activities to precise genomic locations. Through attaching different protein domains to dCas9, this modified version of the CRSISPR/Cas9 RNP can be used to repress gene expression (A), activate gene expression (B), add epigenetic modifiers (C), or tag DNA sequences with a fluorescent protein (D).
Questions for discussion:
What are other uses you could think of for the dCas9 protein?
What are the differences in repressing a gene with CRISPRi versus inactivating a gene product by making a mutation with CRISPR gene editing?
What are possible limitations of Cas9 targeting methods?
Ethical Implications for CRISPR/Cas9 Genome Editing In Humans
Anticipatory guides:
Should CRISPR/Cas9 be used to edit the human genome?
What is a genetically modified organism?
Are CRISPR/Cas9 edited organisms genetically modified organisms?
In a brief time, CRISPR/Cas9 has been modified to create different mutations in a variety of organisms including humans (Supporting Information Fig. S1). As a result, this technology has the potential to treat and cure diseases by editing the DNA associated with a particular disease before a baby is born. For example, Junjiu Huang and colleagues at the Sun Yat‐Sen University in China, demonstrated for the first time that CRISPR/Cas9 can be used to edit the genome of pre‐implantation embryos 52. Huang's group modified the gene responsible for ß‐thalassaemia, a blood disorder resulting in anemia. While this experiment was ground‐breaking and the first step towards a world where scientists could eradicate many detrimental diseases, the editing of human embryos that can be implanted in a uterus raises numerous issues and concerns about the ethics of human genome editing. To avoid some of these issues, the Huang group used “non‐viable triploid embryos.” These embryos were inviable because they were the result of two sperm fertilizing one egg. Due to the ethical concerns, many scientific journals declined to publish their work. An international summit was organized in response to these concerns. As part of the National Academy of Sciences and the National Academy of Medicine's Human Gene‐Editing Initiative, a summit was co‐hosted by the Chinese academy of Sciences and the U.K.'s Royal Society, taking place in Washington, D.C. on December 1–3, 2015. Experts from all around the world gathered to discuss the ethical, scientific, and regulatory issues associated with human genome‐editing research and practice. For example, in less than two years since the summit, a group of scientists working together from all over the world demonstrated that CRISPR/Cas9 can be used to edit viable embryos. Under significant oversight by ethics and regulatory committees, Ma et al. 53, successfully edited embryos that were heterozygous for a mutation in the MYBPC3 gene using CRISPR/Cas9. Mutations in MYBPC3 result in hypertrophic cardiomyopathy, which can cause sudden death. These experiments demonstrate both the power of CRISPR/Cas9 as a gene‐editing technology and the necessity for scientists to continually evaluate and discuss ethical implications as genome‐editing becomes more accessible.
Questions for discussion:
What are the ethical and social ramifications of allowing genome‐editing in embryos?
How do you think CRISPR/Cas9 should be regulated?
What diseases can most likely be treated with genome editing?
Supporting information
Supporting Information
Supporting Information
Acknowledgments
We thank Rachid El Bejjani for helpful discussions and Bryan Thurtle‐Schmidt for critical reading of the manuscript. D.T‐S. thanks Keith Yamamoto for support. Funding is provided by The Life Sciences Research Foundation and the Gordon and Betty Moore Foundation to D.T‐S.
References
- 1. Jinek, M. , East, A. , Cheng, A. , Lin, S. , Ma, E. , Doudna, J. A. (2013) RNA‐programmed genome editing in human cells. Elife 2, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cong, L. , Ran, F. A. , Cox, D. , Lin, S. , Barretto, R. , Habib, N. , Hsu, P. D. , Wu, X. , Jiang, W. , Marraffini, L. A. , Zhang, F. (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mali, P. , Yang, L. , Esvelt, K. M. , Aach, J. , Guell, M. , DiCarlo, J. E. , Norville, J. E. , Church, G. M. (2013) RNA‐guided human genome engineering via Cas9. Science 339, 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bhaya, D. , Davison, M. , Barrangou, R. (2011) CRISPR–Cas systems in bacteria and archaea: Versatile small RNAs for adaptive defense and regulation. Annu. Rev. Genet. 45, 273–297. [DOI] [PubMed] [Google Scholar]
- 5. Jansen, R. , Van Embden, J. D. A. , Gaastra, W. , Schouls, L. M. (2002) Identification of genes that are associated with DNA repeats in prokaryotes. Mol. Microbiol. 43, 1565–1575. [DOI] [PubMed] [Google Scholar]
- 6. Bolotin, A. , Quinquis, B. , Sorokin, A. , Dusko, S. (2005) Ehrlich clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 151, 2551–2561. [DOI] [PubMed] [Google Scholar]
- 7. Barrangou, R. , Fremaux, C. , Deveau, H. , Richards, M. , Boyaval, P. , Moineau, S. , Romero, D. A. , Horvath, P. (2007) CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712. [DOI] [PubMed] [Google Scholar]
- 8. Deltcheva, E. , Chylinski, K. , Sharma, C. M. , Gonzales, K. , Chao, Y. , Pirzada, Z. A. , Eckert, M. R. , Vogel, J. , Charpentier, E. (2011) CRISPR RNA maturation by trans‐encoded small RNA and host factor RNase III. Nature 471, 602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sternberg, S. H. , Redding, S. , Jinek, M. , Greene, E. C. , Doudna, J. A. (2014) DNA interrogation by the CRISPR RNA‐guided endonuclease Cas9. Nature 507, 62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gasiunas, G. , Barrangou, R. , Horvath, P. , Siksnys, V. (2012) Cas9–crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 109, E2579–E2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Anders, C. , Niewoehner, O. , Duerst, A. , Jinek, M. (2014) Structural basis of PAM‐dependent target DNA recognition by the Cas9 endonuclease. Nature 513, 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jinek, M. , Chylinski, K. , Fonfara, I. , Hauer, M. , Doudna, J. A. , Charpentier, E. (2012) A programmable dual‐RNA‐guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wyman, C. and Kanaar, R. (2006) DNA double‐strand break repair: All's well that ends well. Annu. Rev. Genet. 40, 363–383. [DOI] [PubMed] [Google Scholar]
- 14. Bétermier, M. , Bertrand, P. , Lopez, B. S. , Jinks‐Robertson, S. (2014) Is non‐homologous end‐joining really an inherently error‐prone process? PLoS Genet. 10, e1004086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cromie, G. A. , Connelly, J. C. , Leach, D. R. F. (2001) Recombination at double‐strand breaks and DNA ends: Conserved mechanisms from phage to humans. Mol. Cell 8, 1163–1174. [DOI] [PubMed] [Google Scholar]
- 16. Hwang, W. Y. , Fu, Y. , Reyon, D. , Maeder, M. L. , Tsai, S. Q. , Sander, J. D. , Peterson, R. T. , Yeh, J.‐R. J. , Joung, J. K. (2013) Efficient genome editing in zebrafish using a CRISPR–Cas system. Nat. Biotechnol. 31, 227–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ward, J. D. (2015) Rapid and precise engineering of the Caenorhabditis elegans genome with lethal mutation co‐conversion and inactivation of NHEJ repair. Genetics 199, 363–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Haeussler, M. , Schönig, K. , Eckert, H. , Eschstruth, A. , Mianné, J. , Renaud, J.‐B. , Schneider‐Maunoury, S. , Shkumatava, A. , Teboul, L. , Kent, J. , Joly, J.‐S. , Concordet, J.‐P. (2016) Evaluation of off‐target and on‐target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol 17, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Doench, J. G. , Fusi, N. , Sullender, M. , Hegde, M. , Vaimberg, E. W. , Donovan, K. F. , Smith, I. , Tothova, Z. , Wilen, C. , Orchard, R. , Virgin, H. W. , Listgarten, J. , Root, D. E. (2016) Optimized sgRNA design to maximize activity and minimize off‐target effects of CRISPR–Cas9. Nat. Biotechnol. 34, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Doench, J. G. , Hartenian, E. , Graham, D. B. , Tothova, Z. , Hegde, M. , Smith, I. , Sullender, M. , Ebert, B. L. , Xavier, R. J. , Root, D. E. (2014) Rational design of highly active sgRNAs for CRISPR–Cas9–mediated gene inactivation. Nat. Biotechnol. 32, 1262–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wu, X. , Scott, D. A. , Kriz, A. J. , Chiu, A. C. , Hsu, P. D. , Dadon, D. B. , Cheng, A. W. , Trevino, A. E. , Konermann, S. , Chen, S. , Jaenisch, R. , Zhang, F. , Sharp, P. A. (2014) Genome‐wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat. Biotechnol. 32, 670–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang, T. , Wei, J. J. , Sabatini, D. M. , Lander, E. S. (2014) Genetic screens in human cells using the CRISPR–Cas9 system. Science 343, 80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Farboud, B. and Meyer, B. J. (2015) Dramatic enhancement of genome editing by CRISPR/Cas9 through improved guide RNA design. Genetics 199, 959–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gagnon, J. A. , Valen, E. , Thyme, S. B. , Huang, P. , Ahkmetova, L. , Pauli, A. , Montague, T. G. , Zimmerman, S. , Richter, C. , Schier, A. F. , Riley, B. (2014) Efficient mutagenesis by Cas9 protein‐mediated oligonucleotide insertion and large‐scale assessment of single‐guide RNAs. PLoS One 9, 5–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ren, X. , Yang, Z. , Xu, J. , Sun, J. , Mao, D. , Hu, Y. , Yang, S.‐J. , Qiao, H.‐H. , Wang, X. , Hu, Q. , Deng, P. , Liu, L.‐P. , Ji, J.‐Y. , Li, J. B. , Ni, J.‐Q. (2014) Enhanced specificity and efficiency of the CRISPR/Cas9 system with optimized sgRNA parameters in Drosophila . Cell Rep. 9, 1151–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dickinson, D. J. , Ward, J. D. , Reiner, D. J. , Goldstein, B. (2013) Engineering the Caenorhabditis elegans genome using Cas9‐triggered homologous recombination. Nat. Methods 10, 1028–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Waaijers, S. , Portegijs, V. , Kerver, J. , Lemmens, B. B. L. G. , Tijsterman, M. , van den Heuvel, S. , Boxem, M. (2013) CRISPR/Cas9‐targeted mutagenesis in Caenorhabditis elegans . Genetics 195, 1187–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Friedland, A. E. , Tzur, Y. B. , Esvelt, K. M. , Colaiácovo, M. P. , Church, G. M. , a Calarco, J. (2013) Heritable genome editing in C. elegans via a CRISPR–Cas9 system. Nat. Methods 10, 741–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shen, B. , Zhang, J. , Wu, H. , Wang, J. , Ma, K. , Li, Z. , Zhang, X. , Zhang, P. , Huang, X. (2013) Generation of gene‐modified mice via Cas9/RNA‐mediated gene targeting. Cell Res. 23, 720–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lo, T.‐W. , Pickle, C. S. , Lin, S. , Ralston, E. J. , Gurling, M. , Schartner, C. M. , Bian, Q. , Doudna, J. A. , Meyer, B. J. (2013) Precise and heritable genome editing in evolutionarily diverse nematodes using TALENs and CRISPR/Cas9 to engineer insertions and deletions. Genetics 195, 331–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lin, S. , Staahl, B. T. , Alla, R. K. , Doudna, J. A. (2014) Enhanced homology‐directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife 3, e04766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liang, X. , Potter, J. , Kumar, S. , Zou, Y. , Quintanilla, R. , Sridharan, M. , Carte, J. , Chen, W. , Roark, N. , Ranganathan, S. , Ravinder, N. , Chesnut, J. D. (2015) Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J. Biotechnol. 208, 44–53. [DOI] [PubMed] [Google Scholar]
- 33. Aida, T. , Chiyo, K. , Usami, T. , Ishikubo, H. , Imahashi, R. , Wada, Y. , Tanaka, K. F. , Sakuma, T. , Yamamoto, T. , Tanaka, K. (2015) Cloning‐free CRISPR/Cas system facilitates functional cassette knock‐in in mice. Genome Biol. 16, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Paix, A. , Wang, Y. , Smith, H. E. , Lee, C.‐Y. S. , Calidas, D. , Lu, T. , Smith, J. , Schmidt, H. , Krause, M. W. , Seydoux, G. (2014) Scalable and versatile genome editing using linear DNAs with microhomology to Cas9 sites in Caenorhabditis elegans . Genetics 198, 1347–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim, J. M. , Kim, D. , Kim, S. , Kim, J.‐S. (2014) Genotyping with CRISPR–Cas‐derived RNA‐guided endonucleases. Nat. Commun. 5, 1–7. [DOI] [PubMed] [Google Scholar]
- 36. Yang, B. , Wen, X. , Kodali, N. S. , Oleykowski, C. A. , Miller, C. G. , Kulinski, J. , Besack, D. , Yeung, J. A. , Kowalski, D. , Yeung, A. T. (2000) Purification, cloning, and characterization of the CEL I nuclease. Biochemistry 39, 3533–3541. [DOI] [PubMed] [Google Scholar]
- 37. Vouillot, L. , Thelie, A. , Pollet, N. (2015) Comparison of T7E1 and surveyor mismatch cleavage assays to detect mutations triggered by engineered nucleases. G3 (Bethesda) 5, 407–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Evans, T. (2006) Transformation and microinjection. WormBook 1–15.
- 39. a Berkowitz, L. , Knight, A. L. , a Caldwell, G. , a Caldwell, K. (2008) Generation of stable transgenic C. elegans using microinjection. J. Vis. Exp 4–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mali, P. , Aach, J. , Stranges, P. B. , Esvelt, K. M. , Moosburner, M. , Kosuri, S. , Yang, L. , Church, G. M. (2013) CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 31, 833–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shen, B. , Zhang, W. , Zhang, J. , Zhou, J. , Wang, J. , Chen, L. , Wang, L. , Hodgkins, A. , Iyer, V. , Huang, X. , Skarnes, W. C. (2014) Efficient genome modification by CRISPR–Cas9 nickase with minimal off‐target effects. Nat. Methods 11, 399–402. [DOI] [PubMed] [Google Scholar]
- 42. Ran, F. A. , Hsu, P. D. , Lin, C.‐Y. , Gootenberg, J. S. , Konermann, S. , Trevino, A. E. , Scott, D. A. , Inoue, A. , Matoba, S. , Zhang, Y. , Zhang, F. (2013) Double nicking by RNA‐guided CRISPR cas9 for enhanced genome editing specificity. Cell 154, 1380–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shmakov, S. , Abudayyeh, O. O. , Makarova, K. S. , Wolf, Y. I. , Gootenberg, J. S. , Semenova, E. , et al. (2015) Discovery and functional characterization of diverse class 2 CRISPR–Cas systems. Mol. Cell 60, 385–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zetsche, B. , Gootenberg, J. S. , Abudayyeh, O. O. , Slaymaker, I. M. , Makarova, K. S. , Essletzbichler, P. , et al. (2015) Cpf1 Is a single RNA‐guided endonuclease of a class 2 CRISPR–Cas system. Cell 163, 759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gilbert, L. A. , Larson, M. H. , Morsut, L. , Liu, Z. , Brar, G. A. , Torres, S. E. , et al. (2013) CRISPR–mediated modular RNA‐guided regulation of transcription in eukaryotes. Cell 154, 442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. McDonald, J. I. , Celik, H. , Rois, L. E. , Fishberger, G. , Fowler, T. , Rees, R. , et al. (2016) Reprogrammable CRISPR/Cas9‐based system for inducing site‐specific DNA methylation. Biol. Open 5, 866–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xu, X. , Tao, Y. , Gao, X. , Zhang, L. , Li, X. , Zou, W. , et al. (2016) A CRISPR‐based approach for targeted DNA demethylation. Cell Discov. 2, 16009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hilton, I. B. , D'Ippolito, A. M. , Vockley, C. M. , Thakore, P. I. , Crawford, G. E. , Reddy, T. E. , et al. (2015) Epigenome editing by a CRISPR–Cas9‐based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 33, 510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Thakore, P. I. , D'Ippolito, A. M. , Song, L. , Safi, A. , Shivakumar, N. K. , Kabadi, A. M. , et al. (2015) Highly specific epigenome editing by CRISPR–Cas9 repressors for silencing of distal regulatory elements. Nat. Methods 12, 1143–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Heim, R. , Prasher, D. C. , Tsien, R. Y. (1994) Wavelength mutations and posttranslational autoxidation of green fluorescent protein. Proc. Natl. Acad. Sci. 91, 12501–12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen, B. , Gilbert, L. A. , Cimini, B. A. , Schnitzbauer, J. , Zhang, W. , Li, G.‐W. , et al. (2013) Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 155, 1479–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liang, P. , Xu, Y. , Zhang, X. , Ding, C. , Huang, Z. , et al. (2015) CRISPR/Cas9‐mediated gene editing in human tripronuclear zygotes. Protein Cell 6, 363–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ma, H. , Marti‐Gutierrez, N. , Park, S.‐W. , Wu, J. , Lee, Y. , Suzuki, K. , et al. (2017) Correction of a pathogenic gene mutation in human embryos. Nature 548, 413–419. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information