

Abstract
Objective
Mitochondria are multifunctional life-sustaining organelles that represent a potential intersection point between psychosocial experiences and biological stress responses. This article provides a systematic review of the effects of psychological stress on mitochondrial structure and function.
Methods
A systematic review of the literature investigating the effects of psychological stress on mitochondrial function was conducted. The review focused on experimentally controlled studies allowing us to draw causal inference about the effect of induced psychological stress on mitochondria.
Results
A total of 23 studies met the inclusion criteria. All studies involved male laboratory animals, and most demonstrated that acute and chronic stressors influenced specific facets of mitochondrial function, particularly within the brain. Nineteen studies showed significant adverse effects of psychological stress on mitochondria and four found increases in function or size after stress. In humans, only six observational studies were available, none with experimental designs, and most only measured biological markers that do not directly reflect mitochondrial function, such as mitochondrial DNA copy number.
Conclusons
Overall, evidence supports the notion that acute and chronic stressors influence various aspects of mitochondrial biology, and that chronic stress exposure can lead to molecular and functional recalibrations among mitochondria. Limitations of current animal and human studies are discussed. Maladaptive mitochondrial changes that characterize this subcellular state of stress are termed mitochondrial allostatic load. Prospective studies with sensitive measures of specific mitochondrial outcomes will be needed to establish the link between psychosocial stressors, emotional states, the resulting neuroendocrine and immune processes, and mitochondrial energetics relevant to mind-body research in humans.
Keywords: psychological/psychosocial stress, mitochondria, systematic review, psychosomatic medicine, psychoneuroendocrinology
INTRODUCTION
Mitochondria are subcellular organelles that sustain life through energy transformation and intracellular signaling. They have recently emerged as a key component of the stress response (1–3). However, unlike traditional biomarkers that reflect a specific biological process, mitochondria are complex multifunctional organisms that contain their own genome, reproduce, perform hundreds of biochemical reactions, and can adopt specific functions based on which cells they inhabit. Despite the recent rise of mitochondrial research in modern medicine (4), mitochondrial biology is relatively poorly understood compared with other aspects of cellular biology. Nevertheless, research over the past decade has demonstrated that genetic and biochemical mitochondrial defects cause debilitating multisystemic clinical disorders (5), and animal models have confirmed that changes in mitochondria directly influence systemic metabolic regulation (6), brain function (7), immune activation (8), and even the rate of aging and life-span (9–11).
In relation to stress pathophysiology, research has used various indicators of stress (e.g., psychological distress, social or life stressors, negative emotional states, and psychiatric disorders) and has mostly examined physiological processes centered around three systems: the brain, the endocrine, and the immune systems (12,13). The resulting findings from psychosomatic medicine, psychoneuroimmunology, and psychoneuroendocrinology have demonstrated that psychosocial experiences and life exposures influence subcellular processes including immune activation/suppression (14,15), oncogenic behavior (16), gene expression regulation (17), telomere maintenance (18), and epigenetic processes (19). Emotional states—both adverse (e.g., anxiety and depression) and positive (e.g., happiness and optimism)—are thus suspected to trigger broad-acting biological pathways such as inflammation that meaningfully influence morbidity and mortality (20). But how do external exposures make their way “under the skin” and “into the genome”? Do mitochondria contribute to the transduction of stressful experiences into lasting biological consequences that affect human health and disease?
Recent discoveries provide converging evidence implicating mitochondria at two major levels: as a target of stress and as mediator of stress pathophysiology. The latter portion of this model proposes that mitochondria underlie systemic recalibrations associated with stress pathophysiology and allostatic load, and consequently, that dysfunctional mitochondria are harbingers of stress-induced molecular and cellular alterations leading to the biological embedding of stress. For a comprehensive discussion of mitochondrial stress transduction and of the links between key concepts in psychosomatic medicine, psychoneuroendocrinology, psychoneuroimmunology, and mitochondria, the reader is referred to the associated article in this issue of Psychosomatic Medicine (21). Here, we examine evidence about the first step of this model, namely, whether mitochondria respond to psychological stress exposure. We first provide a brief introduction of mitochondrial structures and function relevant to stress pathophysiology.
Mitochondrial Structures
Mitochondria exhibit several structural features of their bacterial origin. This includes a double-membrane organization: the inner and outer mitochondrial membranes, which enclose the mitochondrial matrix (Fig. 1). Under stress, mitochondrial can swell and membranes become distended. The mitochondrial matrix contains enzymes of the Kreb’s cycle (also known as the tricarboxylic acid (TCA) cycle) and β-oxidation pathway that metabolize ingested energy food substrates, including sugars and fats, respectively (27). Another vestige of bacterial heritage is their circular mitochondrial DNA (mtDNA) (Fig. 2). Mitochondria are the only organelle to contain their own genome. In humans and animals, the mtDNA is uniquely inherited from the mother (30) and harbors single-nucleotide polymorphisms that correlate with ethnicity (31,32). The mtDNA does not have loose ends and thus does not contain telomeres, the DNA-protein complexes that cap the end of chromosomes in the nucleus (33). The mtDNA also lacks introns and is more susceptible to damage in comparison to the nuclear genome (34), which may explain its greater vulnerability to damage with aging, and possibly with chronic stress.
FIGURE 1.
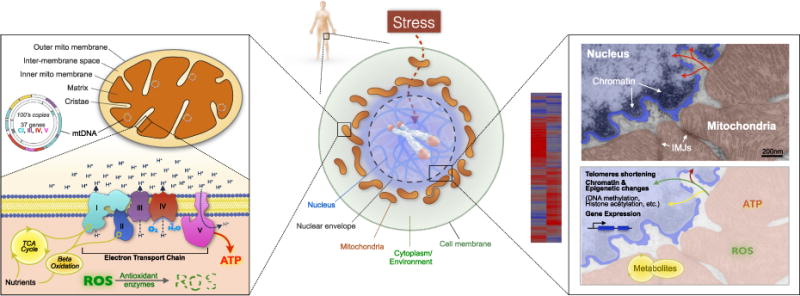
Mitochondrial structures, functions, and their positioning within the cell. (Center) Psychosomatic medicine research aims to understand how psychosocial and behavioral exposures including stress influence biological and physiological processes across the organism, including those inside the cell nucleus where genes are transcriptionally and epigenetically regulated. Mitochondria are positioned in the cell cytoplasm at the interface between psychosocial and behavioral factors and the cell nucleus. (Left) Detailed cartoon of mitochondrial structures, including the electron transport chain (respiratory chain) illustrating the flow of energy from nutrients to the pumping of protons (H+) to generate membrane potential, in part used to synthesize ATP, the energy currency of the cell. This process generates ROS, which are detoxified by specific mitochondrial antioxidant enzymes. (Right) Magnification of the mito-nuclear interface in a pseudo-colored electron micrograph showing the physical proximity between the chromatin in the nucleus and mitochondria (22). Mitochondria also communicate with each other via IMJs, nanotunnels, and other mechanisms (23). Signals derived from mitochondrial metabolism interact with other biochemical factors to coordinate gene expression and telomere maintenance via transcriptional and epigenetic mechanisms (24–26). ROS = reactive oxygen species; IMJs = intermitochondrial junctions; mtDNA = mitochondrial DNA; TCA = tricarboxylic acid; ATP = adenosine triphosphate. Color image is available only in online version (www.psychosomaticmedicine.org).
FIGURE 2.
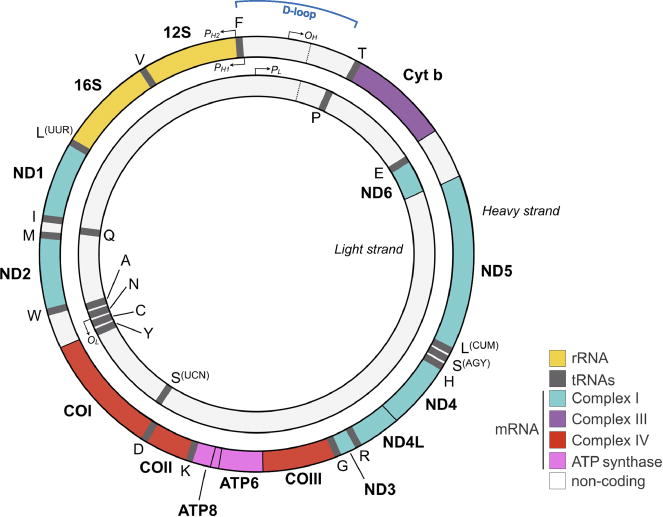
The human mitochondrial genome (mtDNA). The mtDNA contains 16,569 nucleotides and encodes 37 canonical genes, including 2 rRNA and 22 tRNA required for protein synthesis. Encoded proteins constitute part of the respiratory chain and include seven subunits of complex I, one subunit of complex III, three subunits of complex IV, and two subunits of complex V. Other mtDNA-encoded mRNA transcripts giving rise to secreted mitokines have also been described (28). The mtDNA is composed of light (inner circle) and heavy (outer circle) mtDNA strands. The mtDNA is replicated from two origins of replication on the light and heavy strands termed OL and OH, respectively. mtDNA gene expression is initiated from promoters on the heavy strand (PH1 and PH2) and on the light strand (PL). The glucocorticoid receptor and other transcription factors interact with the mtDNA near the D-loop (29). Ethnic differences exist in mtDNA sequence (31,32). Note that colors for each gene are matched to the respiratory chain complexes, of which they encode a subunit in Figure 1. mtDNA = mitochondrial DNA; rRNA = ribosomal RNA; tRNA = transfer RNA; mRNA = messenger RNA; ATP = adenosine triphosphate. Color image is available only in online version (www.psychosomaticmedicine.org).
Mitochondrial Functions
The mitochondrial genome is of prime importance because it encodes essential genes for energy production by the respiratory chain. The respiratory chain, also known as the electron transport chain, is located in the tight folds of the inner membrane called “cristae” and contains five multiprotein enzyme complexes—I, II, III, IV, and V (Fig. 1, left). Most of the mitochondrial proteins (>1500) are encoded in the nuclear genome, except for the 13 encoded by the mtDNA (Fig. 2). All mtDNA-encoded proteins are subunits of the respiratory chain complexes I, III, IV, and V. Only complex II (succinate dehydrogenase) is fully encoded by the nuclear genome. The respiratory chain complexes are large enzymes composed of multiple proteins that transport energy in the form of electrons derived from ingested food substrates, initially catabolized by the TCA cycle and β-oxidation. The terminal respiratory chain complex IV (cytochrome c oxidase, or COX) combines the transported electrons with breathed oxygen (O2), which acts as ultimate electron acceptor. The energy harnessed from this process thus establishes the life-sustaining mitochondrial electrochemical gradient (ΔΨ), akin to a charged battery. Generating the mitochondrial electrochemical gradient is the ultimate reason why living organisms must eat and breathe. The flow of energy through mitochondria distinguishes the living person from the inert body; it enables mitochondrial functions required for life.
Other than energy production, mitochondria perform multiple essential functions that influence gene expression within the cell nucleus (Fig. 1, right) and physiological regulation across the organism (21). Notably, mitochondria are the major producers of reactive oxygen species (ROS) within the cell (35,36), which play signaling and other life-sustaining roles at low levels, but can lead to oxidative stress when they overwhelm antioxidant defense mechanisms, possibly playing key role in neurodegenerative processes (37) and in stress pathophysiology.
The energy harnessed from electron transport stored across the inner mitochondrial membrane as membrane potential is used to drive multiple distinct functions. One of the first to be discovered was the production of adenosine triphosphate (ATP) (38), which fuels most cellular reactions, including gene expression, chromatin remodeling, ion homeostasis, protein and hormone synthesis, secretion, neurotransmitter release and reuptake, and muscle contraction, to name a few. The metaphor of “mitochondria as powerhouse” has thus dominated the imagination of biologists and the public over the past half-century. The ability of mitochondria to use oxygen at the level of the respiratory chain complexes, referred to as mitochondrial oxidative capacity, thus depends on a functional respiratory chain and intact mtDNA. Mitochondrial respiratory capacity can be measured directly in fresh cells by respirometry (39–41), or indirectly from frozen samples by measuring the enzymatic activity of respiratory chain complexes (42). Mitochondrial ATP synthesis can also be directly measured in fresh cells (43).
Quantity Versus Quality
The “quality” or function of mitochondria also varies independently from mitochondrial content and this may influence their responses to stress. For instance, tissues with different metabolic demands contain different types of mitochondria, defined either by their function (44) or by protein composition (45). In neurons, synaptic mitochondria qualitatively differ from mitochondria in the cell body (46)—even if they are from the same cell. Therefore, combining metrics representing both mitochondrial function and content provides a fuller picture of mitochondrial recalibrations. Stress exposure may also independently influence mitochondrial content and function, which may have markedly different effects on cellular and organismal health. This underscores the empirical value to assess in parallel both mitochondrial content or function outcomes to enable an accurate interpretation of stress-induced mitochondrial recalibrations.
Here we present a systematic review evaluating experimental studies of the effects of induced psychological stress on mitochondrial structures and functions.
METHODS
The study selection process for this systematic review was based on PRISMA guidelines (47). Search terms included the following: “mitochondri*” (e.g., mitochondion, mitochondria, and mitochondrial), stress* (e.g., stress, stressful, and stressor), and psycholog* (e.g., psychology and psychological). Date limits were from 1960 to January 1, 2017. Only studies in English or French were included. In categorizing reasons for rejection, in cases where multiple criteria were not met “review” had precedence over “not psychological stress,” which had precedence over “not mitochondrial function,” which had precedence over other categories (Fig. 3).
FIGURE 3.
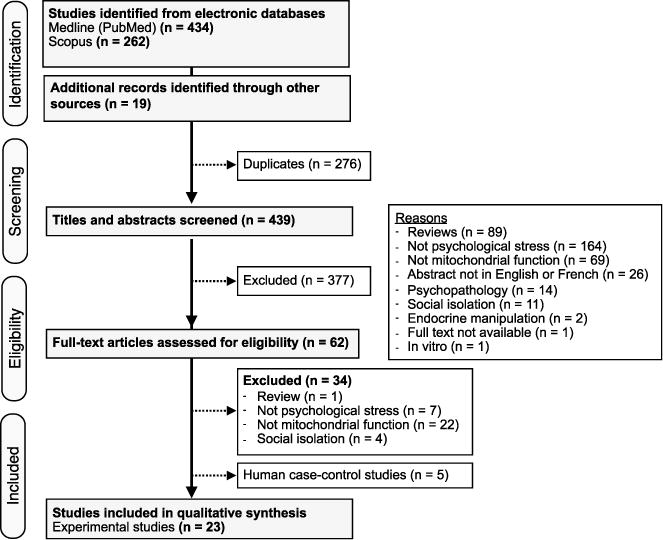
Flow diagram for study selection in the systematic review. See “Methods” for details.
Only studies directly measuring at least one aspect of mitochondrial function (e.g., respiration, respiratory chain enzymatic activity, and ROS production), or electron microscopy studies quantifying changes in mitochondrial morphology or structure, were included. Studies reporting single molecular markers that only provide indirect information on mitochondrial function were not included. Similarly, only studies of psychological stress were retained, whereas studies of physical of metabolic stressors such as food or water restriction were not included. Social isolation studies, which differ in several ways from traditional acute and chronic stress paradigms, were not included in this review, although two reports have also suggested effects on mitochondrial function (48,49).
Notably, after screening titles and abstracts, 22 articles listing “mitochondrial function” as a main outcome in the abstract did not actually assess functional measures and were therefore excluded. These articles, some of which are discussed hereinafter in the section “Additional Findings In Animal Studies,” assessed protein markers, gene expression, or other molecular markers (e.g., metabolites and apoptotic cell death). After exclusion of ineligible studies, 23 articles were retained and included in this systematic review.
RESULTS
Table 1 summarizes data for each study including species, stressor, and main outcome measures. There were no human study with induced psychosocial stress monitoring mitochondrial function, so all studies included are in animal models. Rats were most often studied (58% of studies), followed by the mouse (38%) and one study in gerbils. The stressors most frequently used was chronic restraint stress (e.g., 21 consecutive days of physical restraint, 2 hours per day; 38%), followed by chronic unpredictable stress (e.g., 40 consecutive days with alternating novel environmental exposures on each day; 33%) and acute restraint stress (e.g., a single bout of physical restraint lasting 30–120 minutes; 21%). Other stressors included noise exposure and cage overcrowding. The most studied organs were the brain (67%), the heart (13%), and other specialized tissues (e.g., testes and gut; 21%). Only one study examined more than one tissue, precluding robust conclusions about whether induced stress has organ-specific effects. In 46% of studies, mitochondrial function and integrity were most often measured via respiratory chain enzymatic activity, which can be assessed on frozen tissues (73). Other outcomes included oxygen consumption (33%), quantitative electron microscopy (29%), membrane potential (13%), ATP synthesis (13%), and ROS production (8%). Mitochondrial DNA copy number, which is not a measure of mitochondrial function, was measured in 9% of studies. This distribution of outcome measures reflects in part the technical aspects of mitochondrial studies. Enzymatic activities can measured on frozen tissues, whereas other functional measurements require fresh tissues and are therefore more difficult to implement. This also illustrates the multifunctional nature of mitochondria and the need to empirically assess multiple aspects of their function to define the impact of stress on mitochondrial behavior or phenotype.
TABLE 1.
Results From the Systematic Review of Experimental Studies Evaluating the Effects of Induced Psychological Stress on Mitochondrial Structure and Function
| Author | Year | Reference | Species | Sex | Stressor | Tissue | Mito Outcome | Result |
|---|---|---|---|---|---|---|---|---|
| Andric et al. | 2013 | (50) | Rat | ♂ | ARS (2 h), CRS (2 d, 10 d) | Testes, Leydig cells | Respiration, ΔΨm | ↓ Respiration, ↓ ΔΨm |
| Batandier et al. | 2014 | (51) | Rat | ♂ | ARS (0.5 h) | Brain, forebrain | Respiration, RC activity, ROS production, PTP sensitivity | ↓ CI-driven respiration, ↓ CI activity, ⟷ ROS production, ↓ PTP sensitivity (apoptotic resistance) |
| Cai et al. | 2015 | (52) | Mouse | ♂ | CUS | Liver, saliva, hippocampus, skeletal muscle, ovary | Respiration (liver only), mtDNAcn | ↓ increase in respiration (State 3/State 2), ↑ mtDNAcn |
| Gak et al. | 2015 | (53) | Rat | ♂ | ARS (2 h) CRS (2 d, 10 d) | Testes, Leidig cells | ΔΨm, EM | ↓ ΔΨm at 1 and 2 d, mito swelling, ↑ mitochondrial content at 10 d |
| García-Fernández et al. | 2012 | (54) | Mouse | ♂ | CRS (21 d) | Brain, hippocampus | RC enzyme activity, MnSOD activity | ⟷ COX activity in normal mice, [↓ in malpar1 KO (>60%)] |
| Gesi et al. | 2002 | (55) | Rat | ♂ | Noise (6, 12 h) | Cardiomyocytes, right atrium, and ventricle | EM | 20%–30% of swollen mitochondria (additional data in Gesi et al. 2002) |
| Gong et al. | 2011 | (56) | Mouse | ♂ | CUS | Brain, hippocampus, prefrontal cortex, hypothalamus | Respiration, ΔΨm, EM | ↓ Maximal respiration, ↓ ΔΨm, mito swelling |
| Heinzeller | 1985 | (57) | Gerbil | ♂ | Aggression | Brain, pineal gland | EM | ↓ Mito size with night stress, not day stress |
| Kambe and Miyata | 2015 | (58) | Mouse | ♂ | CRS (14 d) | Brain, forebrain | Respiration | ↓ CI-driven respiration (State 3, 250 μMADP), ⟷ leak respiration (State 4) |
| Li et al. | 2012 | (59) | Mouse | ♂ | ARS (18 h) | Spleen lymphocytes | ΔΨm, Cyt c release | ↓ ΔΨm, ↑ cyt c release, ↑ apoptosis |
| Liu et al. | 2004 | (60) | Rat | ♂ | CRS (21 d) | Heart, ventricle | Respiration, ATP synthesis | ↓ Coupling efficiency (10%–20%), ↓ ATP synthase activity (50%), ↓ ATP |
| Liu and Zhou | 2012 | (61) | Rat | ♂ | CUS (40 d) | Brain, cortex, hippocampus, striatum | RC enzyme activity, ROS production, mtDNAcn | ↑ ROS production, ↓ CI, CIV activity (30%–40%), ↓ mito content (30%), ↓ mtDNAcn (50%–60%) [protection by exercise] |
| Madrigal et al. | 2001 | (62) | Rat | ♂ | CRS (7, 14, 21 d) | Brain, forebrain | RC enzyme activity, respiration, ATP synthesis | ↓ CI, CII activity (50%–70%), ⟷ CIV activity ⟷ respiration, ⟷ ATP synthesis |
| Magarinos et al. | 1997 | (63) | Rat | ♂ | ARS (6 h), CRS (21 d) | Brain, dorsal hippocampus, mossy fibers | EM | ↑ Mitochondrial size (CRS), ⟷ with ARS |
| Martin-Aragon et al. | 2016 | (64) | Mouse | ♂ | CRS (4 d) | Brain, cortex | RC enzyme activity | ↓ CIV activity (30%–50%) [protection by esculetin] |
| Ortmann et al. | 2016 | (65) | Rat | ♂ | CUS (40 d) | Brain, multiple regions | RC enzyme activity | ↑ CI, CII,CIII (20%–100%); ⟷ CIV activity (modified by flavanoid) |
| Rezin et al. | 2008 | (66) | Rat | ♂ | CUS (40 d) | Brain, multiple regions | RC enzyme activity | ↓ CI, CIII, CIV activity (30%–50%) in cerebellum and cortex only, ⟷ CII |
| Rinwa and Kumar | 2012 | (67) | Mouse | ♂ | CUS (28 d) | Brain, whole | RC enzyme activity | ↓ CI, CII, CIII, CIV activity (50%–80%) [protection by curcumin and piperine] |
| Rinwa and Kumar | 2014 | (68) | Mouse | ♂ | CUS (28 d) | Brain, hippocampus | RC enzyme activity | ↓ CI, CII activity (70%–80%) [partial protection by L-NAME and Ginsen] |
| Seo et al. | 2011 | (69) | Mouse | ♂ | CRS (16 d) | Brain, hippocampus | RC enzyme activity | ↓ CIV, ⟷ CI,CII,CS |
| Soldani et al. | 1997 | (70) | Rat | ♂ | Noise (1, 6, 12 h) | Heart, ventricles | EM | Swelling (10%–20%), vacuolization, loss of cristae integrity |
| Vicario et al. | 2012 | (71) | Rat | ♂ | Crowding (15 d) and Rec (1 h–30 d) | Intestinal mucosa | RC enzyme activity + CS, RC protein levels | During recovery ⟷ CII and CIV, ↓ CS (↑ CII/CS and CIV/CS ratios); stress ↓ COXI protein abundance [↑ by CRF] |
| Wen et al. | 2014 | (72) | Rat | ♂ | CUS (28 d) | Brain, raphe nucleus | Respiration, ATP synthesis, SOD and GPX activity | ↑ Respiratory control ratio, ↑ ATP synthesis, ↑ SOD and GPX activity [exercise prevented these effects] |
ARS = acute restraint (immobilization) stress; CRS = chronic restraint (immobilization) stress; ΔΨm = mitochondrial membrane potential; RC = respiratory chain, also electron transport chain; ROS = reactive oxygen species; PTP = mitochondrial permeability transition pore; CI = respiratory chain complex I, NADH dehydrogenase; CUS = chronic unpredictable stress, also known as chronic mild stress; mtDNAcn = mitochondrial DNA copy number; EM = electron microscopy; MnSOD = manganese superoxide dismutase; COX = cytochrome c oxidase; KO = knockout; = ADP = adenosine diphosphate; Cyt c = mitochondrial cytochrome c; ATP = adenosine triphosphate; CIV = respiratory chain complex IV, COX; CII = respiratory chain complex II, succinate dehydrogenase; CIII = respiratory chain complex III; L-NAME = N-nitroarginine methyl ester; CS = citrate synthase; COXI = cytochrome c oxidase, subunit 1; Rec = recovery; CRF = corticotropin releasing factor; SOD = superoxide dismutase; GPX = glutathione peroxidase.
State 2/4 respiration represents oxygen consumption in the absence of ADP (mitochondria not making ATP); State 3 respiration represents oxygen consumption in the presence of ADP (mitochondria actively making ATP); ↑, increase; ↓, decrease; ⟷, unchanged. Items in Results maked by [ ] are interventions/moderators shown to modify the effect of stress on mitochondrial outcomes.
Four main themes emerge from this body of work. First, chronic stress induced through a form of psychosocial stressor decreases mitochondrial energy production capacity and alters mitochondrial morphology. This manifested at multiple levels, in respiratory chain enzymatic activity of various complexes, in the rate or oxygen consumption (i.e., respiration) measured directly from fresh tissues or isolated mitochondria, in mitochondrial membrane potential, or in mitochondrial content (Table 1).
Second, acute and chronic stressors have different and in some cases opposite effects on mitochondrial functions. Acute stress may damage mitochondrial structure within hours and enhance certain aspects of their function. Several studies investigating mitochondrial morphology and ultrastructure by electron microscopy reported acute mitochondrial damage with stress (53,55,57,70,74), whereas other showed increased size of mitochondria with chronic stress (63), possibly reflecting an increase in biogenesis. Although some studies reported gross disruption in mitochondria indicative of dysfunction and pathological swelling, the lack of quantitative analysis of mitochondrial shape and morphology precludes conclusions regarding the effects of stress on mitochondrial shape. Differential effects on mtDNA copy number (mtDNAcn) have also been reported. One study found 50% to 60% decreased mtDNAcn in various brain regions after chronic unpredictable stress (61), whereas another study revealed increased mtDNAcn in peripheral tissues after chronic restraint stress (52). The liver, in particular, showed the highest change after 4 weeks of stress, with a doubling of mtDNAcn (75). The increase in mtDNAcn is in keeping with compensatory up-regulation of biogenesis and greater mtDNA replication in response to energy deficiency when the mtDNA is mutated (76,77). Antioxidant enzymes may also be up-regulated acutely, likely as an adaptive response, but then decrease with chronic restraint stress (78,79). Overall, these data are consistent with an inverted U-shaped relationship linking the duration of psychosocial stress with mitochondrial changes, and mitochondrial allostatic load (MAL) more generally (21).
Third, because parts of the respiratory chain are encoded by mtDNA and other components are not, comparing them allows us to discern functional changes that could arise form mtDNA defects and those arising from other mechanisms, such as changes in biogenesis or regulatory molecular modifications of various proteins. Among the respiratory chain complexes, complex IV (COX), which is encoded by the mtDNA, was the most frequently affected. Studies, mostly conducted with the brain, reported changes in COX activity ranging from 0 to 80% decreased specific COX activity. Unlike other enzymes, no study found increase in COX activity from stress. However, one study found selective decrease in complex I and complex II (succinate dehydrogenase), but not in COX (62).
Fourth, certain factors can influence mitochondrial vulnerability to stress. One study did not find significant alterations of mitochondria function in normal mice, but observed a significant decrease in COX activity in transgenic mice (lacking the Malpar1 gene) (54). Such observation suggests that genetic factors can sensitize or confer a predisposition of mitochondria to the effects of psychological stress. Although not the direct focus of this review, several studies also included an intervention arm where animals either exercised or received specific small-molecule compounds acutely or chronically before stress exposure. As indicated in Table 1 (see results marked with “[ ]”), certain compounds including antioxidants conferred protection against stress-induced mitochondrial dysfunction (64,65,67,68), indicating the existence of stress-sensitizing and stress-buffering factors (i.e., moderators) for the effects of induced stress on mitochondria.
Additional Findings in Animal Studies
This section discusses some studies that did not meet the criteria for inclusion in the present systematic review of the literature because they did not directly measure mitochondrial function, but nevertheless provided strong complimentary indirect evidence that stress alters some aspect of mitochondrial energetics. For example, some studies described herein evaluated specific molecular components of mitochondria (e.g., proteins inside mitochondria), which, although they do not represent function, may reflect some form of molecular recalibration and could constitute the molecular basis for functional defects observed in functional studies. In this section we discuss examples of these complimentary outcomes, which when assessed in parallel with functional outcomes will reinforce the interpretation of future studies.
Some studies analyzed stress biomarkers in rats exposed to chronic mild unpredictable stress using metabolomics. Metabolomics allows for the unbiased detection of hundreds to thousands of metabolites, many of which are directly or indirectly derived from mitochondrial metabolism (80). One study in particular showed circulating metabolic profiles in response to induced stress that are suggestive of altered energy metabolism implicating mitochondrial respiratory chain dysfunction (81). Mice with different anxious-like behavior (e.g., avoidance of open spaces) also have different metabolomic signatures in the brain and blood (82). Interestingly, parallel work in humans has also related various emotional states (e.g., depressive symptoms) to certain metabolomic profiles, such as amino acids, using cross-sectional research designs (20,83).
Other studies have performed detailed analyses of the ensemble of proteins contained in mitochondria using proteomics, finding widespread changes in the protein composition of brain mitochondria under stress (84). The mitochondrial proteome also differs between mice with low- versus high-anxiety–like behaviors (85), suggesting that different mitochondrial phenotypes in the brain may be at the origin of anxious traits and social behavior (86) and influence physiological stress responses (87), as discussed previously.
At the gene level, transcriptomics has been used to reveal that restraint stress alters gene expression widely in the hippocampus (88), but also of mtDNA-encoded genes (87,89). The stressor duration may also have varying effects. In rats, acute stress induces down-regulation of several mtDNA genes, whereas chronic stress causes opposite effects on certain mtDNA transcripts that compose of respiratory chain complex I (89). These changes seem to depend on glucocorticoids and may involve epigenetic changes in mtDNA methylation (89), consistent with the effect of the glucocorticoid receptor (GR) on mtDNA. Accordingly, cortisol treatment of isolated mitochondria enhances respiratory chain efficiency (energy produced per oxygen consumed) and increases complex I–driven respiration, which is partially encoded by the mtDNA, in a mechanism that requires mitochondrial gene expression and protein synthesis (90). In contrast, cortisol does not seem to have an effect on complex II (90), which is entirely encoded by the nuclear DNA, suggesting that glucocorticoids acutely and chronically alter mitochondrial respiratory chain function directly by acting on the mtDNA.
Additional Findings Based on Human Studies
This systematic review yielded six articles reporting mitochondrial function or molecular marker of mitochondrial health in humans exposed to different life stressors. Because of the lack of experimental manipulation of emotional states, which precludes definitive conclusions regarding the directional and causal effects of psychological distress on mitochondria, none of the human studies could be included in the systematic review. Nevertheless, these data form the basis of a new field and deserve discussion here, in the context of evidence presented previously. Table 2 summarizes available human observational studies linking self-reported or interview-based measures of psychosocial stressors and psychological distress, including psychopathology, to some aspects of mitochondrial biology.
TABLE 2.
Studies Investigating the Association of Psychosocial Stressors and Emotional States With Mitochondria in Humans
| Author | Year | Reference | Sex | Stressor | Assessment | Tissue | Mito Outcome | Result |
|---|---|---|---|---|---|---|---|---|
| Boeck et al.a | 2016 | (91) | ♀ | ACE (severity) | Self-report | PBMCs (frozen) | Cellular respiration | Higher cellular respiration in women with severe CTQ scores, positively correlated with inflammatory markers IL-1β, IL-6, and TNFα |
| Cai et al. | 2015 | (52) | ♀ | ACE, SLE, PD (yes, no) | SCID, self-report | Whole blood | mtDNAcn | Higher mtDNAcn with life history of depression and childhood trauma |
| Cai et al. | 2015 | (75) | ♀ | ACE, SLE, PD (yes, no) | SCID, self-report | Whole blood | mtDNA mutations | Higher mtDNA heteroplasmy (mutation load) in depressed individuals versus controls |
| Lindqvist et al. | 2016 | (92) | ♀,♂ | Suicide attempt (yes, no) | SCID | Plasma | ccf-mtDNA | Elevated levels of ccf-mtDNA in suicide attempters versus controls |
| Picard et al.a | 2018 | (93) | ♀ | Caregiving status (yes, no), daily mood (positive, negative) | SCID, self-report | PBMCs | RC enzyme activity and protein, mtDNAcn | Lower MHI in caregivers versus controls, highest effect size for CIV, mediated by positive mood; no difference in mtDNAcn or mitochondrial content |
| Tyrka et al. | 2015 | (94) | ♀,♂ | ACE, PD (yes, no) | SCID, self-report | Whole blood | mtDNAcn | Higher mtDNAcn with both psychopathology (mixed types) and childhood trauma |
ACE = adverse childhood experiences (includes trauma measured by the CTQ and other early life stressors); PBMCs = peripheral blood mononuclear cells; CTQ = Childhood Trauma Questionnaire; IL-1β = interleukin 1β; IL-6 = interleukin 6; TNFα = tumor necrosis factor α; SLE = stressful life events; PD = psychiatric disorders (includes anxiety, major depression, bipolar, posttraumatic stress, and substance abuse); SCID = Structured Clinical Interview for DSM-III/IV; mtDNAcn = mitochondrial DNA copy number; ccf-mtDNA = circulating cell-free mitochondrial DNA; RC = respiratory chain, also electron transport chain; MHI = mitochondrial health index; CIV = respiratory chain complex IV, cytochrome c oxidase.
Denotes studies reporting functional mitochondrial outcomes.
One study found higher mtDNAcn in whole blood from American individuals who had endured either parental loss or childhood maltreatment (adverse childhood experiences, or ACEs) and with psychopathology including major depression, depressive disorders, anxiety disorders, and substance abuse disorders compared with controls without ACE (94). Another study measured whole-blood mtDNA in Chinese women with clinical levels of depressive symptoms, and also found higher mtDNAcn (52) and higher levels of mtDNA mutations (i.e., heteroplasmy) compared with women without depression (75). However, these studies are confounded by the use of whole blood (95,96) (see discussion of limitations hereinafter) and the cross-sectional study design, which cannot tease apart directionality or causality. Whether psychological symptoms (e.g., anhedonia and sadness in major depression) cause changes in mtDNA, or whether changes in mtDNA reflect an underlying pathophysiological state that contributes to psychological symptoms (i.e., reverse causation), remains to be determined.
Another study measured mitochondrial respiration and cellular mitochondrial content in frozen leukocytes of women retrospectively reporting ACEs (91). Using the Childhood Trauma Questionnaire, women were categorized as having “none,” “low/moderate,” or “severe” childhood maltreatment load based on the sum of their ACE frequency. This study found that compared with those who reported no maltreatment load, women with a severe load had blood leukocytes consuming more oxygen under basal (unstimulated) conditions. This result could reflect the combined product of energy demand by the cell and endogenous mitochondrial function, suggesting that early life trauma may reprogram cellular energetics (91). However, it does not specifically reflect mitochondrial dysfunction. Mitochondrial content, assed by citrate synthase (CS) activity, was also similar between individuals with varying levels of childhood trauma and controls. Notably, this research revealed that certain mitochondrial respiratory parameters were correlated with systemic inflammatory markers. Thus, these findings hint at two possibilities: either that stress-induced inflammation influences cell energetics, or that experiencing such psychosocial stressors (and potentially the emotional responses associated with these experiences) causes alterations in cellular or mitochondrial energetics, which in turn induce inflammation. The second possibility is consistent with in vitro work and mitochondrial stress transduction leading to biological embedding of life exposures (21), but further work is needed to establish the directionality of these effects.
One study evaluated the levels of circulating cell-free mtDNA (ccf-mtDNA) in plasma from individuals after nonviolent suicide attempt, an acute stressful event presumably preceded by psychological distress, compared with individuals who did not engage in suicidal behaviors (92). Suicide attempters had strikingly elevated (Cohen d = 2.6) level of ccf-mtDNA compared with controls, possibly reflecting MAL. Because ccf-mtDNA lies outside the cells, circulating in the liquid fraction of blood, it does not contribute to tissue energy production capacity and therefore does not represent mitochondrial function. Current evidence suggests that ccf-mtDNA is a general sign of physiological stress particularly induced by injury and associated with inflammation (21,97). It will therefore be interesting to understand the link between psychological stress, the neuroendocrine and cellular factors that lead to mtDNA release, and both their immediate and long-term physiological effects.
Another study targeted caregiver women who care for a child with autism spectrum disorder, which is a psychopathology that fosters elevated levels of psychological distress in the caregivers given its detrimental impact in various life domains of the child (e.g., persistent deficits in social communication, stereotyped or repetitive behaviors and interests). The authors measured respiratory chain maximal enzymatic activities, mitochondrial content, respiratory chain protein abundance, and mtDNAcn in isolated mixed blood cells (93). Although mitochondrial content was unaltered, mitochondrial respiratory chain activity per mitochondrion–indexed as mitochondrial health–was significantly lower in caregivers compared with controls, with the largest group difference effect size being for COX (which is mtDNA-encoded). This result is similar to findings in animal models which, collectively, implicate mtDNA-encoded components that may decrease the overall energy production capacity of mitochondria in blood cells exposed to chronic stressors. Of relevance to the psycho-somatic model, this study found that morning and evening mood on days preceding blood draw accounted for 12% to 15% of the variance in the mitochondrial health index (MHI) on subsequent days, but not the reverse, suggesting that mitochondria respond to proximal emotional states. Accordingly, lower positive mood was a significant mediator of the effect between the chronic life stressor (i.e., caregiving of a child with autism spectrum disorder versus controls) and poorer mitochondrial health (93), potentially indicating a directional effect of acute psychological states on mitochondrial function.
Overall, there are very few studies in human subjects and most did not directly assess mitochondrial function. Nevertheless, the preliminary data available thus far support a potential association of psychosocial stressors and psychological distress with some aspects of mitochondrial function, but limitations in design and methodology encourage caution in interpreting these results.
DISCUSSION
Studies meeting the inclusion criteria for this systematic review collectively demonstrate four main points. First, chronic stress paradigms cause significant effects on mitochondrial energy production capacity and mitochondrial morphology. Second, acute and chronic stress may have different and possibly opposite effects on mitochondrial functions. Third, specific elements of mitochondrial function may be differentially affected by stress, such as mtDNA-encoded complex IV (COX). Fourth, behavioral, genetic, and dietary factors may influence mitochondrial vulnerability to stress. Collectively, available evidence supports the hypothesis that psychological stress causes MAL and calls for the need for more research investigating this question particularly in humans.
Evidence from metabolomic, proteomic, and transcriptomic studies also suggest additional layers of regulation by which stressful experiences may alter mitochondrial components among rodents. Although these metabolites, protein composition, and gene expression outcomes do not reflect the functionality of mitochondria, when assessed in parallel with functional outcomes, they will help explain and refine our understanding of the mechanisms by which stress influences mitochondrial function and health. In addition, whereas functional changes should be regarded as primary indicators of MAL (21), stress-induced molecular changes within mitochondria may also reflect compensatory mechanisms or recalibrations that contribute to long-term changes in mitochondrial function and to the accumulation of MAL.
Limitations of Animal Studies
Albeit informative, the reviewed animal studies include some major limitations. a) The lack of direct measurement of mitochondrial content to normalize activity metrics makes it impossible to distinguish between overall decrease in mitochondrial content and decreased function or “quality” of individual organelles. This would be resolved by measuring in parallel both markers of mitochondrial content and function in future work. b) Another limitation is the improper methodology to measure mitochondrial membrane potential, which could be improved by the use of ratiometric dyes, and also controlling for mitochondrial content. c) Within each study, analyses were conducted mostly in only one tissue, not enabling to determine the general versus tissue-specific effects of induced stress. d) The studies using prolonged stressors (e.g., restraint for >8 hours) do not always specify whether such duration prevented normal feeding/drinking, which could either counteract or further exaggerate some effects of stress on mitochondrial functions. e) Restraint stress paradigms commonly used could be construed as a “physical” stressor, and consequently, the mitochondrial changes reported may reflect physical rather than psychological stress. It is also apparent that stress induction paradigms and methodologies are not uniform across laboratories, which could contribute to discrepancies in the effects of stress on mitochondrial outcomes outlined in this systematic review. This is also true of assays used to characterize mitochondrial functions, because specific methods favored and available vary from one laboratory to another. Only few studies, apart from those assessing respiratory chain enzymatic activity in brain showing consistent overall decrease maximal enzymatic activity, have been replicated and have provided a satisfactory level of confirmation.
Perhaps the most notable and ubiquitous limitation of existing studies reviewed here is the exclusive use of male animals. This is largely explained by the common assumption in preclinical research that compared with male rodents without an estrous cycle, animal-to-animal variability is greater, and the data therefore are more “messy,” in females. However, this assumption is largely incorrect in regard to behavioral, metabolic, hormonal, and morphological traits (98). However, this still pervasive bias remains a significant obstacle to generalizability and to our ability to translate these data into human research (99). Given substantial sex differences in mitochondrial biology and their responses to perturbations (100) stress-related mitochondrial studies will benefit from equal inclusion of both sexes.
Limitations of Human Studies and Opportunities for Future Research
Existing human studies underscore five main limitations and point the way toward more robust research. The first limitation is the use of single molecular mitochondrial markers, which do not directly reflect mitochondrial function. Certain mitochondrial outcomes such as mtDNAcn, do not directly reflect mitochondrial function. In the absence of functional measures, the physiological meaning of increases and decreases of mtDNAcn is unclear and impossible to conclusively interpret. This is especially true in cases where mtDNAcn is measured from whole blood, which is composed of several different immune and nonimmune cell types with different mtDNA levels. Moving forward, dynamic measures of mitochondrial function could be assessed in parallel with molecular markers of mitochondrial content. Metrics of function and content could also be combined into indices of MHI that may be more sensitive and precise than individual markers alone (93). Improving the sensitivity of mitochondrial outcomes will provide more robust evidence into the link of psychosocial stressors and emotional states with mitochondria.
The second and related limitation is the use of mixed blood cell populations. Blood immune cell composition changes dynamically with various stress indicators (95,96) and undergo substantial circadian (i.e., across the day) oscillations (101). This is relevant because each immune cell type contains different amount of mitochondria that exhibit qualitatively different functions (102). Thus, the relative abundance of different leukocytes subpopulations could substantially alter mitochondrial measures without any real change in mitochondrial function in any cell type. For example, a small change in the proportion of platelets, an enucleated cell type that contributes to coagulation, artificially alters apparent mtDNAcn when measured in whole blood or in peripheral blood mononuclear cells contaminated with platelets (103,104). Future studies would be strengthened by the isolation of specific leukocyte populations. When it is not possible to sort different cell types, avoiding platelet contamination in peripheral blood mononuclear cells (PBMCs), with appropriate platelet depletion steps, would improve the interpretability of mitochondrial function data.
A third limitation of human studies is the exclusive use of blood-derived immune cells. Mitochondria are present in all tissue types, and stress-induced mitochondrial dysfunction could be tissue-specific. Some psychosocial stressors and emotional states may preferentially affect brain mitochondria (contributing to neurological disorders and cognitive decline), whereas others may affect liver and heart mitochondria (contributing to metabolic disorders). Other than blood, biopsies of other tissues are amenable to mitochondrial assessments, including buccal and urinary epithelial cells, as well as fat and skeletal muscle, among others. Certain immune cell subtypes (e.g., monocytes versus granulocytes and memory versus effector lymphocytes) may also be differentially affected. Although studies in leukocytes can be informative of mitochondrial health in immune cells themselves, studies incorporating measures across different accessible tissues and cell types will be insightful to understand the broader relevance—both diagnostic and prognostic—of mitochondrial function and mtDNAcn.
A fourth limitation relates to the retrospective and cross-sectional design of human studies. In comparison to experimental studies such as those reviewed previously where stress exposure is manipulated by experimenters, all human studies so far have compared groups that differ in the experience of psychosocial stressors (e.g., exposure or not to early life stress) or in the levels of psychological distress (e.g., presence versus absence of psychiatric disorders). Such designs limit conclusions about causality and cannot rule out the contribution of potential covariates (e.g., exercise, sleep, and nutrition). Longitudinal and prospective study designs with comprehensive assessment of potential covariates such as physical activity, as well as studies monitoring emotional stress with repeated measures of mitochondrial function, are needed to establish the directionality of effects linking psychosocial stressors, the resulting emotional responses, and mitochondrial functions in humans.
Finally, the fifth limitation noted is the unequal representation of sex in current studies. Contrary to animal studies that exclusively included males, human studies have predominantly included women (see Table 2). Because of known sexual dimorphism in mitochondrial functions (101), research with balanced sex (and gender) composition will be needed to achieve a complete picture of mitochondrial recalibrations in men and women. In addition, so far, human studies have only concentrated on young adults or middle-aged individuals. Considering both sex and age in future research will enable building a more accurate and generalizable understanding of the role of mitochondrial dysfunction in stress pathophysiology in the population and across the life-span. We next discuss processes within mitochondria that may contribute to resilience and possibly explain the effects of stress-buffering interventions on mitochondrial health and downstream stress biomarkers.
Stress-Buffering Interventions: A Role for Mitochondria?
As discussed previously, mitochondrial oxidative stress is an indicator of MAL that contributes to stress pathophysiology (reviewed in Ref. (105)). The corollary is that mitochondrial antioxidant capacity should buffer against the effects of chronic stress. Although the major source of ROS and oxidative stress within cells is generally mitochondria (35), oxidative damage from stress could be of mitochondrial or of other origin. In line with the idea that mitochondrial oxidative stress contributes to cellular dysregulation, enhancing mitochondrial antioxidant capacity by experimentally overexpressing antioxidant enzymes in mouse mitochondria was previously shown to increase resilience to metabolic stress (106,107) and even increase life-span in mice (108). Likewise, oral administration of a mitochondria-targeted antioxidant decreased anxiety-like behavior in inbred mice selected for high-anxious traits (109). A similar effect was observed for depressive-like behaviors with the administration of the mitochondrial metabolite acetyl-L-carnitine (110,111). Similarly, chronic mild stress induces depression-like behavior in normal mice, which is counteracted by the induction of mitochondrial biogenesis in skeletal muscle via the overexpression of peroxisome proliferator–activated receptor-γ co-activator 1α (PGC-1α) (112). Similarly, low sociability usually associated with peripubertal stress is reverted by brain injection of resveratrol, a compound that stimulates mitochondrial biogenesis (113). This body of evidence indirectly supports a role of mitochondrial oxidative stress and mitochondrial function in stress pathophysiology.
However, it remains to be established whether the pathophysiology associated with chronic stress can be prevented or mitigated by mitochondria-targeted interventions, and whether this could be applied in humans. Certain behaviors like physical activity and exercise may have biological stress–buffering effects in humans (114,115). Interestingly, exercise increases several markers of mitochondrial content and function both in humans and in rodents (61,116,117). Furthermore, in one study where stressed sedentary animals experienced a decrease in respiratory chain enzymatic activities, animals trained with treadmill running showed the expected increase in mitochondrial content and function, and in parallel exhibited partial to complete protection against the stress-induced decreases in mitochondrial function and mtDNAcn (61). In humans, acute exercise before a series of mental arithmetic challenges along with social evaluative threat has also been shown to modulate hypothalamic-pituitary-adrenal (HPA) axis reactivity as well as hippocampal and prefrontal cortex activation (118). Because exercise acutely changes mitochondrial function and chronically increases in mitochondrial content and function in various organs, it is possible that the stress-buffering effects of exercise in humans could be mediated by changes of mitochondrial function. This possibility remains to be empirically evaluated.
CONCLUSIONS
Results from this systematic review highlight the need for more interdisciplinary research to map the links between the mind (psyche), mitochondria (mito), and the body (soma). Establishing the effects of psychosocial stress on mitochondria in humans will require the development and application of robust and comprehensive assessments of mitochondrial recalibrations among specific cell types. Moreover, in humans, stressors can vary in their nature (e.g., discrimination, violence, social isolation, job strain/loss, caregiving, and bereavement), can be of changing duration (e.g., acute and chronic), may elicit a wide range of emotional responses (e.g., anxiety, depression, anger, and helplessness) with diverse levels of intensity (e.g., mild, moderate, or severe symptoms), and can occur at different stages of the life-span (early childhood, midlife, or in elderly). Mitochondrial responses to stressors are also likely not linear (21). It will therefore be equally important to operationalize and distinguish between different life stressors and the resulting individualized experiences, and to assess important covariates that can independently influence mitochondrial content or function. Prospective and longitudinal studies combining these variables will be particularly valuable.
Building from established models where psychosomatic processes are known to operate, incorporating mitochondrial measures in study designs should accelerate the progression of our understanding around the interaction of mitochondria with the broad-acting brain-immune-endocrine processes that affect health outcomes. This joining of disciplines under a shared “psycho-mito-somatic” framework, as discussed in the joint article in this issue of Psychosomatic Medicine (21), should simultaneously promote a more holistic understanding of the processes that precipitate dysregulation and disease within the organism, and of the mechanisms that enable healthy individuals to successfully adapt to life challenges.
Acknowledgments
Source of Funding and Conflicts of Interest: Support for this work was provided by the Wharton Fund, National Institutes of Health grants R35GM119793 and R21MH113011 (M.P.), and Hope for Depression Research Foundation (B.S.M.).
The authors are grateful to Claudia Trudel-Fitzgerald for valuable edits to this article and to Robert-Paul Juster for retrieving articles for the systematic review.
Glossary
- ACE
adverse childhood experiences
- ATP
adenosine triphosphate
- ccf-mtDNA
circulating cell-free mtDNA
- Complex I
mitochondrial NADH dehydrogenase
- COX
cytochrome c oxidase, also complex IV
- CS
citrate synthase
- GR
glucocorticoid receptor
- HPA
hypothalamic-pituitary-adrenal
- IL-6
Interleukin 6
- MAL
mitochondrial allostatic load
- mtDNA
mitochondrial DNA
- mtDNAcn
mtDNA copy number
- Nf-kB
nuclear factor kappa B
- PGC-1α
peroxisome proliferator activated receptor gamma co-activator 1 alpha
- ROS
reactive oxygen species
- ROS
reactive oxygen species
- SDH
succinate dehydrogenase, also complex II
Footnotes
The authors have no conflict of interest to report.
References
- 1.Morava E, Kozicz T. Mitochondria and the economy of stress (mal)adaptation. Neurosci Biobehav Rev. 2013;37:668–80. doi: 10.1016/j.neubiorev.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 2.Manoli I, Alesci S, Blackman MR, Su YA, Rennert OM, Chrousos GP. Mitochondria as key components of the stress response. Trends Endocrinol Metab. 2007;18:190–8. doi: 10.1016/j.tem.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 3.Picard M, Juster RP, McEwen BS. Mitochondrial allostatic load puts the ‘gluc’ back in glucocorticoids. Nat Rev Endocrinol. 2014;10:303–10. doi: 10.1038/nrendo.2014.22. [DOI] [PubMed] [Google Scholar]
- 4.Picard M, Wallace DC, Burelle Y. The rise of mitochondria in medicine. Mitochondrion. 2016;30:105–16. doi: 10.1016/j.mito.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gorman GS, Chinnery PF, DiMauro S, Hirano M, Koga Y, McFarland R, Suomalainen A, Thorburn DR, Zeviani M, Turnbull DM. Mitochondrial diseases. Nat Rev Dis Primers. 2016;2:16080. doi: 10.1038/nrdp.2016.80. [DOI] [PubMed] [Google Scholar]
- 6.Morrow RM, Picard M, Derbeneva O, Leipzig J, McManus MJ, Gouspillou G, Barbat-Artigas S, Dos Santos C, Hepple RT, Murdock DG, Wallace DC. Mitochondrial energy deficiency leads to hyperproliferation of skeletal muscle mitochondria and enhanced insulin sensitivity. Proc Natl Acad Sci USA. 2017;114:2705–10. doi: 10.1073/pnas.1700997114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hebert-Chatelain E, Desprez T, Serrat R, Bellocchio L, Soria-Gomez E, Busquets-Garcia A, Pagano Zottola AC, Delamarre A, Cannich A, Vincent P, Varilh M, Robin LM, Terral G, Garcia-Fernandez MD, Colavita M, Mazier W, Drago F, Puente N, Reguero L, Elezgarai I, Dupuy JW, Cota D, Lopez-Rodriguez ML, Barreda-Gomez G, Massa F, Grandes P, Benard G, Marsicano G. A cannabinoid link between mitochondria and memory. Nature. 2016;539:555–9. doi: 10.1038/nature20127. [DOI] [PubMed] [Google Scholar]
- 8.Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science. 2013;342:1242454. doi: 10.1126/science.1242454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Latorre-Pellicer A, Moreno-Loshuertos R, Lechuga-Vieco AV, Sanchez-Cabo F, Torroja C, Acin-Perez R, Calvo E, Aix E, Gonzalez-Guerra A, Logan A, Bernad-Miana ML, Romanos E, Cruz R, Cogliati S, Sobrino B, Carracedo A, Perez-Martos A, Fernandez-Silva P, Ruiz-Cabello J, Murphy MP, Flores I, Vazquez J, Enriquez JA. Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature. 2016;535:561–5. doi: 10.1038/nature18618. [DOI] [PubMed] [Google Scholar]
- 10.Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlof S, Oldfors A, Wibom R, Tornell J, Jacobs HT, Larsson NG. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–23. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 11.Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla TA. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–4. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 12.McEwen BS. Protective and damaging effects of stress mediators: central role of the brain. Dialogues Clin Neurosci. 2006;8:367–81. doi: 10.31887/DCNS.2006.8.4/bmcewen. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kiecolt-Glaser JK, McGuire L, Robles TF, Glaser R. Psychoneuroimmunology and psychosomatic medicine: back to the future. Psychosom Med. 2002;64:15–28. doi: 10.1097/00006842-200201000-00004. [DOI] [PubMed] [Google Scholar]
- 14.Segerstrom SC, Miller GE. Psychological stress and the human immune system: a meta-analytic study of 30 years of inquiry. Psychol Bull. 2004;130:601–30. doi: 10.1037/0033-2909.130.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Irwin MR, Cole SW. Reciprocal regulation of the neural and innate immune systems. Nat Rev Immunol. 2011;11:625–32. doi: 10.1038/nri3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cole SW, Nagaraja AS, Lutgendorf SK, Green PA, Sood AK. Sympathetic nervous system regulation of the tumour microenvironment. Nat Rev Cancer. 2015;15:563–72. doi: 10.1038/nrc3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Slavich GM, Cole SW. The emerging field of human social genomics. Clin Psychol Sci. 2013;1:331–48. doi: 10.1177/2167702613478594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shalev I, Entringer S, Wadhwa PD, Wolkowitz OM, Puterman E, Lin J, Epel ES. Stress and telomere biology: a lifespan perspective. Psychoneuroendocrinology. 2013;38:1835–42. doi: 10.1016/j.psyneuen.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kundakovic M, Champagne FA. Early-life experience, epigenetics, and the developing brain. Neuropsychopharmacology. 2015;40:141–53. doi: 10.1038/npp.2014.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trudel-Fitzgerald C, Qureshi F, Appleton AA, Kubzansky LD. A healthy mix of emotions: underlying biological pathways linking emotions to physical health. Curr Opin Behav Sci. 2017;15 [Google Scholar]
- 21.Picard M, McEwen BS. Psychological stress and mitochondria: a conceptual framework. Psychosom Med. 2018;80:126–40. doi: 10.1097/PSY.0000000000000544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Picard M. Mitochondrial synapses: intracellular communication and signal integration. Trends Neurosci. 2015;38:468–74. doi: 10.1016/j.tins.2015.06.001. [DOI] [PubMed] [Google Scholar]
- 23.Vincent AE, Turnbull DM, Eisner V, Hajnoczky G, Picard M. Mitochondrial Nanotunnels. Trends Cell Biol. 2017;27:787–99. doi: 10.1016/j.tcb.2017.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gut P, Verdin E. The nexus of chromatin regulation and intermediary metabolism. Nature. 2013;502:489–98. doi: 10.1038/nature12752. [DOI] [PubMed] [Google Scholar]
- 25.Picard M, Zhang J, Hancock S, Derbeneva O, Golhar R, Golik P, O’Hearn S, Levy S, Potluri P, Lvova M, Davila A, Lin CS, Perin JC, Rappaport EF, Hakonarson H, Trounce IA, Procaccio V, Wallace DC. Progressive increase in mtDNA 3243A > G heteroplasmy causes abrupt transcriptional reprogramming. Proc Natl Acad Sci USA. 2014;111:E4033–42. doi: 10.1073/pnas.1414028111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shaughnessy DT, McAllister K, Worth L, Haugen AC, Meyer JN, Domann FE, Van Houten B, Mostoslavsky R, Bultman SJ, Baccarelli AA, Begley TJ, Sobol RW, Hirschey MD, Ideker T, Santos JH, Copeland WC, Tice RR, Balshaw DM, Tyson FL. Mitochondria, energetics, epigenetics, and cellular responses to stress. Environ Health Perspect. 2014;122:1271–8. doi: 10.1289/ehp.1408418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nicholls DG, Fergusson SJ. Bioenergetics. 4th. San Diego, CA: Academic Press; 2013. [Google Scholar]
- 28.Lee C, Kim KH, Cohen P. MOTS-c: a novel mitochondrial-derived peptide regulating muscle and fat metabolism. Free Radic Biol Med. 2016;100:182–7. doi: 10.1016/j.freeradbiomed.2016.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Psarra AM, Sekeris CE. Glucocorticoids induce mitochondrial gene transcription in HepG2 cells: role of the mitochondrial glucocorticoid receptor. Biochim Biophys Acta. 2011;1813:1814–21. doi: 10.1016/j.bbamcr.2011.05.014. [DOI] [PubMed] [Google Scholar]
- 30.Giles RE, Blanc H, Cann HM, Wallace DC. Maternal inheritance of human mitochondrial DNA. Proc Natl Acad Sci USA. 1980;77:6715–9. doi: 10.1073/pnas.77.11.6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wallace DC, Chalkia D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb Perspect Med. 2013;3:a021220. doi: 10.1101/cshperspect.a021220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kenney MC, Chwa M, Atilano SR, Falatoonzadeh P, Ramirez C, Malik D, Tarek M, Del Carpio JC, Nesburn AB, Boyer DS, Kuppermann BD, Vawter MP, Jazwinski SM, Miceli MV, Wallace DC, Udar N. Molecular and bioenergetic differences between cells with African versus European inherited mitochondrial DNA haplogroups: implications for population susceptibility to diseases. Biochim Biophys Acta. 2014;1842:208–19. doi: 10.1016/j.bbadis.2013.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan SR, Blackburn EH. Telomeres and telomerase. Philos Trans R Soc Lond B Biol Sci. 2004;359:109–21. doi: 10.1098/rstb.2003.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci USA. 1997;94:514–9. doi: 10.1073/pnas.94.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lambert AJ, Brand MD. Reactive oxygen species production by mitochondria. Methods Mol Biol. 2009;554:165–81. doi: 10.1007/978-1-59745-521-3_11. [DOI] [PubMed] [Google Scholar]
- 36.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–95. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 37.McManus MJ, Murphy MP, Franklin JL. The mitochondria-targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer’s disease. J Neurosci. 2011;31:15703–15. doi: 10.1523/JNEUROSCI.0552-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mitchell P, Moyle J. Chemiosmotic hypothesis of oxidative phosphorylation. Nature. 1967;213:137–9. doi: 10.1038/213137a0. [DOI] [PubMed] [Google Scholar]
- 39.Lanza IR, Nair KS. Mitochondrial metabolic function assessed in vivo and in vitro. Curr Opin Clin Nutr Metab Care. 2010;13:511–7. doi: 10.1097/MCO.0b013e32833cc93d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuznetsov AV, Veksler V, Gellerich FN, Saks V, Margreiter R, Kunz WS. Analysis of mitochondrial function in situ in permeabilized muscle fibers, tissues and cells. Nat Protoc. 2008;3:965–76. doi: 10.1038/nprot.2008.61. [DOI] [PubMed] [Google Scholar]
- 41.Kramer PA, Chacko BK, Ravi S, Johnson MS, Mitchell T, Darley-Usmar VM. Bioenergetics and the oxidative burst: protocols for the isolation and evaluation of human leukocytes and platelets. J Vis Exp. 2014;85 doi: 10.3791/51301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Medja F, Allouche S, Frachon P, Jardel C, Malgat M, Mousson de Camaret B, Slama A, Lunardi J, Mazat JP, Lombes A. Development and implementation of standardized respiratory chain spectrophotometric assays for clinical diagnosis. Mitochondrion. 2009;9:331–9. doi: 10.1016/j.mito.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 43.Booth NE, Myhill S, McLaren-Howard J. Mitochondrial dysfunction and the pathophysiology of myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) Int J Clin Exp Med. 2012;5:208–20. [PMC free article] [PubMed] [Google Scholar]
- 44.Picard M, Hepple RT, Burelle Y. Mitochondrial functional specialization in glycolytic and oxidative muscle fibers: tailoring the organelle for optimal function. Am J Physiol Cell Physiol. 2012;302:C629–41. doi: 10.1152/ajpcell.00368.2011. [DOI] [PubMed] [Google Scholar]
- 45.Glancy B, Balaban RS. Protein composition and function of red and white skeletal muscle mitochondria. Am J Physiol Cell Physiol. 2011;300:C1280–90. doi: 10.1152/ajpcell.00496.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stauch KL, Purnell PR, Fox HS. Quantitative proteomics of synaptic and nonsynaptic mitochondria: insights for synaptic mitochondrial vulnerability. J Proteome Res. 2014;13:2620–36. doi: 10.1021/pr500295n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moher D, Liberati A, Tetzlaff J, Altman DG, Group P Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009;6:e1000097. doi: 10.1371/journal.pmed.1000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhuravliova E, Barbakadze T, Zaalishvili E, Chipashvili M, Koshoridze N, Mikeladze D. Social isolation in rats inhibits oxidative metabolism, decreases the content of mitochondrial K-Ras and activates mitochondrial hexokinase. Behav Brain Res. 2009;205:377–83. doi: 10.1016/j.bbr.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 49.Krolow R, Noschang C, Weis SN, Pettenuzzo LF, Huffell AP, Arcego DM, Marcolin M, Mota CS, Kolling J, Scherer EB, Wyse AT, Dalmaz C. Isolation stress during the prepubertal period in rats induces long-lasting neurochemical changes in the prefrontal cortex. Neurochem Res. 2012;37:1063–73. doi: 10.1007/s11064-012-0709-1. [DOI] [PubMed] [Google Scholar]
- 50.Andric SA, Kojic Z, Bjelic MM, Mihajlovic AI, Baburski AZ, Sokanovic SJ, Janjic MM, Stojkov NJ, Stojilkovic SS, Kostic TS. The opposite roles of glucocorticoid and α1-adrenergic receptors in stress triggered apoptosis of rat Leydig cells. Am J Physiol Endocrinol Metab. 2013;304:E51–9. doi: 10.1152/ajpendo.00443.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Batandier C, Poulet L, Hininger I, Couturier K, Fontaine E, Roussel AM, Canini F. Acute stress delays brain mitochondrial permeability transition pore opening. J Neurochem. 2014;131:314–22. doi: 10.1111/jnc.12811. [DOI] [PubMed] [Google Scholar]
- 52.Cai N, Chang S, Li Y, Li Q, Hu J, Liang J, Song L, Kretzschmar W, Gan X, Nicod J, Rivera M, Deng H, Du B, Li K, Sang W, Gao J, Gao S, Ha B, Ho HY, Hu C, Hu J, Hu Z, Huang G, Jiang G, Jiang T, Jin W, Li G, Li K, Li Y, Li Y, Li Y, Lin YT, Liu L, Liu T, Liu Y, Liu Y, Lu Y, Lv L, Meng H, Qian P, Sang H, Shen J, Shi J, Sun J, Tao M, Wang G, Wang G, Wang J, Wang L, Wang X, Wang X, Yang H, Yang L, Yin Y, Zhang J, Zhang K, Sun N, Zhang W, Zhang X, Zhang Z, Zhong H, Breen G, Wang J, Marchini J, Chen Y, Xu Q, Xu X, Mott R, Huang GJ, Kendler K, Flint J. Molecular signatures of major depression. Curr Biol. 2015;25:1146–56. doi: 10.1016/j.cub.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gak IA, Radovic SM, Dukic AR, Janjic MM, Stojkov-Mimic NJ, Kostic TS, Andric SA. Stress triggers mitochondrial biogenesis to preserve steroidogenesis in Leydig cells. Biochim Biophys Acta. 2015;1853:2217–27. doi: 10.1016/j.bbamcr.2015.05.030. [DOI] [PubMed] [Google Scholar]
- 54.García-Fernández M, Castilla-Ortega E, Pedraza C, Blanco E, Hurtado-Guerrero I, Barbancho MA, Chun J, Rodriguez-de-Fonseca F, Estivill-Torrus G, Santin Nunez LJ. Chronic immobilization in the malpar1 knockout mice increases oxidative stress in the hippocampus. Int J Neurosci. 2012;122:583–9. doi: 10.3109/00207454.2012.693998. [DOI] [PubMed] [Google Scholar]
- 55.Gesi M, Fornai F, Lenzi P, Ferrucci M, Soldani P, Ruffoli R, Paparelli A. Morphological alterations induced by loud noise in the myocardium: the role of benzodiazepine receptors. Microsc Res Tech. 2002;59:136–46. doi: 10.1002/jemt.10186. [DOI] [PubMed] [Google Scholar]
- 56.Gong Y, Chai Y, Ding JH, Sun XL, Hu G. Chronic mild stress damages mitochondrial ultrastructure and function in mouse brain. Neurosci Lett. 2011;488:76–80. doi: 10.1016/j.neulet.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 57.Heinzeller T. Impact of psychosocial stress on pineal structure of male gerbils (Meriones unguiculatus, Cricetidae) J Pineal Res. 1985;2:145–59. doi: 10.1111/j.1600-079x.1985.tb00635.x. [DOI] [PubMed] [Google Scholar]
- 58.Kambe Y, Miyata A. Potential involvement of the mitochondrial unfolded protein response in depressive-like symptoms in mice. Neurosci Lett. 2015;588:166–71. doi: 10.1016/j.neulet.2015.01.006. [DOI] [PubMed] [Google Scholar]
- 59.Li YF, He RR, Tsoi B, Li XD, Li WX, Abe K, Kurihara H. Anti-stress effects of carnosine on restraint-evoked immunocompromise in mice through spleen lymphocyte number maintenance. PLoS One. 2012;7:e33190. doi: 10.1371/journal.pone.0033190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu XH, Qian LJ, Gong JB, Shen J, Zhang XM, Qian XH. Proteomic analysis of mitochondrial proteins in cardiomyocytes from chronic stressed rat. Proteomics. 2004;4:3167–76. doi: 10.1002/pmic.200300845. [DOI] [PubMed] [Google Scholar]
- 61.Liu W, Zhou C. Corticosterone reduces brain mitochondrial function and expression of mitofusin, BDNF in depression-like rodents regardless of exercise preconditioning. Psychoneuroendocrinology. 2012;37:1057–70. doi: 10.1016/j.psyneuen.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 62.Madrigal JL, Olivenza R, Moro MA, Lizasoain I, Lorenzo P, Rodrigo J, Leza JC. Glutathione depletion, lipid peroxidation and mitochondrial dysfunction are induced by chronic stress in rat brain. Neuropsychopharmacology. 2001;24:420–9. doi: 10.1016/S0893-133X(00)00208-6. [DOI] [PubMed] [Google Scholar]
- 63.Magarinos AM, Verdugo JM, McEwen BS. Chronic stress alters synaptic terminal structure in hippocampus. Proc Natl Acad Sci USA. 1997;94:14002–8. doi: 10.1073/pnas.94.25.14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Martin-Aragon S, Villar A, Benedi J. Age-dependent effects of esculetin on mood-related behavior and cognition from stressed mice are associated with restoring brain antioxidant status. Prog Neuropsychopharmacol Biol Psychiatry. 2016;65:1–16. doi: 10.1016/j.pnpbp.2015.08.007. [DOI] [PubMed] [Google Scholar]
- 65.Ortmann CF, Reus GZ, Ignacio ZM, Abelaira HM, Titus SE, de Carvalho P, Arent CO, Dos Santos MA, Matias BI, Martins MM, de Campos AM, Petronilho F, Teixeira LJ, Morais MO, Streck EL, Quevedo J, Reginatto FH. Enriched flavonoid fraction from Cecropia pachystachya Trécul leaves exerts antidepressant-like behavior and protects brain against oxidative stress in rats subjected to chronic mild stress. Neurotox Res. 2016;29:469–83. doi: 10.1007/s12640-016-9596-6. [DOI] [PubMed] [Google Scholar]
- 66.Rezin GT, Cardoso MR, Goncalves CL, Scaini G, Fraga DB, Riegel RE, Comim CM, Quevedo J, Streck EL. Inhibition of mitochondrial respiratory chain in brain of rats subjected to an experimental model of depression. Neurochem Int. 2008;53:395–400. doi: 10.1016/j.neuint.2008.09.012. [DOI] [PubMed] [Google Scholar]
- 67.Rinwa P, Kumar A. Piperine potentiates the protective effects of curcumin against chronic unpredictable stress-induced cognitive impairment and oxidative damage in mice. Brain Res. 2012;1488:38–50. doi: 10.1016/j.brainres.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 68.Rinwa P, Kumar A. Modulation of nitrergic signalling pathway by American ginseng attenuates chronic unpredictable stress-induced cognitive impairment, neuroinflammation, and biochemical alterations. Naunyn Schmiedebergs Arch Pharmacol. 2014;387:129–41. doi: 10.1007/s00210-013-0925-5. [DOI] [PubMed] [Google Scholar]
- 69.Seo JS, Lee KW, Kim TK, Baek IS, Im JY, Han PL. Behavioral stress causes mitochondrial dysfunction via ABAD up-regulation and aggravates plaque pathology in the brain of a mouse model of Alzheimer disease. Free Radic Biol Med. 2011;50:1526–35. doi: 10.1016/j.freeradbiomed.2011.02.035. [DOI] [PubMed] [Google Scholar]
- 70.Soldani P, Pellegrini A, Gesi M, Lenzi P, Cristofani R, Paparelli A. SEM/TEM investigation of rat cardiac subcellular alterations induced by changing duration of noise stress. Anat Rec. 1997;248:521–32. doi: 10.1002/(SICI)1097-0185(199708)248:4<521::AID-AR4>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 71.Vicario M, Alonso C, Guilarte M, Serra J, Martinez C, Gonzalez-Castro AM, Lobo B, Antolin M, Andreu AL, Garcia-Arumi E, Casellas M, Saperas E, Malagelada JR, Azpiroz F, Santos J. Chronic psychosocial stress induces reversible mitochondrial damage and corticotropin-releasing factor receptor type-1 upregulation in the rat intestine and IBS-like gut dysfunction. Psychoneuroendocrinology. 2012;37:65–77. doi: 10.1016/j.psyneuen.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 72.Wen L, Jin Y, Li L, Sun S, Cheng S, Zhang S, Zhang Y, Svenningsson P. Exercise prevents raphe nucleus mitochondrial overactivity in a rat depression model. Physiol Behav. 2014;132:57–65. doi: 10.1016/j.physbeh.2014.04.050. [DOI] [PubMed] [Google Scholar]
- 73.Spinazzi M, Casarin A, Pertegato V, Salviati L, Angelini C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat Protoc. 2012;7:1235–46. doi: 10.1038/nprot.2012.058. [DOI] [PubMed] [Google Scholar]
- 74.Chen YJ, Huang F, Zhang M, Shang HY. Psychological stress alters ultrastructure and energy metabolism of masticatory muscle in rats. J Biomed Biotechnol. 2010;2010:302693. doi: 10.1155/2010/302693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cai N, Li Y, Chang S, Liang J, Lin C, Zhang X, Liang L, Hu J, Chan W, Kendler KS, Malinauskas T, Huang GJ, Li Q, Mott R, Flint J. Genetic control over mtDNA and its relationship to major depressive disorder. Curr Biol. 2015;25:3170–7. doi: 10.1016/j.cub.2015.10.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Giordano C, Iommarini L, Giordano L, Maresca A, Pisano A, Valentino ML, Caporali L, Liguori R, Deceglie S, Roberti M, Fanelli F, Fracasso F, Ross-Cisneros FN, D’Adamo P, Hudson G, Pyle A, Yu-Wai-Man P, Chinnery PF, Zeviani M, Salomao SR, Berezovsky A, Belfort R, Jr, Ventura DF, Moraes M, Moraes Filho M, Barboni P, Sadun F, De Negri A, Sadun AA, Tancredi A, Mancini M, d’Amati G, Loguercio Polosa P, Cantatore P, Carelli V. Efficient mitochondrial biogenesis drives incomplete penetrance in Leber’s hereditary optic neuropathy. Brain. 2014;137:335–53. doi: 10.1093/brain/awt343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yu-Wai-Man P, Sitarz KS, Samuels DC, Griffiths PG, Reeve AK, Bindoff LA, Horvath R, Chinnery PF. OPA1 mutations cause cytochrome c oxidase deficiency due to loss of wild-type mtDNA molecules. Hum Mol Genet. 2010;19:3043–52. doi: 10.1093/hmg/ddq209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Filipovic D, Mandic LM, Kanazir D, Pajovic SB. Acute and/or chronic stress models modulate CuZnSOD and MnSOD protein expression in rat liver. Mol Cell Biochem. 2010;338:167–74. doi: 10.1007/s11010-009-0350-8. [DOI] [PubMed] [Google Scholar]
- 79.Filipovic D, Zlatkovic J, Inta D, Bjelobaba I, Stojiljkovic M, Gass P. Chronic isolation stress predisposes the frontal cortex but not the hippocampus to the potentially detrimental release of cytochrome c from mitochondria and the activation of caspase-3. J Neurosci Res. 2011;89:1461–70. doi: 10.1002/jnr.22687. [DOI] [PubMed] [Google Scholar]
- 80.Johnson CH, Ivanisevic J, Siuzdak G. Metabolomics: beyond biomarkers and towards mechanisms. Nat Rev Mol Cell Biol. 2016;17:451–9. doi: 10.1038/nrm.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li ZY, Zheng XY, Gao XX, Zhou YZ, Sun HF, Zhang LZ, Guo XQ, Du GH, Qin XM. Study of plasma metabolic profiling and biomarkers of chronic unpredictable mild stress rats based on gas chromatography/mass spectrometry. Rapid Commun Mass Spectrom. 2010;24:3539–46. doi: 10.1002/rcm.4809. [DOI] [PubMed] [Google Scholar]
- 82.Filiou MD, Asara JM, Nussbaumer M, Teplytska L, Landgraf R, Turck CW. Behavioral extremes of trait anxiety in mice are characterized by distinct metabolic profiles. J Psychiatr Res. 2014;58:115–22. doi: 10.1016/j.jpsychires.2014.07.019. [DOI] [PubMed] [Google Scholar]
- 83.Tian JS, Xia XT, Wu YF, Zhao L, Xiang H, Du GH, Zhang X, Qin XM. Discovery, screening and evaluation of a plasma biomarker panel for subjects with psychological suboptimal health state using (1)H-NMR-based metabolomics profiles. Sci Rep. 2016;6:33820. doi: 10.1038/srep33820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chakravarty S, Reddy BR, Sudhakar SR, Saxena S, Das T, Meghah V, Brahmendra Swamy CV, Kumar A, Idris MM. Chronic unpredictable stress (CUS)–induced anxiety and related mood disorders in a zebrafish model: altered brain proteome profile implicates mitochondrial dysfunction. PLoS One. 2013;8:e63302. doi: 10.1371/journal.pone.0063302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Filiou MD, Zhang Y, Teplytska L, Reckow S, Gormanns P, Maccarrone G, Frank E, Kessler MS, Hambsch B, Nussbaumer M, Bunck M, Ludwig T, Yassouridis A, Holsboer F, Landgraf R, Turck CW. Proteomics and metabolomics analysis of a trait anxiety mouse model reveals divergent mitochondrial pathways. Biol Psychiatry. 2011;70:1074–82. doi: 10.1016/j.biopsych.2011.06.009. [DOI] [PubMed] [Google Scholar]
- 86.Hollis F, van der Kooij MA, Zanoletti O, Lozano L, Canto C, Sandi C. Mitochondrial function in the brain links anxiety with social subordination. Proc Natl Acad Sci USA. 2015;112:15486–91. doi: 10.1073/pnas.1512653112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Picard M, McManus MJ, Gray JD, Nasca C, Moffat C, Kopinski PK, Seifert EL, McEwen BS, Wallace DC. Mitochondrial functions modulate neuroendocrine, metabolic, inflammatory, and transcriptional responses to acute psychological stress. Proc Natl Acad Sci USA. 2015;112:E6614–23. doi: 10.1073/pnas.1515733112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gray JD, Rubin TG, Hunter RG, McEwen BS. Hippocampal gene expression changes underlying stress sensitization and recovery. Mol Psychiatry. 2014;19:1171–8. doi: 10.1038/mp.2013.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hunter RG, Seligsohn M, Rubin TG, Griffiths BB, Ozdemir Y, Pfaff DW, Datson NA, McEwen BS. Stress and corticosteroids regulate rat hippocampal mitochondrial DNA gene expression via the glucocorticoid receptor. Proc Natl Acad Sci USA. 2016;113:9099–104. doi: 10.1073/pnas.1602185113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Roosevelt TS, Ruhmann-Wennhold A, Nelson DH. Adrenal corticosteroid effects upon rat brain mitochondrial metabolism. Endocrinology. 1973;93:619–25. doi: 10.1210/endo-93-3-619. [DOI] [PubMed] [Google Scholar]
- 91.Boeck C, Koenig AM, Schury K, Geiger ML, Karabatsiakis A, Wilker S, Waller C, Gundel H, Fegert JM, Calzia E, Kolassa IT. Inflammation in adult women with a history of child maltreatment: the involvement of mitochondrial alterations and oxidative stress. Mitochondrion. 2016;30:197–207. doi: 10.1016/j.mito.2016.08.006. [DOI] [PubMed] [Google Scholar]
- 92.Lindqvist D, Fernstrom J, Grudet C, Ljunggren L, Traskman-Bendz L, Ohlsson L, Westrin A. Increased plasma levels of circulating cell-free mitochondrial DNA in suicide attempters: associations with HPA-axis hyperactivity. Transl Psychiatry. 2016;6:e971. doi: 10.1038/tp.2016.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Picard M, Prather AA, Puterman E, Cuillerier A, Coccia M, Aschbacher K, Burelle Y, Epel ES. A mitochondrial health index sensitive to mood and caregiving stress. Biol Psychiatr. 2018 doi: 10.1016/j.biopsych.2018.01.012. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tyrka AR, Parade SH, Price LH, Kao HT, Porton B, Philip NS, Welch ES, Carpenter LL. Alterations of mitochondrial DNA copy number and telomere length with early adversity and psychopathology. Biol Psychiatry. 2016;79:78–86. doi: 10.1016/j.biopsych.2014.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dhabhar FS, McEwen BS. Acute stress enhances while chronic stress suppresses cell-mediated immunity in vivo: a potential role for leukocyte trafficking. Brain Behav Immun. 1997;11:286–306. doi: 10.1006/brbi.1997.0508. [DOI] [PubMed] [Google Scholar]
- 96.Dhabhar FS, Malarkey WB, Neri E, McEwen BS. Stress-induced redistribution of immune cells—from barracks to boulevards to battlefields: a tale of three hormones—Curt Richter Award winner. Psychoneuroendocrinology. 2012;37:1345–68. doi: 10.1016/j.psyneuen.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Boyapati RK, Tamborska A, Dorward DA, Ho GT. Advances in the understanding of mitochondrial DNA as a pathogenic factor in inflammatory diseases. F1000Res. 2017;6:169. doi: 10.12688/f1000research.10397.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Prendergast BJ, Onishi KG, Zucker I. Female mice liberated for inclusion in neuroscience and biomedical research. Neurosci Biobehav Rev. 2014;40:1–5. doi: 10.1016/j.neubiorev.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 99.Klein SL, Schiebinger L, Stefanick ML, Cahill L, Danska J, de Vries GJ, Kibbe MR, McCarthy MM, Mogil JS, Woodruff TK, Zucker I. Opinion: sex inclusion in basic research drives discovery. Proc Natl Acad Sci USA. 2015;112:5257–8. doi: 10.1073/pnas.1502843112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ventura-Clapier R, Moulin M, Piquereau J, Lemaire C, Mericskay M, Veksler V, Garnier A. Mitochondria: a central target for sex differences in pathologies. Clin Sci (Lond) 2017;131:803–22. doi: 10.1042/CS20160485. [DOI] [PubMed] [Google Scholar]
- 101.Born J, Lange T, Hansen K, Molle M, Fehm HL. Effects of sleep and circadian rhythm on human circulating immune cells. J Immunol. 1997;158:4454–64. [PubMed] [Google Scholar]
- 102.Chacko BK, Kramer PA, Ravi S, Johnson MS, Hardy RW, Ballinger SW, Darley-Usmar VM. Methods for defining distinct bioenergetic profiles in platelets, lymphocytes, monocytes, and neutrophils, and the oxidative burst from human blood. Lab Invest. 2013;93:690–700. doi: 10.1038/labinvest.2013.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Urata M, Koga-Wada Y, Kayamori Y, Kang D. Platelet contamination causes large variation as well as overestimation of mitochondrial DNA content of peripheral blood mononuclear cells. Ann Clin Biochem. 2008;45:513–4. doi: 10.1258/acb.2008.008008. [DOI] [PubMed] [Google Scholar]
- 104.Hurtado-Roca Y, Ledesma M, Gonzalez-Lazaro M, Moreno-Loshuertos R, Fernandez-Silva P, Enriquez JA, Laclaustra M. Adjusting MtDNA quantification in whole blood for peripheral blood platelet and leukocyte counts. PLoS One. 2016;11:e0163770. doi: 10.1371/journal.pone.0163770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Schiavone S, Jaquet V, Trabace L, Krause KH. Severe life stress and oxidative stress in the brain: from animal models to human pathology. Antioxid Redox Signal. 2013;18:1475–90. doi: 10.1089/ars.2012.4720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lee HY, Choi CS, Birkenfeld AL, Alves TC, Jornayvaz FR, Jurczak MJ, Zhang D, Woo DK, Shadel GS, Ladiges W, Rabinovitch PS, Santos JH, Petersen KF, Samuel VT, Shulman GI. Targeted expression of catalase to mitochondria prevents age-associated reductions in mitochondrial function and insulin resistance. Cell Metab. 2010;12:668–74. doi: 10.1016/j.cmet.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Picard M, Jung B, Liang F, Azuelos I, Hussain S, Goldberg P, Godin R, Danialou G, Chaturvedi R, Rygiel K, Matecki S, Jaber S, Des Rosiers C, Karpati G, Ferri L, Burelle Y, Turnbull DM, Taivassalo T, Petrof BJ. Mitochondrial dysfunction and lipid accumulation in the human diaphragm during mechanical ventilation. Am J Respir Crit Care Med. 2012;186:1140–9. doi: 10.1164/rccm.201206-0982OC. [DOI] [PubMed] [Google Scholar]
- 108.Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC, Rabinovitch PS. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–11. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- 109.Nussbaumer M, Asara JM, Teplytska L, Murphy MP, Logan A, Turck CW, Filiou MD. Selective mitochondrial targeting exerts anxiolytic effects in vivo. Neuropsychopharmacology. 2016;41:1751–8. doi: 10.1038/npp.2015.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bigio B, Mathe AA, Sousa VC, Zelli D, Svenningsson P, McEwen BS, Nasca C. Epigenetics and energetics in ventral hippocampus mediate rapid antidepressant action: implications for treatment resistance. Proc Natl Acad Sci USA. 2016;113:7906–11. doi: 10.1073/pnas.1603111113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nasca C, Xenos D, Barone Y, Caruso A, Scaccianoce S, Matrisciano F, Battaglia G, Mathe AA, Pittaluga A, Lionetto L, Simmaco M, Nicoletti F. L-acetylcarnitine causes rapid antidepressant effects through the epigenetic induction of mGlu2 receptors. Proc Natl Acad Sci USA. 2013;110:4804–9. doi: 10.1073/pnas.1216100110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Agudelo LZ, Femenia T, Orhan F, Porsmyr-Palmertz M, Goiny M, Martinez-Redondo V, Correia JC, Izadi M, Bhat M, Schuppe-Koistinen I, Pettersson AT, Ferreira DM, Krook A, Barres R, Zierath JR, Erhardt S, Lindskog M, Ruas JL. Skeletal muscle PGC-1α1 modulates kynurenine metabolism and mediates resilience to stress-induced depression. Cell. 2014;159:33–45. doi: 10.1016/j.cell.2014.07.051. [DOI] [PubMed] [Google Scholar]
- 113.Poirier GL, Imamura N, Zanoletti O, Sandi C. Social deficits induced by peripubertal stress in rats are reversed by resveratrol. J Psychiatr Res. 2014;57:157–64. doi: 10.1016/j.jpsychires.2014.05.017. [DOI] [PubMed] [Google Scholar]
- 114.Puterman E, O’Donovan A, Adler NE, Tomiyama AJ, Kemeny M, Wolkowitz OM, Epel E. Physical activity moderates effects of stressor-induced rumination on cortisol reactivity. Psychosom Med. 2011;73:604–11. doi: 10.1097/PSY.0b013e318229e1e0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Puterman E, Lin J, Blackburn E, O’Donovan A, Adler N, Epel E. The power of exercise: buffering the effect of chronic stress on telomere length. PLoS One. 2010;5:e10837. doi: 10.1371/journal.pone.0010837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Steiner JL, Murphy EA, McClellan JL, Carmichael MD, Davis JM. Exercise training increases mitochondrial biogenesis in the brain. J Appl Physiol (1985) 2011;111:1066–71. doi: 10.1152/japplphysiol.00343.2011. [DOI] [PubMed] [Google Scholar]
- 117.Little JP, Safdar A, Benton CR, Wright DC. Skeletal muscle and beyond: the role of exercise as a mediator of systemic mitochondrial biogenesis. Appl Physiol Nutr Metab. 2011;36:598–607. doi: 10.1139/h11-076. [DOI] [PubMed] [Google Scholar]
- 118.Zschucke E, Renneberg B, Dimeo F, Wustenberg T, Strohle A. The stress-buffering effect of acute exercise: evidence for HPA axis negative feedback. Psychoneuroendocrinology. 2015;51:414–25. doi: 10.1016/j.psyneuen.2014.10.019. [DOI] [PubMed] [Google Scholar]