

A recent study by Wiley, Gruenewald, Karlamangla, and Seeman modeling multi-systemic allostatic load (AL) was published in Psychosomatic Medicine (1). The observations reported in this article are consistent with the accumulating evidence published in this journal supporting the role of additive dysregulated biological and physiological processes in a wide range of diseases (2–5). We applaud this article and, here, with the objective of identifying proximal biological processes underlying AL, examine these findings in connection to cellular and mitochondrial biology.
For over two decades, AL has been indexed using numerous stress-related biomarkers (6) and used as a tool to monitor multi-systemic physiological dysregulations and predict disease risk (7). In their analysis of nationally representative data from the Midlife in the United States II (MIDUS) Biomarker Project cohort (N = 1,255), Wiley and colleagues demonstrated that a bifactor model comprising 23 biomarkers loaded simultaneously onto a common AL factor as theorized by the AL model. Moreover, the underlying factor model for AL (i.e., “which biomarker contributes most to AL”) was consistent across a broad age range (34–84 years) and in both women and men. To accomplish this, Wiley et al. analyzed seven unique physiological system-specific factors including i) sympathetic nervous system (SNS), ii) parasympathetic nervous system (PNS), iii) hypothalamic-pituitary-adrenal (HPA) axis, iv) inflammation, v) cardiovascular, vi) glucose, and vii) lipids. Each significantly contributed to the final AL score.
As investigators in psychosomatic medicine aim to integrate psychosocial, biological, and behavioural factors to understand mind-body processes, AL has proven useful as a heuristic model (2). But its acceptance and implementation in various areas of medicine has been hampered by the lack of understanding regarding its underlying biological underpinnings. The question “What does AL actually measure?” is still under debate. This timely refinement of the model by Wiley and colleagues addresses longstanding AL measurement issues (2) and represents a step towards identifying which physiological systems contribute most directly, and most significantly to overall AL.
In tracing the causal chain from systems to cells, we note that physiological functions are in part determined by cellular and sub-cellular processes. For example, studying genetic defects and polymorphisms causing disease teaches us that events at the molecular and sub-cellular level can trickle up to influence the function of complex organ systems such as the HPA axis (8). Thus, sub-cellular factors can contribute to the state of physiological dysregulation that defines AL.
In seeking to integrate knowledge about the underlying biology for individual AL biomarkers, we found it particularly noteworthy that the analysis by Wiley and colleagues revealed that “overall, the largest factor loadings for the common AL factor were from biomarkers from the inflammation, glucose, and lipid systems” (1). This is in contrast to biomarkers that represent the sympathetic nervous system (e.g., epinephrine), parasympathetic nervous system (e.g., heart rate variability), and cardiovascular systems (e.g., systolic blood pressure), which did not load as strongly on an overall AL factor, but loaded instead onto their system-specific factors.
At the sub-cellular level, glucose, lipids, and inflammatory biomarkers share a common origin. Compared to various other AL biomarkers, these are “proximal” and directly linked to a specific cellular component that is essential to energy production and signalling – the mitochondrion. Mitochondria are endosymbiotic organelles with their own genome involved in cellular energy production and signalling. To produce energy, they utilize or “burn” circulating energy substrates (9). This in turn fuels cellular activities such as action potentials in neurons, gene expression, hormone biosynthesis, DNA repair, and cellular replication, among other processes relevant to health and disease.
The ultimate fate of blood glucose and lipids is their oxidation at the level of mitochondria (Figure 1). Glucose is metabolized first by glycolysis and generally followed by oxidative phosphorylation in mitochondria. Mitochondrial oxidation is also the major route to “burn off”, or consume lipids. This in part explains why mitochondrial dysfunction is associated with hyperglycemia and hyperlipidemia in diabetes – and why behaviors like exercise that increase energy metabolism in mitochondria decrease blood glucose and lipids (10). Mitochondria may also secrete signalling peptides that promote glucose homeostasis systemically (11). Thus, the link between mitochondria and glucose and lipid biomarkers that Wiley et al. show contribute substantially to AL, is deeply evolutionary rooted.
Figure 1. Relationship between mitochondrial function and glucose, lipids, and inflammation.
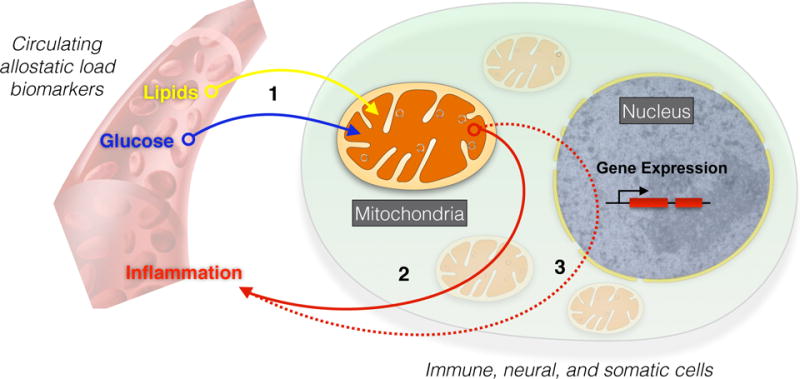
Allostatic load biomarkers are influenced by mitochondrial activities. (1) Glucose and lipids are used as fuels and directly metabolized by mitochondria, effectively removing them from the circulation when mitochondria are functioning normally. Conversely, dysfunctional mitochondria may cause elevated circulating glucose and lipid levels, contributing to allostatic load. Dysfunctional or damaged mitochondria also produce (2) damage-associated molecular pattern molecules (DAMPs) including circulating cell-free mtDNA that directly promote inflammation by activating the immune system, or (3) activate pro-inflammatory gene expression in the cell nucleus, leading to the production of pro-inflammatory cytokines in various cell types.
Likewise, faulty mitochondria can both directly and indirectly promote inflammation (Figure 1). The direct route involves the release of mitochondrial proteins and mtDNA in the blood. Because mitochondria evolved from bacteria and still carry several vestigial features of the bacterial ancestry, mitochondria-derived molecules are recognized as foreign, or immunogenic, by the immune system. Under conditions of stress, including oxidative stress, bacteria-like mitochondrial components can leak into systemic circulation and trigger systemic inflammatory pathways (12). On the other hand, the indirect route involves the release of immunogenic molecules and signalling molecules in the cell cytoplasm, in combination with oxidative stress that activates transcription factors (e.g., NF-kB, HMGB1) inducing pro-inflammatory gene expression programs and the release of cytokines (13). These mechanisms link stress at the level of mitochondria to systemic circulating levels of glucose, lipids, and inflammatory AL biomarkers (14).
Wiley and colleagues’ study (1) also represents an opportunity to reflect on the driving forces and causal pathways underlying AL. The AL model originally centered on stress hormones (e.g., cortisol) as key deregulators of multi-systemic functioning. As we move beyond a cortisol-centric view of AL towards systemic perspectives, three notions must be considered. First, the inter-relationships between individual biomarkers depend on the neuroendocrine and metabolic context in which they are present (15). This includes their direct reciprocal inhibitory/stimulatory effects and/or coupling with other systems. Second, AL indices generally include biomarkers assessed during fasting and resting conditions as originally defined (6). Wiley and colleagues’ inclusion of heart rate variability as part of MIDUS, as well as the inclusion of stress reactive measures in other studies may represent useful additions to capture systems dynamics (16). Such measurements of resting, reactive, or of physiological variability should contribute different information about physiological dysregulation, although it remains largely unclear how to interpret their contributions to AL. Lastly, common sub-cellular factors such as mitochondria may represent convergence points that simultaneously regulate multiple biomarkers (17). For example, studies of genetic manipulation of mitochondrial functions in animal models indicate that mitochondria simultaneously modulate metabolic, inflammatory, HPA and SAM axes, as well as gene expression responses to psychological stress (18). This evidence position mitochondria as modulators of systemic stress responses. More generally, it also suggests that sub-cellular factors regulate multisystemic biobehavioral processes contributing to translate stressors of various nature into variable health trajectories across the lifespan.
This “mitocentric” proposal might seem like yet another reductionist model that narrows complex physiological outcomes to more simple sub-cellular processes, possibly curtailing valuable opportunities to evaluate the health contribution of psychosocial and behavioral factors. To the contrary, we see this as an opportunity to expand psychosomatic medicine. Focusing on energy metabolism and mitochondria represents a new way to apply sensitive and biologically meaningful measures to detect the interactions among biopsychosocial factors. This is because mitochondrial functions i) directly respond to stress-related neuroendocrine mediators, ii) are modulated by behavioral factors such as physical activity and diet, iii) are partially regulated by genetic variants, and iv) exhibit age-related changes that parallel those of telomeres (14). Exploring mitochondrial functions in psychosomatic medicine therefore provides a useful theoretical and biological bridge to study the biobehavioral interactions that take place between individuals and their environment (17).
In summary, this letter outlines a proximal biochemical relationship between mitochondrial function and the biomarkers revealed by Wiley and colleagues (1) to exhibit the strongest statistical associations with a common AL factor, namely glucose, lipids, and inflammation. This association is consistent with the notion that the accumulation of mitochondrial dysfunction as a result of chronic stress – or mitochondrial allostatic load (MAL) – could represent an early event that increases AL and disease risk (14). Examining the biochemical connections linking AL biomarkers to well-defined sub-cellular processes may be critical to refine our understanding of the mechanisms by which chronic stressful experiences are translated into measurable physiological dysregulation. In the long run, this should enable the psychosomatic research community to continue identifying and target novel modifiable pathways to promote resilience to stress and trauma, and thus mitigate stress pathophysiology.
References
- 1.Wiley JF, Gruenewald TL, Karlamangla AS, Seeman TE. Modeling Multisystem Physiological Dysregulation. Psychosom Med. 2016 doi: 10.1097/PSY.0000000000000288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gallo LC, Fortmann AL, Mattei J. Allostatic load and the assessment of cumulative biological risk in biobehavioral medicine: challenges and opportunities. Psychosom Med. 2014;76:478–80. doi: 10.1097/PSY.0000000000000095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gale CR, Batty GD, Cooper SA, Deary IJ, Der G, McEwen BS, Cavanagh J. Reaction time in adolescence, cumulative allostatic load, and symptoms of anxiety and depression in adulthood: the West of Scotland Twenty-07 Study. Psychosom Med. 2015;77:493–505. doi: 10.1097/PSY.0000000000000189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Upchurch DM, Stein J, Greendale GA, Chyu L, Tseng CH, Huang MH, Lewis TT, Kravitz HM, Seeman T. A Longitudinal Investigation of Race, Socioeconomic Status, and Psychosocial Mediators of Allostatic Load in Midlife Women: Findings From the Study of Women’s Health Across the Nation. Psychosom Med. 2015;77:402–12. doi: 10.1097/PSY.0000000000000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Read S, Grundy E. Allostatic load and health in the older population of England: a crossed-lagged analysis. Psychosom Med. 2014;76:490–6. doi: 10.1097/PSY.0000000000000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seeman TE, Singer BH, Rowe J, Horwitz RI, McEwen B. Price of adaptation - allostatic load and its health consequences. Arch Intern Med. 1997;157:2259–68. [PubMed] [Google Scholar]
- 7.McEwen BS, Stellar E. Stress and the individual. Mechanisms leading to disease. Arch Intern Med. 1993;153:2093–101. [PubMed] [Google Scholar]
- 8.Meimaridou E, Hughes CR, Kowalczyk J, Guasti L, Chapple JP, King PJ, Chan LF, Clark AJ, Metherell LA. Familial glucocorticoid deficiency: New genes and mechanisms. Mol Cell Endocrinol. 2013;371:195–200. doi: 10.1016/j.mce.2012.12.010. [DOI] [PubMed] [Google Scholar]
- 9.Nicholls DG, Fergusson SJ. Bioenergetics. 4. Academic Press; 2013. [Google Scholar]
- 10.Picard M, Turnbull DM. Linking the metabolic state and mitochondrial DNA in chronic disease, health, and aging. Diabetes. 2013;62:672–8. doi: 10.2337/db12-1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee C, Zeng J, Drew BG, Sallam T, Martin-Montalvo A, Wan J, Kim SJ, Mehta H, Hevener AL, de Cabo R, Cohen P. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab. 2015;21:443–54. doi: 10.1016/j.cmet.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–7. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu B, Kwan K, Levine YA, Olofsson PS, Yang H, Li J, Joshi S, Wang H, Andersson U, Chavan SS, Tracey KJ. Alpha 7 nicotinic acetylcholine receptor signaling inhibits inflammasome activation by preventing mitochondrial DNA release. Mol Med. 2014 doi: 10.2119/molmed.2013.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Picard M, Juster RP, McEwen BS. Mitochondrial allostatic load puts the ‘gluc’ back in glucocorticoids. Nat Rev Endocrinol. 2014;10:303–10. doi: 10.1038/nrendo.2014.22. [DOI] [PubMed] [Google Scholar]
- 15.McEwen BS. Protective and damaging effects of stress mediators: central role of the brain. Dialogues Clin Neurosci. 2006;8:367–81. doi: 10.31887/DCNS.2006.8.4/bmcewen. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Juster RP, Moskowitz DS, Lavoie J, D’Antono B. Sex-specific interaction effects of age, occupational status, and workplace stress on psychiatric symptoms and allostatic load among healthy Montreal workers. Stress. 2013;16:616–29. doi: 10.3109/10253890.2013.835395. [DOI] [PubMed] [Google Scholar]
- 17.Picard M. Pathways to aging: the mitochondrion at the intersection of biological and psychosocial sciences. J Aging Res. 2011;2011:814096. doi: 10.4061/2011/814096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Picard M, McManus MJ, Gray JD, Nasca C, Moffat C, Kopinski PK, Seifert EL, McEwen BS, Wallace DC. Mitochondrial functions modulate neuroendocrine, metabolic, inflammatory, and transcriptional responses to acute psychological stress. Proc Natl Acad Sci U S A. 2015;112:E6614–23. doi: 10.1073/pnas.1515733112. [DOI] [PMC free article] [PubMed] [Google Scholar]