

Abstract
Ozanimod is a novel, selective, oral sphingosine‐1‐phosphate (1 and 5) receptor modulator in development for multiple sclerosis and inflammatory bowel disease. This randomized, double‐blind, placebo‐controlled, positive‐controlled, parallel‐group thorough QT study characterized the effects of ozanimod on cardiac repolarization in healthy subjects. Eligible subjects were randomized to 1 of 2 groups: ozanimod (escalated from 0.25 to 2 mg over 14 days) or placebo (for 14 days). A single dose of moxifloxacin 400 mg or placebo was administered on days 2 and 17. The primary end point was the time‐matched, placebo‐corrected, baseline‐adjusted mean QTcF (ΔΔQTcF). A total of 113/124 (91.1%) subjects completed the study. The upper limits of the 2‐sided 90% confidence intervals for ΔΔQTcF for both ozanimod 1 and 2 mg were below the 10‐millisecond regulatory threshold. No QTcF >480 milliseconds or postdose change in QTcF of >60 milliseconds was observed. There was no evidence of a positive relationship between concentrations of ozanimod and its active metabolites and ΔΔQTcF. Although ozanimod blunted the observed diurnal increase in heart rate, excursions below predose heart rates were no greater than with placebo. Results demonstrate that ozanimod does not prolong the QTc interval or cause clinically significant bradycardia, supporting ozanimod's evolving favorable cardiac safety profile.
Keywords: cardiac repolarization, inflammatory bowel disease, multiple sclerosis, ozanimod, thorough QT/QTc study
Ozanimod is a novel, orally bioavailable bi‐aryl oxadiazol that selectively and potently activates sphingosine‐1‐phosphate 1 and 5 receptors (S1P1R, S1P5R).1 It is an investigational drug currently in phase 3 clinical development for the treatment of relapsing multiple sclerosis (RMS) and moderate‐to‐severe ulcerative colitis and in phase 2 clinical testing for moderate to severe Crohn disease. Results of a 24‐week phase 2 study of patients with RMS demonstrated that, compared with placebo, once‐daily (QD) oral doses of ozanimod 0.5 mg and 1 mg resulted in significantly fewer gadolinium‐enhancing T1 lesions and new or enlarging T2 lesions detected by magnetic resonance imaging.2 In addition, a phase 2 trial of ozanimod in adults with moderate to severe active ulcerative colitis demonstrated that ozanimod 1 mg/day resulted in a significantly higher proportion of patients in clinical remission compared with placebo; this significance was achieved at week 8 and maintained through week 32 of treatment.3
Ozanimod acts as a functional antagonist of the S1P1R by promoting receptor internalization and degradation, resulting in a reduction of the number of circulating lymphocytes.4, 5 The functional antagonism at S1P1R on lymphocytes renders them insensitive to the S1P signal necessary for release from peripheral lymphoid organs, preventing them from contributing to autoimmune‐mediated inflammation.6 Ozanimod also demonstrates activity, albeit lower, at the S1P5R, which supports oligodendrocyte progenitor process extension and survival7 and contributes to blood‐brain barrier integrity.8
In addition to regulating lymphocyte movement, S1P receptors are important regulators of vascular tone, heart rate (HR), and cardiac repolarization.9 Fingolimod, a nonselective S1P1,3,4,5R agonist approved for treatment of RMS, can cause transient first‐ and second‐degree atrioventricular (AV) blocks10, 11 and prolongation of the corrected QT interval (QTc).12 It has been postulated that this QTc effect may occur via activation of S1P3R expressed in cells of the His‐Purkinje fibers.13 The potential for cardiac adverse events complicates the initiation of treatment with fingolimod and requires cardiac monitoring for at least 6 hours after the first dose. Bradycardia and first‐ and second‐degree AV conduction blocks also have been reported for other S1P receptor agonists in clinical development for multiple sclerosis14, 15, 16, 17, 18, 19 as well as prolongation of the QTc interval.20 This emphasizes the ongoing need to develop therapeutic agents in this class with a more favorable cardiac safety profile. In comparison to fingolimod, ozanimod does not have affinity for the S1P3R, which may signify important pharmacodynamic differences between these 2 compounds that impact cardiac safety and the effect on the QTc interval.
Ozanimod pharmacokinetics (PK) was characterized in healthy volunteers for single oral doses up to 3 mg and multiple oral doses up to 2 mg QD.21 Following oral administration, the median time to maximum concentration (Tmax) was approximately 8.0 hours. Ozanimod exhibited linear PK, with a dose‐proportional increase in exposure and low to moderate intersubject variability; a high steady‐state volume of distribution; a moderate apparent oral clearance; and an elimination half‐life of 17 to 21 hours.21 Ozanimod is eliminated primarily via biotransformation followed by biliary excretion.22 Metabolism studies in animals identified 3 pharmacologically active metabolites (RP101988, RP101075, and RP101442) that have similar S1P selectivity and potency in vitro to ozanimod.22, 23 The mean half‐maximal effective concentrations toward S1P1R for ozanimod, RP101988, RP101075, and RP101442 are 0.44, 0.19, 0.27, and 2.6 nM, respectively, and those toward S1P5R for ozanimod, RP101988, RP101075, and RP101442 are 11.1, 32.8, 5.9, and 171 nM, respectively. The chemical structures for ozanimod and its active metabolites are shown in Figure 1.
Figure 1.
Chemical structures of ozanimod and its active metabolites.
A dose‐dependent, negative chronotropic effect has been reported for S1P receptor modulators. However, this negative chronotropic effect may attenuate over time secondary to S1P desensitization on atrial myocytes.24 In the ozanimod first‐in‐human, single‐ascending‐dose and multiple‐ascending‐dose studies, a transient, dose‐dependent decrease in HR was observed; however, introducing a gradual dose escalation of ozanimod over several days helped to mitigate against larger reductions in HR.22 The use of a dose‐escalation regimen has been carried forward into ozanimod phase 2 and 3 clinical trials.2, 3
This cardiac safety study was conducted to examine the effects of therapeutic and supratherapeutic doses of ozanimod on cardiac repolarization and HR according to the E14 Guidance of the International Conference on Harmonisation.25 The primary objective was to assess whether exposure to ozanimod 1 mg or 2 mg in healthy subjects increased the QTc interval compared with placebo. The secondary objectives were to evaluate the PK of ozanimod and its active metabolites, to assess the safety and tolerability of ozanimod, and to demonstrate assay sensitivity using a positive control (moxifloxacin). An additional study objective was to monitor HR during the dose‐escalation period.
Methods
Study Design and Subjects
The study was approved by an institutional review board (IntegReview, Austin, Texas) and was conducted in accordance with principles set forth in the Declaration of Helsinki and all applicable regulatory requirements. All subjects provided written informed consent before participating in any study procedure. The study was conducted at ICON Development Solutions LLC (San Antonio, Texas) between October 2012 and March 2013.
This phase 1, single‐center, randomized, double‐blind, placebo‐controlled, positive‐controlled, parallel‐group with nested crossover for positive control, thorough QT (TQT) study was conducted in healthy adult subjects. Eligible participants were healthy men and women aged 18 through 45 years with a body mass index of 18 to 29 kg/m2 and body weight of at least 50 kg. Subjects were determined to be in good health by review of their medical history, physical examination, 12‐lead electrocardiogram (ECG), and clinical laboratory tests. Excluded from participation were subjects who had ECG abnormalities, known cardiovascular disorders, a supine HR outside 55 to 90 beats/min (bpm), or Fridericia‐corrected QT (QTcF)26 interval of <320 milliseconds and >450 (men) or >470 milliseconds (women) at screening. Women could not be nursing or pregnant.
The study consisted of a screening period of up to 28 days before dosing, a 1‐day baseline evaluation, a 14‐day treatment period, a washout period, and a follow‐up visit at least 14 days after the last dose of ozanimod or placebo (Figure 2). Eligible subjects checked into the clinical research unit on day −2 and remained in the clinical research unit until discharge on day 19. On day −1 (baseline), a continuous 24‐hour baseline 12‐lead ECG profile was obtained using cardiac telemetry. On day 1, subjects were randomized (1:1) to 1 of 2 treatment groups. In group 1, ozanimod was administered orally, QD, using a 14‐day dose‐escalation regimen of 0.25 mg for 4 days, 0.5 mg for 3 days, 1 mg for 3 days, and 2 mg for 4 days. On days 2 and 17, a single dose of moxifloxacin placebo also was administered. Under the nested crossover design,27 subjects in group 2 received ozanimod placebo QD for 14 days and were randomized (1:1) to receive a single dose of moxifloxacin 400 mg or placebo, alternatively, on days 2 and 17. The reasons for incorporating the nested crossover for the positive‐control moxifloxacin were to provide confirmation of assay sensitivity at the start and the end of the study, as discussed with the US Food and Drug Administration, and to allow a more efficient study design involving only 2 arms (and half the subjects) compared with the conventional 4‐arm TQT design. Ozanimod 2 mg served as the supratherapeutic dose in this TQT study, as it represents approximately 4‐ and 2‐fold multiples of the anticipated ozanimod exposures of the 0.5‐mg and 1‐mg QD regimens, respectively, currently being evaluated in ongoing development programs. All subjects were to have fasted for at least 10 hours before baseline ECG profiling and for 10 hours before dosing on days 1 to 14 and day 17. Discontinuation of a subject from the study was to be considered in the case of the following during treatment: an increase in QT/QTc interval to >500 milliseconds and/or of >60 milliseconds over baseline, any evidence for ventricular or supraventricular arrhythmia with or without hemodynamic compromise or any advanced AV block, or symptomatic persistent bradycardia <35 bpm that required medical intervention.
Figure 2.
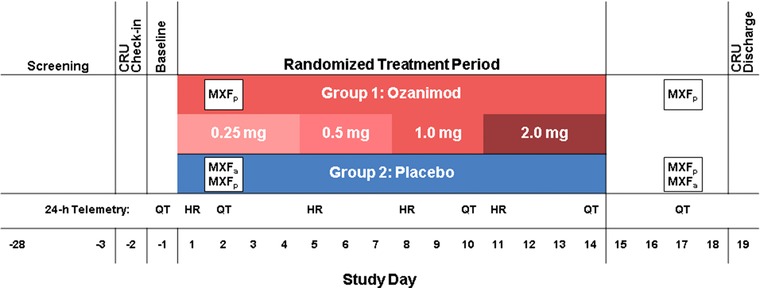
Study design. The ozanimod group also received moxifloxacin placebo on days 2 and 17. Within the placebo group, subjects were randomized (1:1) to receive a single dose of moxifloxacin 400 mg or placebo on days 2 and 17. CRU indicates clinical research unit; HR, 24‐hour cardiac telemetry for heart‐rate analysis; MXF, moxifloxacin (positive control); MXFa, MXF active (400 mg); MXFp, MXF placebo; QT, 24‐hour telemetry for QT assessment.
Study Assessments
Electrocardiograms
ECGs were obtained continuously using 12‐lead Holter devices (H12+, Mortara Instruments, Milwaukee, Wisconsin) on days –1 (baseline), 2 (first moxifloxacin vs placebo assessment), 10 (ozanimod 1‐mg assessment), 14 (ozanimod 2‐mg assessment), and 17 (second moxifloxacin assessment). Triplicate ECGs were extracted at 0, 1, 2, 3, 4, 6, 8, 12, and 24 hours (time‐matched to post‐dose samples on dosing days). On days 2, 10, 14, and 17, extractions were performed at similar time points. Additional extractions were performed at 48, 72, 96, and 120 hours after day 14 dosing. Continuous 12‐lead ECGs were obtained using the Surveyor Central System (Mortara Instruments) and were analyzed (blinded) by a central ECG core laboratory (Bioclinica, Inc, Princeton, New Jersey) using a computer‐assisted (semiautomated) methodology with manual overread (adjudication).
The 24‐hour telemetry monitoring for heart‐rate characterization was performed on the days of each dose increase (ie, days 1, 5, 8, and 11). Minute‐by‐minute telemetry data were processed to derive hourly mean heart‐rate data, except for baseline; baseline for the heart‐rate assessment was defined as the 15‐minute average HR prior to dosing on each day.
Safety and Tolerability
Safety and tolerability were assessed by collection and analysis of adverse events, vital signs, clinical laboratory results, ECGs, and continuous 24‐hour cardiac telemetry. Orthostatic blood pressure changes were performed (supine blood pressure first, followed by standing blood pressure after 2 minutes of standing) each hour for the first 6 hours after receiving study drug on days 1 and 5. A sudden, significant fall in blood pressure (>20 mm Hg) 2 to 5 minutes after standing from the supine position was interpreted as orthostatic hypotension. Adverse events of special interest were prespecified based on previous clinical results with the S1P receptor agonist fingolimod10, 11, 28 and the ozanimod first‐in‐human study21 and included bradycardia, heart conduction abnormalities (eg, AV block of second degree or higher), infection, increases in liver transaminases, and changes in pulmonary function. For identifying adverse events related to changes in pulmonary function, pulmonary function tests (including forced expiratory volume at 1 second and forced vital capacity measurements) were performed at screening and at the follow‐up visit; additional pulmonary function tests were to be conducted for subjects who developed respiratory symptoms during the study.
Pharmacokinetics
Blood samples were collected on days 2, 10, 14, and 17 at time 0 (predose) and at 1, 2, 3, 4, 6, 8, 12, and 24 hours postdose. Additional blood samples were obtained at 48, 72, 96, and 120 hours after the last ozanimod dose on day 14. Plasma moxifloxacin concentrations were measured only if QTc assay sensitivity failed. Plasma concentrations of ozanimod and its active metabolites (RP101988, RP101075, and RP101442) were determined by a bioanalytical core laboratory (ICON Development Solutions, LLC, Whitesboro, New York) using a validated liquid chromatography‒tandem mass spectrometry assay. Ozanimod and its metabolites were extracted from 0.4 mL K2EDTA human plasma by liquid/liquid separation (methanol/water/methyl tert‐butyl ether). Chromatographic separation was achieved on a Waters Acquity UPLC system, running a gradient composed of 0.1% formic acid in water and 0.1% formic acid in methanol at 0.350 mL/min through Waters Acquity UPLC HSS T3, 2.1 × 5.0, 1.8 μm guard column and 2.1 × 100 mm, 1.8 μm analytical column. Quantification was achieved using ratios of analyte peak area to internal standard peak area. Concentrations of the calibration curve standards, quality control samples, and study samples were determined by the method of weighted least‐squares linear regression (1/x2). The bioanalytical method was validated over concentration ranges of 4.0 to 2000 pg/mL for ozanimod and RP101075, 16.0 to 4000 pg/mL for RP101988, and 8.0 to 4000 pg/mL for RP101442. The interassay precision values were ≤5.57% for ozanimod, ≤6.22% for RP101988, ≤9.57% for RP101075, and ≤4.98% for RP101442. The accuracy (percentage bias) values ranged from −4.19% to −3.30% for ozanimod, −2.88% to 2.08% for RP101988, −4.17% to −0.10% for RP101075, and −3.75% to −1.00% for RP101442.
For the plotting of mean plasma concentration‐time profiles, drug concentrations that were below the lower limit of quantification (LLOQ) of the assay were replaced with 0 and included as such in the calculation of the mean values at that particular time point (LLOQ was 4 pg/mL for ozanimod and RP101075, 8 pg/mL for RP101442, and 16 pg/mL for RP101988). The PK parameters of ozanimod, RP101988, RP101075, and RP101442 were estimated using actual PK sample collection times; noncompartmental analysis was performed with Phoenix WinNonlin, version 6.2.1 (Pharsight Corp, St Louis, Missouri). The following PK parameters were derived: maximum observed plasma concentration (Cmax); minimum observed plasma concentration; time to Cmax (Tmax); area under the plasma concentration‐time curve during the dosage interval; and elimination half‐life (t1/2; day 14 only).
Statistical Methods
Sample Size Determination
The planned sample size (120‐132 total subjects) was based on the assumption that the true QTcF prolongation of ozanimod relative to placebo was 3 milliseconds at each of the 9 time points from 0 to 24 hours. The power to achieve an upper bound below the noninferiority margin of 10 milliseconds for the 95% 1‐sided confidence interval (CI) for the placebo‐adjusted change from baseline treatment effect at all time points was estimated to range from 74% to 97%, depending on the correlation between the time points. The minimum power of 74% was based on the assumption of complete independence between the time points. The moxifloxacin QTcF mean effect was expected to be at least 12 milliseconds at its highest point.29 For such an effect, with the above sample size, the power to show superiority (ie, that the lower bound of the 95% 1‐sided test of the observed maximum difference was significantly greater than 5 milliseconds) was approximately 97%.
ECG Primary Analysis
The QT:RR relationship was investigated on an individual subject basis. The performance of 2 standard methods for correcting QT for RR, Bazett and Fridericia, was investigated. For each subject, using data from the drug‐free baseline profile day only (day −1), ln(QT) was regressed against ln(RR), and the slopes of the fitted regressions were retained. The median of these fitted slopes (across all subjects) was denoted as λ and was used to define the study‐specific correction factor, QTcSS = QT/RRλ. Validation of the primary end point QTc variable was performed using the same subset of the data (ie, baseline profile day only), by regressing the standard QTc intervals, QTcF and QTcB, against RR in a similar way (after natural log transformation). If the correction successfully removed the association of QTc with HR the average slope of these regressions should have been close to 0. To test this (separately for QTcF and QTcB), the median slope across all subjects and its 95%CI were derived. The CI for the median was calculated using the binomial‐based method described previously.30 If the 95%CI derived from QTcF included 0, QTcF would be deemed suitable for this study. However, if this CI excluded 0, QTcSS was to be used as the primary end point instead (and QTcF would become a secondary end point).
The time‐matched differences in change from baseline (day −1) in QTcF interval for ozanimod vs placebo (ΔΔQTcF) were calculated using analysis of covariance, separately at each time point, with baseline QTcF as the covariate and with sex and study group as factors. Absence of effects of ozanimod on cardiac repolarization was to be concluded if the upper limit of the 1‐sided 95%CI (or the 2‐sided 90%CI) for ΔΔQTcF was below 10 milliseconds at all time points. These analyses were done separately for the therapeutic dose (day 10 at 0, 1, 2, 3, 4, 6, 8, 12, and 24 hours) and the supratherapeutic dose (day 14 at 0, 1, 2, 3, 4, 6, 8, 12, 24, 48, 72, 96, and 120 hours postdose). At each time point triplicate ECGs were extracted, and the mean value was used for statistical analysis.
For the purpose of demonstrating assay sensitivity, the QTcF data from the time points at 2 and 3 hours were averaged so that only 1 analysis was used for the assessment of assay sensitivity, and no multiplicity correction was required. This analysis used data from group 2 only, regarded as a crossover design with placebo and moxifloxacin as the treatments. The analysis of variance model included treatment (moxifloxacin or placebo), period, and subject as categorical factors. Assay sensitivity was to be deemed as demonstrated if the lower limit of the 1‐sided 95%CI for the difference between moxifloxacin and the placebo group was above 5 milliseconds. In the event that assay sensitivity was not demonstrated with this analysis, sensitivity analyses using different definitions of baseline were also to be presented. In addition to the analysis of the average over the 2‐ and 3‐hour time points, analyses at each individual time point (1, 2, 3, 4, 5, 6, 12, and 24 hours) are also presented.
All statistical analyses were performed using SAS software, version 9.1.3 (SAS Institute, Cary, North Carolina).
Heart‐Rate Analysis
Minute‐by‐minute telemetry data were processed to derive hourly mean heart‐rate data except for the daily predose means, which were based on data over a 15‐minute sampling period performed prior to dosing. The daily minimum hourly HR was calculated for each subject. Hourly heart‐rate and daily minimum heart‐rate data were summarized and presented by treatment group. Frequency tables for the daily minimum hourly HR were presented using the following HR categories: ≥65 bpm, ≥60 to <65 bpm, ≥55 to <60 bpm, ≥50 to <55 bpm, ≥45 to <50 bpm, ≥40 to <45 bpm, ≥35 to <40 bpm, and <35 bpm.
ECG Categorical Analysis
Absolute QTcF values that exceeded 450, 480, and 500 milliseconds as well as QTcF increases from baseline that exceeded 30 and 60 milliseconds were summarized using the frequency count and the percentage of subjects in each category. Frequency tables were restricted to the ECG assessment on days −1 (baseline), 10 (therapeutic dose), and 14 (supratherapeutic dose) because placebo subjects received moxifloxacin at other time points (day 2 or day 17), which would have distorted the comparison between ozanimod and placebo.
ECG Concentration‐Response Analysis
A linear mixed‐effect modeling approach was used to quantify the relationship between the plasma concentrations (ozanimod and its active metabolites) and the time‐matched, baseline‐adjusted change in the QTcF (ΔQTcF). Plasma drug concentrations were set to 0 for placebo subjects, and drug concentrations that were below the LLOQ of the assay were replaced with half the LLOQ (LLOQ was 4 pg/mL for ozanimod and RP101075, 8 pg/mL for RP101442, and 16 pg/mL for RP101988). Predicted QTcF prolongations at the mean Cmax values for each dose level, together with their upper (1‐sided) 95%CIs, are also presented, based on the same fitted line.
The confidence limits for the predicted prolongations were calculated from the variances derived using the standard linear model approach; ie, the variance of the estimator will be calculated as var(y) = bVbʹ where V is the variance‐covariance matrix of the estimated coefficients, b is the vector defining the conditions (eg, the concentration) for which the estimate is required, and var(y) is the variance of the estimate y. If the data showed departure from a linear model (on graphical inspection), a nonlinear model (eg, including quadratic and cubic terms) was considered.
Results
Subject Disposition and Demographics
A total of 124 adult subjects were enrolled and randomized to receive either ozanimod (N = 62) or placebo (N = 62). Of these, 56 ozanimod recipients (90.3%) and 57 placebo recipients (91.9%) completed the study. Reasons for early termination included nonserious adverse events (4 ozanimod, 3 placebo), withdrawal of consent (2 ozanimod, 1 placebo), and noncompliance (1 placebo). More men than women were enrolled (72 men, 52 women). The mean age was 32.0 years, and 71% of the subjects were white. The mean (±SD) weight and body mass index were 71.8 ± 10.9 kg and 25.1 ± 2.4 kg/m2, respectively. Demographic and baseline characteristics were similar for the ozanimod and placebo groups.
ECG Primary Analysis
Regression analysis of (ln)QTcF on (ln)RR resulted in a median slope of 0.001, with the 95%CI including 0 (−0.025, 0.021), confirming QTcF as the appropriate end‐point variable. The upper bounds of the 2‐sided 90%CI for the mean ΔΔQTcF at the therapeutic and supratherapeutic doses of ozanimod were below the 10‐millisecond threshold at all treatment time points (Figures 3A and 3B) and had a maximum value of 8.15 milliseconds (at hour 2 on the therapeutic dose day). Mean ΔΔQTcF ranged from 0.3 to 4.5 milliseconds with the therapeutic dose of ozanimod and from −0.9 to 2.9 milliseconds with the supratherapeutic dose of ozanimod. Assay sensitivity was demonstrated with the positive control moxifloxacin, which caused statistically significant (P < .05) prolongation of QTcF at all time points through 12 hours postdose and produced a maximum ΔΔQTcF prolongation of 11.1 milliseconds (90%CI: 8.6, 13.7) at 3 hours, and was above the 5‐millisecond threshold defined a priori for assay sensitivity (Figure 3C).
Figure 3.
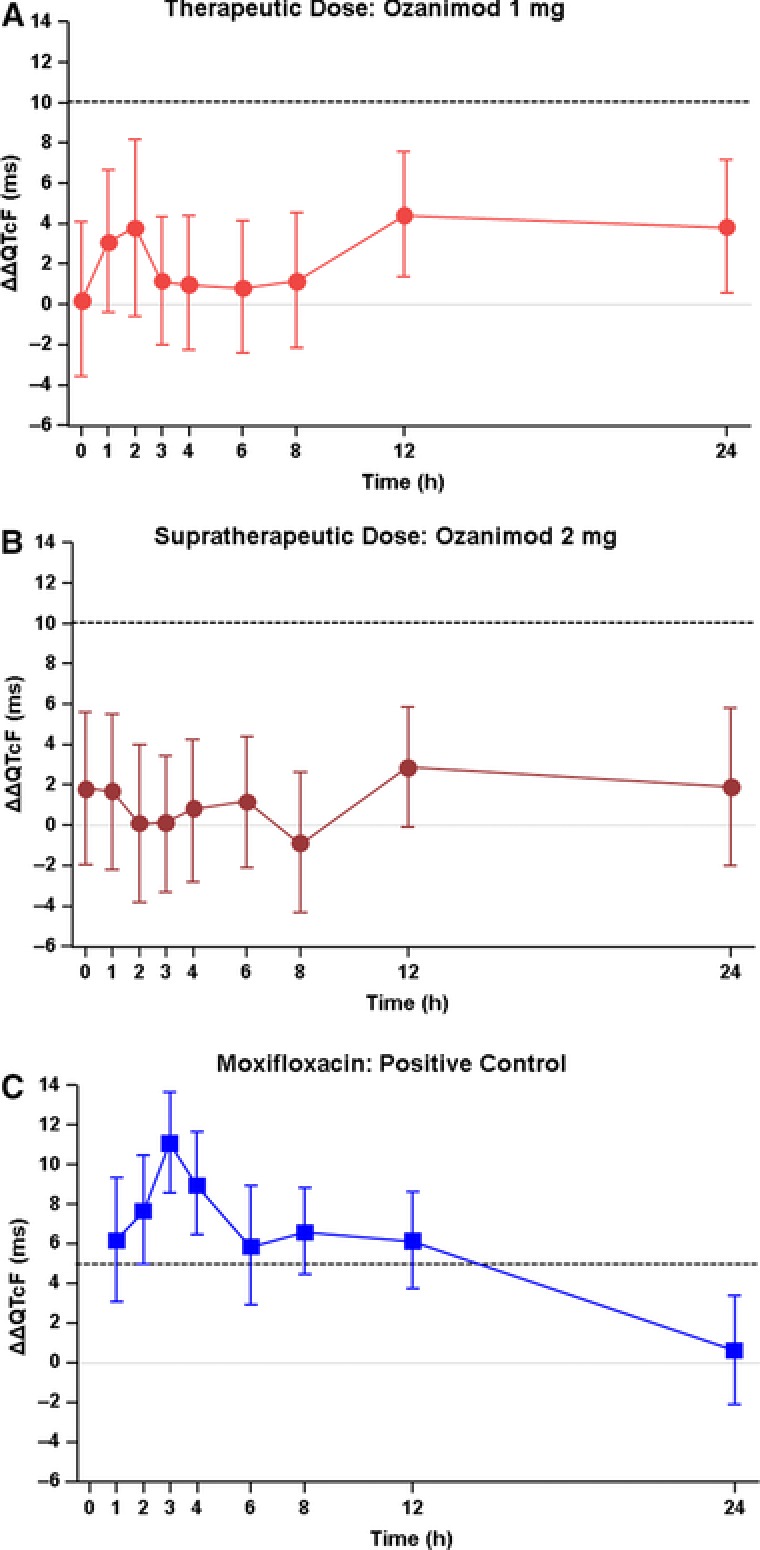
Effect of ozanimod (A and B) and moxifloxacin (C) on ΔΔQTcF. Analyses were conducted on day 10 for ozanimod 1 mg and on day 14 for ozanimod 2 mg; moxifloxacin analyses were conducted on days 2 and 17. Data shown are point estimates ± 90% confidence intervals. Dashed line indicates the regulatory threshold of 10 milliseconds for ozanimod (A and B) and the assay sensitivity lower bound of 5 milliseconds for moxifloxacin (C).
ECG Categorical Analysis
Categorical analysis was used to determine whether any subjects had clinically significant increases in the QTc interval (Table 1). At the supratherapeutic dose, 1 subject in each treatment group had QTcF >450 milliseconds. No subject on either dose of ozanimod had QTcF >480 milliseconds. The number of subjects who experienced a postdose change in QTcF >30 milliseconds was low (<9%) and was balanced between the study groups. One placebo‐treated subject and no ozanimod‐treated subjects had a postdose change in QTcF of >60 milliseconds (Table 1).
Table 1.
QTcF Interval Outlier Categorical Analysis
| Threshold | Day (Dose) | Placebo, n (%) | Ozanimod, n (%) |
|---|---|---|---|
| QTcF >450 ms | 10 (1 mg) | 0 | 0 |
| 14 (2 mg) | 1 (1.8) | 1 (1.8) | |
| QTcF >480 ms | 10 (1 mg) | 0 | 0 |
| 14 (2 mg) | 0 | 0 | |
| QTcF Δ from baseline >30 ms | 10 (1 mg) | 4 (6.9) | 5 (8.8) |
| 14 (2 mg) | 4 (7.0) | 3 (5.4) | |
| QTcF Δ from baseline >60 ms | 10 (1 mg) | 1 (1.7) | 0 |
| 14 (2 mg) | 0 | 0 |
Data denote the number (%) of subjects with ≥1 electrocardiogram reading that exceeded each specified threshold. Placebo group: N = 58 on day 10 and N = 57 on day 14. Ozanimod group: N = 57 on day 10 and N = 56 on day 14.
QTcF indicates Fridericia‐corrected QT interval.
Effect of Ozanimod on HR and Other ECG Variables
Twenty‐four‐hour cardiac telemetry was used to characterize the effect of ozanimod on HR. Daily mean predose baseline HRs were similar for the study groups (ie, between 59 and 71 bpm throughout all dose‐escalation days). The mean hourly HRs during the 24‐hour monitoring period on each dose‐escalation day are presented in Figure 4. Mean HR in the ozanimod‐treated subjects was consistently lower than that in the placebo‐treated subjects. On day 1, the maximum difference from placebo in HR was 8.1 bpm and was observed at hour 10. On day 5, the maximum difference in means was 10.2 bpm and was observed at hour 5. The maximal difference from placebo on the therapeutic and supratherapeutic dose‐escalation days was 13.8 bpm (observed at hour 5) and 12.0 bpm (observed at hour 5), respectively, indicating that no further reduction in HR was observed when the ozanimod dose was escalated up from 1 mg to 2 mg.
Figure 4.
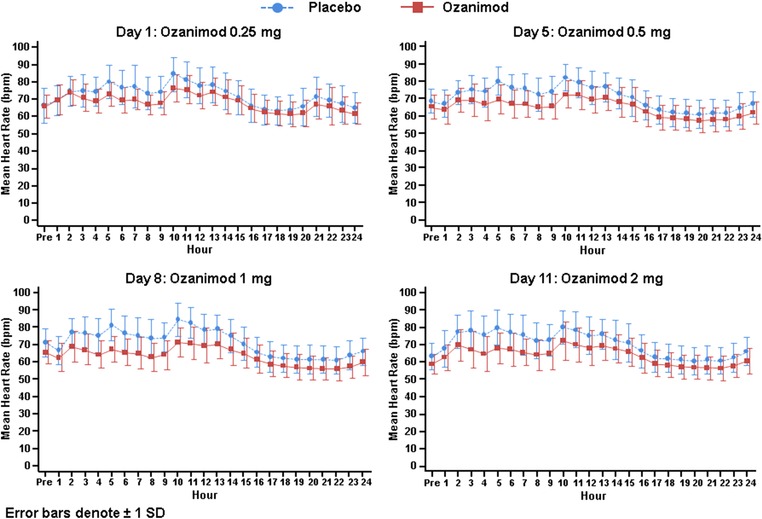
Hourly heart rate during the 24‐hour monitoring period on each dose‐escalation day. Data shown are mean ± SD.
A normal circadian pattern for HR was observed in both study groups, with an increase in HR during the day and a return to baseline at night when subjects were sleeping. The dose‐escalation regimen in this study employed a starting dose of 0.25 mg, which appeared to reduce the “first dose” effect on HR on day 1 compared to what was observed in the phase 1 study when higher fixed doses (eg, 1 mg) were administered directly on day 1. In the ozanimod group there was a smaller increase in HR during the day compared to placebo on day 1, and there were smaller differences relative to placebo during the nighttime hours. During the remaining dose‐escalation period, subjects in the ozanimod group did not experience clinically relevant further decreases in HR after day 1 with dose escalation up to a supratherapeutic dose of 2 mg.
The daily minimum hourly HR generally occurred at night when subjects were sleeping and was similar in both study groups, with mean values for ozanimod subjects on each dose‐escalation day being no more than 5 bpm below those for placebo recipients. In a small number of subjects minimum hourly HRs were less than 45 bpm; a similar number of ozanimod and placebo recipients had values in this range (Table 2). Only 1 subject had a daily minimum hourly HR of 38 bpm, which occurred in the early hours of the morning, at 21 to 22 hours after the administration of a 2‐mg dose of ozanimod on day 11.
Table 2.
Subjects With Minimum Hourly Heart Rate <45 bpm
| Day | Placebo, n (%) | Ozanimod, n (%) |
|---|---|---|
| −1 (predose) | 2 (3.2) | 0 |
| 1 (0.25 mg) | 1 (1.6) | 0 |
| 5 (0.5 mg) | 3 (5.1) | 3 (5.1) |
| 8 (1 mg) | 0 | 2 (3.5) |
| 11 (2 mg) | 2 (3.4) | 1 (1.8) |
Placebo group: N = 61 on day −1 and day 1, N = 59 on day 5, N = 58 on day 8, and N = 57 on day 11. Ozanimod group: N = 62 on day −1 and day 1, N = 61 on day 5, and N = 58 on day 8 and day 11.
bpm indicates beats per minute.
No clinically relevant effects on PR or QRS intervals were observed with ozanimod. The PR interval showed both positive and negative point estimates for the difference between ozanimod and placebo, which were never significantly different from 0. For the QRS intervals, the point estimates were never significantly different from 0 on either treatment day, indicating that there is no evidence for any effect of ozanimod on the QRS duration.
Concentration‐Response QT Analysis
In a linear mixed‐effects model, no relationship was observed between plasma ozanimod concentration and ΔQTcF (slope of 0.0029; not significantly different from 0 [P = .33]) (Figure 5A). Similarly, no relationship was observed between plasma concentrations of RP101988 and RP101075 and ΔQTcF (slopes of 0.0020 and 0.0039, respectively) (Figures 5B and 5C). The majority of RP101442 concentrations were below the LLOQ; therefore, concentration‐response analysis for this metabolite is not meaningful. The upper 1‐sided 95%CI of the predicted value of ΔQTcF at the mean Cmax values for both the therapeutic and supratherapeutic ozanimod dose levels (Table 3) are all substantially below the threshold of regulatory interest (10 milliseconds).
Figure 5.
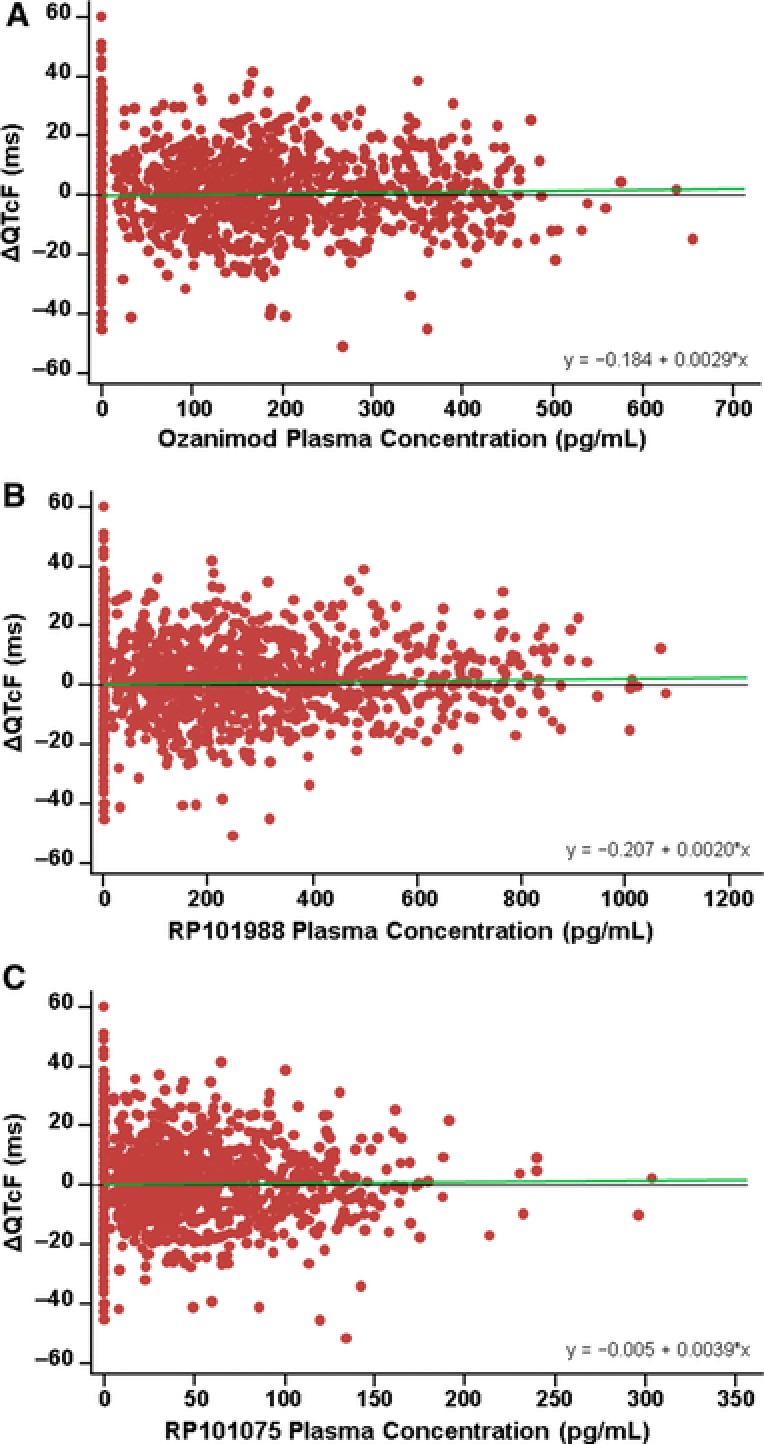
Concentration‐response analysis for ΔQTcF vs ozanimod (A), RP101988 (B), and RP101075 (C). Green line indicates the fitted regression line. R2 values for ozanimod, RP101988, and RP101075 are 0.0109, 0.0126, and 0.00946, respectively.
Table 3.
Summary of Concentration‐QTc Analysis
| Therapeutic Dose: Ozanimod 1 mg | Supratherapeutic Dose: Ozanimod 2 mg | |||||||
|---|---|---|---|---|---|---|---|---|
| Active Moiety | Mean Cmax (pg/mL) | Point Estimate (ms) | Standard Error (ms) | Upper Limit (ms) | Mean Cmax (pg/mL) | Point Estimate (ms) | Standard Error (ms) | Upper Limit (ms) |
| Ozanimod | 204.4 | 0.41 | 0.70 | 1.55 | 400.1 | 0.97 | 1.09 | 2.75 |
| RP101988 | 354.9 | 0.51 | 0.72 | 1.69 | 677.3 | 1.17 | 1.11 | 2.99 |
| RP101075 | 63.9 | 0.24 | 0.70 | 1.39 | 130.5 | 0.51 | 1.09 | 2.30 |
Upper (1‐sided) 95% confidence limits. Cmax, maximum observed plasma concentration.
Pharmacokinetic Analysis
The mean (SD) concentration‐time profiles for ozanimod and the measurable active metabolites are presented in Figure 6. PK parameters for ozanimod, RP101988, and RP101075 are summarized in Table 4. Ozanimod and its active metabolites (RP101988 and RP101075) exhibited similar PK properties. Median Tmax for ozanimod, RP101988, and RP101075 was approximately 6 hours. Dose‐proportional increases in exposure measures (area under the concentration‐time curve, Cmax, and lowest concentration) were observed for ozanimod, RP101988, and RP101075. Mean t1/2 values for ozanimod, RP101988, and RP101075 ranged from approximately 19 to 22 hours. The PK of RP101442 could not be characterized because the majority of RP101442 samples had concentrations below the LLOQ (8 pg/mL).
Figure 6.
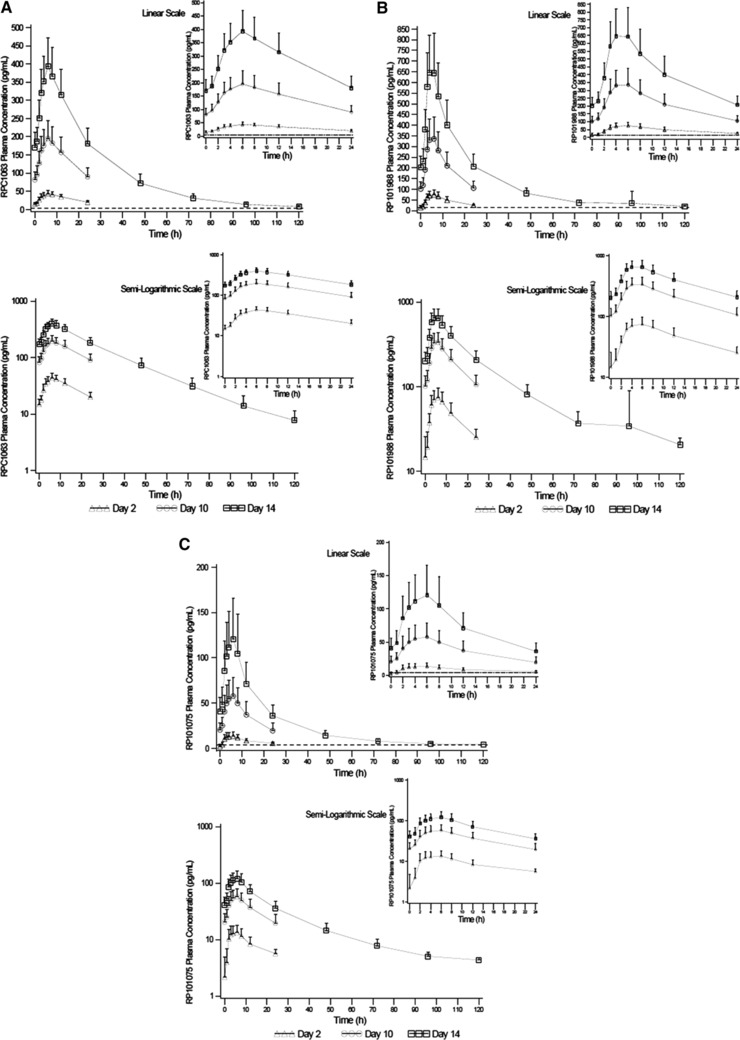
Plasma concentration‐time profiles for ozanimod (A), RP101988 (B), and RP101075 (C). Data shown are mean ± SD.
Table 4.
Pharmacokinetic Parameters of Ozanimod, RP101988, and RP101075
| Ozanimod | RP101988 | RP101075 | ||||
|---|---|---|---|---|---|---|
| Pharmacokinetic Parameter (Unit) | Day 10 (1 mg) | Day 14 (2 mg) | Day 10 (1 mg) | Day 14 (2 mg) | Day 10 (1 mg) | Day 14 (2 mg) |
| Cmax (pg/mL) | 205 (46.0) | 406 (74.3) | 354 (96.0) | 688 (186) | 63.7 (22.3) | 132 (49.5) |
| Cmin (pg/mL) | 81.0 (22.2) | 166 (40.7) | 98.0 (30.7) | 195 (53.1) | 17.3 (6.00) | 33.7 (10.9) |
| Tmax (h) | 6.08 (3.08‐12.08) | 6.08 (2.08‐12.08) | 6.08 (3.08‐8.12) | 4.08 (3.08‐8.08) | 6.08 (2.08‐12.08) | 6.08 (2.08‐8.12) |
| AUCτ (pg·h/mL) | 3405 (7667) | 6768 (1309) | 4930 (1358) | 9441 (2486) | 875 (290) | 1731 (525.6) |
| t1/2 (h) | NC | 20.1 (2.41) | NC | 19.1 (2.98) | NC | 21.5 (5.73) |
Data are presented as mean (SD) except for Tmax, which is expressed as median (range).
AUCτ indicates area under the plasma study drug concentration‐time curve during a dosage interval; Cmax, maximum observed plasma concentration; Cmin, minimum observed plasma concentration; NC, not calculated; Tmax, time to Cmax; t1/2, elimination half‐life.
Safety and Tolerability
The incidence of treatment‐emergent adverse events (TEAEs) was comparable for the ozanimod (48 of 62 subjects; 77.4%) and placebo groups (46 of 62 subjects; 74.2%). All TEAEs were mild or moderate in severity. Among the subjects who experienced TEAEs, most had events that were deemed unrelated to study medication by the investigator (ozanimod, 72.9%; placebo, 76.1%). The number of subjects who discontinued the study due to TEAEs was similar for each group (4 ozanimod subjects [6.5%], 3 placebo subjects [4.8%]). There were no serious TEAEs.
Incidences of the most common TEAEs and cardiac adverse events of special interest are shown in Table 5. The most common TEAE was site reaction caused by the ECG electrode tape. The incidence of specific TEAEs was similar for the study groups, with the exception of orthostatic hypotension, which occurred in 5 ozanimod subjects (8.1%) and 1 placebo subject (1.6%). All 6 events of orthostatic hypotension occurred from 3 to 6 hours after dosing on days 1 (ozanimod 0.25 mg) or 5 (ozanimod 0.5 mg) and were transient and asymptomatic. Cardiac adverse events of special interest were infrequent and of comparable incidence between the study groups. These included short runs of nonsustained ventricular tachycardia (3 placebo subjects, 2 ozanimod subjects), transient and self‐limiting first‐degree AV block (1 ozanimod subject), and transient and self‐limiting second‐degree AV block, Mobitz type 1 (1 subject in each group). None of these cardiac events was temporally associated with the administration of moxifloxacin.
Table 5.
Treatment‐Emergent Adverse Events Experienced by >1 Subject in Either Study Group
| Treatment‐Emergent Adverse Event | Placebo (N = 62), n (%) | Ozanimod (N = 62), n (%) |
|---|---|---|
| Site reaction (ECG electrodes) | 40 (64.5) | 39 (62.9) |
| Headache | 7 (11.3) | 9 (14.5) |
| Orthostatic hypotension | 1 (1.6) | 5 (8.1) |
| Musculoskeletal chest pain | 3 (4.8) | 3 (4.8) |
| Constipation | 2 (3.2) | 3 (4.8) |
| Dizziness | 1 (1.6) | 3 (4.8) |
| Nausea | 3 (4.8) | 0 (0) |
| Abnormal liver function test result | 1 (1.6) | 2 (3.2) |
| Upper respiratory tract infection | 2 (3.2) | 0 (0) |
| Costochondritis | 2 (3.2) | 0 (0) |
| Nasal congestion | 0 (0) | 2 (3.2) |
| Allergic rhinitis | 0 (0) | 2 (3.2) |
| Cardiac adverse events of special interest | ||
| Ventricular tachycardia, nonsustained | 3 (4.8) | 2 (3.2) |
| First‐degree AV block, transient | 0 (0) | 1 (1.6) |
| Second‐degree AV block, transient | 1 (1.6) | 1 (1.6) |
AV indicates atrioventricular; ECG, electrocardiogram.
Other prespecified adverse events of special interest included liver enzyme levels >3 times the upper limit of normal, infection, and decreases in pulmonary function. Few TEAEs of infection or elevated liver enzymes occurred, and they were evenly distributed between the study groups (infection: 3 placebo subjects, 2 ozanimod subjects; elevated liver enzymes: 1 placebo subject, 2 ozanimod subjects). All cases of elevated liver enzymes were detected at follow‐up visits that occurred ≥14 days after the last dose of study medication. All of these cases were transient and considered by the investigator to be unrelated to study medication. No clinically meaningful results of pulmonary function tests were noted as TEAEs for either group. Most laboratory results remained within reference ranges throughout the study. As expected, based on the mechanism of action of ozanimod, there were decreases in lymphocyte counts in ozanimod‐treated subjects with the mean percentage change from baseline of 52.9% at the end of the randomized treatment period (after the 2‐mg dose). Lymphocyte counts returned to 85.1% of baseline by the follow‐up visit (≥14 days after the last dose of study medication).
Discussion
This was a negative thorough QT/QTc study, demonstrating that ozanimod treatment, in doses of up to 2 mg/day, did not have a clinically relevant effect on cardiac repolarization. The upper bound of the 2‐sided 90%CI (or 95% 1‐sided confidence limit) of the ΔΔQTcF was below the 10‐millisecond regulatory threshold at all time points for the therapeutic and supratherapeutic doses of ozanimod. These results were confirmed by demonstrating assay sensitivity with the use of moxifloxacin as a positive control. Consistent with the lack of QTc effect, the concentration‐response analysis showed no effect of ozanimod or its active metabolites (RP101988 and RP101075) on QTcF, with the fitted slopes not differing from 0 and the upper confidence limits of the ΔΔQTcF at the Cmax values for both doses being well below 10 milliseconds. Outlier analyses showed that no subset of subjects had potentially clinically significant increases in the QTc interval following ozanimod administration. Although trough PK samples were not collected for steady‐state confirmation, it is expected that 4‐day dosing (≈5 × t1/2) of the supratherapeutic dose regimen in this study achieved steady‐state concentrations based on the mean t1/2 values for ozanimod and active metabolites of approximately 20 hours. Tmax was approximately 6 hours, at which time ECG extractions also were obtained. The Cmax of ozanimod was 2‐fold higher at the supratherapeutic dose (2 mg) than at the therapeutic dose (1 mg). PK results in this study also showed that the active metabolites (RP101988 and RP101075) exhibit PK properties similar to those of ozanimod.
The International Conference on Harmonisation E14 Guidance recommends assessment of QTc prolongation by the study drug at multiples of the anticipated maximum therapeutic exposure to mimic exposure that may occur under circumstances that might augment potential toxicity (eg, drug‐food or drug‐drug interactions involving metabolic enzymes and drug transporters, or when the patient takes more than the clinical dose prescribed).25 Ozanimod is being evaluated in ongoing development programs of the 0.5‐mg and 1‐mg QD regimens. Because the Cmax of ozanimod at the 2‐mg dose in this study was 2‐fold higher than at the 1‐mg dose, the 2‐mg dose is considered appropriate to cover any increased exposure that might occur in a clinical setting. Food (high‐ or low‐fat meals) did not have any effect on ozanimod exposure.31 Ozanimod is metabolized to form RP101988 and RP101075 via 2 parallel metabolic pathways. Alcohol dehydrogenase and aldehyde dehydrogenase are believed to catalyze a 2‐step conversion of ozanimod to RP101988, and cytochrome P450 (CYP) 3A is the primary human enzyme converting ozanimod to RP101075. Ozanimod is a weak substrate of P‐glycoprotein (P‐gp), whereas RP101988 is a substrate of P‐gp and breast cancer resistance protein (BCRP) drug transporters (data on file). Drug‐drug interaction studies showed that itraconazole, a strong inhibitor of CYP3A and P‐gp, had no effect on ozanimod exposure, whereas cyclosporine, a strong inhibitor of P‐gp and BCRP transporters, increased total agonist exposure (sum of ozanimod, RP101988, and RP101075) by approximately 50% (data on file).
In contrast to ozanimod, fingolimod (an approved nonselective S1P receptor modulator) has produced mild but significant prolongation of the QTc interval.12 Understanding the mechanistic basis of this QT prolongation is important for the development of improved therapeutic agents in the S1P receptor modulator class. In mice, S1P3 receptors are expressed in cardiac Purkinje fibers, and their activation is associated with blockade of ventricular conduction. Fingolimod‐induced cardiac abnormalities are not observed in S1P3 knockout mice and are reversed in wild‐type mice with a selective S1P3 antagonist.13 In human TQT studies, results with fingolimod and 2 other S1P receptor‐targeting agents in development (ponesimod and siponimod) support a potential relationship between activity at S1P3R and QT prolongation. In addition to the observation of QT prolongation with the nonselective agonist fingolimod, such prolongation has occurred in a TQT study of ponesimod, which has only modest selectivity for S1P1R vs S1P3R (≈18‐fold).32 Ponesimod prolonged the QT interval at supratherapeutic doses (40 and 100 mg) in healthy volunteers.20 In contrast, no effect on QT interval was demonstrated with supratherapeutic doses of either siponimod (which has >2500‐fold selectivity for S1P1R vs S1P3R)33, 34 or, in the current study, ozanimod, which has even greater selectivity for S1P1R vs S1P3R (>10,000).1
Transient, dose‐dependent decreases in HR have been observed at treatment initiation of S1P receptor modulators in healthy subjects and in patients with multiple sclerosis. It appears that the negative chronotropic effects of S1P receptor agonists in humans are mediated by S1P1R through activation of G‐protein–coupled inwardly rectifying potassium channels on cardiac myocytes, which reduce excitability and decrease firing rate.16, 33 Receptor desensitization, internalization, and degradation of the cardiac S1P1R have been proposed to explain the time‐related attenuation of heart‐rate effects.1, 17, 33 In a previous phase 1 single‐ascending‐dose and multiple‐ascending‐dose study with ozanimod, a mild dose‐dependent, first‐dose reduction in HR was observed, which was attenuated with an increasing dose‐escalation regimen.21 No further reduction in HR was observed when the ozanimod dose was escalated up from 1 mg to 2 mg in this study, confirming the effectiveness of the dose‐escalation regimen employed in this TQT study. Starting dose escalation at 0.25 mg appeared to attenuate first‐dose effects on HR on day 1 and during the dose‐escalation period when higher doses were administered. Telemetry monitoring in this study was performed to evaluate the heart‐rate effect on each dose‐escalation day to determine whether reduction in HR was attenuated with increasing dose. Drug concentrations and heart‐rate data in this study will be combined with those of other studies (with steady‐state data) for modeling and simulation. Transient and self‐limiting first‐ and second‐degree AV block occurred rarely in the current study and with a similar incidence in the ozanimod and placebo groups.
Conclusions
Results from this TQT study demonstrate that ozanimod does not prolong the QTc interval at therapeutic (1 mg) and supratherapeutic (2 mg) doses. Ozanimod was well tolerated overall, and no apparent safety issues emerged from this study. The incidence of adverse events of special interest, prespecified based on clinical trial results with fingolimod, was similar for ozanimod and placebo. These findings support the continued development of ozanimod for RMS (phase 3 trials: NCT02047734, NCT02294058), moderate to severe ulcerative colitis (phase 3 trial: NCT02435992), and moderate to severe Crohn disease (phase 2 trial: NCT02531113).
The authors received writing and editorial support for manuscript preparation from Nicole Coolbaugh and Philip Sjostedt, BPharm, MPH, from The Medicine Group, which was paid for by Receptos. The authors, however, directed and are fully responsible for all content and editorial decisions for this article.
Acknowledgment
The authors thank Christine Pan and Dennis Chanter for statistical analysis support and Susan Walker for reviewing statistical results of the manuscript.
Funding
This study was supported by Receptos, a wholly owned subsidiary of Celgene Corporation.
Declaration of Conflict of Interest and Financial Disclosure
J.Q. Tran and A. Olson are current employees of Receptos, a wholly owned subsidiary of Celgene Corporation, and are stockholders in Celgene. At the time of the study and analysis, J.P. Hartung, G.A. Timony, M. Boehm, R.J. Peach, S. Gujrathi, and P.A. Frohna were employees of Receptos, a wholly owned subsidiary of Celgene Corporation, and B. Mendzelevski was employed at Bioclinica, Inc. J.P. Hartung is currently employed by JPH Clinical Development, Inc (San Diego, CA). G.A. Timony is currently self‐employed as a pharmaceutical consultant. M.F. Boehm is currently employed by Boehm Projects, LLC (San Diego, CA). R.J. Peach is currently employed by Innate Immunotherapeutics Ltd (Sydney, Australia). S. Gujrathi is currently self‐employed. P.A. Frohna is currently employed by Frohna Biotech Consulting (Solana Beach, CA). B. Mendzelevski is currently employed by Cardiac Safety Consultants Ltd (London, UK).
References
- 1. Scott FL, Clemons B, Brooks J, et al. Ozanimod (RPC1063) is a potent sphingosine‐1‐phosphate receptor‐1 (S1P1) and receptor‐5 (S1P5) agonist with autoimmune disease‐modifying activity. Br J Pharmacol. 2016;173(11):1778‐1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cohen JA, Arnold DL, Comi G, et al. Safety and efficacy of the selective sphingosine1‐phosphate receptor modulator ozanimod in relapsing multiple sclerosis (RADIANCE): a randomised, placebo‐controlled, phase 2 trial. Lancet Neurol. 2016;15(4):373‐381. [DOI] [PubMed] [Google Scholar]
- 3. Sandborn WJ, Feagan BG, Wolf DC, et al. Ozanimod induction and maintenance treatment for ulcerative colitis. N Engl J Med. 2016;374(18):1754‐1762. [DOI] [PubMed] [Google Scholar]
- 4. Brinkmann V, Davis MD, Heise CE, et al. The immune modulator FTY720 targets sphingosine 1‐phosphate receptors. J Biol Chem. 2002;277(24):21453‐21457. [DOI] [PubMed] [Google Scholar]
- 5. Brinkmann V. FTY720 (fingolimod) in multiple sclerosis: therapeutic effects in the immune and the central nervous system. Br J Pharmacol. 2009;158(5):1173‐1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cohen JA, Chun J. Mechanisms of fingolimod's efficacy and adverse effects in multiple sclerosis. Ann Neurol. 2011;69(5):759‐777. [DOI] [PubMed] [Google Scholar]
- 7. Miron VE, Jung CG, Kim HJ, et al. FTY720 modulates human oligodendrocyte progenitor process extension and survival. Ann Neurol. 2008;63(1):61‐71. [DOI] [PubMed] [Google Scholar]
- 8. van Doorn R, Nijland PG, Dekker N, et al. Fingolimod attenuates ceramide‐induced blood‐brain barrier dysfunction in multiple sclerosis by targeting reactive astrocytes. Acta Neuropathol. 2012;124(3):397‐410. [DOI] [PubMed] [Google Scholar]
- 9. Rosen H, Sanna GM, Gonzalez‐Cabrera PJ, Roberts E. The organization of the sphingosine 1‐phosphate signaling system. Curr Top Microbiol Immunol. 2014;378:1‐21. [DOI] [PubMed] [Google Scholar]
- 10. Cohen JA, Barkhof F, Comi G, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362(5):402‐415. [DOI] [PubMed] [Google Scholar]
- 11. Kappos L, Radue E‐W, O'Connor P, et al. A placebo‐controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362(5):387‐401. [DOI] [PubMed] [Google Scholar]
- 12. U.S. Food and Drug Administration. Center for Drug Evaluation and Research. Fingolimod Medical Reviews, NDA 22‐527. 2010. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2010/022527Orig1s000medr.pdf. Accessed September 16, 2016.
- 13. Sanna MG, Vincent KP, Repetto E, et al. Bitopic sphingosine 1‐phosphate receptor 3 (S1P3) antagonist rescue from complete heart block: pharmacological and genetic evidence for direct S1P3 regulation of mouse cardiac conduction. Mol Pharmacol. 2016;89(1):176‐186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Legangneux E, Gardin A, Johns D. Dose titration of BAF312 attenuates the initial heart rate reducing effect in healthy subjects. Br J Clin Pharmacol. 2013;75(3):831‐841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Selmaj K, Li DKB, Hartung HP, et al. Siponimod for patients with relapsing‐remitting multiple sclerosis (BOLD): an adaptive, dose‐ranging, randomised, phase 2 study. Lancet Neurol. 2013;12(8):756‐767. [DOI] [PubMed] [Google Scholar]
- 16. Brossard P, Derendorf H, Xu J, et al. Pharmacokinetics and pharmacodynamics of ponesimod, a selective S1P1 receptor modulator, in the first‐in‐human study. Br J Clin Pharmacol. 2013;76(6):888‐896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brossard P, Scherz M, Halabi A, et al. Multiple‐dose tolerability, pharmacokinetics, and pharmacodynamics of ponesimod, an S1P1 receptor modulator: favorable impact of dose up‐titration. J Clin Pharmacol. 2014;54(2):179‐188. [DOI] [PubMed] [Google Scholar]
- 18. Olsson T, Boster A, Fernandez O, et al. Oral ponesimod in relapsing‐remitting multiple sclerosis: a randomised phase II trial. J Neurol Neurosurg Psychiatry. 2014;85(11):1198‐1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vollmer T, Selmaj K, Bar‐Or A, Zipp F. A double‐blind, placebo‐controlled, phase 2, 26‐week DreaMS Trial of a selective S1P receptor agonist ONO‐4641 in patients with relapsing‐remitting multiple sclerosis [abstract]. Neurology. 2012;79(11):E90. [Google Scholar]
- 20. Hoch M, Darpo B, Brossard P, et al. Effect of ponesimod, a selective S1P1 receptor modulator, on the QT interval in healthy individuals. Basic Clin Pharmacol Toxicol. 2015;116(5):429‐437. [DOI] [PubMed] [Google Scholar]
- 21. Tran JQ, Hartung JP, Peach RJ, et al. Results from the first‐in‐human study with ozanimod, a novel, selective sphingosine‐1‐phosphate (S1P) receptor modulator. J Clin Pharmacol. 2017;57(8):988‐996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Timony G, Brooks J, Hartung J, et al. Pharmacokinetics and pharmacodynamics of RPC1063 and its metabolites in healthy adult volunteers [abstract]. Neurology. 2014;82(10 Supplement):P1.211. [Google Scholar]
- 23. Scott F, Timony G, Brooks J, et al. Metabolites of RPC1063 contribute to in vivo efficacy [abstract]. Neurology. 2013;80(7 Supplement):P05.157. [Google Scholar]
- 24. Kovarik JM, Schmouder R, Barilla D, et al. Multiple‐dose FTY720: tolerability, pharmacokinetics, and lymphocyte responses in healthy subjects. J Clin Pharmacol. 2004;44(5):532‐537. [DOI] [PubMed] [Google Scholar]
- 25. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER); Center for Biologics Evaluation and Research (CBER). Guidance for Industry E14 Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non‐Antiarrhythmic Drugs. 2005. http://www.fda.gov/downloads/RegulatoryInformation/Guidances/ucm129357.pdf. Accessed June 24, 2014.
- 26. Fridericia LS. Die systolendauer im elektrokardiogramm bei normalen menschen und bei herzkranken [The duration of systole in the electrocardiogram of normal subjects and of patients with heart disease]. Acta Med Scand. 1920;53(1):469‐486. [Google Scholar]
- 27. Mendzelevski B, Ausma J, Chanter DO, et al. Assessment of the cardiac safety of prucalopride in healthy volunteers: a randomized, double‐blind, placebo‐ and positive‐controlled thorough QT study. Br J Clin Pharmacol. 2012;73(2):203‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ward MD, Jones DE, Goldman MD. Overview and safety of fingolimod hydrochloride use in patients with multiple sclerosis. Expert Opin Drug Saf. 2014;13(7):989‐998. [DOI] [PubMed] [Google Scholar]
- 29. Bloomfield DM, Kost JT, Ghosh K, et al. The effect of moxifloxacin on QTc and implications for the design of thorough QT studies. Clin Pharmacol Ther. 2008;84(4):475‐480. [DOI] [PubMed] [Google Scholar]
- 30. Mood AMF, Graybill FA. Introduction to the Theory of Statistics. New York, NY: McGraw‐Hill; 1963. [Google Scholar]
- 31. Tran JQ, Hartung JP, C Tompkins, Frohna PA. P752 Effects of high‐ and low‐fat meals on the pharmacokinetics of ozanimod, a novel sphingosine 1‐phosphate receptor modulator [abstract]. Mult Scler. 2016;22(3 suppl):374‐375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Piali L, Froidevaux S, Hess P, et al. The selective sphingosine 1‐phosphate receptor 1 agonist ponesimod protects against lymphocyte‐mediated tissue inflammation. J Pharmacol Exp Ther. 2011;337(2):547‐556. [DOI] [PubMed] [Google Scholar]
- 33. Gergely P, Nuesslein‐Hildesheim B, Guerini D, et al. The selective sphingosine 1‐phosphate receptor modulator BAF312 redirects lymphocyte distribution and has species‐specific effects on heart rate. Br J Pharmacol. 2012;167(5):1035‐1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shakeri‐Nejad K, Aslanis V, Veldandi UK, et al. Effects of therapeutic and supratherapeutic doses of siponimod (BAF312) on cardiac repolarization in healthy subjects. Clin Ther. 2015;37(11):2489‐2505.e2482. [DOI] [PubMed] [Google Scholar]