

Abstract
Phosphorylated proteins play important roles in the regulation of many different cell networks. However, unlike the preparation of proteins containing unmodiffed proteinogenic amino acids, which can be altered readily by site-directed mutagenesis and expressed in vitro and in vivo, the preparation of proteins phosphorylated at predetermined sites cannot be done easily and in acceptable yields. To enable the synthesis of phosphorylated proteins for in vitro studies, we have explored the use of phosphorylated amino acids in which the phosphate moiety bears a chemical protecting group, thus eliminating the negative charges that have been shown to have a negative effect on protein translation. Bis-o-nitrobenzyl protection of tyrosine phosphate enabled its incorporation into DHFR and IκB-α using wild-type ribosomes, and the elaborated proteins could subsequently be deprotected by photolysis. Also investigated in parallel was the re-engineering of the 23S rRNA of Escherichia coli, guided by the use of a phosphorylated puromycin, to identify modified ribosomes capable of incorporating unprotected phosphotyrosine into proteins from a phosphotyrosyl-tRNACUA by UAG codon suppression during in vitro translation. Selection of a library of modified ribosomal clones with phosphorylated puromycin identified six modified ribosome variants having mutations in nucleotides 2600–2605 of 23S rRNA; these had enhanced sensitivity to the phosphorylated puromycin. The six clones demonstrated some sequence homology in the region 2600–2605 and incorporated unprotected phosphotyrosine into IκB-α using a modified gene having a TAG codon in the position corresponding to amino acid 42 of the protein. The purified phosphorylated protein bound to a phosphotyrosine specific antibody and permitted NF-κB binding to a DNA duplex sequence corresponding to its binding site in the IL-2 gene promoter. Unexpectedly, phosphorylated IκB-α also mediated the exchange of exogenous DNA into an NF-κB–cellular DNA complex isolated from the nucleus of activated Jurkat cells.
Graphical Abstract
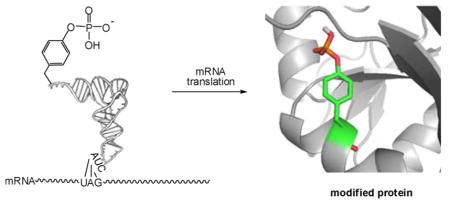
INTRODUCTION
Although the phosphorylation of serine and threonine in cellular proteins has been known since 1954,1 a cellular enzyme capable of protein tyrosine phosphorylation was first described only in 1979.2 This post-translational modification of tyrosine residues has now been well characterized as an important part of regulatory networks in both eukaryotic and prokaryotic systems.3 The later discovery of tyrosine phosphorylation may reflect the cellular abundance of highly active cellular tyrosine phosphatase activities capable of removing the phosphate moiety of phosphotyrosine. For example, phosphotyrosine residues on the EGF receptor have half-lives of only a few seconds and turn over more than 100 times during the early phase of cellular EGF response.4
Phosphotyrosine is also unique in that the immune system can generate antibodies specific to this amino acid, but not to phosphoserine or phosphothreonine.5 Since 1981, such antibodies have been used broadly for the assay of proteins containing phosphotyrosine.6,7 The availability of phospho-specific antibodies has led to the improvement of traditional methods as well as the development of new immunoassay techniques. Western blot and enzyme-linked immunosorbent assay (ELISA) are now common methods used for assessing the phosphorylation state of a protein.8,9 These augment the classical, labor-intensive method of direct protein phosphorylation by incubating whole cells with radiolabeled 32P-orthophosphate, generating cellular extracts, separating the proteins by SDS-PAGE, and exposing the gel to film.10,11 In the field of proteomics, the assessment of protein phosphorylation in complex biological samples, such as cell lysates, can now be realized by mass spectrometry (MS) techniques,12–15 often combined with innovative new strategies.12–15
The foregoing methods can be used for functional studies of phosphorylation events, but the identification of the residues phosphorylated in newly identified phosphoproteins, is still based on sequence comparison with known proteins by sequence conservation analysis.16 For phosphorylation sites that are not positionally conserved, approaches have been developed for the direct study of phosphorylated proteins.17–19 These have included chemical and enzymatic modification of specific amino acid side chains in the proteins or the incorporation of photocaged amino acids during translation. Direct nonsense codon suppression with tRNAs activated with phosphorylated amino acids has been reported in vitro20 in low yields and more recently in cellulo.21
Presently, we describe two complementary approaches for the introduction of phosphotyrosine into proteins by nonsense codon suppression. Bis-o-nitrobenzyl protected phosphotyrosyl-tRNACUA was used to introduce the protected phosphotyrosine into two different proteins in good suppression yields. Photolytic deprotection of the formed proteins proceeded to completion within several minutes, albeit with some loss of enzyme activity. Alternatively, by the use of a library of E. coli harboring plasmids with modified 23S rRNAs, we were able to identify six clones sensitive to phosphorylated puromycin. These clones incorporated phosphotyrosine itself into position 42 of IκB-α. This parallels the success we have had in selecting ribosomes able to incorporate β-amino acids22 and dipeptides23 into proteins. These modified ribosomes have allowed us to prepare IκB-α protein phosphorylated on Tyr42 and to evaluate its activity in activation of NF-κB binding to its template DNA. The elaborated phosphorylated IκB-α was used for the direct study of the interaction of IκB-α and NF-κB.
RESULTS
Incorporation of Phosphorylated Tyrosine Derivatives into Proteins Using Wild-Type Ribosomes
As outlined in Scheme 1, a fully protected derivative of bis(o-nitrobenzyl)phosphotyrosine (4) (Figure 1) was prepared by condensing bis-o-nitrobenzyl N,N-diisopropylphosphoramidite (5)24 with N-pentenoyl-L-tyrosine cyanomethyl ester (6). Product 7 was used to esterify the synthetic dinucleotide pdCpA,25 affording 8 in 22% yield. This aminoacylated pdCpA was then used as a substrate for T4 RNA ligase-mediated condensation with an in vitro tRNACUA transcript lacking the 3′-terminal CpA sequence of tRNA (Scheme 1).26 The resulting tRNACUA activated with amino acid 4 was employed as a substrate for incorporation into position 10 of dihydrofolate reductase (DHFR) and position 42 of IκB-α, as described below. The same suppressor tRNA transcript activated with mono-(o-nitrobenzyl)phosphotyrosine (3) (Figure 1, Scheme S1) and phosphotyrosine (2) (Scheme S2) were prepared analogously. The preparation of tyrosyl-tRNACUA has been described previously.24,27
Scheme 1.

Synthesis of the pdCpA Ester of Bis(o-nitrobenzyl)phosphotyrosine and Bis(o-nitrobenzyl)phosphotyrosyl-tRNACUA
Figure 1.

Structures of tyrosine (1), phosphotyrosine (2), o-nitrobenzylphosphotyrosine (3), and bis(o-nitrobenzyl)phosphotyrosine (4).
The incorporation of amino acids 1–4 into position 10 of E. coli DHFR was carried out using an S-30 preparation containing wild-type ribosomes. This position was chosen because it is postulated to be a surface accessible and relatively unstructured part of the protein, tolerant of numerous substitutions.26 As shown (Figure 2A), though the suppression yield obtained with tyrosyl-tRNACUA was quite high (89%, relative to the yield of wild-type DHFR produced from unmodified mRNA), the incorporation of bis(o-nitrobenzyl)phosphotyrosine (4) was much lower (22%), and the yields of DHFR containing 2 and 3 were 2 and 5%, respectively. A similar result was observed for the incorporation of 2 and 4 into position 42 of IκB-α. The fully protected phosphotyrosine (4) was incorporated with a 23% suppression yield, whereas unprotected phosphotyrosine (2) was incorporated to the extent of only 3%.
Figure 2.
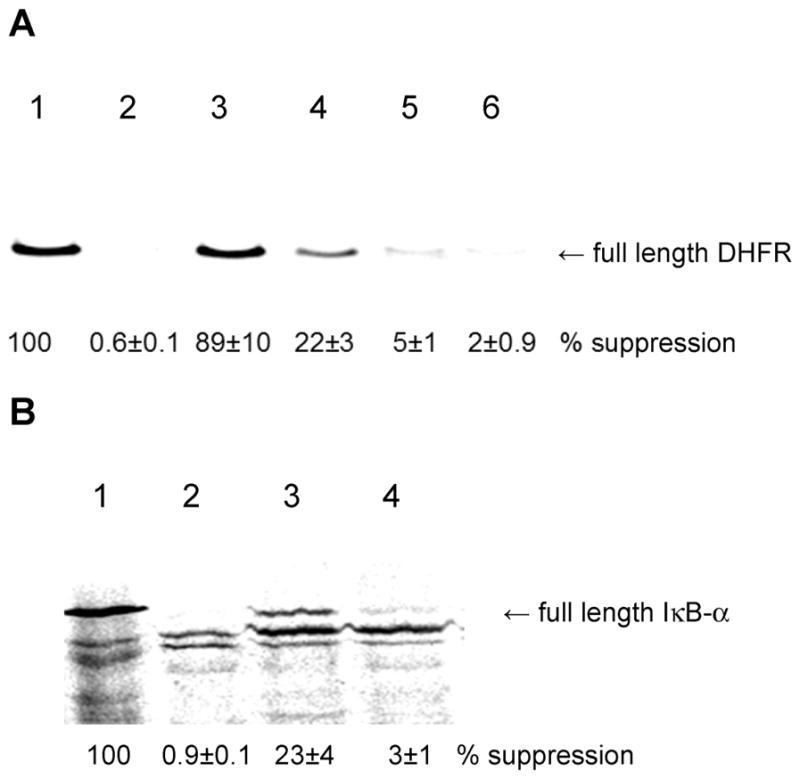
(A) Translation of DHFR from wild-type (lane 1) and modified (TAG codon in protein position 10) genes in the absence (lane 2) and in the presence of tyrosyl-tRNACUA (lane 3), bis(o-nitrobenzyl)phosphotyrosyl-tRNACUA (lane 4), o-nitrobenzylphosphotyrosyl-tRNACUA (lane 5), and phosphotyrosyl-tRNACUA (lane 6). (B) Translation of IκB-α from wild-type (lane 1) and modified (TAG codon in protein position 42) genes in the absence of an activated tRNACUA (lane 2) and in the presence of bis(o-nitrobenzyl)-phosphotyrosyl-tRNACUA (lane 3) and phosphotyrosyl-tRNACUA (lane 4).
The preparation of IκB-α containing 4 at position 42 was scaled up to enable the study of photodeprotection of the phosphate group in the modified IκB-α. Deprotection of the phosphate group was carried out using a 500 W mercury–xenon lamp, which has been used routinely for removal of NVOC (6-nitroveratryloxycarbonyl) protecting groups from the amino acid moiety of aminoacyl-tRNAs prior to protein synthesis.28 As shown in Figure 3A, following irradiation for 3 min, the modified IκB-α reacted with a rabbit polyclonal IgG specific for phosphotyrosine, whereas neither the wild-type protein nor IκB-α containing a protected phosphotyrosine reacted with the antibody. A study of the time course of phosphate group deprotection is shown in Figure 3B, and confirmed that deprotection was complete after 3 min.
Figure 3.
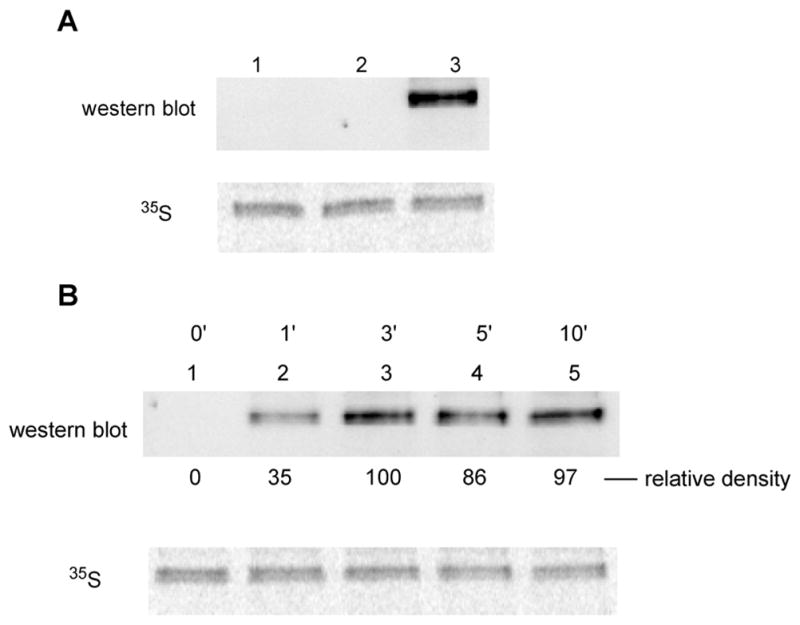
(A) Analysis of wild-type IκB-α (lane 1) and the caged IκB-α protein having a bis-o-nitrobenzyl protected phosphate group before (lane 2) and after (lane 3) 3 min of UV irradiation. The samples were analyzed for reactivity to a polyclonal antibody to phosphotyrosine and for 35S-methionine content. (B) Analysis of the caged IκB-α protein having a bis-o-nitrobenzyl protected phosphate group before, and as a function of the time of UV irradiation. Aliquots were taken at predetermined times and analyzed for reactivity to a polyclonal antibody to phosphotyrosine and for 35S-methionine content.
The use of this strategy for producing phosphorylated proteins for study requires that the elaborated proteins exhibit stability under the conditions needed for photodeprotection. Though phosphorylation of IκB-α at position 42 eliminates its ability to bind tightly to NF-κB,8,29 this property cannot be adapted for the facile quantitative assay of phosphorylated IκB-α. Accordingly, we employed two model proteins, dihydrofolate reductase (DHFR) and green fluorescent protein (GFP), neither of which has been reported to be sensitive to ambient light. As shown in Table 1, both of these proteins retained ~85% of their wild-type activity following 2 min of irradiation with the 500 W mercury–xenon lamp. As expected, longer irradiation times caused a further reduction in activity.
Table 1.
Study of Wild-Type DHFR and GFP Stability During Irradiation
| irradiation time (min) | DHFR (rel. activity) | GFP (rel. emission) |
|---|---|---|
| 0 | 100 | 100 |
| 2 | 85.1 ± 6.2 | 83.6 ± 5.2 |
| 3 | 79.5 ± 5.6 | 82.8 ± 6.7 |
| 4 | 68.3 ± 4.7 | 82.4 ± 14.1 |
| 5 | 56.9 ± 9.6 | 76.8 ± 16.0 |
In parallel with the foregoing study, we also explored a second strategy for introducing phosphotyrosine into predetermined sites in proteins. This involved the selection of modified ribosomes able to recognize and incorporate (unprotected) phosphotyrosine into proteins.
Selection of Modified Ribosomal Clones Able To Recognize Phosphotyrosine and Incorporate This Amino Acid into Protein
A library of E. coli ribosomal clones30 containing modifications in two regions of 23S rRNA corresponding to key elements of the ribosomal peptidyl-transferase center (PTC) was screened with phosphopuromycin to identify sensitive clones. The library described previously was expanded through additional mutations in the regions 2600–2605 and 2606–2611. The key nucleotide in region 2600–2605 is A2602. This nucleotide has been reported to exhibit conformational flexibility within the PTC and to play a key role in PTC rearrangement to accommodate substrate analogues, as well as participating in the catalytic step.31 Region 2606–2611 of the PTC has at least three interesting nucleotides, including C2606, C2609, and U2611, which have been shown to participate in structural networks important for PTC functional architecture.31c,32 C2606 has been shown to interact with U2585, leading to stabilization of the P-site of the PTC. Mutations C2609U and U2611C change the conformation of the PTC, conferring macrolide resistance.
In analogy with the β-puromycins22,30 and dipeptidylpuromycins23 studied previously, we postulated that clones sensitive to phosphopuromycin might contain modified ribosomes capable of recognizing phosphotyrosine and incorporating that amino acid into proteins. The short chemical route used for the synthesis of phosphopuromycin is shown in Scheme 2; it involved condensation of Nα-Fmoc protected phosphotyrosine with puromycin aminonucleoside, followed by removal of the Fmoc protecting group.
Scheme 2.

Synthesis of Phosphopuromycin
We initially surveyed 972 ribosomal clones for sensitivity to phosphopuromycin at a concentration of 100 μg/mL, and identified seven promising clones whose cell growth was inhibited by at least 50% (Table 2). In spite of the general similarity of their sensitivities to 100 μg/mL phosphopuromycin, we observed a larger variation in the IC50 values among these clones using different assay conditions, which ranged from 24 to 97 μg/mL (Figure 4). The extent of inhibition may reflect differences in the levels of the modified ribosomes as well as inherent differences in their sensitivity to phosphopuromycin. It should be noted that these IC50 values were determined in the presence of 2.5 μg/mL of erythromycin, which largely suppressed the activity of the wild-type ribosomes within the clones. Interestingly, the six clones modified in the region 2600–2605 were found to be quite G-rich at positions 2600, 2602, 2604, and 2605, although at positions 2600 and 2602 there were clones having U (Table 3).33
Table 2.
Characterization of rrnB Operon Variants Selected Using Phosphopuromycin
| clone | selection data, % inhibition | sequence in the mutagenized regions
|
|
|---|---|---|---|
| region 1 | region 2 | ||
| 030405 | 87.5 (78.4)a | 2057ATGGTTG2063 | 2600GAGTGA2605 |
| 030449 | 36.4 (56.2)a | 2057ATGGTTG2063 | 2600GTTCGG2605 |
| 030454 | 57.4(52.9)a | 2057ATGGTTG2063 | 2600GAGAGC2605 |
| 040412 | 61.4 (52.2)a | 2057AGCGTGA2063 | 2600 GTTCGG 2605 |
| 040424 | 41.5 (57.3)a | 2057AGCGTGA2063 | 2600TGGTGG2605 |
| 040435 | 79.3 (53.1)a | 2057AGCGTGA2063 | 2600GAGAGG 2605 |
| 030548 | 75.8 (66.4)a | 2057ATGGTTG2063 | 2606GAAGAT2611 |
| wild type | <10% | 2057GAAAGAC2063 | 2600ACAGTT2601 2606CGGTCC2612 |
Data in parentheses are from confirmatory selection experiments.
Figure 4.
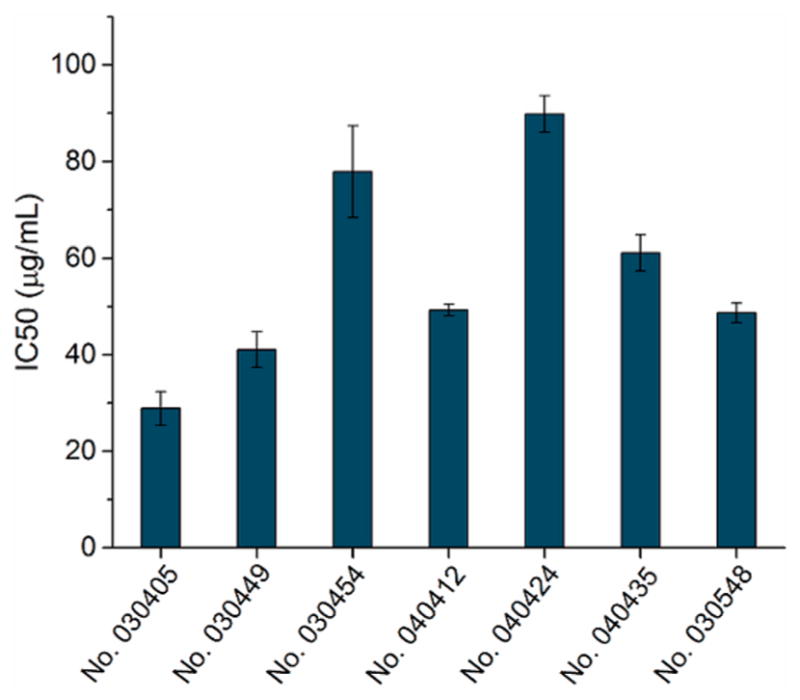
Analysis of the phosphopuromycin sensitivity of BL-21(DE-3) cells having modified ribosomes. The assays were carried out in the presence of 2.5 μg/mL of erythromycin to partially block the activity of endogenous wild-type ribosomes.
Table 3.
Sequence Alignment of Region 2600–2605 of the 23S rRNA of the Modified Ribosomes Selected Using Phosphopuromycin
| nucleotide position | ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| clone | 2600 | 2601 | 2602 | 2603 | 2604 | 2605 |
| 030405 | G | A | G | U | G | A |
| 030449 | G | U | U | C | G | G |
| 030454 | G | A | G | A | G | C |
| 040412 | G | U | U | C | G | G |
| 040424 | U | G | G | U | G | G |
| 040435 | G | A | G | A | G | G |
| wild type | A | C | A | G | U | U |
S-30 preparations were obtained from six clones, omitting 040435 which did not grow well. Four of these six S-30 preparations were able to incorporate phosphotyrosine into position 42 of IκB-α by nonsense codon suppression (in yields up to 24%); the results are summarized in Table 4 and illustrated for two of the clones in comparison with wild type in Figure 5. The efficiency of translation of IκB-α using S-30 preparations from the clones was sometimes (e.g., 030405, 030449, and 040412) roughly correlated with the IC50 values for inhibition by phosphopuromycin (cf Figure 4). However, the S-30 preparation from clone 030454, which had not been inhibited strongly by phosphopuromycin, also afforded an S-30 preparation that produced efficient incorporation of phosphotyrosine (Table 4). Though S-30 preparations from clones 040424 and 030548 produced IκB-α containing phosphotyrosine at position 42 poorly, if at all, they also displayed inefficient incorporation of tyrosine into IκB-α. Because the identification of clones in Table 2 was made based solely on puromycin sensitivity, it is perhaps unsurprising that some of these proved not to be proficient in in vitro protein synthesis. The best incorporation yields of phosphotyrosine were achieved using S-30 preparations from clones 030449 (20%) and 040412 (24%), which had the same sequence (GUUCGG) in the mutagenized region extending between nucleotides 2600 and 2605.
Figure 5.
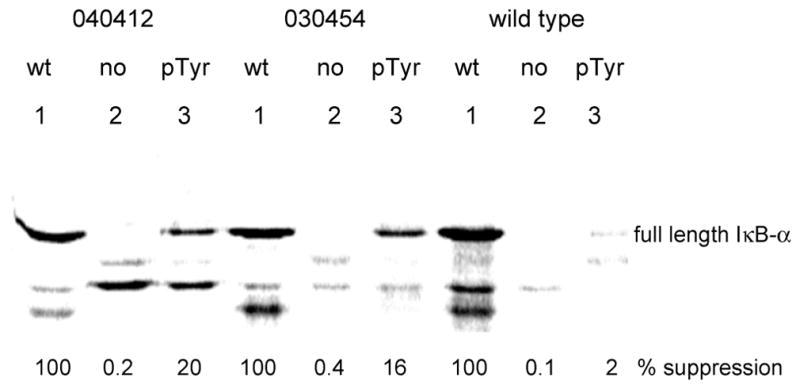
Comparison of the in vitro translation yields of phosphorylated IκB-α obtained using S-30 preparations containing representative modified vs wild-type E. coli ribosomes. The IκB-α samples obtained from wild-type (wt; lanes 1) and modified (TAG codon in protein position 42) genes in the absence (no; lanes 2) and in the presence of phosphotyrosyl-tRNACUA (pTyr; lanes 3) were analyzed by 12% SDS-polyacrylamide gel electrophoresis.
Table 4.
Expression Yields of IκB-α Phosphorylated at Position 42 Using S-30 Preparations Containing Different Modified Ribosomes
| Clone | yield of full length IκB-α, %a
|
||
|---|---|---|---|
| pTyr | Tyr | no activated tRNACUA | |
| wild type | 3 ± 2 | 68 ± 10 | 0.1 ± 0.05 |
| 030405 | 12 ± 2 | 60 ± 4 | 0.8 ± 0.3 |
| 030449 | 20 ± 2 | 93 ± 10 | 0.4 ± 0.2 |
| 030454 | 15 ± 1 | 81 ± 20 | 0.1 ± 0.06 |
| 040412 | 24 ± 3 | 78 ± 6 | 0.15 ± 0.1 |
| 040424 | 5 ± 1 | 28 ± 6 | 0.1 ± 0.05 |
| 030548 | 2 ± 0.8 | 16 ± 3 | 0.2 ± 0.1 |
Yield of modified IκB-α was estimated relative to wild-type IκB-α synthesis in the same experiment.
Purification and Structural Characterization of IκB-α Containing Phosphotyrosine at Position 42
IκB-α prepared by in vitro translation was purified by Ni-NTA chromatography.34 The purified protein had the same mobility as (nonphosphorylated) IκB-α produced from the wild-type gene in vitro when analyzed by 4%–12% SDS-polyacrylamide gradient gel electrophoresis (Figure 6A), albeit not when analyzed on a native gel (vide infra). The presence of phosphotyrosine at position 42 was verified by immunoblotting analysis using an anti-phosphorylated IκB-α (pTyr42) rabbit polyclonal IgG (Figure 6B), which reacted exclusively with the putatively phosphorylated IκB-α. Further characterization was achieved by MS/MS analysis of the tryptic peptides (amino acids 39–47) from wild-type (Figure 7A) and phosphorylated (Figure 7B) IκB-α samples. Both forward peptide fragment ions (y, (M – OH)+) and reverse peptide fragment ions (b, (M + H)+) containing phosphorylated tyrosine at position 42 were identified. Key mass values for the tryptic peptides are summarized in the Supporting Information, Table S1.
Figure 6.
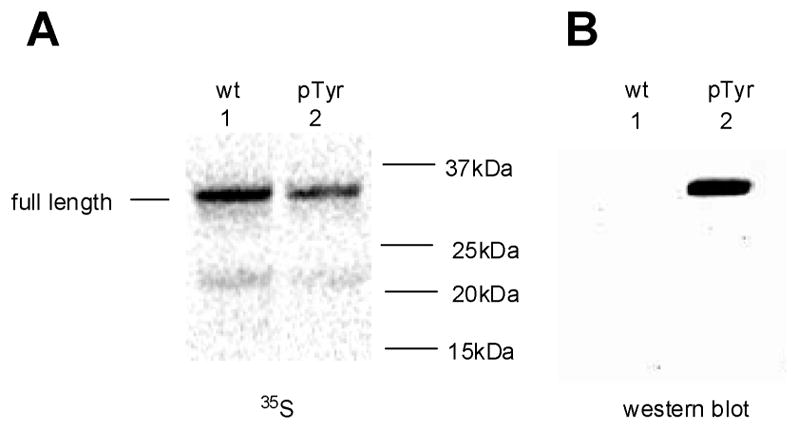
Immunoblotting analysis of IκB-α samples obtained by in vitro translation from the wild-type gene and from the modified (TAG codon in protein position 42) gene in the presence of phosphotyrosyl-tRNACUA (pTyr) and purified by Ni-NTA chromatography. Analysis of samples by 4%–12% SDS-polyacrylamide gradient gel electrophoresis using (A) phosphorimager detection of 35S-methionine and (B) anti-phosphorylated IκB-α (pTyr42) rabbit polyclonal IgG.
Figure 7.

MALDI MS/MS analysis of the tryptic peptides (amino acids 39–47) from (A) wild-type and (B) phosphorylated IκB-α samples.
Biological Characterization of Phosphorylated IκB-α
IκB-α has been reported to prevent the binding of transcription factor NF-κB to its nuclear DNA substrate by masking the nuclear localization signal.8,29,35 Two pathways have been proposed for the release of NF-κB via phosphorylation of IκB-α,8,29,36,37 and both enable binding of NF-κB to its DNA substrate. In the present study, we employed cultured Jurkat cells that were studied either without activation (Figure 8A), or following activation with a combination of PMA (phorbol 12-myristate 13-acetate) and Ca2+ ionophore A23187 (Figure 8B). Cell extracts were purified on DEAE-Sepharose CL-6B columns, and individual fractions were incubated with 32P-labeled double-stranded DNA corresponding to the NF-κB binding site in the IL-2 gene promoter.8 The samples were separated by 6% native polyacrylamide gel electrophoresis and analyzed using a phosphorimager (Figure 8). Lanes 1 in Figure 8A,B contained the crude extract and lanes 2 contained the flow through from the DEAE-Sepharose columns. Lanes 4–6 contained bands postulated to be a complex of NF-κB bound to its 32P-labeled double-stranded DNA substrate; as anticipated, the bands were more intense in the extract derived from the activated cells, reflecting the release of NF-κB from IκB-α upon activation. Quantification of the relative amounts of NF-κB in the unactivated and activated cells was carried out by analyzing equal portions of the eluates results from washing with 300 mM NaCl solutions (lanes 5 in Figure 8A,B). As shown in Figure 8C, the relative densities of the band corresponding to the putative NF-κB–DNA complexes isolated from the unactivated and activated cells were in the ratio 8:100, respectively.
Figure 8.
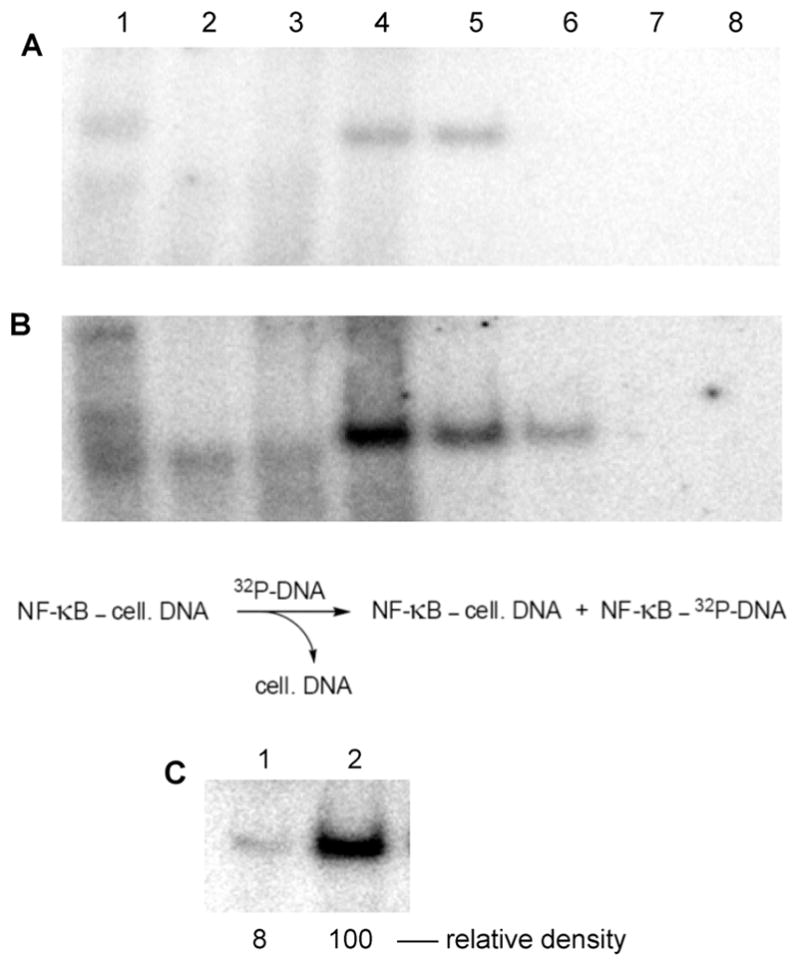
Purification of NF-κB (as a complex with cellular DNA) from extracts of Jurkat cells (A) without initial cell activation and (B) following cell activation with PMA + A23187. The extracts were fractionated using DEAE-Sepharose CL-6B. Each fraction was incubated with 32P-labeled double-stranded DNA corresponding to the NF-κB binding site in the IL-2 gene promoter.8 Samples were separated by 6% native polyacrylamide gel electrophoresis and analyzed using a phosphorimager. Lane 1, crude extract; lane 2, flow through from DEAE-Sepharose CL-6B column; lanes 3–8, eluates resulting from wash with 100, 200, 300, 400, 500, and 600 mM NaCl, respectively. Formation of the NF-κB–32P-labeled DNA complex is envisioned by (partial) chemical exchange for cellular DNA. (C) Comparison of the amount of purified NF-κB isolated from unactivated vs activated Jurkat cells after purification by DEAE-Sepharose CL-6B (300 mM NaCl wash). Samples were separated by 6% native polyacrylamide gel electrophoresis and analyzed using a phosphorimager. Lane 1, purified NF-κB isolated from extracts of unactivated Jurkat cells; lane 2, purified NF-κB isolated from extracts of Jurkat cells activated with PMA + A23187.
In order to determine whether the phosphotyrosine-containing IκB-α, prepared in vitro using our modified ribosomes, was capable of interacting with a preincubation complex of NF-κB and its DNA substrate, we utilized the fractionated nuclear extracts from activated Jurkat cells. Portions of the purified fraction mixed with 5′-32P-labeled 5′-GATCCAAGGGACTTTCCATG-3′ duplex (Figure 9, lane 2) were also treated with in vitro synthesized wild-type IκB-α (Figure 9, lane 3), or with IκB-α expressed with a phosphotyrosine at position 42 (Figure 9, lane 4). As shown in Figure 9, admixture of nonphosphorylated IκB-α to the NF-κB–DNA complex sharply diminished the intensity of the band, consistent with the thesis that IκB-α can replace the DNA substrate and form a (more stable) NF-κB–IκB-α complex. This is entirely consistent with detailed kinetic studies demonstrating that unphosphorylated IκB-α can accelerate the rate of dissociation of NF-κB from the NF-κB–DNA complex.38 Unexpectedly, treatment with phosphorylated IκB-α substantially increased the intensity of the resulting band (lane 4), whereas further treatment with NF-κB (p50) polyclonal rabbit IgG largely reversed the effects of adding phosphorylated IκB-α (lane 5). We postulate that the result observed in lane 4 reflects the presence in the initial mixture of a preponderance of NF-κB bound to (unlabeled) cellular DNA. Admixture of 32P-labeled DNA causes limited exchange (as in lane 2, Figure 9) to afford a mixture of NF-κB, some bound to cellular DNA and some to 32P-labeled DNA. However, in the presence of phosphorylated IκB-α, this protein can also bind transiently to NF-κB, facilitating the exchange of 32P-labeled DNA for cellular DNA. Further admixture of an antibody to the p50 subunit of NF-κB partially blocks this exchange by diminishing available NF-κB.
Figure 9.
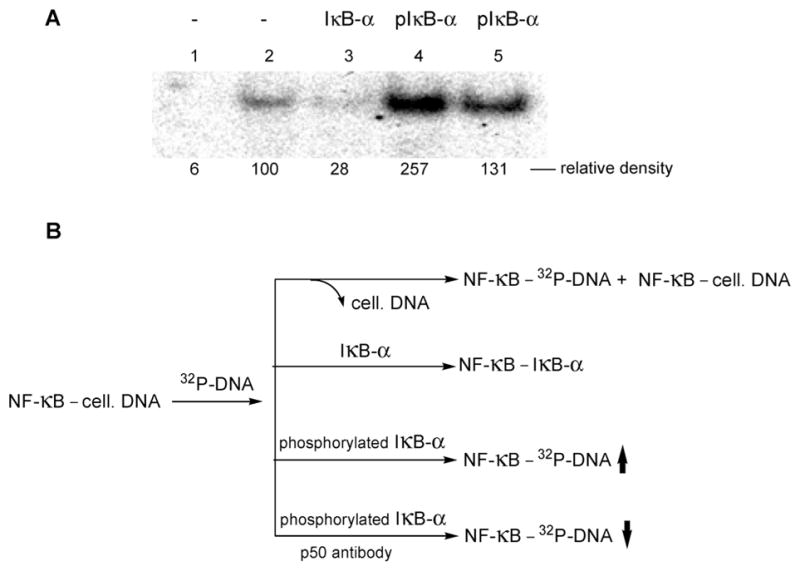
(A) Assay of NF-κB (as a complex with cellular DNA) treated with radiolabeled DNA. Samples were separated by 6% native polyacrylamide gel electrophoresis and analyzed using a phosphor-imager. Lane 1, binding of DNA to NF-κB purified from unactivated Jurkat cells; lane 2, binding of DNA to purified NF-κB from Jurkat cells activated with PMA + A23187; lane 3, binding of DNA to purified NF-κB from Jurkat cells activated with PMA + A23187 in the additional presence of 20 ng of wild-type IκB-α; lane 4, binding of DNA to purified NF-κB from Jurkat cells activated with PMA + A23187 in the additional presence of 20 ng of IκB-α phosphorylated on Tyr42; lane 5, binding of DNA to purified NF-κB from Jurkat cells activated with PMA + A23187 in the additional presence of 20 ng of IκB-α phosphorylated on Tyr42, and 200 ng of NF-κB (p50) polyclonal rabbit IgG. (B) Rationalization of the change in intensity of the observed bands.
In order to test the foregoing hypothesis, two additional experiments were carried out. In the first, we tested the ability of phosphorylated IκB-α to bind to NF-κB. This has not previously been reported, and is contrary to the relief of inhibition of NF-κB binding to its DNA substrate by phosphorylation of IκB-α, which has been reported several times.8,29,36,37 Shown in Figure 10 are the results of an experiment in which we employed 35S-radiolabeled samples of IκB-α, with and without a phosphate moiety at position 42, as substrates for binding to NF-κB without exogenously added DNA. As is clear from Figure 10, unphosphorylated IκB-α bound to NF-κB more avidly, but both forms of the enzyme clearly bound to NF-κB. This supports the possibility that phosphorylated IκB-α can facilitate the equilibration of cellular and 32P-labeled DNA putatively observed in Figure 9. It may be noted also in Figure 10 (lanes 4 and 5) that phosphorylated IκB-α migrated to a lesser extent than IκB-α, on a native gel, consistent with a possible conformational change in the protein upon phosphorylation. In comparison, the phosphorylated and nonphosphorylated proteins showed no difference in mobility on a denaturing gel (Figure 6), as would be expected given the minimal difference in their molecular weights.
Figure 10.
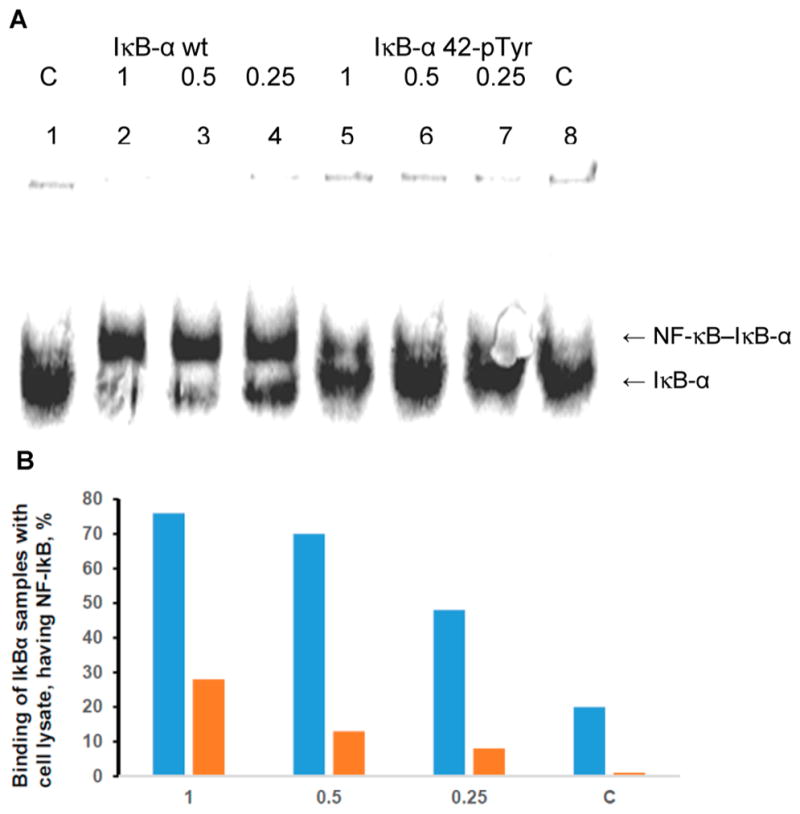
Binding of nonphosphorylated IκB-α (IκB-α wt) and phosphorylated IκB-α (IκB-α 42-pTyr) to NF-κB. (A) Analysis (native 8% polyacrylamide gel) of the purified lysate from activated Jurkat cells (Figure 8B, lane 5) in complex with 35S-labeled wild-type and phosphorylated IκB-α prepared by in vitro protein synthesis. Lanes 1 and 8, 35S-labeled protein not treated with lysate; lanes 2 and 5, 35S-labeled proteins treated with lysate; lanes 3 and 6, 35S-labeled proteins treated with lysate diluted 2-fold with 50 mM Tris–HCl, pH 7.4; lanes 4 and 7, 35S-labeled protein treated with lysate diluted 4-fold with 50 mM Tris–HCl, pH 7.4. (B) Quantification of the binding illustrated in the gel in panel A for 35S-labeled wild-type (blue) and phosphorylated (orange) IκB-α. C, control not treated with lysate; 1, purified lysate present at full strength; 0.5, lysate present at 2-fold dilution; 0.25, lysate present at 4-fold dilution.
A further experiment was carried out under conditions analogous to lane 4 of Figure 9. Admixture of 32P-labeled DNA and phosphorylated IκB-α was effected to the purified NF-κB–cellular DNA complex from activated Jurkat cells; the radiolabeled DNA was in excess relative to phosphorylated IκB-α. The incorporation of radioactivity into a band containing NF-κB was monitored as a function of time; a control lacking phosphorylated IκB-α was run in parallel. As shown in Figure 11, the band containing radiolabeled DNA intensified to a greater extent after 3 min in the presence of phosphorylated IκB-α than in its absence. This phenomenon was also shown to be dependent on the concentration of phosphorylated IκB-α (Figure S1). These findings strongly support the thesis that the IκB-α phosphorylated on position 42 mediates the exchange of exogenous 32P-labeled DNA into the NF-κB–cellular DNA complex.
Figure 11.

Time-dependent binding of NF-κB and 32P end-labeled DNA. (A) Samples were analyzed by 6% native polyacrylamide gel electrophoresis and quantified using a phosphorimager. Upper panel, binding of NF-κB and DNA without phosphorylated IκB-α; lower panel, binding of NF-κB and DNA in the presence of 20 ng of phosphorylated IκB-α. Lane 1, incubation for 0 min; lane 2, incubation for 0.5 min; lane 3, incubation for 1 min; lane 4, incubation for 3 min. (B) Relative intensity of NF-κB–DNA complexes analyzed by 6% native polyacrylamide gel electrophoresis. The intensity of the NF-κB–DNA complex in the reaction of NF-κB and DNA without phosphorylated IκB-α at 0.5 min was defined as 100%.
DISCUSSION
In the present study, we explored two strategies for introducing phosphorylated tyrosine derivatives into proteins. One of these involved the use of a bis-o-nitrobenzyl protected derivative of phosphotyrosine (4), which was introduced onto the 3′-end of a suppressor tRNACUA using a well established methodology (Scheme 1). This protected amino acid could be introduced into position 10 of DHFR and position 42 of IκB-α in ~20–25% yields (Figure 2) by in vitro protein synthesis in a system employing an S-30 preparation, and then deprotection of the elaborated protein by irradiation with a 500 W mercury–xenon lamp. The use of a mono-o-nitrobenzyl protected derivative of phosphotyrosine afforded a suppression yield of only 5% for DHFR (Figure 2). Irradiation of the bis-o-nitrobenzyl protected protein seemed complete after 3 min (Figure 3). To determine whether these conditions would be useable for routine experimentation, we studied the effect of exposing two wild-type proteins (DHFR and GFP) to analogous irradiation for varying lengths of time up to 5 min (Table 1). As expected, the activities of these proteins diminished somewhat as a function of irradiation time, but both retained ~80–85% of their activities after 2–3 min of irradiation. This strategy has the advantage of providing a caged protein initially, which may be of utility for mechanistic studies.20
More satisfactory results were realized using E. coli ribosomes whose 23S rRNA had been modified. Members of a library of clones containing modified ribosomes were tested for sensitivity to a puromycin derivative having a phosphorylated tyrosine moiety (Scheme 2), and seven clones exhibited enhanced sensitivity to this puromycin (Table 2). Analogous to earlier studies using puromycins containing β-amino acids22 and dipeptides,23 the clones sensitive to phosphopuromycin recognized a suppressor tRNACUA activated with phosphotyrosine (Scheme S2). It is interesting that in contrast with the wild-type 23S rRNA, all seven clones were quite G-rich at positions 2600, 2602, 2604, and 2605 (Table 3), each having at least four guanosines. This suggests a common geometry for recognition of phosphotyrosyl-tRNA. Plausibly, the rather basic guanine nucleobases at these positions may enable enhanced binding to the acidic phosphotyrosine moiety of the activated suppressor tRNA.
As shown in Table 4 and Figure 5, S-30 preparations from four of six clones studied were able to facilitate the incorporation of phosphotyrosine into position 42 of IκB-α, giving full length proteins in yields from 12–24%. The two clones that gave lower yields of phosphorylated IκB-α also failed to produce wild-type IκB-α efficiently. The purified phosphorylated IκB-α comigrated with authentic IκB-α produced in vitro (Figure 6A) when analyzed by denaturing PAGE. The presence of a phosphate group at position 42 was verified, both by the use of an anti-phosphorylated IκB-α (pTyr42) rabbit polyclonal IgG (Figure 6B) and by MS/MS analysis of the tryptic peptides (Figure 7).
Given the importance of protein phosphorylation for numerous biochemical regulatory mechanisms, it is not surprising that several strategies have focused on the introduction of this functional group into proteins at specific sites. Nonsense codon suppression in vitro with suppressor tRNAs activated with phosphorylated amino acids has been reported by a few laboratories, albeit in low yields.20,21,24 Several laboratories have used enzymatic methods in conjunction with native chemical ligation to produce phosphorylated proteins in vitro,39 and there have also been modified phosphorylated proteins produced by other combinations of enzymatic and chemical methods.40 Recently, the incorporation of phosphotyrosine into a protein in cellulo has been reported,41 and the incorporation of phosphoserine has also been reported.42 It seems reasonable to anticipate that expression in cells containing the modified ribosomes of the type described here might increase the yield of phosphorylated proteins elaborated in cells.
In the context of protein phosphorylation, IκB-α is of particular interest as it regulates the activity of transcription factor NF-κB by inhibiting its binding to its cellular DNA targets.8,29,35 Two distinct phosphorylation mechanisms involving IκB-α have been reported to relieve its inhibition of NF-κB, enabling it to bind to its DNA substrate in the IL-2 gene promoter.8,29,36,37 IκB-α phosphorylation at Ser32/Ser36 results in NF-κB release as a consequence of the ubiquitination of IκB-α and its subsequent proteasomal degradation.36,37,43 Alternatively, phosphorylation of IκB-α at Tyr42 putatively activates NF-κB without degradation of phosphorylated IκB-α by eliminating its ability to bind to the transcription factor.8,29 Studies of both mechanisms provided convincing analyses of the observed phenomena; most of the experiments involved the analysis of events at a cellular level following cellular activation with any of several different agents, although an insightful in vitro study of NF-κB activation has also appeared.29c It may be noted that many of the conclusions that support the current models of NF-κB activation were necessarily inferential, based on the nature of the experiments carried out. Presently, we have explored the behavior of an authentic sample of IκB-α phosphorylated at position 42.
Cultured Jurkat cells were activated by treatment with a combination of PMA and A23187, in analogy with the seminal study of IκB-α at position 42, in which the investigators achieved cell activation by using pervanadate to induce two tyrosine kinases and inhibit protein tyrosine phosphatases.8 The purified cell lysate enabled isolation of NF-κB bound to cellular DNA. The complex was visualized by admixture of 32P-end labeled DNA having the known target sequence for NF-κB within the IL-2 promoter, in the belief that limited exchange of the cellular and exogenous DNAs would occur over time. In fact, uncomplexed IκB-α has been reported to be able to enter the nucleus and dissociate NF-κB–DNA complexes.8 As anticipated, significantly more of the NF-κB–DNA complex was observed following cell activation (Figure 8) and the difference was quantified (Figure 8C).
Admixture of authentic (unphosphorylated) IκB-α, prepared by in vitro translation, was found to dissociate the NF-κB–DNA complex, consistent with the reported role of IκB-α as an inhibitor of NF-κB-mediated DNA binding and transcriptional activation. Based on the reports that IκB-α phosphorylation diminishes its affinity for NF-κB,8,29,35–37 it was surprising to find that admixture of authentic phosphorylated IκB-α, prepared by in vitro translation, actually increased the amount of 32P-end labeled DNA present within the NF-κB–DNA complexes (cf lanes 2 and 4 in Figure 9). That the complex present in the gel actually included NF-κB was verified by the use of a polyclonal antibody to the p50 subunit of NF-κB, which largely reversed the increase induced by phosphorylated IκB-α (cf lanes 4 and 5 in Figure 9).
The foregoing results seemed consistent with the interpretation that the phosphorylated IκB-α was capable of catalyzing the exchange of 32P-end labeled DNA with the isolated NF-κB–DNA complex. This was studied further, initially by preparing 35S-radiolabeled IκB-α by in vitro translation. As shown in Figure 10, IκB-α lacking pTyr42 bound more avidly to NF-κB than did the phosphorylated IκB-α, but both clearly bound. We next carried out additional experiments of the type involved in lane 4 of Figure 9. As shown, phosphorylated IκB-α mediated an increase in the amount of 32P-labeled DNA–NF-κB complex after 3 min (Figure 11), and the increase was also concentration-dependent (Figure S1).
Though it has been reported that Ser32/Ser36 phosphorylation of IκB-α, in comparison with Tyr42 phosphorylation, results in proteasomal degradation of IκB-α,36,37,42 to the extent that IκB-α phosphorylated at position 42 persists, it seems conceivable that it may serve to mediate the exchange of NF-κB with cellular DNAs, thus facilitating formation of the physiologically relevant complexes between NF-κB and its DNA promoter targets.
CONCLUSIONS
Two strategies have been employed for the in vitro synthesis of proteins containing phosphorylated tyrosine residues at predetermined positions. The first involved the suppression of a UAG codon in the mRNA of the protein of interest, which was realized using a suppressor tRNACUA transcript that had been activated with a phosphotyrosine, the latter of which was chemically protected on the phosphate moiety with one or two o-nitrobenzyl groups. The fully protected phosphotyrosine was incorporated with reasonable (~20–25%) suppression efficiency, and could be deprotected to afford the free phosphate within a few minutes using a high intensity mercury–xenon lamp.
Alternatively, a phosphorylated analogue of puromycin was used to survey clones from a large E. coli library containing bacteria harboring plasmids with the genes for modified 23S rRNAs. Several of the clones were found to be sensitive to the phosphorylated puromycin analogue, implying that the assembled modified ribosomes contained therein had geometries in their peptidyltransferase centers able to recognize phosphotyrosine. In fact, S-30 preparations derived from some of these clones were able to suppress a UAG codon in the mRNA corresponding to position 42 of IκB-α, incorporating phosphotyrosine into the protein from (unprotected) phosphotyrosyl-tRNACUA. The yields of full length protein were in the range of 20–25% for S-30 preparations from the most efficient clones.
The unphosphorylated and phosphorylated IκB-α proteins so prepared were used to study the previously reported interactions of these proteins with the transcription factor NF-κB. In agreement with literature reports, unphosphorylated IκB-α bound tightly to NF-κB and could remove NF-κB from the complex with its cellular DNA target. Unexpectedly, it was found that IκB-α phosphorylated at position 42 retained some affinity for NF-κB, and strongly facilitated the exchange of 32P-end labeled DNA having the same sequence as the NF-κB target in the IL-2 promoter into the NF-κB–cellular DNA complex.
EXPERIMENTAL SECTION
Biochemical Assays of NF-κB and IκB-α
Preparation of Jurkat Nuclear Fraction
Jurkat cells were cultured at 37 °C in a 5% CO2 atmosphere and grown in Gibco RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) containing 50 units/mL of penicillin, 50 μg/mL of streptomycin, and 1 mM sodium pyruvate. When the cells grew to a density of 5 × 105 cells/mL, they were stimulated with PMA (0.3 μM) and calcium ionophore A23187 (1.0 μM) at 37 °C for 1 h. The same number of Jurkat cells without PMA and calcium ionophore A23187 was incubated in parallel as a control.
Jurkat cells with/without stimulation (20 mL each) were harvested by centrifugation at 500g at room temperature for 5 min and washed once with 5 mL of cold PBS. The cell pellets were resuspended in 60 μL of 10 mM Hepes, pH 7.9, containing 10 mM KCl, 1.5 mM MgCl2, 0.5 mM DTT, 0.1% Nonidet P40 and incubated at 4 °C for 10 min. After centrifugation at 1,000g at room temperature for 2 min, supernatants (cytosolic fractions) were removed and the nuclear pellets were resuspended in 90 μL of 10 mM Hepes, pH 7.9, containing 420 mM NaCl, 1.5 mM MgCl2, 0.5 mM DTT, 0.2 mM EDTA, 0.5 mM PMSF, and 25% glycerol. After centrifugation at 10,000g and 4 °C for 10 min, the supernatants (nuclear fractions) were diluted with 135 μL of 20 mM Hepes, pH 7.9, containing 50 mM KCl, 0.5 mM DTT, 0.2 mM EDTA, 0.5 mM PMSF in new tubes and stored at −80 °C.
Purification of NF-κB Using DEAE-Sepharose
Jurkat nuclear fraction (100 μL) was diluted with 300 μL of 20 mM Hepes, pH 7.9, containing 50 mM KCl, 0.5 mM DTT, 0.2 mM EDTA, and 0.5 mM PMSF, and was then loaded onto a 50 μL DEAE-Sepharose CL-6B column. After the crude nuclear fraction had been applied to the column, it was washed with 100 μL of 20 mM Hepes, pH 7.9, containing 50 mM KCl, 0.5 mM DTT, 0.2 mM EDTA, 0.5 mM PMSF, and 5% glycerol containing a step gradient of 100–600 mM NaCl.
NF-κB and IκB-α Binding Assay
The reaction mixture (10 μL) contained 3 μL of purified NF-κB fraction (with its associated cellular DNA), 20 ng of IκB-α, 2 pmol of double-stranded 32P-labeled NF-κB probe (5′-GATCCAAGGGACTTTCCATG-3′), with or without 200 ng NF-κB (p50) polyclonal rabbit IgG, 20 mM Hepes, pH 7.9, 50 mM KCl, 400 mM NaCl, 0.5 mM DTT, 0.2 mM EDTA, 0.5 mM PMSF, and 5% glycerol. The reaction mixtures were incubated at 37 °C for 20 min. The reactions were analyzed by 6% native PAGE at 100 V for 80 min.
Binding of Purified Jurkat Cell Lysate with 35S-Labeled IκB-α Samples
The 35S-labeled IκB-α samples (containing Tyr (wild-type) or phosphoTyr (mutant) at position 42) prepared by in vitro translation and dissolved in 50 mM Tris–HCl buffer, pH 7.4, at a concentration of ~5 ng per μL were used for this experiment. Two μL of fully concentrated, as well as 2- and 4-fold dilutions of Jurkat cell lysate in the same buffer were mixed with 4 μL of 35S-labeled IκB-α samples and incubated for 20 min at 37 °C. Two μL of loading dye (40% glycerol, 0.25% bromophenol blue) was then added, and the samples were analyzed by 10% native polyacrylamide gel electrophoresis then quantified using a phosphorimager.
Supplementary Material
Acknowledgments
This study was supported by Grant GM 103681 from the National Institute of General Medical Sciences, National Institutes of Health.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b05168.
Synthetic procedures and characterization of compounds prepared; biochemical procedures for the expansion of a library of clones containing modified ribosomes, selection of ribosomes recognizing phosphotyrosine, preparation of a suppressor tRNA activated with phosphotyrosine and in vitro synthesis and assay of phosphorylated IκB-α; concentration-dependent binding of NF-κB and DNA (PDF)
References
- 1.Burnett G, Kennedy EP. J Biol Chem. 1954;211:969. [PubMed] [Google Scholar]
- 2.Eckhart W, Hutchinson MA, Hunter T. Cell. 1979;18:925. doi: 10.1016/0092-8674(79)90205-8. [DOI] [PubMed] [Google Scholar]
- 3.(a) Vincent C, Duclos B, Grangeasse C, Vaganay E, Riberty M, Cozzone AJ, Doublet P. J Mol Biol. 2000;304:311. doi: 10.1006/jmbi.2000.4217. [DOI] [PubMed] [Google Scholar]; (b) Pawson T. Cell. 2004;116:191. doi: 10.1016/s0092-8674(03)01077-8. [DOI] [PubMed] [Google Scholar]; (c) Hunter T, Eckhart W. Cell. 2004;116:S35–S39. doi: 10.1016/s0092-8674(04)00049-2. [DOI] [PubMed] [Google Scholar]; (d) Grangeasse C, Cozzone A, Deutscher J, Mijakovic I. Trends Biochem Sci. 2007;32:86. doi: 10.1016/j.tibs.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 4.Kleiman LB, Maiwald T, Conzelmann H, Lauffenburger DA, Sorger PK. Mol Cell. 2011;43:723. doi: 10.1016/j.molcel.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glenney JR, Zokas L, Kamps MP. J Immunol Methods. 1988;109:277. doi: 10.1016/0022-1759(88)90253-0. [DOI] [PubMed] [Google Scholar]
- 6.Czernik AJ, Girault JA, Nairn AC, Chen J, Snyder G, Kebabian J, Greengard P. Methods Enzymol. 1991;201:264. doi: 10.1016/0076-6879(91)01025-w. [DOI] [PubMed] [Google Scholar]
- 7.Mann M, Ong SE, Gronborg M, Steen M, Jensen ON, Pandey A. Trends Biotechnol. 2002;20:261. doi: 10.1016/s0167-7799(02)01944-3. [DOI] [PubMed] [Google Scholar]
- 8.(a) Imbert V, Peyron JF, Farahifar D, Mari B, Auberger P, Rossi B. Biochem J. 1994;297:163. doi: 10.1042/bj2970163. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Imbert V, Rupec RA, Livolsi A, Pahl HL, Traenckner EBM, Mueller-Dieckmann C, Farahifar D, Rossi B, Auberger P, Baeuerle PA, Peyron JF. Cell. 1996;86:787. doi: 10.1016/s0092-8674(00)80153-1. [DOI] [PubMed] [Google Scholar]
- 9.Rijksen G, Adriaansen-Slot SS, Staal GE. Breast Cancer Res Treat. 1996;39:139. doi: 10.1007/BF01806180. [DOI] [PubMed] [Google Scholar]
- 10.de Graauw M, Hensbergen P, van de Water B. Electrophoresis. 2006;27:2676. doi: 10.1002/elps.200600018. [DOI] [PubMed] [Google Scholar]
- 11.Brill LM, Salomon AR, Ficarro SB, Mukherji M, Stettler-Gill M, Peters EC. Anal Chem. 2004;76:2763. doi: 10.1021/ac035352d. [DOI] [PubMed] [Google Scholar]
- 12.Steen H, Kuster B, Fernandez M, Pandey A, Mann M. J Biol Chem. 2002;277:1031. doi: 10.1074/jbc.M109992200. [DOI] [PubMed] [Google Scholar]
- 13.Molina H, Horn DM, Tang N, Mathivanan S, Pandey A. Proc Natl Acad Sci U S A. 2007;104:2199. doi: 10.1073/pnas.0611217104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou H, Watts JD, Aebersold RA. Nat Biotechnol. 2001;19:375. doi: 10.1038/86777. [DOI] [PubMed] [Google Scholar]
- 15.Bodenmiller B, Campbell D, Gerrits B, Lam H, Jovanovic M, Picotti P, Schlapbach R, Aebersold R. Nat Biotechnol. 2008;26:1339. doi: 10.1038/nbt1208-1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rothman DM, Shults MD, Imperiali B. Trends Cell Biol. 2005;15:502. doi: 10.1016/j.tcb.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 17.Jeffery DA, Springer M, King DS, O’Shea EK. J Mol Biol. 2001;306:997. doi: 10.1006/jmbi.2000.4417. [DOI] [PubMed] [Google Scholar]
- 18.Zou K, Cheley S, Givens RS, Bayley H. J Am Chem Soc. 2002;124:8220. doi: 10.1021/ja020405e. [DOI] [PubMed] [Google Scholar]
- 19.Lemke EA, Summerer D, Geierstanger BH, Brittain SM, Schultz PG. Nat Chem Biol. 2007;3:769. doi: 10.1038/nchembio.2007.44. [DOI] [PubMed] [Google Scholar]
- 20.(a) Mamaev SV, Laikhter AL, Arslan T, Hecht SM. J Am Chem Soc. 1996;118:7243. [Google Scholar]; (b) Rothman DM, Petersson EJ, Vázquez ME, Brandt GS, Dougherty DA, Imperiali B. J Am Chem Soc. 2005;127:846. doi: 10.1021/ja043875c. [DOI] [PubMed] [Google Scholar]
- 21.Hoppmann C, Wong A, Yang B, Li S, Hunter T, Shokat KM, Wang L. Nat Chem Biol. 2017;13:842. doi: 10.1038/nchembio.2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.(a) Maini R, Nguyen D, Chen S, Dedkova LM, Roy Chowdhury S, Alcala-Torano R, Hecht SM. Bioorg Med Chem. 2013;21:1088. doi: 10.1016/j.bmc.2013.01.002. [DOI] [PubMed] [Google Scholar]; (b) Maini R, Roy Chowdhury S, Dedkova LM, Roy B, Daskalova SM, Paul R, Chen S, Hecht SM. Biochemistry. 2015;54:3694. doi: 10.1021/acs.biochem.5b00389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.(a) Roy Chowdhury S, Maini R, Dedkova LM, Hecht SM. Bioorg Med Chem Lett. 2015;25:4715. doi: 10.1016/j.bmcl.2015.08.073. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Maini R, Dedkova LM, Paul R, Madathil MM, Roy Chowdhury S, Chen S, Hecht SM. J Am Chem Soc. 2015;137:11206. doi: 10.1021/jacs.5b03135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arslan T, Mamaev SV, Mamaeva NV, Hecht SM. J Am Chem Soc. 1997;119:10877. [Google Scholar]
- 25.Robertson SA, Noren CJ, Anthony-Cahill SJ, Griffith MC, Schultz PG. Nucleic Acids Res. 1989;17:9649. doi: 10.1093/nar/17.23.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karginov VA, Mamaev SV, An H, Van Cleve MD, Hecht SM, Komatsoulis GA, Abelson JA. J Am Chem Soc. 1997;119:8166. [Google Scholar]
- 27.Gao R, Zhang Y, Choudhury AK, Dedkova LM, Hecht SM. J Am Chem Soc. 2005;127:3321. doi: 10.1021/ja044182z. [DOI] [PubMed] [Google Scholar]
- 28.(a) Robertson SA, Ellman JA, Schultz PG. J Am Chem Soc. 1991;113:2722. [Google Scholar]; (b) Lodder M, Golovine S, Hecht SM. J Org Chem. 1997;62:778. doi: 10.1021/jo971692l. [DOI] [PubMed] [Google Scholar]
- 29.(a) Sen R, Baltimore D. Cell. 1986;47:921. doi: 10.1016/0092-8674(86)90807-x. [DOI] [PubMed] [Google Scholar]; (b) Shirakawa F, Mizel SB. Mol Cell Biol. 1989;9:2424. doi: 10.1128/mcb.9.6.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ghosh S, Baltimore D. Nature. 1990;344:678. doi: 10.1038/344678a0. [DOI] [PubMed] [Google Scholar]; (d) Beg AA, Ruben SM, Scheinman RI, Haskill S, Rosen CA, Baldwin AS., Jr Genes Dev. 1992;6:1899. doi: 10.1101/gad.6.10.1899. [DOI] [PubMed] [Google Scholar]
- 30.Dedkova LM, Fahmi NE, Paul R, del Rosario M, Zhang L, Chen S, Feder G, Hecht SM. Biochemistry. 2012;51:401. doi: 10.1021/bi2016124. [DOI] [PubMed] [Google Scholar]
- 31.(a) Bocchetta M, Xiong L, Mankin AS. Proc Natl Acad Sci U S A. 1998;95:3525. doi: 10.1073/pnas.95.7.3525. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schmeing TM, Seila AC, Hansen JL, Freeborn B, Soukup JK, Scaringe SA, Strobel SA, Moore PB, Steitz TA. Nat Struct Biol. 2002;9:225. doi: 10.1038/nsb758. [DOI] [PubMed] [Google Scholar]; (c) Zarivach R, Bashan A, Berisio R, Harms J, Auerbach T, Schluenzen F, Bartels H, Baram D, Pyetan E, Sittner A, Amit M, Hansen HA, Kessler M, Liebe C, Wolff A, Agmon I, Yonath A. J Phys Org Chem. 2004;17:901. [Google Scholar]; (d) Schmeing TM, Huang KS, Kitchen DE, Strobel SA, Steitz TA. Mol Cell. 2005;20:437. doi: 10.1016/j.molcel.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 32.Bashan A, Agmon I, Zarivach R, Schluenzen F, Harms J, Berisio R, Bartels H, Franceschi F, Auerbach T, Hansen HA, Kossoy E, Kessler M, Yonath A. Mol Cell. 2003;11:91. doi: 10.1016/s1097-2765(03)00009-1. [DOI] [PubMed] [Google Scholar]
- 33.The absence of the wild-type nucleotide in any of the modified 23S rRNAs at positions 2600–2605 was a consequence of the selection protocol, which was found30 to facilitate the generation of a library having maximal diversity.
- 34.Janknecht R, de Martynoff G, Lou J, Hipskind RA, Nordheim A, Stunnenberg HG. Proc Natl Acad Sci U S A. 1991;88:8972. doi: 10.1073/pnas.88.20.8972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beg AA, Baldwin AS., Jr Genes Dev. 1993;7:2064. doi: 10.1101/gad.7.11.2064. [DOI] [PubMed] [Google Scholar]
- 36.(a) Miyamoto S, Maki M, Schmitt MJ, Hatanaka M, Verma IM. Proc Natl Acad Sci U S A. 1994;91:12740. doi: 10.1073/pnas.91.26.12740. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Brockman JA, Scherer DC, McKinsey TA, Hall SM, Qi X, Lee WY, Ballard DW. Mol Cell Biol. 1995;15:2809. doi: 10.1128/mcb.15.5.2809. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U. Science. 1995;267:1485. doi: 10.1126/science.7878466. [DOI] [PubMed] [Google Scholar]; (d) Traenckner EBM, Pahl HL, Henkel T, Schmidt KN, Wilk S, Baeuerle PA. EMBO J. 1995;14:2876. doi: 10.1002/j.1460-2075.1995.tb07287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Verma IM, Stevenson JK, Schwarz EM, Van Antwerp D, Miyamoto S. Genes Dev. 1995;9:2723. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- 38.(a) Zabel U, Baeuerle PA. Cell. 1990;61:255. doi: 10.1016/0092-8674(90)90806-p. [DOI] [PubMed] [Google Scholar]; (b) Bergqvist S, Alverdi V, Mengel B, Hoffmann A, Ghosh G, Komives EA. Proc Natl Acad Sci U S A. 2009;106:19328. doi: 10.1073/pnas.0908797106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.(a) Lu W, Gong D, Bar-Sagi D, Cole PA. Mol Cell. 2001;8:759. doi: 10.1016/s1097-2765(01)00369-0. [DOI] [PubMed] [Google Scholar]; (b) Zhang Z, Shen K, Lu W, Cole PA. J Biol Chem. 2003;278:4668. doi: 10.1074/jbc.M210028200. [DOI] [PubMed] [Google Scholar]; (c) Cole PA, Courtney AD, Shen K, Zhang Z, Qiao Y, Lu W, Williams DM. Acc Chem Res. 2003;36:444. doi: 10.1021/ar0201254. [DOI] [PubMed] [Google Scholar]; (d) Zheng W, Zhang Z, Ganguly S, Weller JL, Klein DC, Cole PA. Nat Struct Biol. 2003;10:1054. doi: 10.1038/nsb1005. [DOI] [PubMed] [Google Scholar]; (e) Hahn ME, Muir TW. Angew Chem, Int Ed. 2004;43:5800. doi: 10.1002/anie.200461141. [DOI] [PubMed] [Google Scholar]; (f) Vogel EM, Imperiali B. Protein Sci. 2007;16:550. doi: 10.1110/ps.062549407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.(a) Silversmith RE, Bourret RB. Protein Eng, Des Sel. 1998;11:205. doi: 10.1093/protein/11.3.205. [DOI] [PubMed] [Google Scholar]; (b) Saxl RL, Anand GS, Stock AM. Biochemistry. 2001;40:12896. doi: 10.1021/bi011424o. [DOI] [PubMed] [Google Scholar]; (c) Bernardes GJL, Chalker JM, Errey JC, Davis BG. J Am Chem Soc. 2008;130:5052. doi: 10.1021/ja800800p. [DOI] [PubMed] [Google Scholar]; (d) Rowan FC, Richards M, Bibby RA, Thompson A, Bayliss R, Blagg J. ACS Chem Biol. 2013;8:2184. doi: 10.1021/cb400425t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luo X, Fu G, Wang RE, Zhu X, Zambaldo C, Liu R, Liu T, Lyu X, Du J, Xuan W, Yao A, Reed SA, Kang M, Zhang Y, Guo H, Huang C, Yang P-Y, Wilson IA, Schultz PG, Wang F. Nat Chem Biol. 2017;13:845. doi: 10.1038/nchembio.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.(a) Park HS, Hohn MJ, Umehara T, Guo LT, Osborne EM, Benner J, Noren CJ, Rinehart L, Söll D. Science. 2011;333:1151. doi: 10.1126/science.1207203. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Heinemann IU, Rovner AJ, Aerni HR, Rogulina S, Cheng L, Olds W, Fischer JT, Söll D, Isaacs FJ, Rinehart J. FEBS Lett. 2012;586:3716. doi: 10.1016/j.febslet.2012.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.(a) Rodriguez MS, Michalopoulos I, Arenzana-Seisdedos F, Hay RT. Mol Cell Biol. 1995;15:2413. doi: 10.1128/mcb.15.5.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Scherer DC, Brockman JA, Chen Z, Maniatis T, Ballard DW. Proc Natl Acad Sci U S A. 1995;92:11259. doi: 10.1073/pnas.92.24.11259. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.