

Abstract
Zidovudine (3′-azido-3′-deoxythymidine; AZT) is the most widely used nucleoside reverse transcriptase inhibitor for the treatment of AIDS patients and prevention of mother-to-child transmission of HIV-1. Previously, we demonstrated that AZT had significantly greater growth inhibitory effects upon the human liver carcinoma cell line HepG2 as compared to the immortalized human liver cell line THLE2. We have now used gene expression profiling to determine the molecular pathways associated with toxicity in both cell lines. HepG2 cells were incubated with 0, 2, 20, or 100 μM AZT for 2 weeks; THLE2 cells were treated with 0, 50, 500, or 2,500 μM AZT, concentrations that 2013 were equi-toxic to those used in the HepG2 cells. After the treatment, total RNA was isolated and subjected to microarray analysis. Global analysis of gene expression, with a false discovery rate ≤0.01 and a fold change ≥1.5, indicated that 6- to 70-fold more genes were differentially expressed in a significant concentration-dependent manner in HepG2 cells when compared to THLE2 cells. Comparative analysis indicated that 7 % of these genes were common to both cell lines. Among the common differentially expressed genes, 70 % changed in the same direction, most of which were associated with cell death and survival, cell cycle, cell growth and proliferation, and DNA replication, recombination, and repair. As determined by the uptake of [methyl-3H]AZT, the intracellular levels of total AZT were approximately twofold higher in THLE2 cells than in HepG2 cells. The expression of thymidine kinase 1 (TK1) and UDP-glucuronosyltransferase 2B7 (UGT2B7) genes that regulate the metabolic activation and deactivation of AZT, respectively, was increased in HepG2 cells but decreased in THLE2 cells after treatment with AZT. This differential response in AZT metabolism was confirmed by real-time PCR, western blotting, and/or enzymatic assays. These data indicate that molecular pathways involved with cell death and survival, cell cycle, cell growth and proliferation, and DNA replication, recombination, and repair are involved in the toxicities associated with AZT in both human cell lines, and that the difference in expression of TK1 and UGT2B7 in response to AZT treatment in HepG2 cells and THLE2 cells might explain why HepG2 cells are more sensitive than THLE2 cells to the toxicity of AZT.
Keywords: AZT, Gene expression, Microarray, Thymidine kinase 1, UDP-glucuronosyltransferase 2B7
Introduction
The nucleoside reverse transcriptase inhibitor zidovudine (3′-azido-3′-deoxythymidine; AZT) was approved as the first anti-human immunodeficiency virus-1 (HIV-1) agent in 1987 by the US Food and Drug Administration and is still used extensively for the treatment of patients with acquired immunodeficiency syndrome (AIDS) and for the prevention of the mother-to-child transmission of HIV-1 during pregnancy, labor, and delivery (Fischl et al. 1990; Ingrand et al. 1995; McGowan and Shah 2000; AHFS 2007; PDR 2009). The antiretroviral activity of AZT is dependent upon the formation of AZT 5′-triphosphate, which efficiently inhibits HIV-1 reverse transcriptase by acting as a competitive inhibitor of normal nucleotides and causing proviral DNA termination (Furman et al. 1986; Jones and Bischofberger 1995; Brinkman and Kakuda 2000). In addition to inhibiting viral reverse transcriptase, AZT 5′-triphosphate also is a weak inhibitor of mammalian DNA polymerases α, β, and γ, which catalyze DNA replication in vivo (Huang et al. 1990; Nickel et al. 1992; Brinkman and Kakuda 2000).
AZT is a derivative of thymidine in which the 3′-hydroxy group has been replaced by an azido group. When AZT reaches a cell, it is subjected to successive cellular anabolic phosphorylation that finally converts AZT to the effective 5′-triphosphate form (Lavie et al. 1997). The first phosphorylation step is catalyzed by thymidine kinases (TK) to form AZT 5′-monophosphate. AZT 5′-monophosphate is a very poor substrate for the next kinase, thymidylate kinase (Lavie et al. 1997, 1998). As a result, very high concentrations of AZT 5′-monophosphate can be reached in the cell, whereas concentrations of the effective AZT 5′-triphosphate only account for 2–10 % of total phosphate metabolites of AZT (Barry et al. 1996). Thus, TK is considered to be the key regulatory step in AZT metabolism (Arner and Eriksson 1995). There are two isoforms of TK: cytosolic thymidine kinase 1 (TK1) and mitochondrial thymidine kinase 2 (TK2). TK1 phosphorylates AZT more efficiently than does TK2 (Arner and Eriksson 1995). In addition to its metabolic activation, AZT is eliminated by glucuronidation by UDP-glucuronosyltransferase 2B7 (UGT2B7) (Good et al. 1990; Moore et al. 1995; Barbier et al. 2000; Belanger et al. 2009).
AZT can be incorporated into cellular DNA of mammalian cells (Vazquez-Padua et al. 1990; Brunetti et al. 1990; Tosi et al. 1992; Escobar et al. 2007; Fang et al. 2009b; Fang and Beland 2009; Wu et al. 2011), which may result in the occurrence of irreversible DNA damage. This can subsequently lead to impaired cellular function and contribute to the toxicities of AZT. The toxic effects of AZT are well documented and include the induction of micronuclei, sister chromatid exchange, chromosomal aberrations, mutations, centrosomal abnormalities, and nuclear bud formation (Grdina et al. 1992; Gonzalez Cid and Larripa 1994; Dertinger et al. 1996; Ayers et al. 1996; Borojerdi et al. 2009; Dutra et al. 2010). AZT has been reported to be a transplacental carcinogen in mice and rats (Ayers et al. 1996; Olivero et al. 1997; Zhang et al. 1998; NTP 1999; Torres et al. 2007), and the International Agency for Research on Cancer has classified AZT as a group 2B carcinogen, designated as possibly carcinogenic to humans (IARC 2000). Furthermore, epidemiological studies on the incidence of non-AIDs defining cancers in individuals undergoing combined antiretroviral therapy, of which AZT is a component, have suggested an increased risk for liver cancer (Powles et al. 2009).
AZT has been shown to inhibit growth of a variety of human cancer cells to a much greater extent when compared to non-malignant cells. The differential sensitivity of cancer cells to AZT has been related to a combination of effects, including a delay of cell cycle progression, the induction of apoptosis, and a decrease in telomerase activity (Melana et al. 1998; Collier et al. 2003; Wu et al. 2004; Falchetti et al. 2005; Humer et al. 2008; Fang and Beland 2009). Cancer cells also have higher growth rates than normal cells, with a concomitant higher rate of thymidine turnover, which could contribute to their increased sensitivity to AZT (Melana et al. 1998; Humer et al. 2008). Despite these explanations, the mechanisms underlying the differential responses of normal and cancer cells to AZT are not well understood.
We have demonstrated previously that human liver carcinoma HepG2 cells are more sensitive than the immortalized human liver THLE2 cells to AZT treatment as indicated by much higher concentrations being required to inhibit cell growth in the THLE2 cells (Fang and Beland 2009). As a continuation of our studies to elucidate the mechanisms of toxicity associated with AZT, we have now investigated the transcriptional responsiveness of HepG2 cells and THLE2 cells to AZT using an Agilent whole genome human microarray-based approach. The bioavailability of AZT in these two cell lines and the genes related to AZT metabolism, i.e., TK and UGT2B7, were also investigated.
Materials and methods
Chemicals
AZT was obtained from Cipla Ltd. (Mumbai, India). The purity, as assessed by HPLC, was 99.9 %. [Methyl-3H]AZT (12.7 Ci/mmol, radiochemical purity >99.8 % by HPLC) was obtained from Moravek Biochemicals Inc. (Brea, CA). AZT 5′-monophosphate was purchased from Toronto Research Chemicals Inc. (North York, Ontario, Canada). Williams’ Medium E, phosphoethanolamine, bovine serum albumin, adenosine 5′-triphosphate disodium salt (ATP), and RNase A were acquired from Sigma (St. Louis, MO). Penicillin–streptomycin and 2.5 % trypsin were purchased from Fisher Scientific (Pittsburgh, PA). Dulbecco’s phosphate-buffered saline (minus calcium and magnesium) (PBS), epidermal growth factor (culture grade), and LHC-8 medium were obtained from Invitrogen (Carlsbad, CA). Fetal bovine serum was purchased from Atlanta Biologicals (Lawrenceville, GA). The BCA Protein Assay kit was purchased from Pierce Chemical Co. (Rockford, IL). Complete protease inhibitor cocktail was purchased from Roche Applied Science (Mannheim, Germany). All other chemicals and biochemicals were of analytical grade and used without further purification.
Antibodies
Human UGT2B7 western blotting kits were purchased from BD Biosciences (Bedford, MA). Goat polyclonal antibody to calnexin and monoclonal antibodies to TK1 and β-actin were obtained from Santa Cruz Biotechnology Inc. (Santa Cruz, CA).
Cell culture and treatments
HepG2 cells, a cell line derived from a human hepatocellular carcinoma, and THLE-2 cells, an immortalized normal human liver cell line, were purchased from American Type Culture Collection (Manassas, VA). THLE2 cells express phenotypic characteristics of normal adult liver epithelial cells and retain phase I and II enzyme activities, including the ability to metabolize carcinogens to their ultimate carcinogenic metabolites capable of binding DNA (Pfeifer et al. 1993). HepG2 cells and THLE2 cells were cultured as described previously (Fang and Beland 2009). AZT was dissolved in culture media and stored at 4 °C. Cells in an exponential phase of growth were plated at a density of 5 × 103 cells/cm2 culture surface and exposed to various concentrations of AZT for 2 weeks, the time that gave the maximum responses in HepG2 cells treated with AZT in our previous study (Fang and Beland 2009). When appropriate, fresh medium and AZT were added every other day, with the cells being subcultured weekly. Control cells were fed complete culture medium free of AZT. Each of the incubations was performed three separate times, and all the measurements described below were conducted independently for each of the experiments.
The half inhibitory concentration (IC50) of AZT
The total number of viable cells was determined by an MTT assay, as previously described (Fang and Beland 2009). HepG2 cells were exposed to 0, 7.8, 15.6, 31.2, 62.5, 125, 250, 500, and 1,000 μM AZT for 1 week. THLE2 cells were exposed to 0, 0.094, 0.19, 0.38, 0.75, 1.5, 3.0, and 6.0 mM AZT for 1 week. After the treatment, the absorbance of formazan was determined with a BioTek Synergy 2 reader (BioTek Instruments, Inc., Winooski, VT) at 570 nm, with 620 nm as the reference. The IC50 values were obtained from the cell growth curves using GraphPad Prism 5.0 (GraphPad Software, La Jolla, CA).
RNA isolation and cDNA labeling
After a 2-week AZT treatment, total RNA was isolated from cells using an RNeasy Mini kit (Qiagen, German-town, MD). The yield of the extracted RNA was determined spectrophotometrically by measuring the optical density at 260 nm. The purity and quality of extracted RNA were evaluated using the RNA 6000 LabChip and Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA). RNAs, with RNA integrity numbers greater than 9.0, were used for the microarray analysis. Total RNAs were labeled with cyanine dye Cy3-CTP using a Low RNA Input Linear Amplification Plus kit (Agilent Technologies, Santa Clara, CA).
Hybridization, imaging, and data processing
Cy3-labeled cRNAs were hybridized with Agilent Whole Genome Human Oligo Microarrays containing 44,084 probes following the Agilent One-Color Microarray-Based Gene Expression Analysis Protocol. The processed slides were scanned with a GenePix 4000B scanner. The resulting images were analyzed using Agilent Feature Extraction software (V9.5). The raw intensity values were uploaded into ArrayTrack, a FDA in-house database (Fang et al. 2009a) and then normalized using 75 percentile channel scaling recommended by Agilent before statistical analysis.
Intracellular levels of total AZT
Cells were incubated in complete culture medium, as described above, supplemented with 0, 2, 20, or 100 μM [methyl-3H]AZT (diluted to a specific activity of 10 mCi/mmol with unlabelled AZT) for HepG2 cells or 0, 50, 500, or 2,500 μM [methyl-3H]AZT for THLE2 cells. The incubation times were 10, 30, 60, 120, and 240 min. At the end of incubation, the media was removed and the cells were washed six times with PBS and trypsinized with a 0.125 % trypsin/2 mM EDTA solution. The cells were counted and the cell suspension was centrifuged at 300g for 10 min at 4 °C. The supernatants were discarded, and the cell pellets were re-suspended in 1 ml Tris-buffered saline (25 mM Tris base, 138 mM NaCl, 2.7 mM KCl, pH 7.4). The entire cell suspension was directly measured for radioactivity using a Beckman LS-6500 liquid scintillation counter (Beckman Coulter Inc., Fullerton, CA), with ScintiSafe™ 30 % (Fisher Scientific Inc., Pittsburgh, PA) as the scintillation fluid. The intracellular levels of total [methyl-3H]AZT were expressed as pmol AZT equivalents per 106 cells.
Real-time PCR analysis of thymidine kinase (TK) and UDP-glucuronosyltransferase 2B7 (UGT2B7)
The expression of TK (TK1 and TK2) and UGT2B7 was confirmed using SYBR Green-based quantitative real-time primer assays (Qiagen, Germantown, MD). Briefly, total RNA (1 μg) was reverse transcribed using an Invitrogen SuperScript III First-Strand Synthesis System for RT-PCR, and the cDNAs were amplified in a BioRad iCycler iQ Detection System (Bio-Rad laboratories, Hercules, CA). All samples were run in triplicate. Data were normalized for the endogenous control, 18sRNA, and analyzed using the ΔΔCt method.
Western blot analysis of TK1 and UGT2B7
After a 2-week AZT treatment, cells were trypsinized and washed three times in PBS. Cell lysates were prepared as described previously (Fang et al. 2009b; Fang and Beland 2009). Microsomes were isolated by centrifugation of the whole cell homogenate at 10,500 rpm (9,000g) for 30 min at 4 °C; the supernatant was collected and subsequently centrifuged at 33,500 rpm (105,000g) for 60 min at 4 °C in a SW-55 Ti rotor (Beckman, Palo Alto, CA). The pellets were suspended in Tris-buffered saline and stored at −65 °C in 100 μl aliquots, with total protein concentrations measured using Pierce BCA protein assay kits.
The protein levels of TK1 in cell lysates and UGT2B7 in microsomes were determined using western blot analysis as described previously (Fang et al. 2009b), with minor modifications. Briefly, cell lysates (50 μg) or microsomal proteins (40 μg) were subjected to electrophoresis in a 12 % sodium dodecyl sulfate–polyacrylamide gel. The resolved proteins were electrophoretically transferred onto a polyvinylidine difluoride membrane. Both electrophoresis and blotting were performed with a Mini-PROTEAN® 3 electrophoresis system (Bio-Rad). Blots were blocked with 5 % milk in PBS/Tween-20 and probed with anti-TK1 (1:1,000 dilution), anti-UGT2B7 (1:2,000 dilution), anti-β-actin (1:2,500 dilution), or anti-calnexin (1:500 dilution), followed by a secondary antibody to IgG conjugated with horseradish peroxidise. The blots were then detected by chemiluminescence using Immobilon Western Horseradish Peroxidise Substrate (Millipore Corporation, Billerica, MA) and a UVP BioSpectrum AC Imaging System (UVP LLC, Upland, CA). The intensity of each band was quantified with ImageJ (National Institute of Health, Bethesda, MD). The relative protein levels were calculated using β-actin and calnexin as a loading control for cell lysates and microsomes, respectively.
Analysis of TK activity
The activity of TK in whole cell lysates was determined as described by Jacobsson et al. (Jacobsson et al. 1995). After the treatment, the cells were trypsinized, washed three times in PBS, suspended in a lysis buffer consisting of 10 mM Tris–HCl (pH 7.5), 10 mM KCl, 1 mM MgCl2, and 1 mM dithiothreitol, and subjected to three rounds of freeze–thaw (liquid nitrogen/37 °C) before gentle homogenization. Cell lysates were centrifuged at 10,000g for 10 min to sediment the particulate material. The protein concentrations of cell lysates were determined using a Pierce BCA protein assay kit, and cell lysates were stored at −65 °C.
TK activity was determined using the following conditions: cell lysates (30 μg of protein) were diluted in test buffer (1 mM DTT, 0.5 mg BSA/ml, 50 mM Tris–HCl, pH 7.6) to a volume of 40 μl, and incubated with 5 mM MgCl2, 100 mM KCl, 1 mM DTT, 15 mM NaF, 5 mM ATP, 20.7 μM [methyl-3H]AZT (specific activity 430 mCi/mmol) in a total volume of 60 μl at 37 °C for 30 min with shaking at 300 rpm. The reaction was terminated by the addition of an equal volume of methanol. Precipitates were removed by centrifugation at 13,000 rpm (16,200g) at 4 °C for 30 min and supernatants were analyzed for the formation of [methyl-3H]AZT 5′-monophosphate using a reversed-phase HPLC with online radiochemical detection. The HPLC system consisted of a Waters 600 Controller, a Waters 996 Photodiode Array detector, a Waters 717 plus autosampler (Waters Corporation, Milford, MA), and a Packard 500TR Flow Scintillation analyzer equipped with a 0.5-ml liquid flow cell. The samples were injected onto a 5-μM Phenomenex C18 column (250 × 4.6 mm) (Phenomenex, Torrance, CA). Separations were conducted using 7.5 mM ammonium acetate (pH 6.5) containing 30 % methanol. The HPLC flow rate was 0.65 ml/min, and the scintillation fluid flow rate was 0.65 ml/min. The column was washed with 100 % methanol for 15 min and equilibrated after every HPLC run with 7.5 mM ammonium acetate (pH 6.5) containing 30 % methanol for at least 20 min. Peaks corresponding to [methyl-3H]AZT 5′-monophosphate were identified by retention time. The activity of TK was defined as the amount of [methyl-3H]AZT 5′-monophosphate that was formed from [methyl-3H]AZT by 1 mg protein per minute under the above assay conditions. The intra-assay variation was 3.8 % (15.9 ± 0.6 pmol/mg/min; n = 8), as determined by assessing TK activity in HepG2 cell lysates within 1 day; the inter-assay variation was 5.4 % (18.5 ± 1.0 pmol/mg/min; n = 8), as determined by assessing TK activity in HepG2 cell lysates for eight consecutive days.
Data analysis
Differentially expressed genes were identified using one-way analysis of variance (ANOVA), with a cutoff at false discovery rate (FDR) ≤0.01 and fold change ≥1.5. Principal component analysis (PCA) was conducted within ArrayTrack. Additional calculations were performed within JMP 7.0 (SAS Institute, Cary, NC). The pathway analyses were generated through the use of Ingenuity Pathway Analysis (IPA) software (Ingenuity Systems, www.ingenuity.com). The significance of pathways was determined using Fisher’s Exact test at p < 0.05.
Data for the intracellular levels of total AZT, the relative expression levels of genes and proteins, and enzymatic activity are expressed as mean ± standard deviation. Comparisons among concentrations were conducted by one-way ANOVA, with multiple comparisons versus control group being performed by Dunnett’s method. When necessary, the data were log-transformed to maintain an equal variance or normal data distribution. The results were considered significant at p < 0.05.
Results
The concentration of AZT inhibiting cell growth by 50 % (IC50)
AZT inhibited the cell growth of HepG2 cells and THLE2 cells in a dose-dependent manner, with an IC50 of 89.2 μM for HepG2 cells and 2.2 mM for THLE2 cells (Fig. 1). The IC50 value in THLE2 cells is approximately 25 times higher than that in HepG2 cells.
Fig. 1.
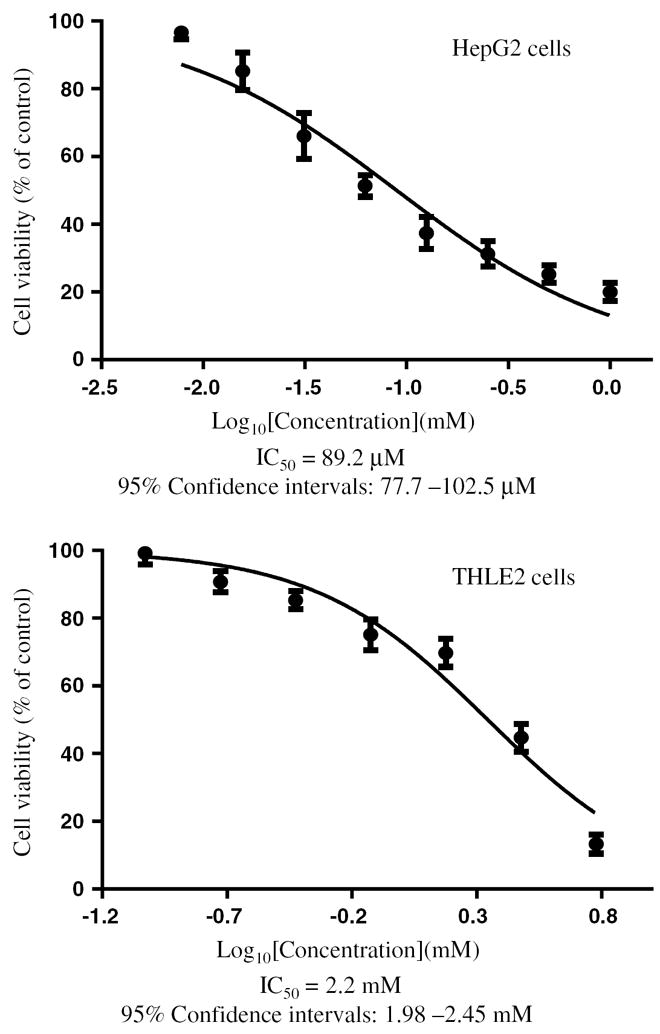
The half inhibitory concentration (IC50) of AZT. HepG2 cells and THLE2 cells were exposed to various concentrations of AZT (0–1 mM for HepG2 cells; 0–6 mM for THLE2 cells) for 1 week. After the treatment, the total number of viable cells was determined by an MTT assay. The IC50 values were obtained from the cell growth curves using GraphPad Prism 5.0
Gene expression profiles
The gene expression profiles from HepG2 cells and THLE2 cells treated with various concentrations of AZT for 2 weeks were determined using a human genome-wide gene expression microarray assay. There was one control group for each cell line (complete medium free of AZT) and three treatment groups for HepG2 cells (2, 20, and 100 μM AZT) and THLE2 cells (50, 500, and 2,500 μM AZT), with each group containing 5 biological replicates. Thus, a total of 40 microarrays were performed.
To explore the treatment effects and determine the relationships among the samples based on the expression profiles, unsupervised PCA was employed. The resulting cluster analysis is displayed in Fig. 2. The analysis clearly showed that there were two large clusters, and that there was a clear difference in gene expression between the two cell lines, with or without drug treatment. In the cluster formed by the HepG2 cells, clear separation was observed for higher concentration groups (20 and 100 μM AZT) compared to the control cells (0 μM AZT) and the lowest concentration group (2 μM AZT). Less separation was seen in THLE2 cells incubated with AZT.
Fig. 2.
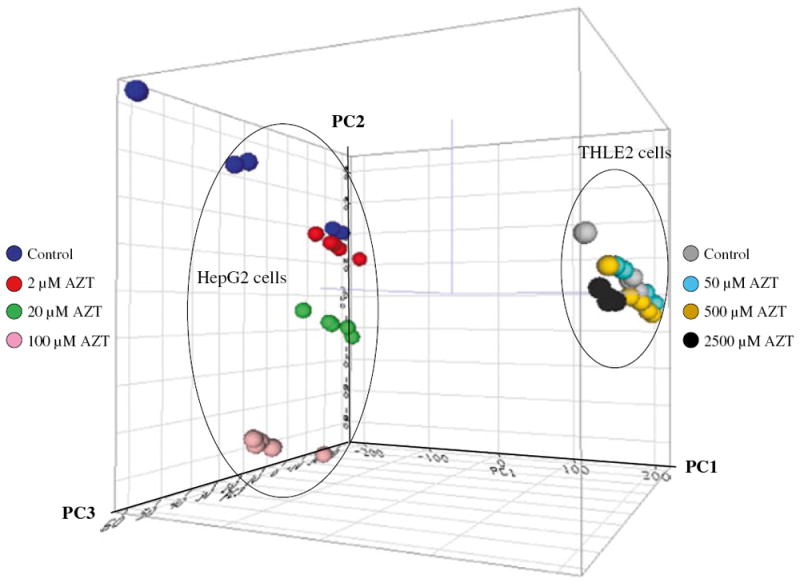
Principal Component Analysis (PCA) of gene expression profiles for control and AZT-treated HepG2 cells and THLE2 cells. The intensity of the entire gene set was used; no specific cutoff was applied for the analysis. The auto scaled method was used for the PCA. PC1, PC2, and PC3 represent the first, second, and third principal components, respectively
Using the criteria of a fold change ≥1.5 (up or down) and an FDR ≤0.01 in comparison to the control, differentially expressed genes were identified. AZT treatment led to a larger number of changes in gene expression in HepG2 cells than in THLE cells: 1,047, 3,748, and 5,458 genes were identified in HepG2 cells exposed to 2, 20, and 100 μM AZT, respectively (Fig. 3a), while only 15, 168, and 892 genes were identified in THLE2 cells exposed to 50, 500, and 2,500 μM AZT, respectively (Fig. 3b). There was a total of 71 up-regulated and 22 down-regulated genes with a fold change >10, at an FDR <0.01. Each of these differentially expressed genes was observed only in HepG2 cells.
Fig. 3.
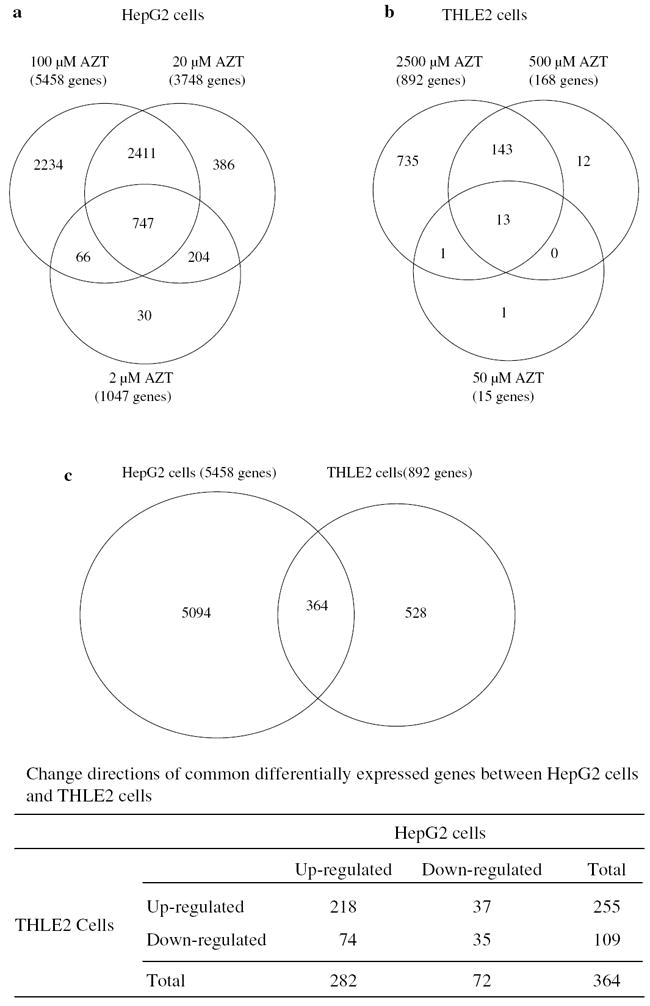
The number of genes altered by AZT treatment in HepG2 cells (a), THLE2 cells (b), and comparison of genes altered by 100 μM AZT in HepG2 cells and 2,500 μM AZT in THLE2 cells (c). A gene was identified as differentially expressed if the fold change was ≥1.5 (up- or down-regulated) and the false discovery rate was ≤0.01 in comparison to its respective control group
As displayed in the Venn diagrams (Fig. 3a, b), the majority of the genes altered in the lower concentration groups were also found in the higher concentration groups. In HepG2 cells, out of 1,047 genes altered in the lowest concentration group (2 μM AZT), 813 genes (78 %) overlapped with those identified in the highest dose group (100 μM AZT). In THLE2 cells, more than 93 % genes altered in the 50 and 500 μM AZT groups overlapped with those identified in the 2,500 μM AZT group (Fig. 3a, b). Thus, we focused further analyses, including gene functions and pathways, primarily on those genes altered in the highest concentration groups (100 μM AZT in HepG2 cells and 2,500 μM AZT in THLE2 cells).
The number of genes altered by 100 μM AZT in HepG2 cells and 2,500 μM AZT in THLE2 cells is shown in Fig. 3c. Out of 5,458 differentially expressed genes in HepG2 cells, 364 genes (7 %) overlapped with the genes in THLE2 cells. Focusing on the intersection of the 364 AZT-modulated genes in common to both cell lines, 253 differentially expressed genes (70 %) responded in same direction, which consisted of 218 genes up-regulated and 35 genes down-regulated in both cell lines; another 111 differentially expressed genes (30 %) were expressed in opposite directions in each cell line, including 74 genes up-regulated in HepG2 cells and 37 genes up-regulated in THLE2 cells (Fig. 3c).
IPA software was used to identify the most relevant molecular and cellular function pathways. Differentially expressed genes were mapped to the IPA database for the pathway analysis and p values were then calculated to determine the significance of the pathways. Overall, genes altered by AZT treatment in both cell lines are involved in more than 60 functional pathways; the top 12 functional pathways in HepG2 cells and THLE2 cells are listed in Table 1. The 253 common genes altered by the equi-toxic concentrations of AZT in the same direction with both cell lines are mainly associated with cell death and survival (60 genes), cell cycle (37 genes), cellular growth and proliferation (68 genes), DNA replication, recombination, and repair (33 genes), and lipid metabolism (10 genes) (Table 2).
Table 1.
Top 12 functional pathways altered by AZT in HepG2 cells and THLE2 cells
| Pathway | p value | # of genes |
|---|---|---|
| HepG2 cells | ||
| Cell death and survival | 2.27E–07 to 3.49E–02 | 879 |
| Protein degradation | 3.84E–06 to 3.24E–02 | 122 |
| Protein synthesis | 3.84E–06 to 3.69E–02 | 197 |
| Cellular movement | 1.31E–05 to 3.46E–02 | 506 |
| Post-translational modification | 4.77E–05 to 3.24E–02 | 150 |
| Cellular growth and proliferation | 1.25E–04 to 3.45E–02 | 886 |
| Cell cycle | 5.59E–04 to 3.85E–02 | 339 |
| Cell-to-cell signaling and interaction | 5.59E–04 to 3.93E–02 | 154 |
| Amino acid metabolism | 5.62E–04 to 3.08E–02 | 52 |
| Lipid metabolism | 5.62E–04 to 3.75E–02 | 248 |
| Molecular transport | 5.62E–04 to 2.86E–02 | 137 |
| Small molecule biochemistry | 5.62E–04 to 3.90E–02 | 336 |
| THLE2 cells | ||
| Cell cycle | 1.39E–13 to 2.25E–02 | 126 |
| Cellular growth and proliferation | 7.60E–13 to 2.25E–02 | 239 |
| Cellular development | 1.58E–09 to 2.25E–02 | 184 |
| Cellular movement | 4.24E–09 to 2.25E–02 | 154 |
| Cellular assembly and organization | 1.78E–08 to 2.52E–02 | 111 |
| DNA replication, recombination, and repair | 1.73E-08 to 2.25E-02 | 72 |
| Cell death and survival | 3.29E–06 to 2.23E–02 | 192 |
| Cellular function and maintenance | 8.74E-05 to 2.25E-02 | 66 |
| Cellular compromise | 1.24E–04 to 2.25E–02 | 5 |
| Cell-to-cell signaling and interaction | 4.21E-04 to 1.77E-02 | 62 |
| Cell morphology | 1.15E–03 to 1.75E–02 | 17 |
| Molecular transport | 1.54E–03 to 2.25E–02 | 13 |
The represented pathways with the lowest p values from IPA’s “Function Pathways” database for each cell line are reported, along with the number of differentially expressed genes within the pathway. Pathways common to both cell lines are bolded
Table 2.
Top 5 functional pathways altered by AZT at the equi-toxic concentration
| Pathway | p value | # of genes |
|---|---|---|
| Cell death and survival | 5.62E–07 to 2.75E–02 | 60 (ABCA3, ANKRD1, APOE, AURKB, BBC3, BIRC5, BTG2, CA9, CCND2, CD24, CDKN1A, CDKN2C, CEBPD, CREB3L2, CRYAB, CSTA, CXCR4, DDB2, FAS, FDXR, GDNF, GPC1, GSN, HEY1, HMGB2, HS1BP3, ID4, IL11, IP6K3, IRF5, L1CAM, LRIG1, MDM2, MFGE8, NT5 M, NUF2, PACS2, PHLDA3, PHLPP1, PIDD, PLK3, POLH, PON3, RASD1, RRAD, RRM2B, SEMA3B, SKP2, SLC47A1, SMC1A, SOCS3, SULF2, TIMP1, TNFRSF14, TP53I3, TP53INP1, TPP1, TTK, UBD, XPC) |
| Cell cycle | 1.74E–05 to 2.75E–02 | 37 (APOE, AURKB, BIRC5, BTG2, BUB1, CCND2, CD24, CDKN1A, CDKN2B, CDKN2C, CDS1, CEBPD, CENPA, DDB2, FAS, GPC1, IL11, IRF5, KIF15, LRIG1, MDM2, NUF2, PLK3, PLK4, POLH, RRAD, RRM2B, SKP2, SMC1A, SOCS3, TBX2, TMEM8B, TMPO, TP53INP1, TTK, UBD, XPC) |
| Cellular growth and proliferation | 2.51E–05 to 2.75E–02 | 68 (AKR1B10, ANKRD1, APOE, AURKB, BBC3, BIRC5, BTG2, BUB1, CA9, CCND2, CD24, CDKN1A, CDKN2B, CDKN2C, CEBPD, CES2, COL6A1, CSMD3, CXCR4, DDB2, DIRAS1, ELOVL7, EPCAM, FAS, FBLN5, FBXO2, FDXR, FOLR1, GDNF, GPC1, GSN, HEY1, HMGB2, HNRNPR, HYAL1, IGFBP2, IL11, KIF15, KIF20A, L1CAM, LRIG1, MDM2, MLXIPL, NEU1, PHLPP1, PLK3, PLK4, POLH, PTPRU, RASD1, RRAD, SEMA3B, SESN1, SKP2, SLC12A4, SLC22A18, SLC2A11, SLC52A1, SMC1A, SOCS3, SPINK1, TBX2, TIMP1, TNFRSF14, TRIM22, TTK, XPC, ZMAT3) |
| Lipid metabolism | 2.51E–05 to 2.75E–02 | 10 (ALDH5A1, ARSA, CDS1, ELOVL7, FAS, GAD1, GM2A, GSN, PLA2G4C, TBXAS1) |
| DNA replication, recombination, and repair | 5.91E–05 to 2.75E–02 | 33 (APOE, AURKB, BIRC5, BTG2, BUB1, CCND2, CDKN1A, CDKN2B, CDKN2C, CENPA, DDB2, FBLN5, FOLR1, GDNF, HMGB2, IGFBP2, MDM2, NT5 M, NUF2, PHLDA3, PLK3, PLK4, POLH, RPS27L, RRAD, SESN1, SKP2, SMC1A, TK2, TMPO, TTK, UBD, XPC) |
The represented pathways with the lowest p values from IPA’s “Function Pathways” database for the common 253 differentially expressed genes altered by AZT at the equi-toxic concentration to both cell lines are reported, along with the number of differentially expressed genes within the pathway. Genes in bold indicate a significant down-regulation of expression by AZT treatment
Intracellular levels of AZT and its metabolites
Ninety-four transporter genes were altered in HepG2 cells treated with 100 μM AZT, of which 67 genes were up-regulated and 27 down-regulated (Supplementary Table S1). In THLE2 cells, only 19 transporter genes (15 up-regulated and 4 down-regulated) were differentially expressed by treatment with 2,500 μM AZT.
The gene expression levels of transporters that are involved in influx and efflux of AZT are summarized in Table 3. All eight influx transporters were expressed at a low magnitude in both cell lines. The background levels of SLC22A6 and SLC28A3 were two- to fourfold higher in HepG2 cells as compared to THLE2 cells, and the levels of SLC22A7 were slightly induced by AZT treatment in HepG2 cells. The background levels of efflux transporter ABCG2 in HepG2 cells were approximately eightfold higher than those in THLE2 cells. The expression of efflux transporter gene ABCG2 was down-regulated by 60 % in THLE2 cells treated with 2,500 μM AZT while there was no change in HepG2 cells.
Table 3.
Gene expression levels of AZT transporters in HepG2 cells and THLE2 cells treated with various concentrations of AZT
| HepG2 cells
|
THLE2 cells
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Control | AZT 2 μM | AZT 20 μM | AZT 100 μM | Control | AZT 50 μM | AZT 500 μM | AZT 2,500 μM | |
| SLC22A1 | 10.4 ± 2.6 | 10.0 ± 2.2 | 11.1 ± 2.8 | 13.1 ± 2.3 | 8.2 ± 3.0 | 7.0 ± 2.8 | 6.7 ± 1.0 | 7.2 ± 2.6 |
| SLC22A6 | 5.1 ± 1.1 # | 5.0 ± 2.1 | 4.0 ± 2.3 | 3.6 ± 1.6 | 2.4 ± 0.5 | 3.2 ± 1.5 | 4.4 ± 2.4 | 2.5 ± 0.8 |
| SLC22A7 | 26.0 ± 2.8 | 36.8 ± 5.6* | 33.0 ± 3.8* | 42.4 ± 6.0* | 21.4 ± 4.6 | 20.0 ± 4.5 | 18.6 ± 4.8 | 17.8 ± 2.6 |
| SLC22A8 | 2.4 ± 0.4 | 2.4 ± 0.2 | 2.4 ± 0.3 | 2.3 ± 0.3 | 2.5 ± 0.2 | 2.2 ± 0.3 | 2.5 ± 0.3 | 2.4 ± 0.2 |
| SLC22A11 | 5.2 ± 1.4 | 5.2 ± 2.3 | 6.2 ± 1.0 | 7.2 ± 2.8 | 3.9 ± 1.5 | 3.9 ± 1.4 | 4.3 ± 2.7 | 7.2 ± 4.9 |
| SLC28A1 | 6.7 ± 3.2 | 8.1 ± 1.1 | 5.9 ± 3.8 | 5.0 ± 2.3 | 3.4 ± 1.7 | 4.6 ± 3.7 | 2.9 ± 1.1 | 3.4 ± 2.0 |
| SLC28A3 | 18.3 ± 4.4 # | 19.9 ± 1.5 | 27.5 ± 1.9* | 18.6 ± 9.1 | 3.7 ± 0.9 | 4.2 ± 1.7 | 5.1 ± 0.9* | 4.6 ± 1.9 |
| SLC29A2 | 36.8 ± 6.2 | 38.9 ± 3.3 | 39.0 ± 4.2 | 38.8 ± 4.8 | 36.1 ± 5.3 | 35.5 ± 5.4 | 34.5 ± 4.5 | 36.8 ± 3.4 |
| ABCC4 | 2.1 ± 0.2 | 2.1 ± 0.1 | 2.1 ± 0.3 | 2.1 ± 0.2 | 2.3 ± 0.2 | 2.2 ± 0.5 | 2.3 ± 0.2 | 2.2 ± 0.2 |
| ABCG2 | 513 ± 113 # | 571 ± 52.8 | 587 ± 42.3 | 587 ± 61.9 | 62.3 ± 11.0 | 51.8 ± 7.1 | 51.8 ± 6.1 | 36.7 ± 1.3* |
The levels of gene expression were measured by Agilent human genome-wide gene expression microarray assays. Data are expressed as the normalized intensity (mean ± standard deviation) for each gene of five biological replicates.
Significantly (p < 0.05) different from the control group of THLE2 cells;
Significant difference (p< 0.05) compared to the control group within each cell line
This difference in the expression of transporter genes might impact the intracellular levels of AZT and its metabolites. Therefore, the intracellular levels of AZT and its metabolites were determined in an experiment in which HepG2 cells and THLE2 cells were incubated with various concentrations of [methyl-3H]AZT for up to 240 min. [Methyl-3H]AZT was found to be rapidly taken up by HepG2 cells and THLE2 cells in a concentration-dependent manner (Fig. 4). The levels of [methyl-3H]AZT and its metabolites reached a plateau levels after 30 min in the HepG2 cells. With the THLE2 cells, a plateau was obtained after 30 min with 50 and 500 μM AZT and after 60 min with 2,500 μM AZT. Although the concentrations in THLE2 cells were 25 times higher than those in HepG2 cells, the intracellular levels of [methyl-3H]AZT and its metabolites in THLE2 cells at 50, 500, and 2,500 μM AZT were only 10.3-, 2.4-, and 2.1-fold higher than those in HepG2 cells at 2, 20, and 100 μM AZT. These data suggest that the transport of AZT into the cells is more efficient in the HepG2 cells.
Fig. 4.
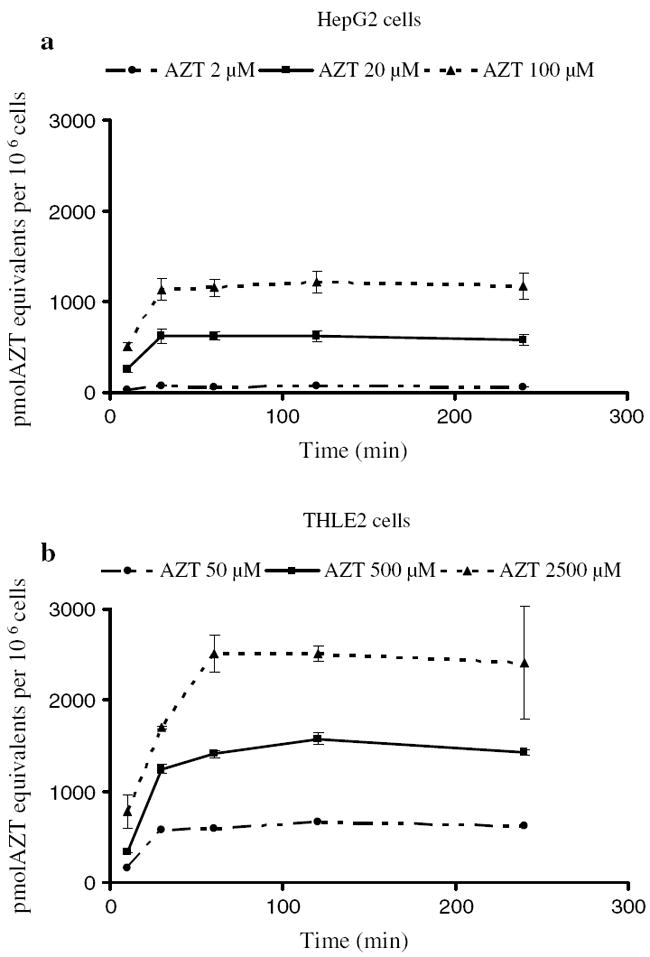
Time course of the intracellular uptake of [methyl-3H]AZT by HepG2 cells (a) and THLE2 cells (b). HepG2 cells and THLE2 cells were incubated with [methyl-3H]AZT for 10, 30, 60, 120, and 240 min. The intracellular uptake of [methyl-3H]AZT was determined as described in “Materials and methods.” The radioactivity in control cells, which were fed with complete culture medium free of [methyl-3H]AZT, was same as the background; therefore, the intracellular uptake of [methyl-3H]AZT in control was given a nominal value of 0.0. The data represent means and standard deviations for three separate experiments
Gene expression levels of TK and UGT2B7
Since the concentration of AZT was 25-fold lower in HepG2 cells compared to THLE2 cells, but the amount of AZT taken up was only twofold lower, the genes involved in AZT metabolism, i.e., TK1, TK2, and UGT 2B7, were further investigated. As determined by the human microarray analysis, the expression of the TK1 gene showed a concentration-related increase in expression in HepG2 cells treated with AZT, with the increase being significant at all concentrations of AZT. In contrast, there was a concentration-related decrease in TK1 gene expression in THLE2 cells, with the decrease being significant at 2,500 μM AZT (Fig. 5). There were no changes in the expression of the TK2 gene in HepG2 cells. With the THLE2 cells, there was a concentration-related increase in the expression of the TK2 gene, with the increase being slightly elevated after a 2-week treatment with 500 and 2,500 μM AZT. The expression of the UGT2B7 gene showed a concentration-related increase in HepG2 cells, with the increase being significant at 20 and 100 μM AZT. There was no change in UGT2B7 gene expression in THLE2 cells. Results from real-time PCR analyses in HepG2 cells were consistent with the microarray analyses: there was increased expression of TK1 and UGT2B7 and no change in expression of TK2 (Fig. 5). In THLE2 cells, real-time PCR analyses revealed a decrease in the expression of the TK1 and UGT2B7 genes at 500 and 2,500 μM AZT and no change in TK2 expression with any of the AZT concentrations (Fig. 5). These data indicated that AZT treatment modulated the gene expression of TK1 and UGT2B7 in opposite directions in HepG2 cells and THLE2 cells.
Fig. 5.
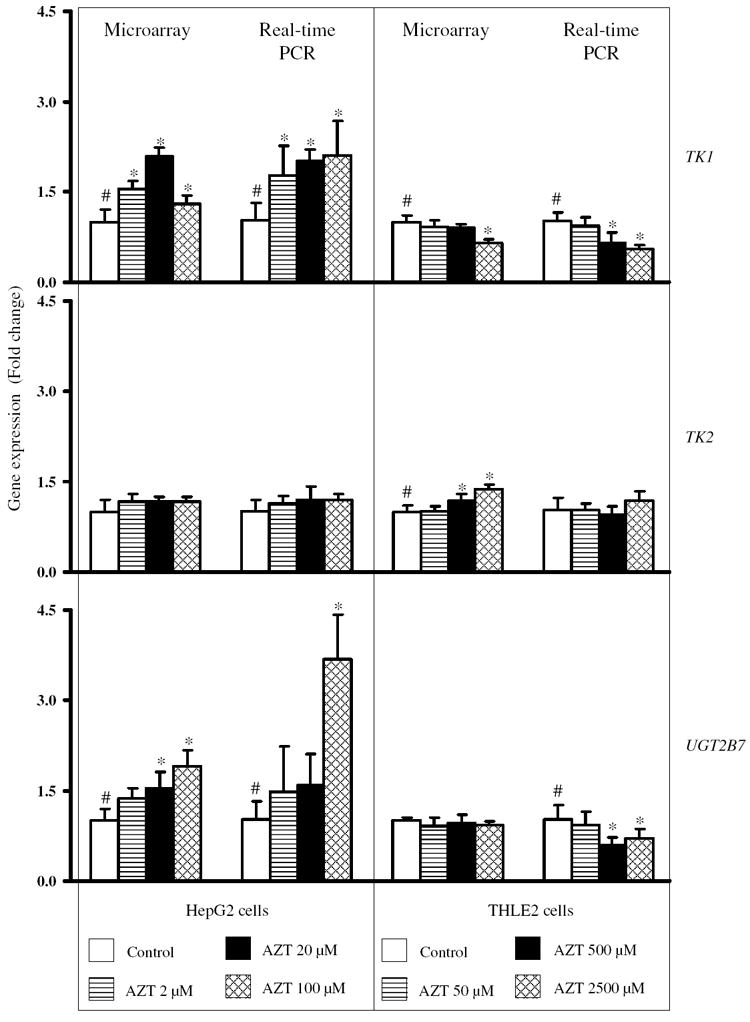
Relative gene expression levels of TK1, TK2 and UGT2B7 in HepG2 cells and THLE2 cells treated with various concentrations of AZT. The levels of gene expression were measured by Agilent human genome-wide gene expression microarray assays and real-time PCR assays. Data are expressed as the fold change (mean ± standard deviation) for each gene as determined from five biological replicates (microarray assays) and three separate experiments (real-time PCR assays). #Significant concentration-related trend (p < 0.05); *significant difference (p < 0.05) compared to the control group
Protein levels of TK1 and UGT2B7
The effects of AZT on the protein levels of TK1 and UGT2B7 were analyzed by Western blotting. In HepG2 cells treated with AZT, there was a concentration-related increase in the relative levels of TK1 protein, with the increase being significant at all concentrations of AZT (Fig. 6a). In THLE2 cells, there was a concentration-related decrease in the relative levels of TK1 protein, with the decrease being significant at 500 and 2,500 μM AZT (Fig. 6a).
Fig. 6.
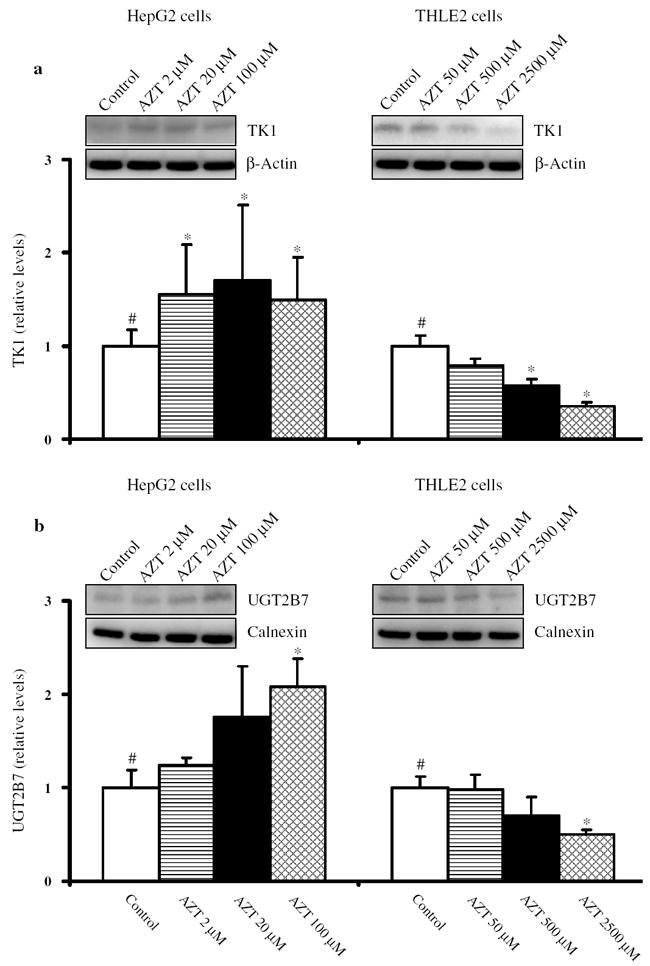
Western blotting of TK1 (a) and UGT2B7 (b) in HepG2 cells and THLE2 cells treated with AZT for 2 weeks. After treatment, whole cell lysates were prepared and equal amounts of protein (50 μg) were loaded in each well. Immunoblotting for each protein was done in triplicate. β-Actin was used as a loading control. The data are normalized to the control value. Columns and bars are means and standard deviations for three separate experiments. #Significant (p < 0.05) concentration-related trend; *significant difference (p < 0.05) compared to the control group
Western blot analysis of HepG2 cells showed a concentration-related increase in the relative protein levels of UGT2B7, with the increase being significant at 100 μM AZT (Fig. 6b). In contrast, there was a concentration-related decrease in the relative protein levels of UGT2B7, with the decrease being significant at 2,500 μM AZT (Fig. 6b).
These data indicated that AZT treatment modulated the protein levels of TK1 and UGT2B7 in opposite directions between HepG2 cells and THLE2 cells.
Enzymatic activity of TK
The activity of cellular TK that is responsible for 5′-phosphorylation of AZT was further determined using a reverse-phase HPLC with online radiochemical detection. As shown in Fig. 7, the activity of TK was elevated two-, five-, and threefold in HepG2 cells treated with 2, 20, and 100 μM AZT, respectively, for 2 weeks when compared to that of the control cells. In contrast, exposure of THLE2 cells to AZT resulted in a concentration-related decrease in the enzymatic activity of TK, with the decrease being significant at 500 and 2,500 μM AZT (Fig. 7). The enzymatic activities of TK in THLE2 cells treated with 500 and 2,500 μM AZT were 55 and 23 % of the control, respectively. These data indicated that AZT treatment modulated the enzymatic activity of TK in opposite directions between HepG2 cells and THLE2 cells.
Fig. 7.
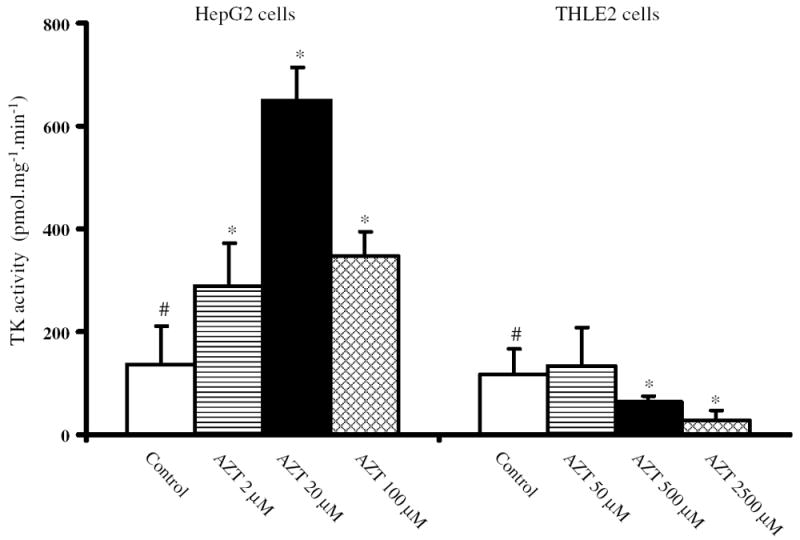
Analysis of TK activity in HepG2 cells and THLE2 cells treated with AZT for 2 weeks. The activity of TK was determined using a reverse-phase HPLC with online radiochemical detection. The activity of TK was defined as the amount of [methyl-3H]AZT 5′-monophosphate formed from [methyl-3H]AZT by one mg protein per minute under the standard conditions as described in “Materials and methods.” Columns and bars are means and standard deviations for three separate experiments. #Significant (p < 0.05) concentration-related trend; *significant difference (p < 0.05) compared to the control group
Discussion
In this study, we have demonstrated that the IC50 for AZT in human hepatoma HepG2 cells was approximately 25 times lower than that for the immortalized normal liver THLE2 cells. This is consistent with our previous report that 25-fold higher levels of AZT are required in THLE2 cells to have the same cytotoxicity as observed in HepG2 cells (Fang and Beland 2009). At comparable IC50 values, 6–70 times as many genes were differentially expressed in HepG2 cells as compared to THLE2 cells even though the concentration of AZT was 25-fold lower in the HepG2 cells.
AZT can be incorporated into replicating DNA in place of thymidine and cause damage to the DNA. The incorporation of AZT into the DNA of HepG2 cells and THLE2 cells has clearly been demonstrated previously, with approximately 25-fold more AZT being required to give the same level of AZT incorporation in THLE2 cells as compared to HepG2 cells (Fang and Beland 2009). The resulting damaged DNA might either delay cell cycle progression until the damage is repaired or cause apoptotic cell death (Melana et al. 1998; Falchetti et al. 2005; Humer et al. 2008; Fang and Beland 2009; Fang et al. 2009b). In addition, recent studies have demonstrated that XPC, via the nucleotide excision repair pathway, is essential for the repair of AZT-induced DNA damage in human HepG2 cells and THLE2 cells (Wu et al. 2011, 2013) In the current study, molecular and cellular function pathway analysis indicated that most of the 253 common genes altered in the same direction by AZT at the equi-toxic concentrations in both HepG2 cells and THLE2 cells were involved in cell death and survival, cellular growth and proliferation, cell cycle, and DNA replication, recombination, and repair (Table 2), which is in consistent with previous studies (Fang and Beland 2009; Collier et al. 2003; Wu et al. 2004; Melana et al. 1998; Roskrow and Wickramasinghe 1990; Falchetti et al. 2005; Humer et al. 2008).
Emerging evidence suggests that transporters, including the ATP-binding cassette (ABC) and solute carrier (SLC) transporters, play significant roles in the absorption and disposition of drugs and xenobiotics (Beringer and Slaughter 2005; Halestrap 2012; Li et al. 2012). Several studies have addressed the expression and activity of transporters of nucleoside/nucleotide reverse transcriptase inhibitors and their association with intracellular drug levels (Beringer and Slaughter 2005; Purcet et al. 2006; Pan et al. 2007; Huber-Ruano and Pastor-Anglada 2009; Kis et al. 2010). AZT influx involves the gene families SLC22 (SLC22A1, 6, 7, 8, and 11), SLC28 (SLC28A1 and 3), and SLC29A2 (Huber-Ruano and Pastor-Anglada 2009; Kis et al. 2010; Purcet et al. 2006), and efflux transporters ABCG2 and ABCC4 actively transport AZT out of cells (Beringer and Slaughter 2005; Kis et al. 2010; Pan et al. 2007). In our study, with exception of ABCG2 in HepG2 cells, the baseline levels of the transporter genes were low in both cell lines. AZT treatment did lead to a slight increase in the levels of SLC22A7 in HepG2 cells and the expression level of efflux transporter gene ABCG2 was down-regulated by 1.7-fold in THLE2 cells; nonetheless, these alterations of AZT-associated transporters do not provide a clear explanation for the more efficient uptake of AZT into HepG2 cells compared to THLE2 cells.
AZT is phosphorylated intracellularly by successive cellular kinases to its mono-, di-, and tri-phosphates, with AZT 5′-triphosphate being the active metabolite. The first phosphorylation step is initiated by thymidine kinases and is considered to be the rate-limiting step in the pathway of activation of AZT (Arner and Eriksson 1995). It has been reported that cells may become resistant to AZT or cross-resistant to thymidine and deoxycytidine analogs partially through inactivation of TK1 after AZT exposure (Avramis et al. 1993; Groschel et al. 2000, 2002; Han et al. 2004). Our data indicate that the expression of TK1 gene and protein as well as its enzymatic activity is inhibited in THLE2 cells and elevated in HepG2 cells following AZT treatment. The suppression of TK1 results in a decreased activation of AZT, and consequently, decreased AZT incorporation into DNA (Fang and Beland 2009; Olivero et al. 2010). Importantly, it remains to be established whether the observed induction of TK1 expression may lead to an increase in the intracellular concentration of AZT 5′-triphosphate. In addition, the mechanism underlying the opposite changes of TK1 by AZT in HepG2 cells and THLE2 cells remains unknown. AZT has been reported to induce hypermethylation of the TK promoter (Wu et al. 1995), which could explain the reduction in TK activity observed in THLE2 cells after treatment with AZT.
Glucuronidation catalyzed by UDP-glucuronosyltransferase (UGT) is one of the major steps in the metabolism of endogenous substances and xenobiotics. UGT2B7 is primarily responsible for AZT glucuronidation (Barbier et al. 2000; Belanger et al. 2009). It has been demonstrated that AZT treatment decreases UGT2B7 protein expression and enzyme activities in JEG-3 cells (cultured with or without serum) and villous placental cultures (first trimester and term) (Collier et al. 2003), and the inhibition of UGT2B7 by fluconazole and valproic acid has been correlated with an increase in plasma AZT levels (Lertora et al. 1994; Sahai et al. 1994). A decrease in UGT2B7 gene and protein expression was observed in THLE2 cells following AZT treatment. Conversely, UGT2B7 gene and protein expression increased in HepG2 cells exposed to AZT. The mechanism underlying the opposite changes of UGT2B7 by AZT in HepG2 cells and THLE2 cells is unknown. Nonetheless, because the enzymatic activity of UGT2B7 in HepG2 cells and THLE2 cells was too low to be detected using a reversed-phase HPLC with online radiochemical detection to analyze the formation of [methyl-3H]AZT glucuronide, the role of UGT2B7 changes in AZT-induced toxicity is probably limited.
In conclusion, we have demonstrated that molecular pathways involved with cell death and survival, cell cycle, cell growth and proliferation, and DNA replication, recombination, and repair are involved in the toxicities associated with AZT in HepG2 and THLE2 cells. Furthermore, the differential response in AZT metabolism between HepG2 cells and THLE2 cells may explain why HepG2 cells are more sensitive than THLE2 cells to the toxicity of AZT.
Supplementary Material
Acknowledgments
Qiangen Wu was supported by an appointment to the Postgraduate Research Program in the Division of Biochemical Toxicology at the National Center for Toxicological Research administered by Oak Ridge Institute for Science Education through an interagency agreement between the US Department of Energy and the FDA. This research was supported through an interagency agreement between the National Center for Toxicological Research, US Food and Drug Administration and the National Toxicology Program, National Institute of Environmental Health Sciences. (FDA IAG: 224-07-0007; NIH Y1ES1027).
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00204-013-1169-3) contains supplementary material, which is available to authorized users.
The views presented in this article do not necessarily reflect those of the US Food and Drug Administration.
Conflict of interest The authors declare that there are no conflicts of interest.
Contributor Information
Jia-Long Fang, Division of Biochemical Toxicology, National Center for Toxicological Research, Jefferson, AR 72079, USA.
Tao Han, Division of Systems Biology, National Center for Toxicological Research, Jefferson, AR 72079, USA.
Qiangen Wu, Division of Biochemical Toxicology, National Center for Toxicological Research, Jefferson, AR 72079, USA.
Frederick A. Beland, Division of Biochemical Toxicology, National Center for Toxicological Research, Jefferson, AR 72079, USA
Ching-Wei Chang, Division of Bioinformatics and Biostatistics, National Center for Toxicological Research, Jefferson, AR 72079, USA.
Lei Guo, Division of Biochemical Toxicology, National Center for Toxicological Research, Jefferson, AR 72079, USA.
James C. Fuscoe, Division of Systems Biology, National Center for Toxicological Research, Jefferson, AR 72079, USA
References
- AHFS. Nucleoside and nucleotide reverse transcriptase inhibitors, zidovudine. In: McEvoy GK, editor. American hospital formulary service drug information 8.18.08.20. American Society of Health-System Pharmacists, Inc.; Bethesda, MD: 2007. [Google Scholar]
- Arner ES, Eriksson S. Mammalian deoxyribonucleoside kinases. Pharmacol Ther. 1995;67(2):155–186. doi: 10.1016/0163-7258(95)00015-9. [DOI] [PubMed] [Google Scholar]
- Avramis VI, Kwock R, Solorzano MM, Gomperts E. Evidence of in vitro development of drug resistance to azidothymidine in T-lymphocytic leukemia cell lines (Jurkat E6-1/AZT-100) and in pediatric patients with HIV-1 infection. J Acquir Immune Defic Syndr. 1993;6(12):1287–1296. [PubMed] [Google Scholar]
- Ayers KM, Clive D, Tucker J, Walter E, Hajian G, de Miranda P. Nonclinical toxicology studies with zidovudine: genetic toxicity tests and carcinogenicity bioassays in mice and rats. Fundam Appl Toxicol. 1996;32(2):148–158. doi: 10.1006/faat.1996.0118. [DOI] [PubMed] [Google Scholar]
- Barbier O, Turgeon D, Girard C, et al. 3′-azido-3′-deoxythimidine (AZT) is glucuronidated by human UDP-glucuronosyltransferase 2B7 (UGT2B7) Drug Metab Dispos. 2000;28(5):497–502. [PubMed] [Google Scholar]
- Barry MG, Khoo SH, Veal GJ, et al. The effect of zidovudine dose on the formation of intracellular phosphorylated metabolites. AIDS. 1996;10(12):1361–1367. doi: 10.1097/00002030-199610000-00008. [DOI] [PubMed] [Google Scholar]
- Belanger AS, Caron P, Harvey M, Zimmerman PA, Mehlotra RK, Guillemette C. Glucuronidation of the antiretroviral drug efavirenz by UGT2B7 and an in vitro investigation of drug–drug interaction with zidovudine. Drug Metab Dispos. 2009;37(9):1793–1796. doi: 10.1124/dmd.109.027706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beringer PM, Slaughter RL. Transporters and their impact on drug disposition. Ann Pharmacother. 2005;39(6):1097–1108. doi: 10.1345/aph.1E614. [DOI] [PubMed] [Google Scholar]
- Borojerdi JP, Ming J, Cooch C, et al. Centrosomal amplification and aneuploidy induced by the antiretroviral drug AZT in hamster and human cells. Mutat Res. 2009;665(1–2):67–74. doi: 10.1016/j.mrfmmm.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman K, Kakuda TN. Mitochondrial toxicity of nucleoside analogue reverse transcriptase inhibitors: a looming obstacle for long-term antiretroviral therapy? Curr Opin Infect Dis. 2000;13(1):5–11. doi: 10.1097/00001432-200002000-00002. [DOI] [PubMed] [Google Scholar]
- Brunetti I, Falcone A, Calabresi P, Goulette FA, Darnowski JW. 5-Fluorouracil enhances azidothymidine cytotoxicity: in vitro, in vivo, and biochemical studies. Cancer Res. 1990;50(13):4026–4031. [PubMed] [Google Scholar]
- Collier AC, Helliwell RJA, Keelan JA, Paxton JW, Mitchell MD, Tingle MD. 3′-Azido-3′-deoxythymidine (AZT) induces apoptosis and alters metabolic enzyme activity in human placenta. Toxicol Appl Pharmacol. 2003;192(2):164–173. doi: 10.1016/s0041-008x(03)00274-6. [DOI] [PubMed] [Google Scholar]
- Dertinger SD, Torous DK, Tometsko KR. Induction of micronuclei by low doses of azidothymidine (AZT) Mutat Res. 1996;368(3–4):301–307. doi: 10.1016/s0165-1218(96)90072-5. [DOI] [PubMed] [Google Scholar]
- Dutra A, Pak E, Wincovitch S, John K, Poirier MC, Olivero OA. Nuclear bud formation: a novel manifestation of Zidovudine genotoxicity. Cytogenet Genome Res. 2010;128(1–3):105–110. doi: 10.1159/000298794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobar PA, Olivero OA, Wade NA, et al. Genotoxicity assessed by the comet and GPA assays following in vitro exposure of human lymphoblastoid cells (H9) or perinatal exposure of mother-child pairs to AZT or AZT-3TC. Environ Mol Mutagen. 2007;48(3–4):330–343. doi: 10.1002/em.20285. [DOI] [PubMed] [Google Scholar]
- Falchetti A, Franchi A, Bordi C, et al. Azidothymidine induces apoptosis and inhibits cell growth and telomerase activity of human parathyroid cancer cells in culture. J Bone Miner Res. 2005;20(3):410–418. doi: 10.1359/JBMR.041123. [DOI] [PubMed] [Google Scholar]
- Fang J-L, Beland FA. Long-term exposure to zidovudine delays cell cycle progression, induces apoptosis, and decreases telomerase activity in human hepatocytes. Toxicol Sci. 2009;111(1):120–130. doi: 10.1093/toxsci/kfp136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang H, Harris SC, Su Z, et al. ArrayTrack: an FDA and public genomic tool. Methods Mol Biol. 2009a;563:379–398. doi: 10.1007/978-1-60761-175-2_20. [DOI] [PubMed] [Google Scholar]
- Fang J-L, McGarrity LJ, Beland FA. Interference of cell cycle progression by zidovudine and lamivudine in NIH 3T3 cells. Mutagenesis. 2009b;24(2):133–141. doi: 10.1093/mutage/gen059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischl MA, Richman DD, Hansen N, et al. The safety and efficacy of zidovudine (AZT) in the treatment of subjects with mildly symptomatic human immunodeficiency virus type 1 (HIV) infection. A double-blind, placebo-controlled trial The AIDS Clinical Trials Group. Ann Intern Med. 1990;112(10):727–737. doi: 10.7326/0003-4819-112-10-727. [DOI] [PubMed] [Google Scholar]
- Furman PA, Fyfe JA, St Clair MH, et al. Phosphorylation of 3′-azido-3′-deoxythymidine and selective interaction of the 5′-triphosphate with human immunodeficiency virus reverse transcriptase. Proc Natl Acad Sci USA. 1986;83(21):8333–8337. doi: 10.1073/pnas.83.21.8333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez Cid M, Larripa I. Genotoxic activity of azidothymidine (AZT) in vitro systems. Mutat Res. 1994;321(1–2):113–118. doi: 10.1016/0165-1218(94)90127-9. [DOI] [PubMed] [Google Scholar]
- Good SS, Koble CS, Crouch R, Johnson RL, Rideout JL, de Miranda P. Isolation and characterization of an ether glucuronide of zidovudine, a major metabolite in monkeys and humans. Drug Metab Dispos. 1990;18(3):321–326. [PubMed] [Google Scholar]
- Grdina DJ, Dale P, Weichselbaum R. Protection against AZT-induced mutagenesis at the HGPRT locus in a human cell line by WR-151326. Int J Radiat Oncol Biol Phys. 1992;22(4):813–815. doi: 10.1016/0360-3016(92)90530-u. [DOI] [PubMed] [Google Scholar]
- Groschel B, Miller V, Doerr HW, Cinatl J., Jr Activity of cellular thymidine kinase 1 in PBMC of HIV-1-infected patients: novel therapy marker. Infection. 2000;28(4):209–213. doi: 10.1007/s150100070037. [DOI] [PubMed] [Google Scholar]
- Groschel B, Kaufmann A, Hover G, et al. 3′-Azido-2′,3′-dideoxythymidine induced deficiency of thymidine kinases 1, 2 and deoxycytidine kinase in H9 T-lymphoid cells. Biochem Pharmacol. 2002;64(2):239–246. doi: 10.1016/s0006-2952(02)01109-7. [DOI] [PubMed] [Google Scholar]
- Halestrap AP. The monocarboxylate transporter family: structure and functional characterization. IUBMB Life. 2012;64(1):1–9. doi: 10.1002/iub.573. [DOI] [PubMed] [Google Scholar]
- Han T, Fernandez M, Sarkar M, Agarwal RP. 2′, 3′-Dideoxycytidine represses thymidine kinases 1 and 2 expression in T-lymphoid cells. Life Sci. 2004;74(7):835–842. doi: 10.1016/j.lfs.2003.07.023. [DOI] [PubMed] [Google Scholar]
- Huang P, Farquhar D, Plunkett W. Selective action of 3′-azido-3′-deoxythymidine 5′-triphosphate on viral reverse transcriptases and human DNA polymerases. J Biol Chem. 1990;265(20):11914–11918. [PubMed] [Google Scholar]
- Huber-Ruano I, Pastor-Anglada M. Transport of nucleoside analogs across the plasma membrane: a clue to understanding drug-induced cytotoxicity. Curr Drug Metab. 2009;10(4):347–358. doi: 10.2174/138920009788499030. [DOI] [PubMed] [Google Scholar]
- Humer J, Ferko B, Waltenberger A, Rapberger R, Pehamberger H, Muster T. Azidothymidine inhibits melanoma cell growth in vitro and in vivo. Melanoma Res. 2008;18(5):314–321. doi: 10.1097/CMR.0b013e32830aaaa6. [DOI] [PubMed] [Google Scholar]
- IARC. Zidovudine (AZT) IARC monographs on the evaluation of carcinogenic risks to humans. Vol. 76. IARC; Lyon, France: 2000. Some antiviral and antineoplastic drugs, and other pharmaceutical agents; pp. 73–127. [Google Scholar]
- Ingrand D, Weber J, Boucher CA, et al. Phase I/II study of 3TC (lamivudine) in HIV-positive, asymptomatic or mild AIDS-related complex patients: sustained reduction in viral markers. The Lamivudine European HIV Working Group. AIDS. 1995;9(12):1323–1329. doi: 10.1097/00002030-199512000-00004. [DOI] [PubMed] [Google Scholar]
- Jacobsson B, Britton S, He Q, Karlsson A, Eriksson S. Decreased thymidine kinase levels in peripheral blood cells from HIV-seropositive individuals: implications for zidovudine metabolism. AIDS Res Hum Retroviruses. 1995;11(7):805–811. doi: 10.1089/aid.1995.11.805. [DOI] [PubMed] [Google Scholar]
- Jones RJ, Bischofberger N. Minireview: nucleotide prodrugs. Antiviral Res. 1995;27(1–2):1–17. doi: 10.1016/0166-3542(95)00011-a. [DOI] [PubMed] [Google Scholar]
- Kis O, Robillard K, Chan GN, Bendayan R. The complexities of antiretroviral drug–drug interactions: role of ABC and SLC transporters. Trends Pharmacol Sci. 2010;31(1):22–35. doi: 10.1016/j.tips.2009.10.001. [DOI] [PubMed] [Google Scholar]
- Lavie A, Schlichting I, Vetter IR, Konrad M, Reinstein J, Goody RS. The bottleneck in AZT activation. Nat Med. 1997;3(8):922–924. doi: 10.1038/nm0897-922. [DOI] [PubMed] [Google Scholar]
- Lavie A, Ostermann N, Brundiers R, et al. Structural basis for efficient phosphorylation of 3′-azidothymidine monophosphate by Escherichia coli thymidylate kinase. Proc Natl Acad Sci USA. 1998;95(24):14045–14050. doi: 10.1073/pnas.95.24.14045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lertora JJ, Rege AB, Greenspan DL, et al. Pharmacokinetic interaction between zidovudine and valproic acid in patients infected with human immunodeficiency virus. Clin Pharmacol Ther. 1994;56(3):272–278. doi: 10.1038/clpt.1994.137. [DOI] [PubMed] [Google Scholar]
- Li Y, Lu J, Paxton JW. The role of ABC and SLC transporters in the pharmacokinetics of dietary and herbal phytochemicals and their interactions with xenobiotics. Curr Drug Metab. 2012;13(5):624–639. doi: 10.2174/1389200211209050624. [DOI] [PubMed] [Google Scholar]
- McGowan JP, Shah SS. Prevention of perinatal HIV transmission during pregnancy. J Antimicrob Chemother. 2000;46(5):657–668. doi: 10.1093/jac/46.5.657. [DOI] [PubMed] [Google Scholar]
- Melana SM, Holland JF, Pogo BG. Inhibition of cell growth and telomerase activity of breast cancer cells in vitro by 3′-azido-3′-deoxythymidine. Clin Cancer Res. 1998;4(3):693–696. [PubMed] [Google Scholar]
- Moore KH, Raasch RH, Brouwer KL, et al. Pharmacokinetics and bioavailability of zidovudine and its glucuronidated metabolite in patients with human immunodeficiency virus infection and hepatic disease (AIDS Clinical Trials Group protocol 062) Antimicrob Agents Chemother. 1995;39(12):2732–2737. doi: 10.1128/aac.39.12.2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickel W, Austermann S, Bialek G, Grosse F. Interactions of azidothymidine triphosphate with the cellular DNA polymerases α, δ, and ε and with DNA primase. J Biol Chem. 1992;267(2):848–854. [PubMed] [Google Scholar]
- NTP. Toxicology and carcinogenesis studies of AZT (CAS No. 30516-87-1) and AZT/α-interferon A/D B6C3F1 mice (gavage studies) Natl Toxicol Program Tech Rep Ser. 1999;469:1–361. [PubMed] [Google Scholar]
- Olivero OA, Anderson LM, Diwan BA, et al. Transplacental effects of 3′-azido-2′,3′-dideoxythymidine (AZT): tumorigenicity in mice and genotoxicity in mice and monkeys. J Natl Cancer Inst. 1997;89(21):1602–1608. doi: 10.1093/jnci/89.21.1602. [DOI] [PubMed] [Google Scholar]
- Olivero OA, Vazquez IL, Cooch CC, et al. Long-term AZT exposure alters the metabolic capacity of cultured human lymphoblastoid cells. Toxicol Sci. 2010;115(1):109–117. doi: 10.1093/toxsci/kfq023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan G, Giri N, Elmquist WF. Abcg2/Bcrp1 mediates the polarized transport of antiretroviral nucleosides abacavir and zidovudine. Drug Metab Dispos. 2007;35(7):1165–1173. doi: 10.1124/dmd.106.014274. [DOI] [PubMed] [Google Scholar]
- PDR. Physicians’ desk reference Physicians’. Vol. 63. Desk Reference Inc.; Montvale: 2009. Retrovir® (GlaxoSmithKline) (zidovudine) tablets, capsules, and syrup; pp. 1574–1582. [Google Scholar]
- Pfeifer AM, Cole KE, Smoot DT, et al. Simian virus 40 large tumour antigen-immortalized normal human liver epithelial cells express hepatocyte characteristics and metabolize chemical carcinogens. Proc Natl Acad Sci USA. 1993;90:5123–5127. doi: 10.1073/pnas.90.11.5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powles T, Robinson D, Stebbing J, et al. Highly active antiretroviral therapy and the incidence of non-AIDS-defining cancers in people with HIV infection. J Clin Oncol. 2009;27(6):884–890. doi: 10.1200/JCO.2008.19.6626. [DOI] [PubMed] [Google Scholar]
- Purcet S, Minuesa G, Molina-Arcas M, et al. 3′-Azido-2′,3′-dideoxythymidine (zidovudine) uptake mechanisms in T lymphocytes. Antivir Ther. 2006;11(6):803–811. [PubMed] [Google Scholar]
- Roskrow M, Wickramasinghe SN. Acute effects of 3′-azido-3′-deoxythymidine on the cell cycle of HL60 cells. Clin Lab Haematol. 1990;12(2):177–184. [PubMed] [Google Scholar]
- Sahai J, Gallicano K, Pakuts A, Cameron DW. Effect of fluconazole on zidovudine pharmacokinetics in patients infected with human immunodeficiency virus. J Infect Dis. 1994;169(5):1103–1107. doi: 10.1093/infdis/169.5.1103. [DOI] [PubMed] [Google Scholar]
- Torres SM, Walker DM, Carter MM, et al. Mutagenicity of zidovudine, lamivudine, and abacavir following in vitro exposure of human lymphoblastoid cells or in utero exposure of CD-1 mice to single agents or drug combinations. Environ Mol Mutagen. 2007;48(3–4):224–238. doi: 10.1002/em.20264. [DOI] [PubMed] [Google Scholar]
- Tosi P, Calabresi P, Goulette FA, Renaud CA, Darnowski JW. Azidothymidine-induced cytotoxicity and incorporation into DNA in the human colon tumor cell line HCT-8 is enhanced by methotrexate in vitro and in vivo. Cancer Res. 1992;52(15):4069–4073. [PubMed] [Google Scholar]
- Vazquez-Padua MA, Starnes MC, Cheng YC. Incorporation of 3′-azido-3′-deoxythymidine into cellular DNA and its removal in a human leukemic cell line. Cancer Commun. 1990;2(1):55–62. doi: 10.3727/095535490820874740. [DOI] [PubMed] [Google Scholar]
- Wu S, Liu X, Solorzano MM, Kwock R, Avramis VI. Development of zidovudine (AZT) resistance in Jurkat T cells is associated with decreased expression of the thymidine kinase (TK) gene and hypermethylation of the 5′ end of human TK gene. J Acquir Immune Defic Syndr Hum Retrovirol. 1995;8(1):1–9. [PubMed] [Google Scholar]
- Wu YW, Xiao Q, Jiang YY, Fu H, Ju Y, Zhao YF. Synthesis, in vitro anticancer evaluation, and interference with cell cycle progression of N-phosphoamino acid esters of zidovudine and stavudine. Nucleosides, Nucleotides Nucleic Acids. 2004;23(11):1797–1811. doi: 10.1081/NCN-200034057. [DOI] [PubMed] [Google Scholar]
- Wu Q, Beland FA, Chang C-W, Fang J-L. XPC is essential for nucleotide excision repair of zidovudine-induced DNA damage in human hepatoma cells. Toxicol Appl Pharmacol. 2011;251(2):155–162. doi: 10.1016/j.taap.2010.12.009. [DOI] [PubMed] [Google Scholar]
- Wu Q, Beland FA, Chang C-W, Fang J-L. Role of DNA repair pathways in response to zidovudine-induced DNA damage in immortalized human liver THLE2 cells. Int J Biomed Sci. 2013;9(1):18–25. [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Diwan BA, Anderson LM, et al. Skin tumorigenesis and Ki-ras and Ha-ras mutations in tumors from adult mice exposed in utero to 3′-azido-2′,3′-dideoxythymidine. Mol Carcinog. 1998;23(1):45–51. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.