

Summary
Angiogenesis, the formation of new blood vessels by endothelial cells (EC), is an adaptive response to oxygen/nutrient deprivation orchestrated by vascular endothelial growth factor (VEGF) upon ischemia or exercise. Hypoxia is the best-understood trigger of VEGF expression via the transcription factor HIF1α. Nutrient deprivation is inseparable from hypoxia during ischemia, yet its role in angiogenesis is poorly characterized. Here, we identified sulfur amino acid restriction as a proangiogenic trigger, promoting increased VEGF expression, migration and sprouting in EC in vitro, and increased capillary density in mouse skeletal muscle in vivo, via the GCN2/ATF4 amino acid starvation response pathway independent of hypoxia or HIF1α. We also identified a requirement for cystathionine-γ-lyase in VEGF-dependent angiogenesis via increased hydrogen sulfide (H2S) production. H2S mediated its proangiogenic effects in part by inhibiting mitochondrial electron transport and oxidative phosphorylation, resulting in increased glucose uptake and glycolytic ATP production.
Graphical Abstract
Restricting dietary sulfur can trigger angiogenesis and improve vascular health
Introduction
Angiogenesis is the formation of new blood vessels from existing ones through sprouting, proliferation and migration of endothelial cells (EC). In adult mammals, angiogenesis is an adaptive response to normal and pathophysiological conditions characterized by inadequate supply of oxygen and nutrients, ranging from tissue ischemia upon vessel occlusion or tumorigenesis to endurance exercise.
Hypoxia is the best-understood trigger of angiogenesis, stabilizing the oxygen-sensitive transcription factor hypoxia inducible factor (HIF)-1α in multiple cell types and promoting expression of the master regulator of angiogenesis, vascular endothelial growth factor (VEGF). VEGF expression can also be induced by the transcriptional co-activator PGC1α upon nutrient deprivation through an ERR-α-dependent, HIF-1α independent pathway in muscle cells but not EC (Arany et al., 2008), as well as by the ATF4 transcription factor downstream of the integrated stress response (ISR) triggered by either ER stress or amino acid (AA) deprivation (Abcouwer et al., 2002).
VEGF acts via binding to EC-specific cell-surface tyrosine kinase receptors (VEGFR2), triggering an orchestrated cascade of signal transduction via the PI3K and MAPK pathways involving critical second messengers nitric oxide (NO) and cyclic GMP (cGMP) and changes in gene expression facilitating EC migration, proliferation and vessel formation (Olsson et al., 2006). VEGF-mediated angiogenesis is potentiated by the NAD+-dependent deacetylase SIRT1, which deacetylates and inactivates FOXO transcription factors (Potente et al., 2007) involved in negative regulation of EC migration and tube formation (Potente et al., 2005). VEGF signalling also triggers changes in cellular energy metabolism, namely increased glucose uptake and glycolysis necessary to provide rapid energy for EC migration (De Bock et al., 2013).
Hydrogen sulfide (H2S) is a proangiogenic gas (Cai et al., 2007; Szabo, 2007) produced in EC upon VEGF stimulation (Papapetropoulos et al., 2009) primarily by the transsulfuration enzyme cystathionine-γ-lyase (CGL aka CTH or CSE) (Wang, 2012). Like NO, which in addition to activating cGMP synthesis functions through post-translational modification (S-nitrosylation) of target proteins (Fukumura et al., 2006), H2S promotes angiogenesis through S-sulfhydration and activation of proximal signal transduction components including VEGFR2 (Tao et al., 2013) and eNOS (Altaany et al., 2014; Coletta et al., 2012). Angiogenesis is compromised upon genetic CGL deficiency in aorta explant assays ex vivo (Papapetropoulos et al., 2009) and arterial ligation in vivo (Kolluru et al., 2015). However, mechanisms of CGL regulation in EC and the relative contribution of H2S vs. NO in angiogenesis remain unclear (Katsouda et al., 2016).
Dietary restriction (DR), defined as reduced nutrient/energy intake without malnutrition, is best known for its ability to extend lifespan, improve metabolic fitness and increase stress resistance (Colman et al., 2009; Fontana et al., 2010; Hine et al., 2015). DR regimens, which vary widely, can emphasize either restriction of total food intake (calorie restriction, CR) or dilution of specific nutrients in the diet, such as the sulfur amino acids (SAA) methionine (M) and cysteine (C) (methionine restriction, MR) (Miller et al., 2005; Orentreich et al., 1993). We recently reported that CR increases hepatic CGL expression, endogenous H2S production capacity and resistance to hepatic ischemia reperfusion injury, and that these are abrogated by dietary C excess (Hine et al., 2015). CR also promotes revascularization and recovery from femoral artery ligation in rodents (Kondo et al., 2009), and maintains vascular health in rodents and non-human primates in part by preserving capillary density in skeletal muscle (Omodei and Fontana, 2011). Interestingly, SIRT1 is activated in some tissues upon DR (Cantó and Auwerx, 2009; Wang, 2014) and required for VEGF-dependent angiogenesis (Potente et al., 2007). However, the effects of DR on angiogenesis and the potential role of H2S remain unknown. Here, we identified SAA restriction as a proangiogenic trigger in EC in vitro and in skeletal muscle in mice in vivo.
Results
SAA restriction induces endothelial VEGF expression in vitro and functional angiogenesis in vivo
We tested the potential of isolated nutrient restriction independent of ischemia or hypoxia to impact angiogenesis in vitro using a model of SAA restriction (Hine et al., 2015). Human umbilical vein endothelial cell (HUVEC) cultured overnight in media lacking SAA (-M&C) displayed increased VEGF mRNA expression and protein secretion into the media (Fig. 1A). This correlated with increased proangiogenic potential, including migration across a scratch (Fig. 1B); formation of capillary-like structures (tube formation, Fig. 1C); and increased sprout length in 3-dimensional HUVEC spheroid cultures, an effect that was abrogated by the specific VEGFR2 inhibitor SU5416 (Fig. 1D). Inhibiting SIRT1 activity with Ex-527 significantly reduced HUVEC tube formation (Fig. 1C) and branch point number (Fig. S1A) upon -M&C, suggesting that the proangiogenic pathway triggered by -M&C is dependent on both VEGF and SIRT1 activity.
Figure 1. SAA restriction induces endothelial VEGF expression in vitro and functional angiogenesis in vivo.

(A) VEGF mRNA levels (left, n=4 experiments/group) and secreted protein concentration in the media (right, n=6 experiments/group) of HUVEC cultured in control (Ctrl) or SAA deficient (- M&C) media for 16hr; error bars indicate SEM. (B) Migration assay: Representative migration across a scratch (left, 10X mag at t=20hr; dotted lines indicate boundary of the scratch at t= 0hr) and area under the curve (AUC, right, n=7–10 data points/condition, with each data point representing the mean of multiple measures within a single well in a representative experiment) from HUVEC cultured in the indicated media. (C) Tube formation assay: Representative capillary-like structures (left, 40X mag) and quantification of tube length/field in arbitrary units (AU, right; n=8–10 data points/condition) in HUVEC incubated in the indicated media +/−SIRT1 inhibitor Ex-527 for 18hr. (D) Spheroid assay: Representative images (left, 40X mag) and quantification (right, in triplicate) of sprouting HUVEC spheroids in the indicated media +/− VEGFR2 inhibitor SU5416 for 24hr; blue, DNA (DAPI); red, F-actin (phalloidin). (E) Representative transverse sections (left, 40X mag) and quantification (right) of gastrocnemius muscle stained for endothelial marker CD31 in mice fed 2wk on Ctrl or MR diet +/−VEGFR2 inhibitor axitinib; n=6–8 mice/group. (F) Longitudinal Doppler imaging of blood flow in WT mice preconditioned 1mo on Ctrl or MR diet prior to femoral artery ligation (I, ischemic; NI, non-ischemic). Left: representative infrared images on the indicated day after ligation. Right: quantification of blood flow recovery with individual animal AUCs used for statistical comparison; n=7–8 mice/group. (G) Representative transverse sections (left, 40X mag) and quantification (right) of CD31-stained gastroc 10d after ligation from (F); n=4 mice/group. Error bars indicate SD unless otherwise noted; asterisks indicate the significance of the difference by Student’s T test or 1-way ANOVA with Sidak’s MCT between diets in vivo or SAA deprivation in vitro; *P<0.05, **P<0.01, ***P<0.001. See also Fig. S1.
To test the impact of dietary SAA restriction on angiogenesis in vivo, mice were given ad libitum access to an MR diet containing a limiting amount of M and lacking C (Miller et al., 2005; Orentreich et al., 1993). Young adult wildtype (WT) mice on MR for 2mo maintained a lower body weight despite normal food intake relative to mice fed a control diet containing normal M and C levels (Fig. S1B).
Strikingly, MR resulted in increased vascular density in skeletal muscle as determined by immunostaining (Fig. 1E) and flow cytometric analysis (Fig. S1C) for the EC marker CD31. Consistent with VEGF dependence, this effect was blocked by axitinib, one of the best characterized VEGF receptor inhibitors in vivo with demonstrated antiangiogenic activity in the context of tumor neovascularization (Ma and Waxman, 2008) (Fig. 1E, S1C). Interestingly, although expression of VEGF mRNA was not consistently affected upon MR in whole gastrocnemius muscle (Fig. S1D), there was a trend toward increased VEGF protein in gastrocnemius muscle extracts (Fig. S1E). VEGF and CD31 co-localized in gastrocnemius muscle by IHC (Fig. S1F) consistent with EC as the source of VEGF upon MR in vivo as observed upon -M&C in vitro (Fig. 1A).
Functional significance was tested in the context of femoral artery ligation in mice preconditioned on MR or control diets for 1mo prior to surgical occlusion, and returned to a complete diet after surgery (Fig. S1G). Although blood flow was similarly interrupted in both diet groups immediately after ligation (d0), return of blood flow indicative of neovascularization was accelerated in MR mice, with significant improvement by d3 after ligation (Fig. 1F). CD31 immunohistochemistry of muscle sections confirmed a relative increase in capillary density in both ischemic and non-ischemic legs of MR vs. control mice despite a return to a complete diet for 10d (Fig. 1G). Functional improvement was also observed in mice preconditioned on a different DR regimen, 40% calorie restriction (CR), for 1mo prior to femoral ligation (Fig. S1H–I). In addition to improved return of blood flow (Fig. S1H), CR mice also demonstrated improved treadmill exercise endurance testing on d4 after ligation (Fig. S1I). Together, these data suggest neovascularization induced by DR (in the form of CR or MR) as a contributing factor in the improved physiological response to acute blood flow cessation.
GCN2-dependent, hypoxia-independent regulation of VEGF and angiogenesis upon SAA restriction
Although HIF1α upon hypoxia is the best-characterized trigger of VEGF expression in multiple cell types including EC, VEGF expression upon -M&C was unaffected by HIF1α RNAi knockdown (KD) (Fig. 2A, S2A) and coincided with a trend towards reduced HIF1α protein expression (Fig. 2B, S2B). PGC1α can also induce VEGF independently of HIF1α upon total nutrient/growth factor deprivation in myocytes but not EC (Arany et al., 2008). Consistent with this, endogenous PGC1α mRNA expression in HUVEC was very low as judged by Ct value (data not shown) and unaffected by -M&C (Fig. S2C), while exogenous PGC1α overexpression in HUVEC failed to modulate VEGF expression (Fig. S2D–E).
Figure 2. GCN2-dependent, hypoxia-independent regulation of VEGF and angiogenesis upon SAA restriction.

(A) Relative VEGF mRNA expression in HUVEC 2d after transfection with HIF1α siRNA or control scrambled (Sble) siRNA and cultured in control (Ctrl) or SAA deficient (-M&C) media for 16hr; n=5 experiments/group; error bars indicate SEM. (B) Immunoblots of HIF1α, eIF2α (p- Ser51, total) and ATF4 in HUVEC cultured as indicated for 16hr. (C) Relative VEGF mRNA expression in HUVEC 2d after transfection with ATF4 or Sble siRNA and cultured as indicated for 16hr; n=4 experiments/group; SEM. (D, E) Relative HUVEC VEGF mRNA expression (D, n=3 experiments/group; SEM) and secreted VEGF protein concentration in media (E, n=3–6 experiments/group; SEM) 2d after transfection with ATF4 overexpression (ATF4OE) or control construct (Empty). (F, G) VEGF mRNA expression (F) and spheroid formation (G) in WT and GCN2KO primary mouse EC from n=3 mice/genotype cultured as indicated for 16hr. For sprouting assay (G), representative images (left, 40X mag) and quantification (right) of WT and GCN2KO EC spheroids cultured in the indicated media for 24hr; blue, DNA (DAPI); red, F-actin (phalloidin). (H) Representative transverse sections (left, 40X mag) and quantification (right) of CD31-stained gastroc in WT or GCN2KO mice fed for 2–4wk on Ctrl or MR diets; n=5–6 mice/group. (I) VEGF mRNA in MDF, MEF or C2C12 myotubes cultured as indicated for 16hr; n=4–6 experiments/group; SEM. (J) VEGF mRNA expression in WT and GCN2KO primary mouse skeletal myotubes (n=5 mice/genotype tested at 2 different passages) cultured as indicated for 16hr. (K) Immunoblots of HIF1α, PGC1α, eIF2α (p-Ser51, total) and ATF4 in C2C12 myotubes cultured as indicated for 16hr. Error bars indicate SD unless otherwise noted; asterisks indicate the significance of the difference by Student’s T test or 1-way ANOVA with Sidak’s MCT between diets in vivo or SAA deprivation in vitro; *P<0.05, **P<0.01, ***P<0.001. See also Fig. S2.
The AA starvation response (AASR), a branch of the ISR involving binding of uncharged cognate tRNAs to the general control nonderepressible 2 (GCN2) kinase, phosphorylation of eukaryotic translation initiation factor 2α (eIF2α) and translational derepression of ATF4 (Kilberg et al., 2005; Wek et al., 1995), has been implicated in DR-mediated resistance to ischemia reperfusion injury (Peng et al., 2012), but has not been assessed in EC. In HUVEC, - M&C increased eIF2α phosphorylation, ATF4 protein expression and transcription of the ATF4 target, Asns (Fig. 2B, S2B, F). ATF4 siRNA (Fig. S2G) reduced VEGF and Asns transcriptional upregulation upon -M&C (Fig. 2C, S2F), while ATF4 overexpression increased VEGF and Asns mRNA expression (Fig. 2D, S2G–I) and VEGF secretion into the media (Fig. 2E) independent of nutrient deprivation.
The requirement for GCN2 was tested in primary EC isolated from WT and GCN2KO mice (Fig. S2J). Similar to HUVEC, -M&C significantly increased VEGF and Asns mRNA expression (Fig. 2F, S2K) and sprout length (Fig. 2G) in WT but not GCN2KO EC. In vivo, GCN2KO mice failed to increase vascular density upon 2–4wk of MR compared to controls (Fig. 2H).
-M&C also increased VEGF expression in primary mouse dermal fibroblasts (MDF), immortalized mouse embryonic fibroblasts (MEF) and C2C12 myotubes (Fig. 2I). In primary skeletal myotubes, VEGF induction upon -M&C required GCN2 (Fig. 2J). In MDF, ATF4 shRNA prevented the increase in VEGF mRNA by -M&C (Fig. S2L). In C2C12 myotubes, VEGF induction coincided with increased eIF2α phosphorylation, ATF4 expression and reduced HIF1α protein levels (Fig. 2K). Notably, VEGF induction upon -M&C in C2C12 myotubes was unaffected by HIF1α RNAi KD under normoxic (20%) or hypoxic (<1%) oxygen tensions (Fig. S2M–O). PGC1α RNAi KD also failed to dampen VEGF induction upon -M&C in C2C12 myotubes (Fig. S2P).
Taken together, these data reveal SAA restriction and the GCN2/ATF4-dependent AASR as a novel trigger of increased VEGF expression and angiogenesis independent of hypoxia, HIF1α or PGC1α.
VEGF signalling and AASR converge on endothelial H2S production by CGL
VEGF promotes angiogenesis in part by stimulating CGL-dependent production of the proangiogenic gas H2S in EC (Papapetropoulos et al., 2009), however mechanisms of CGL regulation in EC and the importance of H2S in angiogenesis remain poorly understood. H2S production capacity (Fig. 3A) and endogenous H2S levels (Fig. 3B) were similarly increased by - M&C and exogenous VEGF addition, and sensitive to the CGL inhibitor propargylglycine (PAG). -M&C also boosted endogenous H2S production in primary hepatocytes, while VEGF did not because hepatocytes lack VEGFR2 (Fig. S3A).
Figure 3. VEGF signalling and AASR converge on endothelial H2S production by CGL.

(A) Representative H2S production capacity as indicated by black lead sulfide formation from HUVEC cultured in media +/−M&C or VEGF (50ng/mL) in the presence or absence of the CGL inhibitor PAG (100μM) as indicated for 16hr. (B) Representative (left) endogenous H2S levels (blue, H2S (P3 fluorescence); red, DNA (DRAQ5)) and quantification of P3 intensity (right) in HUVEC upon VEGF or -M&C treatment; n=4 wells/treatment with 4–6 images/well; 1-way ANOVA with Sidak’s MCT vs. Control (asterisks) or +/−PAG within treatment (carets). (C) CGL mRNA expression in WT and GCN2KO primary mouse EC cultured from n=3 mice/genotype in control (Ctrl) or -M&C media for 16hr. (D) CGL mRNA expression in HUVEC 2d after transfection with ATF4 or control scrambled (Sble) siRNA and cultured in the indicated media for 16hr; n=4 experiments/group; SEM. (E) CGL mRNA expression in HUVEC 2d after transfection with ATF4 overexpression or control (empty) plasmid; n=3 experiments/group; SEM. (F, G) Representative images (left, 40X mag) and quantification (right, in triplicate) of spheroids cultured from (F) HUVEC +/−M&C for 24hr in the presence of vehicle (Veh) or PAG, and (G) WT or CGLKO primary EC sprouts in control or -M&C media for 24hr; blue, DNA (DAPI); red, F-actin (phalloidin). Unless otherwise indicated, error bars indicate SD, and asterisks indicate the significance of the difference between diets in vivo or SAA levels in vitro by Student’s T test or 1-way ANOVA with Sidak’s MCT; *P<0.05, **P<0.01, ***/^^^P<0.001. See also Fig. S3.
CGL is an ATF4 target downstream of the ISR triggered either by ER stress in MEF (Dickhout et al., 2012) or cysteine deprivation in HepG2 (Lee et al., 2008). In primary mouse EC, CGL mRNA was strongly induced upon -M&C. (Fig. 3C). This effect was abrogated in EC lacking GCN2 (Fig. 3C) or in HUVEC upon ATF4 knockdown (Fig. 3D, S2F), while ATF4 overexpression increased CGL independent of SAA deprivation (Fig. 3E). Importantly, VEGF mRNA was not affected by the absence of CGL (Fig. S3B) in vitro, consistent with CGL and VEGF as independent downstream targets of the AASR. Interestingly, exogenous VEGF increased CGL mRNA (Fig. S3C) and protein (Fig. S3D) expression in EC independent of nutrient deprivation, suggestive of a positive feedback loop between VEGF and CGL expression, but without affecting expression of the two other H2S-generating enzymes, CBS or 3-MST.
The functional relevance of endothelial CGL in angiogenic potential in vitro was assessed in the EC spheroid assay. Increased sprout length of EC spheroids upon -M&C was prevented by the CGL inhibitor PAG in HUVEC (Fig. 3F) and in CGLKO mouse EC (Fig. 3G). Thus, CGL is required for angiogenesis induced by -M&C in vitro.
CGL required for angiogenesis in vivo
We next tested the requirement for CGL-derived H2S in angiogenesis triggered by MR in vivo. Consistent with CGL as the major H2S producer in EC in vivo (Wang, 2012), P3 fluorescence indicative of endogenous H2S production co-localized with CD31+ cells in fresh-frozen sections of gastrocnemius muscle from WT but not CGLKO mice (Fig. 4A). Quantification of P3 intensity in WT and CGLKO mice fed Ctrl vs. MR diets for 2wk confirmed a CGL-dependent increase in EC H2S production upon MR in vivo (Fig. 4B). Coincident with failure to increase H2S, CGLKO mice failed to increase capillary density upon MR in vivo relative to WT mice (Fig. 4C). Furthermore, the CGL inhibitor PAG partially prevented 40% CR from improving recovery from femoral ligation (Fig. S1H–I). Taken together, these data are consistent with a requirement for CGL-derived H2S for angiogenesis triggered by nutrient deprivation.
Figure 4. CGL required for angiogenesis in vivo.

(A) Representative transverse sections (20X mag) of gastrocnemius muscle from WT and CGLKO mice stained for CD31 and endogenous H2S. (B) Quantification of endogenous H2S in CD31+ EC in the gastrocnemius muscle of WT and CGLKO mice fed for 2wk on Ctrl or MR diets as indicated; n=4–5 mice/group, with quantification of 3–10 images/mouse. (C) Representative transverse sections (left, 40X mag) and quantification (right) of CD31-stained gastroc from WT and CGLKO mice fed for 2wk on Ctrl or MR diets as indicated; n=4 mice/group. (D) Representative transverse sections of CD31-stained gastroc (left, 40X mag) and quantification (right) 2wk after Ad-Null or Ad-CGL injections; n=3–4 mice/group; Student’s T test. (E) Representative transverse sections (left, 40X mag) and quantification (right) of CD31-stained gastroc from WT and CGLKO mice subjected to low intensity running (exercised) vs. control (sedentary) for 1mo; n=4–5 mice/group. (F) Representative transverse sections of CD31-stained gastroc (left, 40X mag) and quantification (right) from WT and CGLKO mice 6d after the final intramuscular injection of control (Ad-Null) or VEGF165 -expressing (Ad-VEGF) adenovirus; n=4 mice/group. Error bars indicate SD; asterisks indicate the significance of the difference between diets or treatments within genotype by 1-way ANOVA with Sidak’s MCT unless otherwise noted; *P<0.05, **P<0.01, ***P<0.001. See also Fig. S4.
We next asked if CGL-derived H2S is sufficient to promote angiogenesis independent of SAA restriction. To this end, we injected CGL-expressing adenovirus into the gastrocnemius muscle of WT mice via intra-muscular injection, likely resulting in CGL overexpression in both myotubes and EC due to the ability of the Ad5 serotype to infect multiple cell types. Local CGL viral transduction increased muscle H2S production capacity (Fig. S4A) and vascular density (Fig. 4D) independent of any other proangiogenic stimulus, suggesting that an increase in CGL-derived H2S is sufficient to trigger angiogenesis.
To test this apparent general requirement for CGL in angiogenesis independent of the upstream stimulus, we induced angiogenesis by either treadmill exercise training or by VEGF overexpression via intra-muscular injection of VEGF-overexpressing (ad-VEGF165) adenovirus. Exercise training increased endogenous VEGF mRNA expression in WT mice (Fig. S4B), while both exercise training (Fig. 4E) and local VEGF overexpression via adenoviral gene delivery (Fig. 4F) increased capillary density in WT but not CGLKO mice. Taken together, these data indicate that CGL is necessary for VEGF-mediated neovascularization in vivo independent of the upstream proangiogenic stimulus.
H2S promotes glucose uptake and ATP generation by glycolysis for EC migration
Because CGL is a promiscuous enzyme that can convert cystathionine to C as part of the transsulfuration pathway, but can also use C to produce H2S and serine, we sought more direct evidence of H2S as the CGL metabolite relevant to angiogenesis. H2S addition to standard EC media in the form of NaHS increased proliferation (Fig. S5A) and migration across a scratch wound (Fig. 5A). This latter effect was only partially blocked with mitomycinC (MitoC), consistent with migration as a critical factor in H2S-induced proangiogenic potential. In support of this, HUVEC overexpressing CGL (Ad-CGL) formed lamellipodial projections over larger areas (Fig. 5B) coincident with increased migration speed and greater cell body displacement (Fig. 5C).
Figure 5. H2S promotes glucose uptake and ATP generation by glycolysis for EC migration.

(A) Representative migration across scratch (left, 10X mag) and quantification (right) of HUVEC +/−100μM NaHS in the presence of vehicle or mitomycin C (MitoC, 1μg/mL) to inhibit proliferation; n=12 wells each from cells at 2 different passages; 1-way ANOVA with Sidak’s MCT between control and NaHS within vehicle or MitoC treatment group. (B, C) Representative images (B) and quantification (C) of migration speed (left, n=5–7 cells/condition) and distance (right, n=5–7 cells/condition in x and y directions) from time-lapse video imaging of GFP+ HUVEC infected with control (Ad-Null) or CGL adenovirus (Ad-CGL) as indicated; Student’s T test. (D) Relative glucose uptake in HUVEC pretreated with NaHS or 50ng/mL VEGF for 1hr; n=3–6 experiments/group; 1-way ANOVA with Dunnett’s MCT. (E) Extracellular acidification rate (ECAR) in WT and CGLKO primary mouse EC pretreated for 1hr with VEGF or NaHS; 10 technical replicates from EC pooled from 6 mice/genotype; 1-way ANOVA with Sidak’s MCT as indicated. (F) Glycolytic flux in HUVEC pretreated for 3hr with NaHS or VEGF; 1-way ANOVA with Dunnett’s MCT. Representative experiment of 6 with n=3/group; 1-way ANOVA with Sidak’s MCT. (G) Time dependent ATP production in HUVEC pretreated with NaHS or 1mM 2DG at t=0; n=4 experiments each for NaHS and VEGF and 2 for 2DG; error bars indicate SEM; 2-way ANOVA with Dunnett’s MCT relative to t=0 (asterisk, NaHS; caret, VEGF; pound sign, 2DG). (H, I) Representative migration (left, 10X mag) and quantification (right, AUC) of HUVEC treated +/−NaHS (H, n=11 technical replicates/condition) or infected with a control (Ad-Null) or CGL adenovirus (Ad-CGL) at a multiplicity of infection of 50 (I, n=5–6 technical replicates/condition), in the presence of vehicle or 2DG; 1-way ANOVA with Sidak’s MCT between control and NaHS within 2DG or vehicle treatment group. (J) Log2 fold change of C13- labelled metabolites in HUVEC measured by mass spectrometry after 1hr of NaHS pretreatment compared to control; red dots, metabolites with FDR adjusted P<0.05 and absolute value of log2 fold change>1.2; blue dots, metabolites with FDR adjusted P>0.05 and/or absolute value of log2 fold change<1.2. (K) Plot of the first two components of orthogonal partial least squares discriminant analysis on unlabeled metabolite levels in HUVEC after 15min, 2hr or 4hr treatment with NaHS or -M&C. Ellipses represent 99% confidence bound for treatment groups. (L) Average metabolite log2 fold changes after 15min of NaHS or -M&C. All metabolites that significantly (P<0.05) changed in the same direction in both treatment groups are shown. Error bars indicate SD unless otherwise indicated; */^P<0.05, **P<0.01, ***/###P<0.001, ####P<0.0001. See also Fig. S5.
Cell migration requires rapid ATP generation to facilitate actin cytoskeleton rearrangement, which in EC is met by increasing glycolytic metabolism (De Bock et al., 2013; Schoors et al., 2014). We thus examined glucose uptake and glycolytic ATP production as a function of genetic and pharmacological H2S modulation. Treatment for 30min with NaHS increased glucose uptake in HUVEC similar to VEGF (Fig. 5D). Interestingly, H2S-induced glucose uptake was independent of SIRT1 in mouse EC (Fig. S5B).
Glycolytic activity was assessed using several methods. Extracellular acidification rate (ECAR), a surrogate marker of glycolysis, was increased by NaHS treatment in HUVEC (Fig. S5C). In primary EC from WT mice, NaHS and VEGF increased ECAR to a similar level (Fig. 5E, S5D), while in CGLKO EC, ECAR was significantly reduced at baseline, and increased by NaHS but not VEGF administration (Fig. 5E). Interestingly, accumulation of the end product of aerobic glycolysis, lactate, in the media following NaHS treatment did not reach the level of statistical significance (Fig. S5E). We thus measured glycolysis directly using the release of 3H2O from C5-3H-glucose and found a significant increase upon NaHS or VEGF treatment (Fig. 5F) beginning after 30min (Fig. S5F).
Consistent with increased glycolysis, H2S boosted intracellular ATP levels over a rapid time course similar to exogenous VEGF (Fig. 5G). Both exogenous H2S addition and CGL overexpression also increased EC migration, and this was sensitive to competitive inhibition of glycolysis by 2-deoxy-D-glucose (2DG; Fig. 5H–I). Together, these data support the functional relevance of CGL-derived H2S in activation of glycolytic ATP generation necessary for proangiogenic migratory behaviour.
Steady-state flux analysis was employed to better understand glucose disposal upon NaHS treatment. Addition of 13C1,2-glucose to the media for the final 15min of a 2hr NaHS treatment revealed significant changes in labeling of glycolytic intermediates, including an increase in glyceraldehyde-3-phosphate, dihydroxy-acetone-phosphate and fructose-phosphate (Fig. 5J). Interestingly, a significant increase was also observed in pentose phosphate pathway (PPP) and purine biosynthetic intermediates, including sedoheptulose-7P, phosphoribosyl-1- pyrophosphate and IMP (Fig. 5J). A significant increase in total unlabelled glycolytic and nucleotide metabolites was also observed (Fig. S5G). Increased glucose flux through the PPP is required for angiogenesis (Bierhansl et al., 2017; Vizan et al., 2009) and could also contribute to increased ECAR via CO2 release during the oxidative phase of the reaction (Fig. 5E, S5C), but is not mutually exclusive with the observed increase in glycolytic ATP generation.
Finally, we asked if NaHS and M&C removal triggered similar metabolic responses as predicted if endogenous H2S production upon -M&C is important for its proangiogenic action. To this end, we performed unbiased metabolomic analyses of HUVEC cultured under standardized media conditions (complete DMEM supplemented with dialyzed FBS and EC growth factors) for 1hr before addition of NaHS addition or M&C removal. A comparison of global profiles over a time course following treatment revealed a time-dependent shift upon -M&C away from the control and a striking convergence after 4hr with the 15min NaHS profile (Fig. 5K). An analysis of all significant changes in the same direction between the 15min NaHS and -M&C groups included glycolytic and PPP intermediates (Fig. 5L).
H2S shifts oxidative/glycolytic balance concomitant with inhibition of mitochondrial OXPHOS
By what mechanism does H2S promote glucose uptake and disposal in EC? Increased ECAR by NaHS was unaffected by axitinib or the eNOS competitive antagonist L-nitroarginine methyl ester (L-NAME) (Fig. S5H). Similarly, NaHS-induced EC migration was unaffected by L-NAME (Fig. S5I) or genetic eNOS knockdown (Fig. S5J–K), together suggesting a mechanism of action either independent or downstream of proximal VEGFR2 signalling.
In many cell types, inhibition of mitochondrial oxidative phosphorylation (OXPHOS) and transient ATP depletion initiates adaptive responses including AMPK activation that boost glucose uptake and glycolytic ATP production (Hardie et al., 2016). At high concentrations, H2S can inhibit complex IV of the mitochondrial ETC and prevent ATP generation by OXPHOS (Smith et al., 1977), however whether this mechanism of action contributes to its proangiogenic effect is unknown. Despite the fact that EC rely predominantly on glycolysis (De Bock et al., 2013), we observed significant changes in glucose flux through the citric acid cycle (reduced citrate/isocitrate and oxaloacetate, Fig. 5J) as well as reduced total citrate (Fig. S5G) suggesting a block in mitochondrial OXPHOS without an increase in cell death (Fig. S5L). Interestingly, labeled and total malate were increased in the same analyses, possibly due to an increase in the cytoplasmic pool, which cannot be distinguished from the mitochondrial pool in whole cell lysates.
CGLKO EC displayed increased oxygen consumption rate (OCR) under basal conditions relative to WT cells (Fig. 6A), consistent with the ability of endogenous H2S to inhibit mitochondrial ETC activity. Similarly, exogenous H2S decreased OCR concomitant with an increase in ECAR to the same levels as the Complex V inhibitor oligomycin (Fig. 6B). To confirm the predicted effects of H2S on Complex IV activity, OCR was measured in isolated HUVEC mitochondria in the presence of NaHS or the Complex IV inhibitor KCN. In addition to the expected decrease in OCR in the presence of the Complex IV substrate TMPD-ascorbate, NaHS also reduced oxygen consumption fueled by pyruvate, consistent with additional ETC inhibition at Complex I (Fig. 6C).
Figure 6. H2S shifts oxidative/glycolytic balance concomitant with inhibition of mitochondrial OXPHOS.

(A) Basal oxygen consumption rate (OCR) in WT and CGLKO primary mouse EC; n=10 technical replicates from EC pooled from 6 mice/genotype; Student’s T test. (B) OCR (left) and extracellular acidification rate (ECAR, right) in HUVEC pretreated for 2hr with 100μM NaHS followed by oligomycin (oligo, 2.5μM) injection at the indicated time; representative experiment with n=10 technical replicates/treatment. (C) Mitochondrial complex activity in permeabilized HUVEC pretreated for 1hr with NaHS or 10μM KCN ; representative experiment with n=6 technical replicates; 1-way ANOVA with Sidak’s MCT within complex activity group. (D, E) Immunoblots of ACC (pSer79, total) and AMPK (Thr172, total) in HUVEC treated with NaHS (D) or KCN (E) for the indicated time. (F) ECAR in HUVEC pretreated for 1hr with NaHS +/−10μM Compound C (Comp C, an AMPK inhibitor) as indicated; n=10 technical replicates from EC pooled from 6 mice/genotype; 1-way ANOVA with Sidak’s MCT between NaHS treatment within Comp C treatment group. (G) Relative glucose uptake in HUVEC pretreated with the indicated agent for 1–3hr; n=2–8 experiments/group; error bars indicate SEM; 1-way ANOVA with Dunnett’s MCT. (H) ECAR in HUVEC pretreated for 2hr with KCN, 2μM oligo or phenformin (Phen, 500μM) expressed as a percent of control over time after addition of 10mM glucose; n=12–19 technical replicates; 1-way ANOVA with Dunnett’s multiple comparison test. (I) Migration across scratch expressed as fold change relative to control in HUVEC treated with oligomycin (2μM), phenformin or KCN +/−2DG (1mM) as indicated; n=5–12 AUC values/group each from cells at different passages; 1-way ANOVA with Sidak’s MCT vs. control without 2DG treatment. Error bars indicate SD unless otherwise noted; *P<0.05, **P<0.01, ***P<0.001. See also Fig. S6.
We next asked if Complex IV inhibition was sufficient to trigger AMPK activation in EC. AMPK was activated within 5min of NaHS or KCN treatment (Fig. 6D–E) and returned to baseline after 30min. Treatment with an AMPK inhibitor (Compound C) prevented the increase in ECAR (Fig. 6F), glucose uptake and migration (Fig. S6A–B). Consistent with the rapid and transient nature of AMPK activation, 10min NaHS treatment was sufficient to promote EC migration during the subsequent 12hr period, and this was blocked by Compound C (Fig. S6C–D).
To determine if energy stress was the proximal trigger to AMPK activation, we measured ATP/ADP ratio over a time course following NaHS addition, as well as energy charge 15min after NaHS addition, but observed no significant changes (Fig. S6E–F). AMPK can also be activated by upstream kinases such as CamKKβ (Lee et al., 2012) or reactive oxygen species (ROS) upon hypoxia (Emerling et al., 2009) or nutrient/energy deprivation (Li et al., 2013). However, NaHS-mediated glucose uptake was unaffected by the CamKKβ inhibitor STO-609 (Fig. S6G), while mitochondrial ROS failed to increase upon NaHS or KCN treatment (Fig. S6H).
Nonetheless, if inhibition of mitochondrial ATP synthesis is the trigger by which H2S promotes AMPK activation, glycolytic ATP production and angiogenesis, then any ETC inhibitor should have similar proangiogenic effects. Consistent with this hypothesis, multiple ETC inhibitors targeting different complexes, as well as the uncoupler FCCP, increased glucose uptake (Fig. 6G), albeit with slightly different kinetics (Fig. S6I). ECAR (Fig. 6H, S6J) and glycolytic flux (Fig. S6K) were also stimulated by OXPHOS inhibition. Finally, multiple ETC inhibitors stimulated 2DG-sensitive EC migration (Fig. 6I, S6L). Together, these data are consistent with a mechanism of H2S action in angiogenesis involving transient inhibition of mitochondrial respiration by direct inhibition of ETC, resulting in increased glucose uptake and a shift from oxidative phosphorylation to glycolysis and PPP.
Discussion
SAA restriction as a proangiogenic trigger
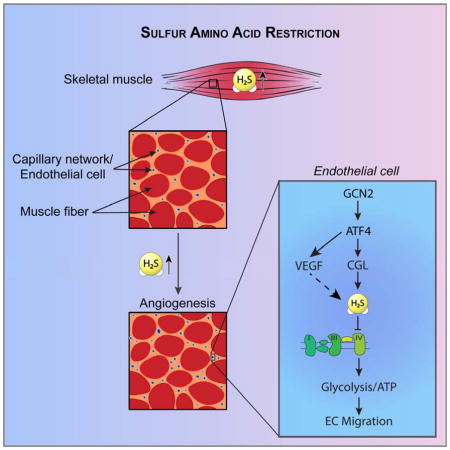
A model for the proangiogenic pathway activated by SAA restriction and controlled by the GCN2/ATF4-dependent AASR independent of hypoxia, HIF1α or PGC1α in EC is presented in Fig. 7. While ATF4 regulation of VEGF expression has been reported in the context of the ISR activated by AA deprivation or ER stress in a human retinal pigmented epithelial cell line in vitro (Abcouwer et al., 2002) or by mutation of tRNA synthetase genes in zebrafish in vivo (Castranova et al., 2016), it has not been previously linked to SAA deprivation via GCN2 in EC. ATF4 can also activate CGL and H2S production, which can feed back to increase eIF2α phosphorylation and further activate the ISR (Yadav et al., 2017). Future studies will be required to determine the impact of AA deprivation on other proangiogenic factors such as FGF, and if there is any specificity to SAA. Furthermore, while ATF4 can activate VEGF and CGL expression in multiple different primary cell types in vitro, it remains to be determined which cell types in vivo are critical for the proangiogenic effects of SAA restriction.
Figure 7. Model for regulation of angiogenesis by AA restriction.

GCN2/ATF4 pathway activation in EC by AA restriction induces VEGF expression as well as CGL-mediated H2S production with effects on glucose uptake and utilization via glycolysis and PPP required for EC migration and proliferation.
Although H2S can also be generated by other enzymes including CBS or 3-MST, we found a genetic requirement for CGL in angiogenesis triggered by nutrient deprivation, exercise or VEGF injection (Fig. 4). CGL is also critical for de novo C biogenesis via TSP, linking methionine cycle metabolites upstream to glutathione and taurine production downstream, perturbation of any of which could potentially contribute to the observed effects. Finally, we acknowledge the apparent paradox of increased endogenous H2S production in response to restriction of its substrate, C. However, as the source of free C for H2S generation by CGL is not currently known, it could come from pools distinct from diet-derived or de novo-produced C. For example, cytoplasmic glutathione levels which are in the mM range could be utilized as a source of free C, as could products of proteosomal protein degradation or lysosomal autophagy. In support of this latter notion, the increase in H2S production induced by growth factor (serum) withdrawal in cultured cells is partially abrogated by genetic or pharmacological inhibition of autophagy (Hine et al., 2017), while the benefits of MR on longevity in yeast require autophagy (Ruckenstuhl et al., 2014).
In an accompanying manuscript, Das et al. report an interaction between H2S and SIRT1 in the regulation of angiogenesis as evidenced by the ability of H2S/NMN supplementation in old mice to reverse age-associated loss of muscle vascular density and improved exercise performance (Das et al.). Although the mechanism by which H2S augments SIRT1 function remains to be elucidated, these data suggest that H2S and SIRT1 function in a critical axis regulating angiogenesis with the potential to mitigate or reverse oxidative stress-induced and aging-related changes in vascular health using pharmacological agents.
Mechanisms of H2S regulation of energy metabolism in EC
H2S at physiological levels is thought to exert its biological activities through non-mutually exclusive mechanisms, including post-translational modification of target proteins via S-sulfhydration of surface-exposed C residues (Mishanina et al., 2015; Mustafa et al., 2009); direct or indirect antioxidant action (Whiteman et al., 2004; Whiteman et al., 2005); or ATP generation via transfer of electrons to the mitochondrial SQR protein (Goubern et al., 2007; Yong and Searcy, 2001). There is clearly precedent for the potential of H2S to regulate angiogenesis via S-sulfhydration of target proteins in multiple pathways implicated in angiogenesis, ranging from VEGFR2 signalling (Altaany et al., 2014; Coletta et al., 2012; Tao et al., 2013) to cellular energy metabolism via activation of the rate-limiting glycolytic enzyme GAPDH (Mustafa et al., 2009).
H2S can also inhibit terminal electron transfer to oxygen (Nicholls and Kim, 1982) at supraphysiological levels, estimated to be greater than 20μM upon acute treatment of intact human colon carcinoma epithelial cells (Leschelle et al., 2005). Our data are consistent with the potential relevance this mechanism in regulation of EC oxidative/glycolytic energy balance, although whether levels of endogenously-generated H2S upon SAA deprivation here or in other contexts reach this inhibitory concentration remains unclear (Cooper and Brown, 2008). Consistent with our findings in EC, an increase in H2S in pancreatic β cells exposed to ER stress promotes aerobic glycolysis associated with decreased OXPHOS and S-sulfhydration of enzymes involved in energy metabolism (Gao et al., 2015). Interestingly, a number of tumors and cancer cells lines also upregulate GCN2 (Lehman et al., 2015; Wang et al., 2013) or H2S production (Bhattacharyya et al., 2013; Sen et al., 2015; Sonke et al., 2015; Szabo et al., 2013), possibly contributing to the Warburg effect through inhibition of mitochondrial respiration.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, James R. Mitchell (jmitchel@hsph.harvard.edu)
EXPERIMENTAL MODELS
Mice
All experiments were performed with the approval of the Harvard Medical Area or Boston University Institutional Animal Care and Use Committee (IACUC). 8–14wk old male or female C57BL/6 mice (The Jackson Laboratory, Bar Harbor, ME) were used for all experiments unless otherwise indicated. Male and female CGL WT and KO mice on a mixed 129/C57BL/6 background (Yang et al., 2008) and GCN2KO and control mice on a C57BL/6 background (Peng et al., 2012) were bred at our facility. Except where indicated, animals were maintained under standard group housing conditions with ad libitum access to food (Purina 5058) and water, 12-hr light/12-hr dark cycles, temperature between 20–23°C with 30–70% relative humidity.
Experimental diets were based on Research Diets D12450B with approximately 18% of calories from protein (hydrolyzed casein or individual crystalline amino acids (Ajinomoto) in the proportions present in casein), 10% from fat and 72% from carbohydrate. MR diets containing 1.5g methionine (M)/kg food and lacking cysteine (C) (Miller et al., 2005) in the context of a 14% protein/ 76% carbohydrate calorie diet were provided AL. In MR experiments with WT and CGLKO mice, the control diet was supplemented with 4.3g C/kg food to compensate for the inability of CGLKO mice to make C. AL food intake/g body weight was monitored daily for several days and used to calculate calorie restriction (CR) based on initial animal weights. Animals were fed daily with fresh food between 6–7pm.
Where indicated, axitinib was supplemented at a daily dose of ~30mg/kg/d in the food as previously described (Alonso et al., 2010; Ma and Waxman, 2008); and PAG was dosed once daily i.p. (10mg/kg) for the indicated time.
Cell Lines and Primary Tissue Culture Studies
Pooled human umbilical vein endothelial cells (HUVEC) were obtained from Lonza (C2519A, Lonza) and used between passage 1 and 7. HUVEC were cultured in endothelial basal medium (EBM-2) supplemented with 2%FBS and endothelial growth medium SingleQuots (Clonetics, Lonza) at 37°C in a humidified, multigas incubator (Napco Series 8000 WJ, Thermo Scientific) at 5%CO2 and 3%O2 achieved by displacing air with nitrogen gas.
Primary mouse endothelial cells were isolated from the lung by collagenase digestion (Liberase, Roche) followed by sequential affinity selection method using Dynabeads™ goat anti-rat conjugated to rat-anti mouse CD31 (BD Biosciences, San Jose, CA), and cultured in endothelial basal medium (EBM-2) supplemented with 2%FBS and endothelial growth medium SingleQuots (Clonetics, Lonza) at 37°C, 5%CO2 and 3%O2. At least 3 independent primary mouse EC cultures/genotype was tested per experiment.
Primary mouse hepatocytes were isolated by collagenase digestion, Percoll (GE Healthcare) gradient centrifugation and cultured in William’s E media (Sigma) with 5%FBS at 37°C, 5%CO2 and 3%O2.
Primary mouse dermal fibroblasts (MDF) were obtained from tail skin of wildtype mice following collagenase digestion and cultured in DMEM with 10%FBS at 37°C, 5%CO2 and 3%O2.
Primary mouse skeletal myotubes were isolated from leg skeletal muscle following collagenase/dispase digestion and cultured in Ham’s F12 with 20%FBS and 10ng/mL bFGF for approximately one week, then switched to Ham’s F12 supplemented with 4% horse serum but lacking bFGF for 7d at 37°C in 5%CO 2 at normoxia (20%O2).
C2C12 myoblasts were cultured in DMEM with 10%FBS until they reached 90% confluence, then switched to DMEM supplemented with 4% horse serum for 7d at 37°C in 5%CO2 at normoxia (20%O2).
Immortalized mouse embryonic fibroblasts (MEF) were cultured in DMEM with 10%FBS at 37°C in 5%CO 2 at normoxia (20%O2).
The following methionine and cysteine deprivation conditions (-M&C) were used to simulate methionine restriction in vitro. For primary EC, when cultures reached confluence, the media was removed and replaced either with complete DMEM +/−M&C plus Glutamax (no pyruvate) (Sigma) supplemented with the same amount (2%) of dialyzed FBS and endothelial growth medium SingleQuots (Clonetics, Lonza) for 1 to 24hr. For hepatocytes, MDF, primary or C2C12 myotubes and MEF cultures, the media was removed and replaced with complete DMEM +/−M&C (plus Glutamax, no pyruvate; Sigma) supplemented with dialyzed FBS (10% for hepatocytes, MDF and MEF; 4% for the primary skeletal muscle and C2C12 myotubes)
Where indicated, media was supplemented with L-NAME (100μM), NaHS (100μM), PAG (100μM), SU5416 (20μM), axitinib (10μM), Ex527 (10μM), 2DG (1mM or 50mM), Compound C (10μM) or STO-609 (5μg/mL). Hypoxia was induced via air displacement with nitrogen gas.
METHOD DETAILS
Intramuscular adenoviral-mediated gene delivery
Local overexpression of CGL or VEGF in gastrocnemius was accomplished by intramuscular injection of 40μl containing a total of 109 PFU of an adenovirus-type 5 (dE1/E3) containing the CMV promoter driving expression of the mouse CGL gene (Ad-mCTH/CGL, Genbank RefSeq BC019483, ADV-256305 Vector Biolabs) or the human VEGF gene (Ad-hVEGFA165 Genbank RefSeq NM_001171626, Vector Biolabs) or the negative control virus Ad-CMV-Null (1300 Vector Biolabs) once weekly for 2wk.
Hindlimb ischemia model
12wk old C57BL/6 WT mice were anaesthetized with isoflurane and body temperature maintained on a circulating heated water pad. Following a 1cm groin incision, the neurovascular pedicle was visualized under a microscope (LW Scientific, Z2 Zoom Stereoscope) and the femoral nerve carefully dissected out. The femoral vein (located medially) was separated from the femoral artery (located laterally) allowing electrocoagulation of the left common femoral artery, proximal to the bifurcation of superficial and deep femoral artery while sparing the vein and nerve. Once the artery was occluded, the surgical site was inspected for any residual bleeding (Hoefer et al., 2004; Mirabella et al., 2011).
Laser Doppler perfusion imaging
Laser Doppler perfusion imaging (LDPI) was performed as described previously (Hoefer et al., 2004; Mirabella et al., 2011). Briefly, mice were kept under isoflurane anesthesia, and body temperature maintained on a circulating heated water pad. Blood flow recovery was monitored at d 0 (immediately post-surgery), d1, d3, and d10 using an LDPI analyzer (Moor Instruments, Inc. DE). The LDPI intensity of the ischemic foot was normalized to the contralateral foot and represented as relative blood flow of the ischemic limb (Ischemic/Non-ischemic ratio). AUCs from I/NI ratios from each animal over time were used for statistical comparisons between groups.
Treadmill exercise training
Twelve wk old male WT and CGLKO mice were randomized into sedentary or exercise groups. Mice were acclimatized to the treadmill (Columbus Instruments 6 lane treadmill) at 8 m/min for 5min for 3d prior to exercise training. Mice ran 30min/d at 5° incline at 12m/min for the first wk of training. Mice continued running 30min/d at 5° incline at 14m/min for an additional 3wk to reach 1mo total of exercise training. Sedentary controls and exercised animals were co-housed. Mice were euthanized 1hr after the final exercise bout (Narkar et al., 2008).
Treadmill exercise test
2 and 3d after ischemic injury, mice were acclimatized to the treadmill (Columbus Instruments 6 lane treadmill) at 8 m/min for 5min prior to exercise training. At d4, mice were run until exhaustion at 5° incline, 8m/min for 10min then 10m/min for 5min, with a 2m/min increase in speed every 5min (Narkar et al., 2008).
Gene expression analysis by qPCR
Total RNA was isolated from tissues and cells using RNeasy Mini Kit (Qiagen) and cDNA synthesized by random hexamer priming with the Verso cDNA kit (Thermo). qRT-PCR was performed with SYBR green dye (Lonza) and TaqPro DNA polymerase (Denville). Fold changes were calculated by the ΔΔCt method (Livak and Schmittgen, 2001) using Hprt, 18S and/or β-Actin genes as standards, and normalized to the experimental control. Human primer sequences are indicated in the Key Resources Table, and mouse primer sequences are additionally found in Table S1.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-CGL (Anti-Cystathionase) | Abcam | Cat# Ab151769 |
| Anti-CBS | Abcam | Cat# Ab135626 |
| Anti-3MST (Anti-MPST) | Sigma | Cat# HPA001240 |
| Anti-ATF4 (Anti-CREB-2) | Santa Cruz | Cat# Sc-200 |
| Anti-beta Tubulin | Cell Signaling | Cat# 2128 |
| Anti-Actin | Cell Signaling | Cat# 4970 |
| HRP conjugated anti-rabbit | Dako | Cat# P044801-2 |
| Anti-CD31 | BD Bioscience | Cat# 557355 |
| Anti-HIF1a | Cayman Chemical | Cat# 10006421 |
| Anti-p-eIF2α Ser51 | Cell Signaling | Cat# 9712S |
| Anti-total eIF2α | Cell Signaling | Cat# 9722S |
| Anti-Tubulin | Cell Signaling | Cat# 2146S |
| Anti-CD31-APC | Biolegend | Cat# 102410 |
| Alexa Fluor 555 phalloidin | ThermoFisher | Cat# A34055 |
| Bacterial and Virus Strains | ||
| Ad-CMV-CGL (Ad-mCTH) | Vector Biolabs | Cat# ADV-256305 |
| Ad-CMV-Null | Vector Biolabs | Cat# 1300 |
| Ad-h-VEGFA165 | Vector Biolabs | Cat# ADV-227457 |
| Ad-m-ATF4-shRNA | Vector Biolabs | Cat# shADV-253208 |
| Ad-GFP | Vector Biolabs | Cat# 1060 |
| Biological Samples | ||
| Livers (frozen) taken from experimental mouse strains listed in the Experimental Models: Organisms/Strains section | See Experimental Models: Organisms/Strains section | See Experimental Models: Organisms/Strains section |
| Serum/Plasma (frozen) taken from experimental mouse strains listed in the Experimental Models: Organisms/Strains section | See Experimental Models: Organisms/Strains section | See Experimental Models: Organisms/Strains section |
| Skeletal muscle (frozen) taken from experimental mouse strains listed in the Experimental Models: Organisms/Strains section | See Experimental Models: Organisms/Strains section | See Experimental Models: Organisms/Strains section |
| Chemicals, Peptides, and Recombinant Proteins | ||
| NaHS | Sigma | Cat# 161527 |
| GYY4137 | Sigma | Cat# SML0100 |
| DL-Propargylglycine | Sigma | Cat# P7888 |
| Passive Lysis Buffer (5x) | Promega | Cat# E1941 |
| PLP (Pyridoxal 5′-phosphate) | Sigma | Cat# P9255 |
| L-cysteine | Sigma | Cat# C7352 |
| Lead (II) acetate trihydrate | Sigma | Cat# 316512 |
| P3 H2S Detection Probe | From the lab of Prof. K.H. Ahn | Singha et al., 2015 |
| SU5146 | Tocris | Cat. No. 3037 |
| Ex-527 | Cayman Chemical | Cat# 10009798 |
| VECTASHIELD Antifade Mounting Medium with DAPI | Vector laboratories | Cat# H-1200 |
| Potassium Cyanide | Sigma | Cat# 11813 |
| Phenformin | Sigma | Cat# P7045 |
| 2-Deoxy-D-Gucose | Sigma | Cat# D8375 |
| Oligomycin A | Sigma | Cat# 75351 |
| FCCP (Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone) | Sigma | Cat# C2920 |
| Antimycin A | Sigma | Cat# A8674 |
| MitomycinC | Sigma | Cat# M4287-2MG |
| Compound C (Dorsomorphin) | Abcam | Cat# Ab120843 |
| Axitinib | Selleckchem | Cat# S1005 |
| L-NAME (hydrochloride) | Cayman Chemical | Cat# 80210 |
| Recombinant human VEGF165 | Peprotech | Cat# 100-20 |
| Recombinant murine VEGF165 | Peprotech | Cat# 450-32 |
| Propidium Iodine | ThermoFisher Scientific | Cat# P3566 |
| Annexin V | ThermoFisher Scientific | Cat# A13201 |
| Critical Commercial Assays | ||
| Mouse VEGF ELISA | Peprotech | Cat# 900-K99 |
| Mouse VEGF Quantikine ELISA kit | R&D Systems | Cat# MMV00 |
| Human VEGF Quantikine ELISA kit | R&D Systems | Cat# DVE00 |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| Primary mouse endothelial cells prepared from C57BL/6, CGL KO, GCN2 KO and SIRT1 inducible KO mice (freshly isolated in the lab of Dr. James Mitchell or Dr. David Sinclair for each experiment) | Jackson Laboratories and laboratory of Dr. James R. Mitchell | Cat# 000664; this paper; Das et al. in this issue |
| Primary mouse myotubes from GCN2 WT and KO mice | From the laboratory of Dr. James R. Mitchell | This paper |
| Primary CGL WT and KO mouse tail dermal fibroblasts | From the laboratory of Dr. James R. Mitchell | This paper |
| Primary mouse hepatocytes from WT mice | From the laboratory of Dr. James R. Mitchell | This paper |
| Immortalized MEF | From the laboratory of Dr. James R. Mitchell | This paper |
| HUVEC | Lonza | Cat# CC-2519 |
| C2C12 | ATCC | Cat# CRL-1772 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6 male and female Mice | Jackson Laboratories | Cat# 000664 |
| WT and KO CGL male and female mice on mixed 129/C57BL/6 background | Strain originally from Dr. Rui Wang and bred in the lab of Dr. James R. Mitchell | Yang et al., 2008; Hine et al., 2015 |
| GCN2 WT and KO male and female mice | Strain originally from Dr. David Ron and bred in the lab of Dr. James R. Mitchell | Munn et al., 2005; Peng et al., 2012 |
| Oligonucleotides | ||
| si-Scramble (Selective negative control No.1 siRNA) | ThermoFisher Scientific | Cat# 4390843 |
| Mouse si-PGC1a (PPARGC1A) | ThermoFisher Scientific | Cat# n253420 |
| Mouse si-HIF1a | ThermoFisher Scientific | Cat# s67530 |
| Human si-ATF4 | ThermoFisher Scientific | Cat# s1704 |
| Human si-PGC1alpha (PPARGC1A) | ThermoFisher Scientific | Cat# s21394 |
| Human si-HIF1a (HIFA) | ThermoFisher Scientific | Cat# s6539 |
| Human si-eNOS (NOS3) | ThermoFisher Scientific | Cat# s9623 |
| human ACTIN/ACTB F: GTTGTCGACGACGAGCG R: GCACAGAGCCTCGCCTT | N/A | N/A |
| human ASNS F: GCGGAGTGCTTCAATGTAAC R: CCAATAAGAAAGTGTTCCTGGG | N/A | N/A |
| human ATF4 F: CTATACCCAACAGGGCATCC R: GTCCCTCCAACAACAGCAAG | N/A | N/A |
| For a full list of all primers used, please see Table S1 | ||
| Recombinant DNA | ||
| prK-ATF4 overexpression plasmid | Addgene | 26114 |
| Software and Algorithms | ||
| ImageJ | National Institutes of Health | https://imagej.nih.gov/ij/download.html |
| GraphPad Prism | GraphPad | Version 7.0 |
| FlowJo | FlowJo LLC | https://www.flowjo.com/solutions/flowjo |
| Fiji software | GPL v2, Fiji | http://fiji.sc/Fiji |
| Matlab R2017A | MathWorks | https://www.mathworks.com/programs/trials/trial_request.html?prodcode=ML |
| Other | ||
Immunoblotting
Cells were homogenized with passive lysis buffer (Promega), normalized for protein content, boiled with SDS loading buffer and separated by SDS-PAGE. Proteins were transferred to PVDF membrane (Whatman) and blotted for CGL (ab151769 Abcam), HIF1α (10006421 Cayman Chemical), p-eIF2α Ser51 (9712S Cell Signaling), total eIF2α(9722S Cell Signaling), ATF4 (11815 Cell Signaling), Actin (13E5 Cell Signaling) and Tubulin (2146S Cell Signaling) and secondarily with HRP-conjugated anti-rabbit antibody (Dako).
VEGF ELISAs
Mouse and human VEGF ELISA kits were purchased from Peprotech (900-K99) and R&D System (DEV00), respectively, and assays performed according to manufacturer’s instructions on 100μl of plasma or cell culture media per analysis. For analysis of VEGF protein in muscle, approximately 100mg of frozen gastrocnemius muscle was pulverized using a mortar and pestle, and the powder transferred to a 1.5mL microcentrifuge tube containing 150μL of PBS. The tissue was further disrupted using a mechanical tissue homogenizer (Kimble Kontes Pellet Pestle, Fisher Scientific). After three cycles of freezing and thawing, the tissue suspension was microcentrifuged at max speed for 10min and the supernatant recovered and stored at −80 °C. Lysates were adjusted to 0.5μg/μL in PBS and run using a mouse VEGF ELISA kit from R&D systems (MMV00) was used.
Immunohistochemistry and capillary density analysis
IHC was performed on frozen sections of unfixed gastrocnemius muscle (50μm for CD31 quantification; 20μm for CD31/VEGF/IB4 co-staining). After 5min fixation in PFA 4% and rinsing in PBS, immunostaining was performed as previously described (Longchamp et al., 2014). Primary antibodies included anti-mouse CD31 (BD Bioscience), anti-mouse VEGF (Novus Biologicals) and Isolectin B4 (Life Technologies) at a dilution of 1:100. For capillary density measurements, CD31 area was quantified from randomly photographed 10μm stack sections (6 images per section, 4 sections per muscle per mouse) using Fiji software (http://fiji.sc/Fiji). All quantifications were performed blindly.
CD31 FACS analysis
Following enzymatic digestion of muscle with collagenase/dispase mix, cells were blocked with mouse FcR blocking reagent (Miltenyl Biotech) and stained for 30min at 4 °C in the dark with CD31APC at 1:100 (BioLegend). Cells were washed and acquired immediately on an LSR II flow cytometer (BD Biosciences) and analyzed with FlowJo. CD31 positive endothelial cells are expressed as a percentage of total cells recovered from the enzymatically digested muscle cells as assessed by forward and side scatter.
H2S measurements
Detection of H2S production capacity in live cultures by lead sulfide method
For detection of H2S production in live cells, growth media was supplemented with 10mM cysteine and 10μM pyridoxal 5’-phosphate hydrate (PLP, Sigma), and a lead 6x4 inch pieces of lead acetate paper, made by soaking 703 size blotting paper (VWR) in 20mM lead acetate (Sigma) and then vacuum drying, was placed over the plate for 2–24hr of further incubation in a CO2 incubator at 37°C until lead sulfide was detected but not saturated.
Detection of endogenous H2S with fluorescent P3 probe in cultured cells or frozen tissue sections
For detection of endogenous H2S production in live adherent cells, growth media was supplemented with 10μM P3 probe (Singha et al., 2015) for 30min prior to fixation. Quantification was performed by calculating the average P3 signal intensity per cell. Cell areas were automatically segmented using brightfield images. Values presented are average P3 intensity per cell, corrected for total cell area. Analyses were performed using Matlab R2017A. For detection of endogenous H2S production in tissue sections, 50μm frozen sections of unfixed gastrocnemius muscle were incubated with 20μM P3 probe for 5min and washed 2X with PBS. Sections were then fixed for 5min in 4% PFA prior to immunostaining using a 2 photon microscope (Zeiss LSM780 w/ Mai Tai HP 2-photon laser (Spectra Physics) at 880nm excitation and 520–550nm emission (Singha et al., 2015).
Genetic manipulations in cultured cells
siRNA knockdown
siRNA knockdown of human activating transcription factor 4 (ATF4), human endothelial nitric oxide synthase (eNOS), human hypoxia-inducible factor 1-α (HIF1α), PGC1α (PPARGC1A) in HUVEC as well as mouse HIF1α and PGC1α in C2C12 myoblasts was performed using lipofectamine RNAiMAX (Life Technologies) and 30nM siRNA purchased from Ambion (Ambion, ThermoFisher) as described previously (Hine et al., 2015). All experiments were performed 2 d after transfection. Knockdown was confirmed by immunoblot and/or qPCR.
shRNA knockdown of ATF4: MDF were infected overnight in complete medium and collected 2d later using Ad-m-ATF4-shRNA or the negative control virus Ad-CMV-Null adenovirus amplified and purified by Vector Biolabs (Philadelphia, PA, U.S.A.).
ATF4 overexpression
ATF4 overexpression in HUVEC was performed using Lipofectamine 2000 (Life Technologies) and 1ug of prK-ATF4 plasmid per well (12-well format) overnight. All experiments were performed 2d after transfection. Overexpression was confirmed by immunoblot and/or qPCR.
Adenoviral-mediated CGL overexpression
HUVEC were infected overnight in complete medium and collected 2 d later using Ad-m-CTH or the negative control virus Ad-CMV-Null adenovirus amplified and purified by Vector Biolabs (Philadelphia, PA, U.S.A.).
Angiogenesis assays in vitro
Migration assay
EC were seeded at 100,000 cells per well in 24-well plate in EGM with 2% serum and growth factors (Lonza). ~12hr later, media was switched to EGM without serum/growth factors, and in some cases mitotically arrested (1μg/mL MitoC). A single scratch wound was created using a sterile p200 pipette tip on a confluent field of EC. Floating cells were washed away and EGM (or DMEM +/−M&C media) with dialyzed serum and growth factors replaced, including treatments. Repopulation/migration across the scratch wound was recorded by phase-contrast microscopy every 4hr for up to 20hr using a digital camera. Wound closure (gap area at t=Xhr relative to t=0hr) was determined at each time point from digital images using ImageJ software.
Tube formation
Formation of tube networks was assessed as described previously (Borradaile and Pickering, 2009). HUVEC were seeded at 10,000 cells per well in a 24-well plate (Corning) coated with 150 μL Cultrex reduced growth factor basement membrane extract (Trevigen). Following an 18 h-incubation, resulting tube networks were analyzed by light microscopy (Nikon Eclipse TiE). The total length of tubule networks and the number of branch points were quantified by ImageJ software.
Spheroid capillary sprouting assay
Hanging drops of HUVEC or primary mouse EC in EGM2 (De Bock et al., 2013) were embedded in Matrigel® (Corning) and cultured in the indicated media for 24hr to induce sprouting. Compounds were added at the indicated concentrations during the gel culture step, using corresponding vehicle concentrations as control. Spheroid cultures were stained with phalloidin diluted 1:500 in PBST for 1hr at RT and counterstained with DAPI. Images were captured with a Zeiss LSM 510 Meta NLO confocal microscope (oil objectives: x 40 with NA 1.3, x 63 with NA1.4, x 100 with NA 1.3; Carl Zeiss, Munich, Germany) or a Leica laser-scanning SP5 confocal microscope (Leica, Manheim, Germany). Analysis of the sprout length was performed using ImageJ software.
Proliferation
HUVEC were cultured to 60% confluency in a 12-well plate on glass coverslips, washed with PBS and incubated for 24hrs in EGM2 containing 0.1mM BrdU. Immunostaining was performed on cells washed and fixed for 5min in −20°C acetone, air -dried, rinsed in PBS and permeabilized for 1 h in PBS supplemented with 2% BSA and 0.1% Triton X-100. BrdU positive nuclei were automatically detected using the ImageJ software and normalized to the total number of DAPI-positive nuclei.
Glucose uptake
For VEGF and NaHS treatment +/− Compound C (10μM) (Fig. 5D, Fig. S6A), HUVEC were pretreated for 1hr with 50ng/ml VEGF (Peprotech) or 100μM NaHS. Cells were then depleted in Krebs-Ringer Bicarbonate Buffer (KRB; NaH2PO4/Na2HPO4 10mM, NaCl 136mM, KCl 4.7mM, MgSO4 1.25mM, CaCl2 1.25mM, pH7.4), without glucose and serum for 30min and then incubated for 6min in a solution containing 0.5μCi 3H-2DG. On ice, cells were then washed in cold PBS 3 times, lysed, and sample counted in a liquid scintillation counter. Samples were normalized to protein content as measured from the same cells by BCA. For remaining glucose uptake experiments (Fig. S5B, S6E, I), EC were treated with NaHS (100μM), KCN (10μM), Antimycin A (2.5μM), Oligomycin (2μM), or FCCP (1.5μM) for 45, 75 or 195min in EGM-2. When indicated, inhibitors were added 30min before the addition of the tracer. Fifteen min before the termination of the experiment, 0.4μCi of 3H-2DG was added to each well (1mL final volume, 12-well format). At the end of the incubation the plate was rapidly transferred on ice, media removed and washed 4 times with PBS + BSA 0.1%. Finally, cell lysis was performed with NaOH 0.2% + SDS 0.5%. Lysate (500uL) was mixed with scintillation fluid (5mL) and sample radioactivity measured in a scintillation counter (Beckman).
Glycolytic flux analysis
HUVEC were cultured with the standard media (EGM-2), and stimulated for up to 3hr with test compounds (NaHS (100μM), VEGF (50ng/mL), Antimycin A (2.5μM), Oligomycin (2μM), or 2DG (1mM). 15min before the end of the incubation, 5uCi D-[5-3H(N)]-glucose was added to 1.5mL media (12-well format), then the plate was rapidly transferred on ice, and the media (1.5mL) transferred into a 5 mL vial. One PBS wash (1.5mL) was performed and liquid combined with media. The vial was inserted into a 50mL tube pre-filled with 3mL water. Tube was tightly capped and evaporation was performed at room temperature for 48hr. Finally, 2mL of water was collected from the 50mL tube and mixed with 10mL of scintillation fluid. Sample radioactivity measured in a scintillation counter (Beckman).
ATP and ATP/ADP ratio
Cells in a 96-well format were treated with NaHS, VEGF or 2DG in a reverse time course. ATP was measured by addition of 70μL of Cell TITER-Glo (Promega), plate incubated in the dark for 10min and luminescence quantified by a plate reader (BioTek instruments). ADP/ATP ratio was calculated after measuring ADP and ATP by a commercially available enzymatic assay (ApoSENSOR, BioVision), according to the manufacturer’s instructions.
Energy charge
Steady-state mass spec measurements of AMP, ADP and ATP were used to calculate energy charge using the following equation: (ATP +1/2ADP)/(ATP + ADP + AMP).
Lactate
Lactate in HUVEC culture media was measured after 30min and 2hr of treatment with NaHS (100μM) or -M&C. The assay was performed using a plate-based colorimetric assay (Cell Biolabs Inc.) according to the manufacturer’s protocol.
Cell death and ROS
Apoptosis and cell death was measured after 12hr treatment with NaHS (100μM) or – M&C using a FACS-based annexin V/propidium iodide assay according to the manufacturer’s protocol (BD Biosciences).
Reactive oxygen species were measured after 15min, 1hr and 2hr incubations with NaHS (100μM), FCCP (2μM) or KCN (10μM). Menadione (10μM) was used as a positive control. Cells were stained for 15min with CellROX Green dye in the treatment media, fixed with 2% paraformaldehyde and measured using FACS.
Seahorse
Cellular oxygen consumption and extracellular acidification rate was measured using the Seahorse Cell Metabolism Analyzer XF96 (Seahorse Biosciences). Cells were plated at a density of 12,000 cells and untreated or pretreated with 100μM NaHS, 10μM KCN or 500μM phenformin for 2hr. After 24hr, media was changed to unbuffered XF assay media with 0 or 11mM glucose, 0 or 2mM glutamine and pyruvate at pH7.4 and basal OCR and ECAR measured for 5 blocks of 2min mixing and 5min measuring. Glucose (10mM final), 2DG (50mM final) and Oligomycin (2.5μM final) were injected at indicated times. All plates were normalized to protein content as measured from the same cells after Seahorse by BCA.
Mitochondrial respiration in permeabilized cells using complex-specific substrates was measured in a Seahorse Cell Metabolism Analyzer XF24 (Seahorse Biosciences) as previously described (Salabei et al., 2014). Briefly, cells were plated at a density of 100,000 cells in EGM- 2. Four hr later, cells were washed with MAS buffer, then incubated with MAS buffer containing 10mM pryruvate, 2mM malate, 4uM FCCP and 25ug/mL Saponin for 1hr, untreated or pretreated with 100μM NaHS or 10μM KCN. OCR was measured for 10 blocks of 1min mixing and 2min measuring. Rotenone (1μM final), Succinate (10mM final), Antimycin A (20μM final), and TMPD/Ascorbate (0.5mM/2mM final) were injected in order.
Metabolite profiling for glucose flux analyses
To determine the relative levels of intracellular metabolites, extracts were prepared and analyzed by LC/MS/MS. Triplicate 15-cm confluent plates were incubated in EGM-2 media in presence or absence of 100μM NaHS 105min prior to extraction. For D-[1,2-13C ]-glucose flux studies, cells were washed once with serum- and glucose free DMEM and then incubated in DMEM containing a 10mM 1:1 mixture of D-[1,2-13C ]-glucose and unlabeled D-glucose for 15min. Metabolites were extracted on dry ice with 4-mL 80% methanol (−80°C), as described previously (Ben-Sahra et al., 2013). Insoluble material was pelleted by centrifugation at 3000g for 5min, followed by two subsequent extractions of the insoluble pellet with 0.5-mL 80% methanol, with centrifugation at 16,000g for 5min. The 5mL metabolite extract from the pooled supernatants was dried down under nitrogen gas using an N-EVAP (Organomation Associates, Inc.).
Dried pellets were resuspended using 20μL HPLC grade water for mass spectrometry. 10μL were injected and analyzed using a 5500 QTRAP triple quadrupole mass spectrometer (AB/SCIEX) coupled to a Prominence UFLC HPLC system (Shimadzu) via selected reaction monitoring (SRM). Some metabolites were targeted in both positive and negative ion mode for a total of 287 SRM transitions using pos/neg polarity switching. ESI voltage was +4900V in positive ion mode and –4500V in negative ion mode. The dwell time was 3ms per SRM transition and the total cycle time was 1.55 seconds. Approximately 10–14 data points were acquired per detected metabolite. Samples were delivered to the MS via normal phase chromatography using a 4.6mm i.d. x 10cm Amide Xbridge HILIC column (Waters Corp.) at 350μL/min. Gradients were run starting from 85% buffer B (HPLC grade acetonitrile) to 42% B from 0–5min; 42% B to 0% B from 5–16min; 0% B was held from 16–24min; 0% B to 85% B from 24–25min; 85% B was held for 7min to re-equilibrate the column. Buffer A was comprised of 20mM ammonium hydroxide/20mM ammonium acetate (pH=9.0) in 95:5 water:acetonitrile. Peak areas from the total ion current for each metabolite SRM transition were integrated using MultiQuant v2.0 software (AB/SCIEX). For stable isotope labeling experiments, custom SRMs were created for expected 13C incorporation in various forms for targeted LC/MS/MS. Samples were quantile normalized and log transformed and metabolites were pareto scaled prior to analysis. Analyses were performed using R version 3.3.2.
Global metabolite profiling
HUVEC grown in EBM-2 media with BulletKit were switched to DMEM with 2% dialyzed FBS all non-serum BulletKit components. After a 1hr equilibration period, cells were switched to treatment media (control, -M&C, NaHS 100μM). After the treatment period (15min, 2hr, 4hr) cells were collected and analyzed for total metabolite profile by mass spectrometry using the methods detailed in the preceding section.
QUANTIFICATION and STATISTICAL ANALYSIS
Data are displayed as means +/− standard deviation (SD) and statistical significance assessed in GraphPad Prism using Student’s T test, 1-way or 2-way ANOVA unless otherwise specified. A P-value of 0.05 or less was deemed statistically significant.
Supplementary Material
(A) Tube formation assay: Quantification of tube branch points per field of view in HUVECs seeded onto growth factor-replete Matrigel and incubated in the indicated media +/−SIRT1 inhibitor Ex-527 for 18hr; n=8–10 data points/condition. (B) Daily body weight (left, n=5 mice/group) and food intake expressed as grams of food eaten per gram of mouse body weight (right, n=2 cages/group) of mice given ad libitum access to methionine restricted (MR) vs. control (Ctrl) diet for 2mo in a representative experiment. (C) Representative flow cytometric analysis of CD31 positive cells from gastrocnemius muscle of mice fed for 2wk on Ctrl or MR diets with or without VEGFR2 inhibition (axitinib, +A) expressed as a percentage of total cells recovered from the enzymatically digested muscle tissue; quantification at right (n=3–6 mice/group). (D) Expression of VEGF mRNA in gastrocnemius muscle of mice subject to MR for 2–8wk expressed relative to Ctrl diet group; n=22–23/mice group. (E) VEGF protein levels in extracts of muscle from mice on the indicated diet for at least 2wk as determined by ELISA; n=11–13/group. (F) Representative transverse sections of gastrocnemius muscle stained for VEGF (left, 40X mag), IsolectinB4 (IB4, middle, 40X mag) and merged (VEGF red, IB4 green, DAPI blue; right, insert 160X mag) in mice preconditioned for 2mo on control (Ctrl) or methionine restricted (MR) diets. (G) Schematic representation of the hindlimb ischemia procedure. (H, I) Longitudinal Doppler imaging of blood flow (H) and distance run on treadmill exercise test (I) following ischemia in WT mice preconditioned on a complete diet fed ad libitum (AL) or calorie restricted daily by 40% of the AL amount (CR) for 1mo prior to femoral artery ligation, +/− propargylglycine (PAG) daily for 1wk prior to ligation and 2wk after as indicated. (H) Left: representative infrared images on the indicated after ligation (I, ischemic; NI, non-ischemic). Right: quantification of blood flow recovery with individual animal AUCs used for statistical comparisons; n=6 mice/group. (I) Distance run on treadmill exercise test on d4 after ligation of the indicated groups from (H). Error bars indicate SD; asterisks indicate the significance of the difference by Student’s T test or 1-way ANOVA with Sidak’s MCT between diets in vivo or sulfur amino acid levels in vitro; *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
(A) Immunoblot of HIF1α protein in HUVEC 2 d after transfection with HIF1α siRNA or control scrambled (Sble) siRNA. (B) Quantification of immunoblots from Fig. 2B of the indicated protein from HUVEC extracts after culturing in control (Ctrl) or M&C deficient media. (C) PGC1α mRNA levels of HUVEC cultured in the indicated media for 16hr; n=4 experiments/group, error bars indicate SEM. (D, E) PGC1α (D) and VEGF (E) mRNA levels in HUVEC, 2 d after transfection with a PGC1α overexpressing construct, and expressed relative to cells transfected with a control AdGFP vector; n=3 experiments/group, SEM. (F) Asns (left) and ATF4 (right) mRNA expression in HUVEC 2 d after transfection with ATF4 siRNA or control scrambled (Sble) siRNA and cultured in the indicated media for 16hr; n=4 experiments/group, SEM. (G–I) Immunoblot of ATF4 protein (G), mRNA expression of ATF4 (H) and Asns (I) in HUVEC 2d after transfection with a CMV-driven ATF4 over-expressing construct (ATF4OE) or empty vector (Empty). (J) Representative images of primary WT mouse ECs stained for CD31 (red), Actin (green) and DAPI (blue) at 40X mag; n=3 experiments/group, SEM. (K) Asns mRNA in WT and GCN2KO primary mouse EC from n=5–6 mice per genotype cultured as indicated for 16hs. (L) VEGF mRNA in MDF 2d after infection with shATF4 or control scrambled adenovirus and cultured for the final 16hr as indicated; n=5–8 experiments/group, SEM. (M) HIF1α mRNA expression in C2C12 cells 2d after transfection with HIF1α or control scrambled (Sble) siRNA and then cultured for an additional 16hr in control media; n=11–12 experiments/group, SEM. (N, O) C2C12 HIF1α protein level (N) and VEGF mRNA expression (O) 2d after transfection with HIF1α siRNA or control scrambled (Sble) siRNA, and then cultured for an additional 24hr under 20% or <1% oxygen tension in presence or absence of M&C; n=3–6 experiments/group, SEM. (P) Relative PGC1α (left) and VEGF (right) mRNA expression in HUVEC 2d after transfection with PGC1α siRNA or control scrambled siRNA and cultured in the indicated media for 16hr; n=3 experiments/group, SEM. Unless otherwise indicated, error bars indicate SD; asterisks indicate the significance of the difference between sulfur amino acid levels or genetic interventions by Student’s T test; or between sulfur amino acid levels within genotype/intervention/condition by 1-way ANOVA with Sidak’s MCT; *P<0.05, **P<0.01, ***P<0.001.
(A) Representative images (10X mag) of H2S-specific probe (P3) fluorescence in primary mouse hepatocytes treated with VEGF (50ng/ml) or deprived of all amino acids as well as serum (-Nutrients) for 16hr. (B) VEGF mRNA in WT and CGLKO primary mouse ECs (n=3 mice/group) cultured in M&C deficient media relative to control (Ctrl) media for 16hr; error bars indicate SD; 1-way ANOVA with Sidak’s MCT between SAA levels. (C, D) CBS, CGL and 3-MST mRNA expression (C) and protein level (D) in HUVEC 16hr after treatment with VEGF; n=3 experiments/group, error bars indicate SEM; Student’s T test. **P<0.01, ***P<0.001.
(A) H2S production capacity of gastrocnemius muscle extracts of Ad-Null or Ad-CGL infected mice 1wk after the final intramuscular injection. (B) Expression of VEGF mRNA in the gastrocnemius muscle of mice subjected to low intensity running (exercised) vs. control (sedentary) for 1mo; n=4 mice/group. Error bars indicate SD; asterisks indicate the significance of the difference between exercise groups by Student’s T test; **P<0.01.
(A) HUVEC proliferation over a 24hr period +/−NaHS (100uM) as assessed by radiolabelled thymidine incorporation; n=5–9 experiments/group, error bars indicate SEM. (B). Glucose uptake in SIRT1KO primary mouse ECs treated in triplicate as indicated. (C) Extracellular acidification rate (ECAR) in HUVEC pretreated for 2hr with NaHS (100μM); n=8 technical replicates from each of 2 experiments. (D) Extracellular acidification rate (ECAR) over time in mouse ECs pretreated for 2hr with VEGF (50ng/ml) or NaHS (100μM) and then treated with 10mM Glucose, 2.5μM Oligomycin and 50mM 2DG injection as indicated by the arrows; representative experiment shown. (E) Fold change of lactate concentration in the media of HUVECs treated with NaHS (100μM) for 1.5–6 hr vs. control; n=3 experiments per group; SEM. (F) Glycolytic flux in HUVEC at the indicated time after NaHS addition; n=3 technical replicates/time point; 1-way ANOVA with Dunnett’s MCT vs. t=0. (G). Log2 fold change of unlabeled metabolites in HUVECs treated for 1hr with NaHS (100μM) vs. control. (H) ECAR over time in HUVEC pre-treated for 2hr with axitinib (10μM) or L-NAME (100μM) and then treated with NaHS or 2DG as indicated by the arrows; n=15 technical replicates/time point (I) Quantification of HUVEC migration across scratch +/−NaHS (100μM) or eNOS inhibitor L-NAME (100μM); n=5–6 AUC values/group each from cells at different passage number. (J) Immunoblot of eNOS in HUVEC 2d after transfection with eNOS or control scramble (Sble) siRNA. (K) Quantification of HUVEC migration across scratch 2d after transfection with eNOS siRNA or control scramble (Sble) siRNA +/−NaHS (100μM) as indicated; n=6 AUC values/group each from cells at different passage number. (L) Gating percentages based on annexin V and propidium iodide stains in HUVECs treated with NaHS (100μM) or –M&C for 12hr. Unless otherwise indicated, error bars indicate SD and asterisks indicate the significance of the difference between SAA levels by Student’s T test or between SAA levels within genotype/condition by 1-way ANOVA with Sidak’s MCT; *P<0.05, **P<0.01, ***P<0.001.
(A) Glucose uptake in HUVEC treated with NaHS for 1hr +/−10μM Compound C (Comp C); n=6 experiments/group, error bars indicate SEM; 1-way ANOVA with Sidak’s MCT between +/−NaHS within Comp C treatment group. (B) Migration across scratch in HUVEC treated with NaHS +/− Comp C; n=6 AUC values/group each from cells at different passage number; 1-way ANOVA with Sidak’s MCT between +/−NaHS within Comp C treatment group. (C, D) Schematic of the experiment (C) and quantification of migration across scratch (D) in HUVEC treated with NaHS for 10min in the presence of vehicle or Comp C; n=5–6 AUC values/group each from cells at different passage number; 1-way ANOVA with Sidak’s MCT between +/−NaHS within Comp C treatment group. (E) Glucose uptake in HUVEC treated with NaHS or the CamKKβ inhibitor STO-609; n=3–9 values pooled from 2 independent experiments; 1-way ANOVA with Sidak’s MCT between. (F) ADP/ATP ratio over time in HUVEC treated with the indicated compounds; representative of 2 experiments; n=4–12 values/time point. (G) Energy charge calculated from ATP, ADP and AMP measurements by mass spec in HUVEC treated for 15min with NaHS; n=6 values pooled from 2 independent experiments; Student’s T test. (H) ROS levels expressed as mean fluorescent intensity (MFI) in HUVEC treated over time with NaHS (100μM), KCN (10μM), FCCP (2μM) or menadione (MD, 10μM) as indicated; n=2 values/time point. (I) Time dependent glucose uptake in HUVEC treated with NaHS, KCN or antimycin as indicated; each time point is in triplicate. (J) Extracellular acidification rate (ECAR) over time in HUVEC pretreated for 2hr with 10μM cyanide, 2μM oligomycin (Oligo) or 500μM phenformin (Phen) and then treated with 10mM glucose, 2.5μM oligomycin and 50mM 2DG as indicated by the arrows; n=20 technical replicates pooled from 2 independent experiments; (K) Glycolytic flux in HUVEC pretreated for 1–3hr with oligomycin, antimycin A or 2DG as indicated; n=2–6 experiments/group; 1-way ANOVA with Sidak’s MCT relative to control. (L) Representative images of migration across a scratch (10X mag at t=20hr; dotted lines indicate boundary of the scratch at t=0hr) from HUVEC cultured in the presence of 10μM KCN (left), 500μM phenformin (Phen, middle) or 2μM oligomycin (Oligo, right) +/−1mM 2DG as indicated. Unless otherwise noted, error bars indicate SD; **P<0.01, ***P<0.001.
Highlights.
Sulfur amino acid (SAA) restriction triggers angiogenesis independent of hypoxia or HIF1α
GCN2/ATF4 pathway regulates VEGF and CGL expression upon SAA restriction in ECs
CGL is required for skeletal muscle angiogenesis activated by diet or exercise
H2S triggers glucose uptake, glycolysis and PPP concomitant with OXPHOS inhibition in ECs
Acknowledgments
We thank Florant Allagnat for critical discussions and reading the manuscript; Andrew Thompson, Nandan Nurukar and Rohan Kulkarni for technical assistance; Gokhan Hotamisligil for the use of the Seahorse; John Asara for assistance with metabolomics; and Constance Cepko for the use of the 2-photon microscope. This work was supported by grants from the Swiss National Science Foundation (P1LAP3_158895) to A.L.; National Science Foundation (NSF-DGE1144152) to L.E.B.; the Canadian Institutes of Health Sciences to R.W.; NIH (EB00262) to C.S.C.; American Heart Association (12GRNT9510001, 12GRNT1207025), Lea Carpenter du Pont Vascular Surgery Fund, and Carl and Ruth Shapiro Family Foundation to C.K.O.; and NIH (AG036712, DK090629) and Charoen Pokphand Group to J.R.M.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Author Contributions
Conceptualization: A.L., T.M., A.A., M.R.M., A.D., C.H., C.S.C., C.K.O., J.R.M.; Methodology: A.L., T.M., A.A., M.R.M., J.H.T.-V., C.H., I.B.-S., N.H.K., L.E.B.; Investigation: A.L., T.M., A.A., M.R.M., A.D., J.H.T.-V., C.H., I.B.-S., N.H.K., L.E.B., J.R., P.M., M.T., G.S; Resources: R.W., J.- M.C., J-A.H., K.H.A, C.-H.L., B.D.M., D.A.S., C.S.C., C.K.O., J.R.M.; Writing, A.L., J.R.M.; Funding Acquisition, A.L., C.K.O., J.R.M.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abcouwer SF, Marjon PL, Loper RK, Vander Jagt DL. Response of VEGF expression to amino acid deprivation and inducers of endoplasmic reticulum stress. Invest Ophthalmol Vis Sci. 2002;43:2791–2798. [PubMed] [Google Scholar]
- Alonso F, Krattinger N, Mazzolai L, Simon A, Waeber G, Meda P, Haefliger JA. An angiotensin II- and NF-kappaB-dependent mechanism increases connexin 43 in murine arteries targeted by renin-dependent hypertension. Cardiovasc Res. 2010;87:166–176. doi: 10.1093/cvr/cvq031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altaany Z, Ju Y, Yang G, Wang R. The coordination of S-sulfhydration, Snitrosylation, and phosphorylation of endothelial nitric oxide synthase by hydrogen sulfide. Sci Signal. 2014;7:ra87. doi: 10.1126/scisignal.2005478. [DOI] [PubMed] [Google Scholar]
- Arany Z, Foo SY, Ma Y, Ruas JL, Bommi-Reddy A, Girnun G, Cooper M, Laznik D, Chinsomboon J, Rangwala SM, et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature. 2008;451:1008–1012. doi: 10.1038/nature06613. [DOI] [PubMed] [Google Scholar]
- Ben-Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science. 2013;339:1323– 1328. doi: 10.1126/science.1228792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya S, Saha S, Giri K, Lanza IR, Nair KS, Jennings NB, Rodriguez-Aguayo C, Lopez-Berestein G, Basal E, Weaver AL, et al. Cystathionine beta-synthase (CBS) contributes to advanced ovarian cancer progression and drug resistance. PLoS One. 2013;8:e79167. doi: 10.1371/journal.pone.0079167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierhansl L, Conradi LC, Treps L, Dewerchin M, Carmeliet P. Central Role of Metabolism in Endothelial Cell Function and Vascular Disease. Physiology (Bethesda) 2017;32:126–140. doi: 10.1152/physiol.00031.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borradaile NM, Pickering JG. Nicotinamide phosphoribosyltransferase imparts human endothelial cells with extended replicative lifespan and enhanced angiogenic capacity in a high glucose environment. Aging Cell. 2009;8:100–112. doi: 10.1111/j.1474-9726.2009.00453.x. [DOI] [PubMed] [Google Scholar]
- Cai WJ, Wang MJ, Moore PK, Jin HM, Yao T, Zhu YC. The novel proangiogenic effect of hydrogen sulfide is dependent on Akt phosphorylation. Cardiovasc Res. 2007;76:29–40. doi: 10.1016/j.cardiores.2007.05.026. [DOI] [PubMed] [Google Scholar]
- Cantó C, Auwerx J. Caloric restriction, SIRT1 and longevity. Trends Endocrinol Metab. 2009;20:325–331. doi: 10.1016/j.tem.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castranova D, Davis AE, Lo BD, Miller MF, Paukstelis PJ, Swift MR, Pham VN, Torres-Vazquez J, Bell K, Shaw KM, et al. Aminoacyl-Transfer RNA Synthetase Deficiency Promotes Angiogenesis via the Unfolded Protein Response Pathway. Arterioscler Thromb Vasc Biol. 2016;36:655–662. doi: 10.1161/ATVBAHA.115.307087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coletta C, Papapetropoulos A, Erdelyi K, Olah G, Módis K, Panopoulos P, Asimakopoulou A, Gerö D, Sharina I, Martin E, et al. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc Natl Acad Sci U S A. 2012;109:9161–9166. doi: 10.1073/pnas.1202916109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, Allison DB, Cruzen C, Simmons HA, Kemnitz JW, et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325:201–204. doi: 10.1126/science.1173635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper CE, Brown GC. The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: chemical mechanism and physiological significance. J Bioenerg Biomembr. 2008;40:533–539. doi: 10.1007/s10863-008-9166-6. [DOI] [PubMed] [Google Scholar]
- De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, Quaegebeur A, Ghesquiere B, Cauwenberghs S, Eelen G, et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell. 2013;154:651–663. doi: 10.1016/j.cell.2013.06.037. [DOI] [PubMed] [Google Scholar]
- Dickhout JG, Carlisle RE, Jerome DE, Mohammed-Ali Z, Jiang H, Yang G, Mani S, Garg SK, Banerjee R, Kaufman RJ, et al. Integrated stress response modulates cellular redox state via induction of cystathionine gamma-lyase: cross-talk between integrated stress response and thiol metabolism. J Biol Chem. 2012;287:7603–7614. doi: 10.1074/jbc.M111.304576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerling BM, Weinberg F, Snyder C, Burgess Z, Mutlu GM, Viollet B, Budinger GR, Chandel NS. Hypoxic activation of AMPK is dependent on mitochondrial ROS but independent of an increase in AMP/ATP ratio. Free Radic Biol Med. 2009;46:1386–1391. doi: 10.1016/j.freeradbiomed.2009.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana L, Partridge L, Longo VD. Extending healthy life span--from yeast to humans. Science. 2010;328:321–326. doi: 10.1126/science.1172539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumura D, Kashiwagi S, Jain RK. The role of nitric oxide in tumour progression. Nat Rev Cancer. 2006;6:521–534. doi: 10.1038/nrc1910. [DOI] [PubMed] [Google Scholar]
- Gao XH, Krokowski D, Guan BJ, Bederman I, Majumder M, Parisien M, Diatchenko L, Kabil O, Willard B, Banerjee R, et al. Quantitative H2S-mediated protein sulfhydration reveals metabolic reprogramming during the integrated stress response. Elife. 2015;4:e10067. doi: 10.7554/eLife.10067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goubern M, Andriamihaja M, Nubel T, Blachier F, Bouillaud F. Sulfide, the first inorganic substrate for human cells. FASEB J. 2007;21:1699–1706. doi: 10.1096/fj.06-7407com. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Schaffer BE, Brunet A. AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends Cell Biol. 2016;26:190–201. doi: 10.1016/j.tcb.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hine C, Harputlugil E, Zhang Y, Ruckenstuhl C, Lee BC, Brace L, Longchamp A, Trevino-Villarreal JH, Mejia P, Ozaki CK, et al. Endogenous hydrogen sulfide production is essential for dietary restriction benefits. Cell. 2015;160:132–144. doi: 10.1016/j.cell.2014.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hine C, Kim HJ, Zhu Y, Harputlugil E, Longchamp A, Matos MS, Ramadoss P, Bauerle K, Brace L, Asara JM, et al. Hypothalamic-Pituitary Axis Regulates Hydrogen Sulfide Production. Cell Metab. 2017;25:1320–1333. e1325. doi: 10.1016/j.cmet.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoefer IE, van Royen N, Rectenwald JE, Deindl E, Hua J, Jost M, Grundmann S, Voskuil M, Ozaki CK, Piek JJ, et al. Arteriogenesis proceeds via ICAM-1/Mac- 1- mediated mechanisms. Circ Res. 2004;94:1179–1185. doi: 10.1161/01.RES.0000126922.18222.F0. [DOI] [PubMed] [Google Scholar]
- Katsouda A, Bibli SI, Pyriochou A, Szabo C, Papapetropoulos A. Regulation and role of endogenously produced hydrogen sulfide in angiogenesis. Pharmacol Res. 2016;113:175–185. doi: 10.1016/j.phrs.2016.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilberg MS, Pan YX, Chen H, Leung-Pineda V. Nutritional control of gene expression: how mammalian cells respond to amino acid limitation. Annu Rev Nutr. 2005;25:59–85. doi: 10.1146/annurev.nutr.24.012003.132145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolluru GK, Bir SC, Yuan S, Shen X, Pardue S, Wang R, Kevil CG. Cystathionine γ-lyase regulates arteriogenesis through NO dependent monocyte recruitment. Cardiovasc Res. 2015 doi: 10.1093/cvr/cvv198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo M, Shibata R, Miura R, Shimano M, Kondo K, Li P, Ohashi T, Kihara S, Maeda N, Walsh K, et al. Caloric restriction stimulates revascularization in response to ischemia via adiponectin-mediated activation of endothelial nitric-oxide synthase. J Biol Chem. 2009;284:1718–1724. doi: 10.1074/jbc.M805301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Mariappan MM, Feliers D, Cavaglieri RC, Sataranatarajan K, Abboud HE, Choudhury GG, Kasinath BS. Hydrogen sulfide inhibits high glucoseinduced matrix protein synthesis by activating AMP-activated protein kinase in renal epithelial cells. J Biol Chem. 2012;287:4451–4461. doi: 10.1074/jbc.M111.278325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JI, Dominy JE, Jr, Sikalidis AK, Hirschberger LL, Wang W, Stipanuk MH. HepG2/C3A cells respond to cysteine deprivation by induction of the amino acid deprivation/integrated stress response pathway. Physiol Genomics. 2008;33:218–229. doi: 10.1152/physiolgenomics.00263.2007. [DOI] [PubMed] [Google Scholar]
- Lehman SL, Ryeom S, Koumenis C. Signaling through alternative Integrated Stress Response pathways compensates for GCN2 loss in a mouse model of soft tissue sarcoma. Sci Rep. 2015;5:11781. doi: 10.1038/srep11781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leschelle X, Goubern M, Andriamihaja M, Blottiere HM, Couplan E, Gonzalez-Barroso MD, Petit C, Pagniez A, Chaumontet C, Mignotte B, et al. Adaptative metabolic response of human colonic epithelial cells to the adverse effects of the luminal compound sulfide. Biochim Biophys Acta. 2005;1725:201–212. doi: 10.1016/j.bbagen.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Li L, Chen Y, Gibson SB. Starvation-induced autophagy is regulated by mitochondrial reactive oxygen species leading to AMPK activation. Cell Signal. 2013;25:50–65. doi: 10.1016/j.cellsig.2012.09.020. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Longchamp A, Alonso F, Dubuis C, Allagnat F, Berard X, Meda P, Saucy F, Corpataux JM, Déglise S, Haefliger JA. The use of external mesh reinforcement to reduce intimal hyperplasia and preserve the structure of human saphenous veins. Biomaterials. 2014;35:2588–2599. doi: 10.1016/j.biomaterials.2013.12.041. [DOI] [PubMed] [Google Scholar]
- Ma J, Waxman DJ. Modulation of the antitumor activity of metronomic cyclophosphamide by the angiogenesis inhibitor axitinib. Mol Cancer Ther. 2008;7:79–89. doi: 10.1158/1535-7163.MCT-07-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RA, Buehner G, Chang Y, Harper JM, Sigler R, Smith-Wheelock M. Methionine-deficient diet extends mouse lifespan, slows immune and lens aging, alters glucose, T4, IGF-I and insulin levels, and increases hepatocyte MIF levels and stress resistance. Aging Cell. 2005;4:119–125. doi: 10.1111/j.1474-9726.2005.00152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirabella T, Teodelinda M, Cilli M, Michele C, Carlone S, Sebastiano C, Cancedda R, Ranieri C, Gentili C, Chiara G. Amniotic liquid derived stem cells as reservoir of secreted angiogenic factors capable of stimulating neo-arteriogenesis in an ischemic model. Biomaterials. 2011;32:3689–3699. doi: 10.1016/j.biomaterials.2011.01.071. [DOI] [PubMed] [Google Scholar]
- Mishanina TV, Libiad M, Banerjee R. Biogenesis of reactive sulfur species for signaling by hydrogen sulfide oxidation pathways. Nat Chem Biol. 2015;11:457–464. doi: 10.1038/nchembio.1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa AK, Gadalla MM, Sen N, Kim S, Mu W, Gazi SK, Barrow RK, Yang G, Wang R, Snyder SH. H2S signals through protein S-sulfhydration. Sci Signal. 2009;2:ra72. doi: 10.1126/scisignal.2000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narkar VA, Downes M, Yu RT, Embler E, Wang YX, Banayo E, Mihaylova MM, Nelson MC, Zou Y, Juguilon H, et al. AMPK and PPARdelta agonists are exercise mimetics. Cell. 2008;134:405–415. doi: 10.1016/j.cell.2008.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls P, Kim JK. Sulphide as an inhibitor and electron donor for the cytochrome c oxidase system. Can J Biochem. 1982;60:613–623. doi: 10.1139/o82-076. [DOI] [PubMed] [Google Scholar]
- Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling - in control of vascular function. Nat Rev Mol Cell Biol. 2006;7:359–371. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- Omodei D, Fontana L. Calorie restriction and prevention of age-associated chronic disease. FEBS Lett. 2011;585:1537–1542. doi: 10.1016/j.febslet.2011.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orentreich N, Matias JR, DeFelice A, Zimmerman JA. Low methionine ingestion by rats extends life span. J Nutr. 1993;123:269–274. doi: 10.1093/jn/123.2.269. [DOI] [PubMed] [Google Scholar]
- Papapetropoulos A, Pyriochou A, Altaany Z, Yang G, Marazioti A, Zhou Z, Jeschke MG, Branski LK, Herndon DN, Wang R, et al. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc Natl Acad Sci U S A. 2009;106:21972–21977. doi: 10.1073/pnas.0908047106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng W, Robertson L, Gallinetti J, Mejia P, Vose S, Charlip A, Chu T, Mitchell JR. Surgical stress resistance induced by single amino acid deprivation requires Gcn2 in mice. Sci Transl Med. 2012;4:118ra111. doi: 10.1126/scitranslmed.3002629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potente M, Ghaeni L, Baldessari D, Mostoslavsky R, Rossig L, Dequiedt F, Haendeler J, Mione M, Dejana E, Alt FW, et al. SIRT1 controls endothelial angiogenic functions during vascular growth. Genes Dev. 2007;21:2644–2658. doi: 10.1101/gad.435107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potente M, Urbich C, Sasaki K, Hofmann WK, Heeschen C, Aicher A, Kollipara R, DePinho RA, Zeiher AM, Dimmeler S. Involvement of Foxo transcription factors in angiogenesis and postnatal neovascularization. J Clin Invest. 2005;115:2382–2392. doi: 10.1172/JCI23126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruckenstuhl C, Netzberger C, Entfellner I, Carmona-Gutierrez D, Kickenweiz T, Stekovic S, Gleixner C, Schmid C, Klug L, Sorgo AG, et al. Lifespan extension by methionine restriction requires autophagy-dependent vacuolar acidification. PLoS Genet. 2014;10:e1004347. doi: 10.1371/journal.pgen.1004347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salabei JK, Gibb AA, Hill BG. Comprehensive measurement of respiratory activity in permeabilized cells using extracellular flux analysis. Nat Protoc. 2014;9:421–438. doi: 10.1038/nprot.2014.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoors S, De Bock K, Cantelmo AR, Georgiadou M, Ghesquière B, Cauwenberghs S, Kuchnio A, Wong BW, Quaegebeur A, Goveia J, et al. Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab. 2014;19:37–48. doi: 10.1016/j.cmet.2013.11.008. [DOI] [PubMed] [Google Scholar]
- Sen S, Kawahara B, Gupta D, Tsai R, Khachatryan M, Roy-Chowdhuri S, Bose S, Yoon A, Faull K, Farias-Eisner R, et al. Role of cystathionine β-synthase in human breast Cancer. Free Radic Biol Med. 2015;86:228–238. doi: 10.1016/j.freeradbiomed.2015.05.024. [DOI] [PubMed] [Google Scholar]
- Singha S, Kim D, Moon H, Wang T, Kim KH, Shin YH, Jung J, Seo E, Lee SJ, Ahn KH. Toward a selective, sensitive, fast-responsive, and biocompatible two-photon probe for hydrogen sulfide in live cells. Anal Chem. 2015;87:1188–1195. doi: 10.1021/ac503806w. [DOI] [PubMed] [Google Scholar]
- Smith L, Kruszyna H, Smith RP. The effect of methemoglobin on the inhibition of cytochrome c oxidase by cyanide, sulfide or azide. Biochem Pharmacol. 1977;26:2247–2250. doi: 10.1016/0006-2952(77)90287-8. [DOI] [PubMed] [Google Scholar]
- Sonke E, Verrydt M, Postenka CO, Pardhan S, Willie CJ, Mazzola CR, Hammers MD, Pluth MD, Lobb I, Power NE, et al. Inhibition of endogenous hydrogen sulfide production in clear-cell renal cell carcinoma cell lines and xenografts restricts their growth, survival and angiogenic potential. Nitric Oxide. 2015;49:26–39. doi: 10.1016/j.niox.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C. Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov. 2007;6:917–935. doi: 10.1038/nrd2425. [DOI] [PubMed] [Google Scholar]
- Szabo C, Coletta C, Chao C, Módis K, Szczesny B, Papapetropoulos A, Hellmich MR. Tumor-derived hydrogen sulfide, produced by cystathionine-β-synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer. Proc Natl Acad Sci U S A. 2013;110:12474–12479. doi: 10.1073/pnas.1306241110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao BB, Liu SY, Zhang CC, Fu W, Cai WJ, Wang Y, Shen Q, Wang MJ, Chen Y, Zhang LJ, et al. VEGFR2 functions as an H2S-targeting receptor protein kinase with its novel Cys1045-Cys1024 disulfide bond serving as a specific molecular switch for hydrogen sulfide actions in vascular endothelial cells. Antioxid Redox Signal. 2013;19:448– 464. doi: 10.1089/ars.2012.4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizan P, Sanchez-Tena S, Alcarraz-Vizan G, Soler M, Messeguer R, Pujol MD, Lee WN, Cascante M. Characterization of the metabolic changes underlying growth factor angiogenic activation: identification of new potential therapeutic targets. Carcinogenesis. 2009;30:946–952. doi: 10.1093/carcin/bgp083. [DOI] [PubMed] [Google Scholar]
- Wang R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev. 2012;92:791–896. doi: 10.1152/physrev.00017.2011. [DOI] [PubMed] [Google Scholar]
- Wang Y. Molecular Links between Caloric Restriction and Sir2/SIRT1 Activation. Diabetes Metab J. 2014;38:321–329. doi: 10.4093/dmj.2014.38.5.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ning Y, Alam GN, Jankowski BM, Dong Z, Nör JE, Polverini PJ. Amino acid deprivation promotes tumor angiogenesis through the GCN2/ATF4 pathway. Neoplasia. 2013;15:989–997. doi: 10.1593/neo.13262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wek SA, Zhu S, Wek RC. The histidyl-tRNA synthetase-related sequence in the eIF-2 alpha protein kinase GCN2 interacts with tRNA and is required for activation in response to starvation for different amino acids. Mol Cell Biol. 1995;15:4497– 4506. doi: 10.1128/mcb.15.8.4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteman M, Armstrong JS, Chu SH, Jia-Ling S, Wong BS, Cheung NS, Halliwell B, Moore PK. The novel neuromodulator hydrogen sulfide: an endogenous peroxynitrite ‘scavenger’? J Neurochem. 2004;90:765–768. doi: 10.1111/j.1471-4159.2004.02617.x. [DOI] [PubMed] [Google Scholar]
- Whiteman M, Cheung NS, Zhu YZ, Chu SH, Siau JL, Wong BS, Armstrong JS, Moore PK. Hydrogen sulphide: a novel inhibitor of hypochlorous acid-mediated oxidative damage in the brain? Biochem Biophys Res Commun. 2005;326:794–798. doi: 10.1016/j.bbrc.2004.11.110. [DOI] [PubMed] [Google Scholar]
- Yadav V, Gao XH, Willard B, Hatzoglou M, Banerjee R, Kabil O. Hydrogen sulfide modulates eukaryotic translation initiation factor 2alpha (eIF2alpha) phosphorylation status in the integrated stress-response pathway. J Biol Chem. 2017;292:13143–13153. doi: 10.1074/jbc.M117.778654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S, et al. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322:587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong R, Searcy DG. Sulfide oxidation coupled to ATP synthesis in chicken liver mitochondria. Comp Biochem Physiol B Biochem Mol Biol. 2001;129:129–137. doi: 10.1016/s1096-4959(01)00309-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Tube formation assay: Quantification of tube branch points per field of view in HUVECs seeded onto growth factor-replete Matrigel and incubated in the indicated media +/−SIRT1 inhibitor Ex-527 for 18hr; n=8–10 data points/condition. (B) Daily body weight (left, n=5 mice/group) and food intake expressed as grams of food eaten per gram of mouse body weight (right, n=2 cages/group) of mice given ad libitum access to methionine restricted (MR) vs. control (Ctrl) diet for 2mo in a representative experiment. (C) Representative flow cytometric analysis of CD31 positive cells from gastrocnemius muscle of mice fed for 2wk on Ctrl or MR diets with or without VEGFR2 inhibition (axitinib, +A) expressed as a percentage of total cells recovered from the enzymatically digested muscle tissue; quantification at right (n=3–6 mice/group). (D) Expression of VEGF mRNA in gastrocnemius muscle of mice subject to MR for 2–8wk expressed relative to Ctrl diet group; n=22–23/mice group. (E) VEGF protein levels in extracts of muscle from mice on the indicated diet for at least 2wk as determined by ELISA; n=11–13/group. (F) Representative transverse sections of gastrocnemius muscle stained for VEGF (left, 40X mag), IsolectinB4 (IB4, middle, 40X mag) and merged (VEGF red, IB4 green, DAPI blue; right, insert 160X mag) in mice preconditioned for 2mo on control (Ctrl) or methionine restricted (MR) diets. (G) Schematic representation of the hindlimb ischemia procedure. (H, I) Longitudinal Doppler imaging of blood flow (H) and distance run on treadmill exercise test (I) following ischemia in WT mice preconditioned on a complete diet fed ad libitum (AL) or calorie restricted daily by 40% of the AL amount (CR) for 1mo prior to femoral artery ligation, +/− propargylglycine (PAG) daily for 1wk prior to ligation and 2wk after as indicated. (H) Left: representative infrared images on the indicated after ligation (I, ischemic; NI, non-ischemic). Right: quantification of blood flow recovery with individual animal AUCs used for statistical comparisons; n=6 mice/group. (I) Distance run on treadmill exercise test on d4 after ligation of the indicated groups from (H). Error bars indicate SD; asterisks indicate the significance of the difference by Student’s T test or 1-way ANOVA with Sidak’s MCT between diets in vivo or sulfur amino acid levels in vitro; *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
(A) Immunoblot of HIF1α protein in HUVEC 2 d after transfection with HIF1α siRNA or control scrambled (Sble) siRNA. (B) Quantification of immunoblots from Fig. 2B of the indicated protein from HUVEC extracts after culturing in control (Ctrl) or M&C deficient media. (C) PGC1α mRNA levels of HUVEC cultured in the indicated media for 16hr; n=4 experiments/group, error bars indicate SEM. (D, E) PGC1α (D) and VEGF (E) mRNA levels in HUVEC, 2 d after transfection with a PGC1α overexpressing construct, and expressed relative to cells transfected with a control AdGFP vector; n=3 experiments/group, SEM. (F) Asns (left) and ATF4 (right) mRNA expression in HUVEC 2 d after transfection with ATF4 siRNA or control scrambled (Sble) siRNA and cultured in the indicated media for 16hr; n=4 experiments/group, SEM. (G–I) Immunoblot of ATF4 protein (G), mRNA expression of ATF4 (H) and Asns (I) in HUVEC 2d after transfection with a CMV-driven ATF4 over-expressing construct (ATF4OE) or empty vector (Empty). (J) Representative images of primary WT mouse ECs stained for CD31 (red), Actin (green) and DAPI (blue) at 40X mag; n=3 experiments/group, SEM. (K) Asns mRNA in WT and GCN2KO primary mouse EC from n=5–6 mice per genotype cultured as indicated for 16hs. (L) VEGF mRNA in MDF 2d after infection with shATF4 or control scrambled adenovirus and cultured for the final 16hr as indicated; n=5–8 experiments/group, SEM. (M) HIF1α mRNA expression in C2C12 cells 2d after transfection with HIF1α or control scrambled (Sble) siRNA and then cultured for an additional 16hr in control media; n=11–12 experiments/group, SEM. (N, O) C2C12 HIF1α protein level (N) and VEGF mRNA expression (O) 2d after transfection with HIF1α siRNA or control scrambled (Sble) siRNA, and then cultured for an additional 24hr under 20% or <1% oxygen tension in presence or absence of M&C; n=3–6 experiments/group, SEM. (P) Relative PGC1α (left) and VEGF (right) mRNA expression in HUVEC 2d after transfection with PGC1α siRNA or control scrambled siRNA and cultured in the indicated media for 16hr; n=3 experiments/group, SEM. Unless otherwise indicated, error bars indicate SD; asterisks indicate the significance of the difference between sulfur amino acid levels or genetic interventions by Student’s T test; or between sulfur amino acid levels within genotype/intervention/condition by 1-way ANOVA with Sidak’s MCT; *P<0.05, **P<0.01, ***P<0.001.
(A) Representative images (10X mag) of H2S-specific probe (P3) fluorescence in primary mouse hepatocytes treated with VEGF (50ng/ml) or deprived of all amino acids as well as serum (-Nutrients) for 16hr. (B) VEGF mRNA in WT and CGLKO primary mouse ECs (n=3 mice/group) cultured in M&C deficient media relative to control (Ctrl) media for 16hr; error bars indicate SD; 1-way ANOVA with Sidak’s MCT between SAA levels. (C, D) CBS, CGL and 3-MST mRNA expression (C) and protein level (D) in HUVEC 16hr after treatment with VEGF; n=3 experiments/group, error bars indicate SEM; Student’s T test. **P<0.01, ***P<0.001.
(A) H2S production capacity of gastrocnemius muscle extracts of Ad-Null or Ad-CGL infected mice 1wk after the final intramuscular injection. (B) Expression of VEGF mRNA in the gastrocnemius muscle of mice subjected to low intensity running (exercised) vs. control (sedentary) for 1mo; n=4 mice/group. Error bars indicate SD; asterisks indicate the significance of the difference between exercise groups by Student’s T test; **P<0.01.
(A) HUVEC proliferation over a 24hr period +/−NaHS (100uM) as assessed by radiolabelled thymidine incorporation; n=5–9 experiments/group, error bars indicate SEM. (B). Glucose uptake in SIRT1KO primary mouse ECs treated in triplicate as indicated. (C) Extracellular acidification rate (ECAR) in HUVEC pretreated for 2hr with NaHS (100μM); n=8 technical replicates from each of 2 experiments. (D) Extracellular acidification rate (ECAR) over time in mouse ECs pretreated for 2hr with VEGF (50ng/ml) or NaHS (100μM) and then treated with 10mM Glucose, 2.5μM Oligomycin and 50mM 2DG injection as indicated by the arrows; representative experiment shown. (E) Fold change of lactate concentration in the media of HUVECs treated with NaHS (100μM) for 1.5–6 hr vs. control; n=3 experiments per group; SEM. (F) Glycolytic flux in HUVEC at the indicated time after NaHS addition; n=3 technical replicates/time point; 1-way ANOVA with Dunnett’s MCT vs. t=0. (G). Log2 fold change of unlabeled metabolites in HUVECs treated for 1hr with NaHS (100μM) vs. control. (H) ECAR over time in HUVEC pre-treated for 2hr with axitinib (10μM) or L-NAME (100μM) and then treated with NaHS or 2DG as indicated by the arrows; n=15 technical replicates/time point (I) Quantification of HUVEC migration across scratch +/−NaHS (100μM) or eNOS inhibitor L-NAME (100μM); n=5–6 AUC values/group each from cells at different passage number. (J) Immunoblot of eNOS in HUVEC 2d after transfection with eNOS or control scramble (Sble) siRNA. (K) Quantification of HUVEC migration across scratch 2d after transfection with eNOS siRNA or control scramble (Sble) siRNA +/−NaHS (100μM) as indicated; n=6 AUC values/group each from cells at different passage number. (L) Gating percentages based on annexin V and propidium iodide stains in HUVECs treated with NaHS (100μM) or –M&C for 12hr. Unless otherwise indicated, error bars indicate SD and asterisks indicate the significance of the difference between SAA levels by Student’s T test or between SAA levels within genotype/condition by 1-way ANOVA with Sidak’s MCT; *P<0.05, **P<0.01, ***P<0.001.
(A) Glucose uptake in HUVEC treated with NaHS for 1hr +/−10μM Compound C (Comp C); n=6 experiments/group, error bars indicate SEM; 1-way ANOVA with Sidak’s MCT between +/−NaHS within Comp C treatment group. (B) Migration across scratch in HUVEC treated with NaHS +/− Comp C; n=6 AUC values/group each from cells at different passage number; 1-way ANOVA with Sidak’s MCT between +/−NaHS within Comp C treatment group. (C, D) Schematic of the experiment (C) and quantification of migration across scratch (D) in HUVEC treated with NaHS for 10min in the presence of vehicle or Comp C; n=5–6 AUC values/group each from cells at different passage number; 1-way ANOVA with Sidak’s MCT between +/−NaHS within Comp C treatment group. (E) Glucose uptake in HUVEC treated with NaHS or the CamKKβ inhibitor STO-609; n=3–9 values pooled from 2 independent experiments; 1-way ANOVA with Sidak’s MCT between. (F) ADP/ATP ratio over time in HUVEC treated with the indicated compounds; representative of 2 experiments; n=4–12 values/time point. (G) Energy charge calculated from ATP, ADP and AMP measurements by mass spec in HUVEC treated for 15min with NaHS; n=6 values pooled from 2 independent experiments; Student’s T test. (H) ROS levels expressed as mean fluorescent intensity (MFI) in HUVEC treated over time with NaHS (100μM), KCN (10μM), FCCP (2μM) or menadione (MD, 10μM) as indicated; n=2 values/time point. (I) Time dependent glucose uptake in HUVEC treated with NaHS, KCN or antimycin as indicated; each time point is in triplicate. (J) Extracellular acidification rate (ECAR) over time in HUVEC pretreated for 2hr with 10μM cyanide, 2μM oligomycin (Oligo) or 500μM phenformin (Phen) and then treated with 10mM glucose, 2.5μM oligomycin and 50mM 2DG as indicated by the arrows; n=20 technical replicates pooled from 2 independent experiments; (K) Glycolytic flux in HUVEC pretreated for 1–3hr with oligomycin, antimycin A or 2DG as indicated; n=2–6 experiments/group; 1-way ANOVA with Sidak’s MCT relative to control. (L) Representative images of migration across a scratch (10X mag at t=20hr; dotted lines indicate boundary of the scratch at t=0hr) from HUVEC cultured in the presence of 10μM KCN (left), 500μM phenformin (Phen, middle) or 2μM oligomycin (Oligo, right) +/−1mM 2DG as indicated. Unless otherwise noted, error bars indicate SD; **P<0.01, ***P<0.001.