

Abstract
The biological consequences of exposure to piperidine nitroxides is a concern, given their widespread use in manufacturing processes and their potential use in clinical applications. Our previous study reported that TEMPO (2,2,6,6-tetramethylpiperidine-1-oxyl), a low molecular weight free radical, possesses pro-oxidative activity in L5178Y cells. In this study, we investigated and characterized the role of reactive oxygen species (ROS) in TEMPO-induced toxicity in L5178Y cells. We found that TEMPO induced time-and concentration-dependent intracellular ROS production and glutathione depletion. TEMPO also induced apoptosis as demonstrated by increased caspase-3/7 activity, an increased proportion of annexin V stained cells, and decreased expression of anti-apoptotic proteins including Bcl-2, Bcl-xL and Mcl-1. N-acetylcysteine, a ROS scavenger, attenuated the ROS production and apoptosis induced by TEMPO. Moreover, Western blot analyses revealed that TEMPO activated γ-H2A.X, a hallmark of DNA damage, and c-Jun N-terminal kinases (JNK), a key member in the mitogen-activated protein kinase (MAPK) signaling pathway. Addition of SP600125, a JNK-specific inhibitor, blocked TEMPO-mediated JNK phosphorylation and also attenuated TEMPO-induced apoptosis. These findings indicate that both ROS production and JNK activation are involved in TEMPO-induced apoptosis, and may contribute to the toxicity of TEMPO in L5178Y cells.
Keywords: Nitroxide, TEMPO, Reactive oxygen species, Apoptosis, MAPK pathway
1. Introduction
Piperidine nitroxides are a group of low molecular weight, cell-permeable, and stable organic free radicals [1–3]. They have been proposed as a new class of antioxidants due to their ability to prevent oxidative damage in various biological systems, both in vivo and in vitro [4–6]. TEMPO (2,2,6,6-tetramethylpiperidine-1-oxyl, C9H18NO) is one of the most frequently used piperidine nitroxide catalysts in organic and pharmaceutical syntheses. TEMPO has shown its protective properties in a variety of pathological situations related to oxidative stress, including radiation injury [7], protein oxidation [8], and ischemia/reperfusion injury [9]. Recently, a nitroxide radical-containing nanoparticle, which incorporates TEMPO in its core, has been synthesized for therapeutic purposes [10,11]. The widespread use of TEMPO in the manufacturing process and potential clinical applications raise safety concerns; however, mechanistic studies evaluating the toxicity of TEMPO are limited [12–14].
TEMPO, like all other antioxidants, demonstrates pro-oxidative activity under certain conditions, conditions that are likely associated with toxicity and genotoxicity [2]. In a previous study, we evaluated TEMPO-induced genotoxicity and also investigated the intracellular production of reactive oxygen species (ROS). We found that there was oxidative stress in TEMPO-treated cells [15]. Oxidative stress, due to overproduction of ROS and depletion of antioxidant capacity, is reported to have destructive effects on lipids, DNA, RNA, and proteins [16].
Generation of ROS in response to various stimuli may trigger apoptosis under both physiological and pathological conditions [17]. The B-cell lymphoma-2 (Bcl-2) protein family, composed of pro-apoptotic members such as Bax, Bak, Bid, Bik, Bim, and Puma, and anti-apoptotic members such as Bcl-2, Bcl-xL, and Mcl-1, are well-characterized factors in regulating apoptotic cell death [18,19]. The balance of expression between pro- and anti-apoptotic signals determines cell fate, i.e., cell death from apoptosis, from necrosis, or survival [20]. ROS, generated in mitochondria and other sources, have been shown to be mediators of apoptosis, cell proliferation, and cell death [17]. Several studies have reported that perturbation in the expression of Bcl-2 family proteins mediates ROS-induced apoptosis, which occurs through various pathways [21–24].
Mitogen-activated protein kinase (MAPK) is one of the reported signaling pathways associated with ROS and apoptosis [25,26]. MAPKs are essential for transducing extracellular signals to the intracellular machinery that regulates cellular processes [27]. Mammalian cells have three well-defined subgroups of MAPKs: the extracellular signal regulated kinases (ERKs), the c-Jun N-terminal kinases (JNKs), and the p38 MAP kinases. The last two subgroups can be activated by apoptosis signal-regulating kinase 1 (ASK1), a member of the MAP3K family. MAPKs can be activated by phosphorylation of their specific substrates at serine and/or threonine residues in response to a wide range of environmental stimuli, including DNA damage, ischemia, UV irradiation, and oxidative stress [28]. A close correlation has been reported between MAPK signaling pathway activation and oxidative stress in chemical-induced toxicity; and recent studies have demonstrated that ROS-induced DNA damage as an upstream event that may cause JNK activation through various pathways [29].
Previously, we have demonstrated that TEMPO can induce oxidative stress in mouse lymphoma L5178Y cells [15]. Extending this earlier work, the present study investigates the underlying mechanisms of TEMPO-associated toxicity, particularly the roles of ROS and MAPK signaling. Our results demonstrate that TEMPO induces oxidative stress and apoptosis in a concentration-dependent manner. These toxic effects could be reduced by the addition of an ROS scavenger or a JNK-specific inhibitor, thereby demonstrating that TEMPO-induced apoptosis and cytotoxicity are, at least in part, mediated by oxidative stress and activation of JNK in the MAPK pathway.
2. Materials and methods
2.1. Materials and reagents
2,2,6,6-Tetramethylpiperidine 1-oxyl (TEMPO, C9H18NO, CAS #2564-83-2), dimethyl sulfoxide (DMSO), and N-acetyl-L-cysteine (NAC) were purchased from Sigma (St. Louis, MO). MnTBAP chloride {4-[10,15,20-tris(4-Carboxyphenyl)porphyrin-22,24-diid-5-yl]benzoic acid manganese(III) chloride} was purchased from Abcam (Cambridge, MA). Fischer’s medium was obtained from Quality Biological (Gaithersburg, MD). 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA), and all other cell culture supplies were acquired from Invitrogen Life Technologies (Carlsbad, CA). For Western blotting assay, the primary antibodies against Bcl-2 (#2870), Bcl-xL (#2764), Mcl-1 (#5453), Bax (#2772), JNK (#9258), phospho-JNK (p-JNK) (#4668), p38 (#8690), phospho-p38 (p-p38) (#4511), ERK1/2 (#4695), phospho-ERK1/2 (p-ERK1/2, #4370), phospho-histone H2A.X (γ-H2A.X, #2577), caspase-3 (#9665), and cleaved caspase-3 (#9664) were purchased from Cell Signaling Technology (Danvers, MA). GAPDH (#sc-365062) and β-actin (#A2228) were obtained from Santa Cruz (Dallas, TX) and Sigma, respectively. The Pierce BCA Protein Assay kit and RIPA buffer were obtained from Thermo Scientific (Rockford, IL).
2.2. Cell culture and treatment with TEMPO
The L5178Y/Tk+/− 3.7.2C mouse lymphoma cell line was used for this study. The cells were cultured in Fischer’s medium for leukemic cells of mice containing L-glutamine and supplemented with penicillin (100 units/mL), streptomycin (100 μg/mL), pluronic F68 (0.1%), sodium pyruvate (1 mM), and heat-inactivated horse serum (10%). TEMPO working solutions (100×) were prepared just prior to use by dissolving TEMPO in DMSO. Unless stated otherwise, 6 × 106 cells in a total volume of 10 mL medium with 5% horse serum were exposed to different concentrations of TEMPO or vehicle control (DMSO) for 4 h. The final concentration of DMSO in the medium was 1%. After exposure, the cells were harvested immediately for use in the following assays.
2.3. Cell viability assay
The alamarBlue assay (Invitrogen Life Technologies, Carlsbad, CA) uses the reducing power of living cells to quantify cell viability and proliferation. Following a 4-h exposure, 100 μL of cells treated with TEMPO at various concentrations were seeded in the wells of 96-microwell plates in quadruplicates, and 10 μL alamarBlue reagent were added to each well. The plates were incubated at 37 °C in a humidified incubator with 5% CO2 in air for 1 h. Fluorescence was measured using wavelengths of 530 nm for excitation and 590 nm for emission in a Synergy H4 Hybrid multimode microplate reader (BioTek, Winooski, VT).
2.4. Measurement of cellular ATP levels and caspase 3/7 activities
The CellTiter-Glo Luminescent Cell Viability Assay and the Caspase-Glo 3/7 Assay (Promega, Madison, WI) were used to determine ATP levels and caspase 3/7 activities, respectively, in TEMPO-treated L5178Y cells. According to the manufacturer’s manual, after the 4-h treatment, 3 × 104 or 1 × 104 cells (for the ATP assay or caspase 3/7assay, respectively) were dispensed into the wells of a 96-well white flat-bottomed plate in quadruplicate. Fifty microliters of reaction reagent were added into each well. Following a short incubation, luminescence was recorded with a Synergy H4 Hybrid microplate reader.
2.5. Detection of mitochondria membrane potential
JC-1dye was used to determine changes in mitochondrial membrane potential (MMP). JC-1 is a membrane-permeable lipophilic cationic fluorochrome. Its accumulation in mitochondria is membrane-potential-dependent. Under high mitochondrial potential, JC-1 forms a red-fluorescent aggregate (dimer) spontaneously [30]. When the mitochondrial membrane depolarizes, JC-1 cannot be taken up by the mitochondria and remains in the cytoplasm as monomers (green), resulting in a decrease in the red/green ratio. Following treatment, cells were harvested, resuspended in basic medium containing 2.5 μg/mL JC-1dye, and incubated in the dark for 30 min at 37 °C. Then the cells were washed twice and resuspended in PBS, and fluorescence was read using a Synergy H4 Hybrid microplate reader. The ratio of JC-1 dimers to monomers was calculated as a measure of MMP.
2.6. The apoptosis and necrosis assay
The fractions of apoptotic and necrotic cells were quantified using the FITC Annexin V Apoptosis Detection Kit I (BD Biosciences, San Jose, CA) following the 4-h treatment. Briefly, cells were collected and washed twice with cold PBS, and then resuspended in binding buffer at a concentration of 1 × 106 cells/mL. One hundred microliters of cell suspension were incubated with 5 μL FITC Annexin V and 5 μL propidium iodide (PI) for 15 min at room temperature in the dark. The stained cells were diluted with 400 μL of binding buffer and analyzed on a FACSCantoII flow cytometer (BD Biosciences).
2.7. Measurement of ROS
Intracellular ROS production was assessed using H2DCF-DA staining [15]. Cells were treated with 5 μM H2DCF-DA for 30 min at 37 °C in the dark. The cells were washed with PBS to remove unincorporated dye, and then treated with 0.1–5 mM TEMPO in phenol-red-free medium. Subsequently, the treated cells were transferred into a 96-well plate at a concentration of 1 × 104-cells/well, using four replicate wells per treatment condition. Initially, fluorescence was measured within 10 min of treatment using wavelengths of 485 nm for excitation and 528 nm for emission. The plate was incubated at 37 °C with 5% CO2 and the fluorescence intensity was measured at 1-h and 4-h time points on Synergy reader.
2.8. Measurement of glutathione levels
The intracellular reduced glutathione (GSH) levels were measured immediately after the 4-h treatment of L5178Y cells with different concentrations of TEMPO using GSH-Glo Assay kit (Promega) as described previously [15]. Total glutathione, including GSH and oxidized glutathione (GSSG), and GSH/GSSG ratios in treated cells were measured using GSH/GSSG-Glo Assay kit (Promega) as described by the manufacturer.
2.9. Western blot analysis
Western blot analysis was performed as described previously [25]. Briefly, following the 4-h treatment, cells were washed with cold PBS and lysed with RIPA buffer containing 1× Halt Protease Inhibitor Cocktail (Thermo Fisher Scientific, Rockford, IL). A Pierce BCA protein assay kit (Thermo Fisher Scientific) was used to determine the protein concentration in cell lysates. The primary antibodies were against Bcl-2, Bcl-xL, Mcl-1, Bax, JNK, p-JNK, p38, p-p38, ERK1/2, p-ERK1/2, γ-H2A.X, caspase-3, and cleaved caspase-3. GAPDH and β-actin were used as internal controls. Following an incubation with the horseradish peroxidase-conjugated secondary antibody (goat anti-mouse or anti-rabbit monoclonal IgG), the protein signals were detected by chemiluminescence and imaged with FluorChem E System (ProteinSimple, San Jose, CA). The intensity of each band was also quantified with FluorChem E System.
2.10. Data analysis
All data are presented as mean ± standard deviation (SD) from at least three independent experiments. Statistical analyses were performed using SigmaPlot 11.0 (Systat Software, San Jose, CA). Differences between groups were evaluated by one-way analysis of variance (ANOVA) followed by Dunnett’s test for pairwise comparisons to vehicle control. A t-test was used for paired comparison of two treatment groups. The differences were considered statistically significant when the p value was <0.05.
3. Results
3.1. TEMPO induces cytotoxicity in L5178Y cells
The cytotoxicity of TEMPO was assessed by cell proliferation measurement using the reducing power of living cells (alamarBlue assay); and in parallel, ATP levels were determined in order to evaluate energy metabolism. Treatment of TEMPO for 4 h decreased cellular ATP content significantly, with the effect starting at 2 mM and decreasing in a concentration-dependent manner (Fig. 1A). A significant decrease in cell viability was observed only at 5 mM, the highest concentration tested in this study (Fig. 1B). These results indicate that ATP depletion occurs prior to a reduction in L5178Y cell proliferation, which implies that mitochondrial dysfunction is an early effect of TEMPO, because ATP is produced in the mitochondria. Accordingly, we tested whether or not TEMPO causes mitochondrial disruption. The decrease in the ratio of red/green fluorescence with increasing concentration of TEMPO indicates that TEMPO decreases mitochondrial membrane potential (Fig. 1C).
Fig. 1.
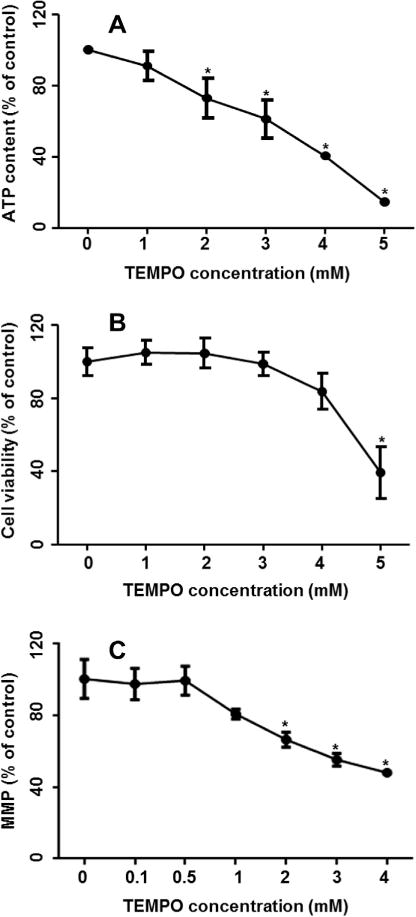
Effect of TEMPO on ATP content, cell viability, and mitochondrial membrane potential in L5178Y cells. Cells were exposed to various concentrations of TEMPO for 4 h; the cellular ATP content (A) and cell viability (B) were measured by the CellTiter-Glo luminescent assay and the alamarBlue assay, respectively. (C) Mitochondrial membrane potential (MMP) was determined by JC-1 staining. The data points represent the mean ± 1 SD from three independent experiments with 4 parallel samples per concentration in each experiment. * indicates p < 0.05 compared to the vehicle control.
3.2. TEMPO induces apoptosis in L5178Y cells with reduction of anti-apoptotic proteins
Disruption of mitochondrial function is one of the distinctive characteristics of apoptosis, so it was of interest to test if TEMPO induces apoptosis. A battery of apoptosis tests was employed, including flow cytometric analyses, caspase-3/7 activity measurements, and the determination of caspase-3, pro-apoptotic and anti-apoptotic proteins. Flow cytometric analysis demonstrated that the percentage of apoptotic cells increased in TEMPO-treated cultures in a concentration-related manner (Fig. 2A). Following a 4-h treatment, 48% and 72% of L5178Y cells underwent apoptosis when exposed to 4 and 5 mM TEMPO, respectively (Fig. 2B). Concentrations of 3 mM TEMPO or less did not induce significant apoptosis in treated cells.
Fig. 2.
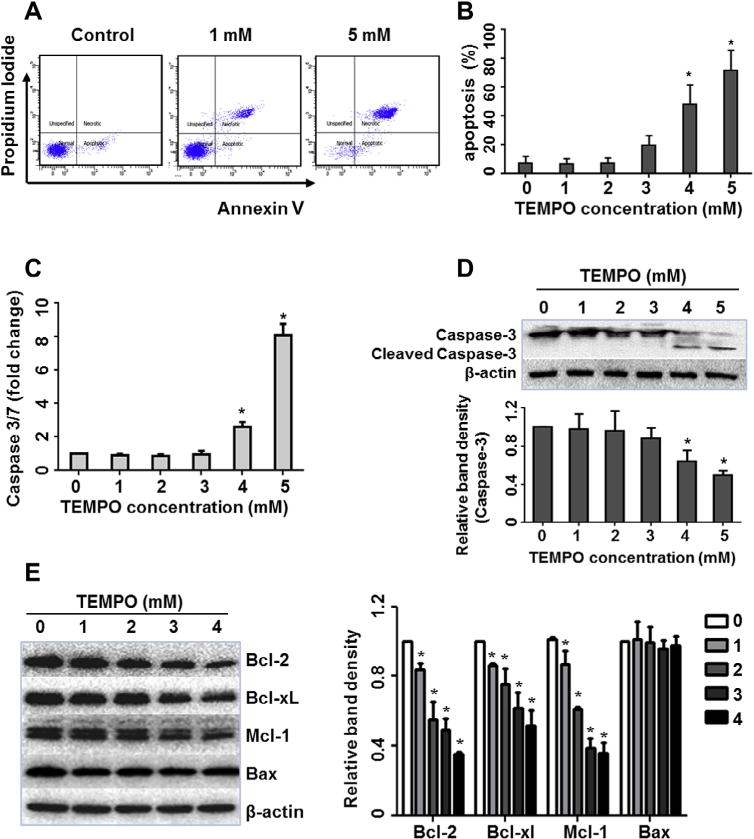
TEMPO induced apoptosis in L5178Y cells. Cells were exposed to different concentrations of TEMPO for 4 h. (A) Representative flow cytometry plots are shown from one experiment. Cells were stained with Annexin V/PI and apoptotic cells were quantified by flow cytometric analysis. (B) The bar graph depicts the mean percentages of apoptotic cells (±1 SD) from three independent experiments, as a function of TEMPO concentration. (C) Histogram shows the caspase 3/7 activity as fold-increase relative to the vehicle control groups from three independent experiments (mean ± SD). (D and E) Western blot shows the protein expression of caspase-3 and cleaved caspase-3 (D), and Bcl-2 family members (E) in treated cells. β-Actin was used as a loading control. The bar graphs represent the densitometric analysis of caspase-3 (D) and Bcl-2 family members (E) from three independent experiments (mean ± SD). Intensities of bands were normalized to the amount of β-actin. * indicates p < 0.05 compared to the vehicle control.
To examine whether or not caspase activation is involved in the apoptotic effect of TEMPO, we measured the enzymatic activity of caspase-3/7 and determined the protein expression of caspase-3 and cleaved caspase-3. Caspase-3/7 activity was significantly increased at 4 mM TEMPO; and at 5 mM, TEMPO induced an 8-fold increase of caspase-3/7 activity as compared to the vehicle control (Fig. 2C). In parallel, by Western blot analysis, the cleaved form of caspase-3 was markedly increased at ≫4 mM TEMPO, with a concentration-dependent caspase-3 decrease in treated cells (Fig. 2D).
Next we examined the expression of Bcl-2 family proteins because the balance between the pro- and anti-apoptotic members of Bcl-2 family regulates caspase activation and determines cellular fate [31]. Western blot analysis showed that TEMPO induced concentration-dependent decreases in the expression of three anti-apoptotic proteins (Bcl-2, Bcl-xL and Mcl-1), whereas no change was observed in pro-apoptotic Bax expression (Fig. 2E), indicating that TEMPO-induced apoptosis is likely due to reduced expression of anti-apoptotic members.
3.3. TEMPO exhibits pro-oxidant activities
Our previous study demonstrated that significant increases in intracellular ROS production and decreases in GSH levels were observed in L5178Y cells exposed to 1–3 mM TEMPO [15]. To investigate whether or not TEMPO induces oxidative stress at lower concentrations, we expanded the treatment range of TEMPO concentrations from 0.1 mM to 5 mM (Fig. 3). The results showed that TEMPO induced time- and concentration-dependent ROS production (Fig. 3A) and GSH depletion over the concentration-range tested (Fig. 3B). ROS levels increased significantly within 10 min of treatment with 1 mM TEMPO. The intracellular ROS level in high concentration treatments (3 and 5 mM) increased dramatically, reaching a peak level (about 7-fold) after a 1-h treatment. Following a 4-h treatment, ROS levels were increased significantly in cells exposed to ≫0.5 mM TEMPO (Fig. 3A) and reached the maximal induction level (5-fold) with 1 mM TEMPO treatment. The induction declined slightly for the 3 mM and 5 mM treatments, presumably due to reduced cell growth (Fig. 1). In parallel, GSH content was measured and significant concentration-dependent GSH depletion was observed at concentrations ≫1 mM (Fig. 3B).
Fig. 3.
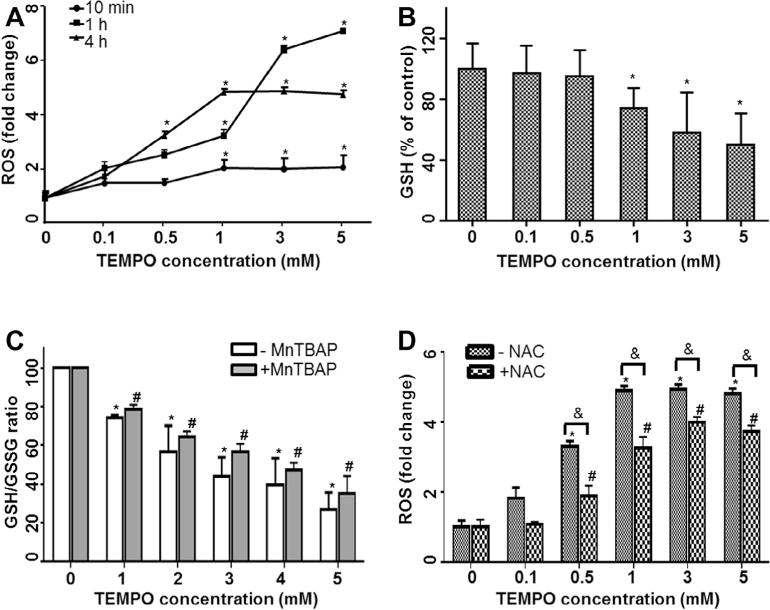
TEMPO induced ROS generation and GSH depletion in L5178Y cells. (A) The reactive oxygen species (ROS) values were measured at 10 min, 1 h, and 4 h after adding different concentrations of TEMPO to L5178Y cells. (B) Intracellular glutathione (GSH) levels were determined after a 4-h treatment with TEMPO. (C) GSH/GSSG ratios were calculated after 4-h TEMPO treatments with and without 1-h pretreatment of 50 μM MnTBAP. (D) Intracellular ROS levels were measured after a 4-h TEMPO treatment with and without 1-h pretreatment of 5 mM NAC. The data points represent the mean ± 1 SD from at least three independent experiments with four parallel samples per concentration in each experiment. * and # indicate p < 0.05 compared to the vehicle control without or with antioxidant pretreatment, respectively. & indicates p < 0.05 between the treatments at the same concentration of TEMPO with and without antioxidant pretreatment.
In order to clarify whether GSH depletion is due to oxidation or conjugation with TEMPO, we determined GSH/GSSG ratios in cells exposed to TEMPO both with and without 1-h pretreatment with 50 μM MnTBAP, a cell-permeable superoxide dismutase mimetic agent. In agreement with the GSH content measurement, GSH/GSSG ratios in cells treated with TEMPO declined in a concentration-dependent manner, and antioxidant MnTBAP slightly increased the ratios at concentrations tested, but no significant difference was observed between cell cultures treated with the same concentrations of TEMPO (Fig. 3C).
The effects of the ROS scavenger NAC on intracellular ROS production also were assessed in cells exposed to TEMPO for 4 h. Pretreatment with 5 mM NAC for 1 h significantly reduced the ROS levels observed in cultures treated with TEMPO at concentrations of 0.5–5 mM. The maximum ROS level was increased less than 4-fold with NAC pretreatment, while a greater than 5-fold increase was observed with TEMPO alone (Fig. 3D).
3.4. Effects of NAC on cell viability, apoptosis, and Bcl-2 protein expression in treated cells
To further examine the role of ROS in TEMPO-induced apoptosis, cells were pretreated with NAC for 1 h prior to treatment with TEMPO. The ROS scavenger significantly blocked the decline in ATP levels (Fig. 4A), suppressed the activation of caspase-3/7 (Fig. 4B); also, it reduced the fraction of apoptotic cells as measured by flow cytometric analysis (Fig. 4C) and cell death in cultures treated with high concentrations of TEMPO (Supplementary Fig. 1). In addition, the decrease in Bcl-2 expression caused by TEMPO was partially reversed by 5 mM NAC (Fig. 4D).
Fig. 4.
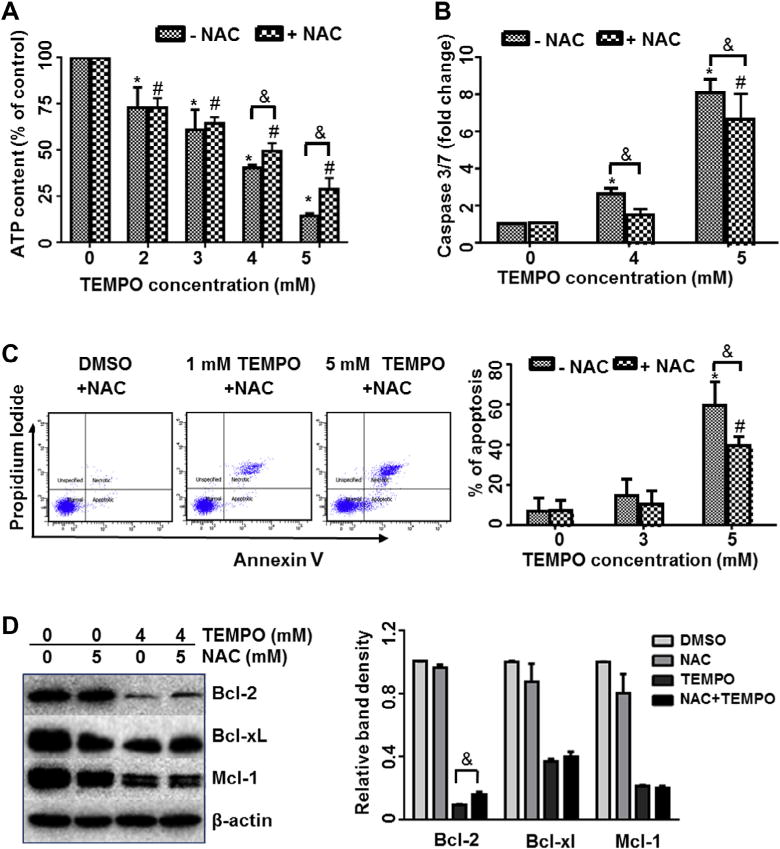
Effects of NAC on ATP content (A), caspase 3/7 activity (B), apoptosis (C), and anti-apoptotic Bcl-2 family protein expressions (D) in TEMPO-treated cells. After 1-h pretreatment with or without 5 mM NAC, cells were treated with TEMPO at different concentrations for 4 h. The data points represent the mean ± 1 SD from at least three independent experiments with four parallel samples per concentration in each experiment. Representative flow cytometry plots are shown from one experiment (C). The bar graph (D) represents the densitometric analysis of Bcl-2 family members from three independent experiments. Intensities of bands were normalized to the amount of β-actin. * and # indicate p < 0.05 compared to the vehicle control without or with 5 mM NAC pretreatment, respectively. & indicates p < 0.05 between the treatments at the same concentration of TEMPO with and without NAC pretreatment.
3.5. ROS are responsible for JNK activation by TEMPO
Many studies have shown that ROS overproduction leads to activation of multiple signaling pathways, including the MAPK pathway [32,33]. We examined the activation (phosphorylation) of three key members of the MAPK signaling pathway (i.e., JNK, p38, and ERK1/2) in L5178Y cells treated with TEMPO. Phosphorylated-JNK (p-JNK) was significantly increased in TEMPO-treated cultures in a concentration-dependent manner; and phosphorylated p38 (p-p38) also showed a significant increase with increasing TEMPO concentration but not as much as p-JNK. There was no discernable activation of ERK1/2; in fact, phosphorylated ERK1/2 was not detected in treated cells (Fig. 5A). These results indicate that TEMPO may regulate the MAPK signaling pathway, with JNK being a major effector.
Fig. 5.
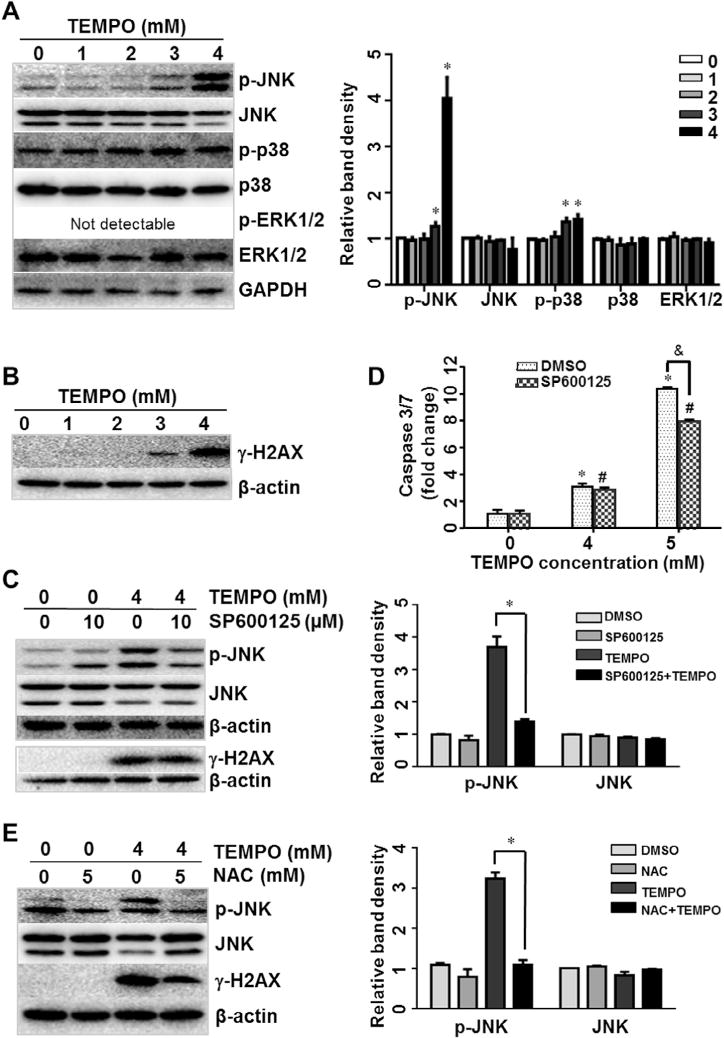
Expression of MAPK signaling pathway proteins and effects of SP600125 on TEMPO-induced caspase 3/7 activity in L5178Y cells. (A and B) Total cellular proteins were extracted after a 4-h exposure to TEMPO. The levels of activated JNK (phospho-JNK), p38 (phospho-p38), ERK1/2 (phospho-ERK1/2) (A), and γ-H2AX (B) were determined by Western blot analyses. GAPDH or β-actin was analyzed as a loading control. (C) The cells were pretreated for 2 h with 10 μM SP600125, a JNK-specific inhibitor, and levels of phospho-JNK were measured in TEMPO treated cells. (D) Effects of SP600125 on apoptosis were evaluated by caspase 3/7 activities. The bar graphs show the mean ± 1 SD of three experiments. * and # indicate p < 0.05, compared to the vehicle control without or with 2-h pretreatment of 10 μM SP600125, respectively. & indicates p < 0.05 between the TEMPO treatments with and without SP600125 pretreatment. (E) Effects of NAC on the expression of phospho-JNK and γ-H2AX were determined by Western blot analyses. The bar graphs (A, C, and E) represent the densitometric analysis of protein expression from three independent experiments. Intensities of bands were normalized to the amount of loading control. ⁄p < 0.05.
Recent studies revealed that physical DNA breakage may lead to JNK activation and downstream consequences [29]. We examined the expression of γ-H2A.X protein, a hallmark of DNA damage. Following a 4-h treatment, TEMPO significantly increased the expression of γ-H2A.X at concentrations starting at 3 mM (Fig. 5B).
A specific inhibitor of JNK (SP600125) was further used to investigate the role of JNK activation in TEMPO-associated toxicity and its relationship to different cellular events, such as MAPK pathway signaling and apoptosis. As expected, pretreatment with 10 μM SP600125 caused a remarkable suppression of JNK activation (Fig. 5C). SP600125 significantly inhibited the caspase-3/7 activation in TEMPO-treated cells, suggesting JNK has a role in TEMPO-induced apoptosis (Fig. 5D). However, SP600125 did not significantly change the expression of γ-H2A.X (Fig. 5C), probably due to SP600125 did not inhibit DNA damage formation. On the other hand, pretreatments with a ROS scavenger NAC reversed TEMPO-induced DNA damage and JNK phosphorylation (Fig. 5E), demonstrating an interaction between ROS, DNA damage, and JNK activation. Addition of the JNK inhibitor did not significantly block TEMPO-induced ATP depletion (Supplementary Fig. 2) or its effect on cell viability (Supplementary Fig. 3). This suggests that other cellular events, in addition to DNA damage and MAPK signaling pathway, contribute to TEMPO’s toxicity.
4. Discussion
The anti-oxidative properties of TEMPO and its derivatives are well-accepted and their ability to scavenge ROS has been evaluated in in vitro and in vivo models for potential medical applications, such as prevention and treatment of cancer, hypertension, and glaucoma [34–36]. However, its pro-oxidative activities may compromise TEMPO’s beneficial effects [15,37,38]. In the current study, we investigated the role of ROS in TEMPO-induced toxicity and the underlying molecular mechanisms of toxicity, with the aim of providing essential information for in-depth understanding the adverse effects of TEMPO and eventually developing strategies to minimize the adverse effects and enable the safe use of TEMPO.
The present study showed that a 4-h treatment with 0.1 mM TEMPO induced a 2-fold increase in ROS level over control (Fig. 3A), and 0.5 mM TEMPO produced a measurable significant increase in intracellular ROS, with no other observable effects (i.e., changes in GSH level, cell proliferation, apoptosis, or protein expression) (Figs. 1–3). One millimolar TEMPO induced significant concentration-dependent GSH depletion and increased ROS levels, while ATP depletion was observed at concentrations ≫2 mM. These results indicate that ROS generation is an early event in cells treated with TEMPO. This is in agreement with a previous report showing that, in human leukemia U937 cells treated with TEMPO for 30 min, 10 mM TEMPO causes an early transient elevation of and a late induction of only , coupled with a slight decrease in GSH and 30–50% reductions in ATP levels [39]. ROS formation and GSH depletion are involved in chemically induced apoptosis in a variety of cell types [40–43]. Our study demonstrated a close relationship between TEMPO-induced ROS formation and apoptosis, evidenced by the fact that NAC pretreatment significantly reduced ROS production, caspase 3/7 activation, and the fraction of apoptotic cells (Figs. 3D, 4B and C). Next we investigated whether cellular GSH was depleted via oxidation or conjugation with TEMPO, because TEMPO has been shown to form conjugations and adducts, especially with cysteine residue [44], one of the components in GSH. We found that the declined GSH/GSSG ratios in TEMPO-treated cells were slightly reversed by pretreatment with MnTBAP (Fig. 3C), a cell-permeable superoxide dismutase mimetic agent, indicating that both ROS oxidation and chemical conjugation may account for the depletion of GSH in treated cells.
The Bcl-2 protein family members are critical regulators of apoptosis, acting as upstream regulators of caspases and mitochondrial function [18,19]. Maintaining a high Bcl-2/Bax ratio is important for cell survival [20]. The increased expression of pro-apoptotic proteins such as Bax is reportedly involved in apoptosis [45]; however, our Western blot results showed that the expression of Bax was not affected by TEMPO (Fig. 2E). In contrast, the expression of three anti-apoptotic proteins, Bcl-2, Bcl-xL, and Mcl-1, was inhibited by TEMPO in a concentration-dependent manner (Fig. 2E). These results suggest that the suppression of anti-apoptotic members rather than activation pro-apoptotic members of the Bcl-2 family proteins is responsible for the subsequent caspase-3 activation and induction of apoptosis [46]. Down-regulation of Bcl-2, the key protein in anti-apoptotic family, is particularly important in ROS-related apoptosis induced by TEMPO, because the expression of Bcl-2 was partially reversed by NAC (Fig. 4D). Bcl-2 cysteine oxidation caused by ROS overproduction, as well as TEMPO conjunction with Bcl-2 cysteine residue, may serve as potential mechanisms of Bcl-2 down-regulation in apoptosis [22,47].
It is known that ROS initiates apoptosis and activates various signaling pathways, with the cascaded reactions and crosstalk among multiple cellular events contributing to toxicity associated with certain chemicals [25,48–54]. In this present study, we found that two members of the MAPK signaling pathway, JNK and p38, were activated at high and moderate levels, respectively (Fig. 5A). This is consistent with a previous report showing that in human breast cancer cells, a treatment with 10 mM TEMPO resulted in a concentration-dependent significant increase in the level of phosphorylated stress-activated protein kinase SAPK/JNK and no change in ERK1 activity [14]. In many conditions involving the induction of apoptosis, high level of JNK activity and suppression of ERK activity have been reported [55,56]. The dynamic balance between stress-activated JNK and p38 and growth-activated ERK pathways determines a cell’s fate (survival, apoptosis, or necrosis) [57]. In our study, strong induction of JNK, mild induction of p38, and no activation of ERK1 were observed, suggesting that JNK may be the major effector in TEMPO-induced toxicity. The involvement of JNK in TEMPO-induced apoptosis was further supported by analysis of JNK inhibition, which demonstrated that TEMPO-induced apoptosis was partially attenuated by the JNK-specific inhibitor SP600125 (Fig. 5D). Since the activation of JNK is reported to be responsible for Bcl-2/Bcl-xL phosphorylation in various cell lines with Bcl-2 as a key target [58,59], our results (Figs. 2, 4 and 5) suggested that TEMPO induces apoptosis in L5178Y cells likely through JNK-mediated Bcl-2/Bcl-xL pathway.
Overproduction of ROS can result in DNA damage, and physical DNA breakage has been linked to JNK activation [29]. To further explore the relationship between ROS, DNA damage, the MAPK pathway, and apoptosis, we examined protein expression of γ-H2A.X and the effects of a ROS scavenger (NAC) and a JNK inhibitor (SP600125) on the expression of γ-H2A.X and JNK. TEMPO significantly increased expression of γ-H2A.X in a concentration-dependent manner (Fig. 5B). NAC significantly reduced the expression of both γ-H2A.X and JNK (Fig. 5E) because of its anti-oxidative property. However, SP600125 only caused a remarkable suppression of JNK activation and did not change the expression of γ-H2A.X (Fig. 5C), probably due to SP600125 cannot reduce TEMPO-induced DNA damage. These results are consistent with the observation that NAC significantly increased ATP levels (Fig. 4A) and cell viability (Supplementary Fig. 1) in TEMPO-treated cell cultures, but SP600125 did not (Supplementary Figs. 2 and 3). Taken together, the MAPK pathway is involved in ROS-mediated TEMPO-induced apoptosis, although other signaling pathways also may participate in TEMPO-induced cell death.
The present study addressed the relationship between the pro-oxidative properties of TEMPO and apoptosis, as well as changes in the levels of regulators involved in this process. Pretreatment with NAC provided protection against TEMPO-induced ROS generation, ATP depletion, phosphorylation of JNK, caspase-3/7 activation, down-regulation of Bcl-2 anti-apoptotic protein, and apoptosis in L5178Y cells. Fig. 6 depicts the relationships between the multiple interrelated cellular responses induced by TEMPO. Following treatment of TEMPO, early accumulation of ROS triggers GSH depletion, which subsequently may induce more ROS generation. ROS signaling, likely combined with GSH depletion, can induce apoptosis either directly or via activation of MAPK pathways, especially JNK. Down-regulation of anti-apoptotic Bcl-2 is the most likely contributor to TEMPO-induced apoptosis.
Fig. 6.
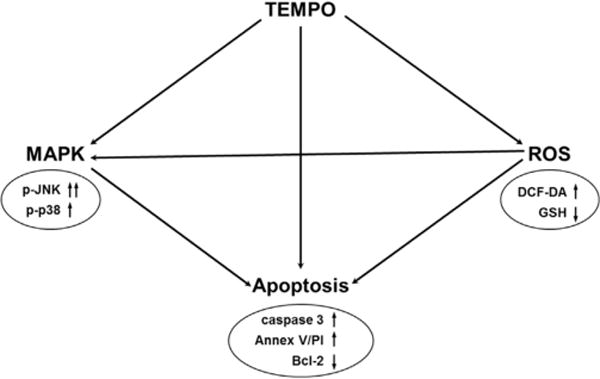
Proposed mechanism for ROS-mediated TEMPO-induced apoptosis in L5178Y cells.
It is worth mentioning that our previous study showed that TEMPO was genotoxic in the micronucleus assay and in the mouse lymphoma assay at concentrations of 1.4 mM and 2.5 mM, respectively [15]. The present study demonstrated increases in ROS generation at concentrations as low as 0.1 mM and apoptosis at concentrations of 4 mM and above. These observations indicate that TEMPO may cause genotoxicity at concentrations lower than those at which it can induce apoptosis. Previously, TEMPO was proposed as a novel antineoplastic agent because of its ability to induce apoptosis and cell cycle arrest in prostate carcinoma cells in vitro models and to inhibit tumor growth in LNCaP tumor-bearing mice [60]. Specifically, 24–48 h treatments with up to 5 mM TEMPO caused significant increases in the number of apoptotic cells and decreased cell viability in prostate carcinoma DU-145, PC-3, and LNCaP cells, and increased caspase-3 and caspase-9 activities in LNCaP cells. Our study raises the question of whether TEMPO is an appropriate candidate for cancer therapy. Although different cell lines possess different sensitivities and responses, our study suggests that TEMPO may be relatively safe as an antioxidant at low concentrations (probably < 0.1 mM), where no significant effects on cell viability, GSH level, or expression of apoptotic signaling pathway molecules are observed. However, at high concentrations, TEMPO induces significant genotoxicity [15], ROS generation, apoptosis, and cell death.
In summary, the present study demonstrates that TEMPO induces apoptosis in L5178Y cells at least partially via a ROS-mediated DNA damage-JNK pathway. Due to the potential widespread use of TEMPO as an anti-oxidant, awareness of TEMPO-induced genotoxicity, oxidative stress, and apoptosis is important for its safe application in the future.
Supplementary Material
Acknowledgments
This research was partly supported by an appointment (S.C. and Z.Z.) to the Postdoctoral Research Program at the National Center for Toxicological Research (NCTR) administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U.S. Department of Entergy and the U.S. Food and Drug Administration (FDA). We thank Drs. Barbara L. Parsons and Page B. Mckinzie for their helpful discussions and comments. The information in this paper is not a formal dissemination of information by the U.S. FDA and does not represent the agency position or policy.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cbi.2015.04.009.
Footnotes
The information in this manuscript is not a formal dissemination of information by the U.S. Food and Drug Administration and does not represent the agency position or policy.
Conflict of Interest
The authors declare that there are no conflicts of interest.
Transparency Document
The Transparency document associated with this article can be found in the online version.
References
- 1.Dragutan I, Mehlhorn RJ. Modulation of oxidative damage by nitroxide free radicals. Free Radical Res. 2007;41:303–315. doi: 10.1080/10715760601089356. [DOI] [PubMed] [Google Scholar]
- 2.Glebska J, Skolimowski J, Kudzin Z, Gwozdzinski K, Grzelak A, Bartosz G. Pro-oxidative activity of nitroxides in their reactions with glutathione. Free Radical Biol Med. 2003;35:310–316. doi: 10.1016/s0891-5849(03)00306-x. [DOI] [PubMed] [Google Scholar]
- 3.Krishna MC, Samuni A. Nitroxides as antioxidants. Methods Enzymol. 1994;234:580–589. doi: 10.1016/0076-6879(94)34130-3. [DOI] [PubMed] [Google Scholar]
- 4.Risso-de Faverney C, Lafaurie M, Girard JP, Rahmani R. The nitroxide stable radical tempo prevents metal-induced inhibition of CYP1A1 expression and induction. Toxicol Lett. 2000;111:219–227. doi: 10.1016/s0378-4274(99)00183-6. [DOI] [PubMed] [Google Scholar]
- 5.Udassin R, Haskel Y, Samuni A. Nitroxide radical attenuates ischaemia/reperfusion injury to the rat small intestine. Gut. 1998;42:623–627. doi: 10.1136/gut.42.5.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xavier S, Yamada K, Samuni AM, Samuni A, DeGraff W, Krishna MC, Mitchell JB. Differential protection by nitroxides and hydroxylamines to radiation-induced and metal ion-catalyzed oxidative damage. Biochim Biophys Acta. 2002;1573:109–120. doi: 10.1016/s0304-4165(02)00339-2. [DOI] [PubMed] [Google Scholar]
- 7.Cuscela D, Coffin D, Lupton GP, Cook JA, Krishna MC, Bonner RF, Mitchell JB. Protection from radiation-induced alopecia with topical application of nitroxides: fractionated studies. Cancer J Sci Am. 1996;2:273–278. [PubMed] [Google Scholar]
- 8.Pattison DI, Lam M, Shinde SS, Anderson RF, Davies MJ. The nitroxide TEMPO is an efficient scavenger of protein radicals: cellular and kinetic studies. Free Radical Biol Med. 2012;53:1664–1674. doi: 10.1016/j.freeradbiomed.2012.08.578. [DOI] [PubMed] [Google Scholar]
- 9.Gelvan D, Saltman P, Powell SR. Cardiac reperfusion damage prevented by a nitroxide free radical. Proc Natl Acad Sci USA. 1991;88:4680–4684. doi: 10.1073/pnas.88.11.4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nagasaki Y. Nitroxide radicals and nanoparticles: a partnership for nanomedicine radical delivery. Ther Delivery. 2012;3:165–179. doi: 10.4155/tde.11.153. [DOI] [PubMed] [Google Scholar]
- 11.Yoshitomi T, Nagasaki Y. Nitroxyl radical-containing nanoparticles for novel nanomedicine against oxidative stress injury. Nanomedicine. 2011;6:509–518. doi: 10.2217/nnm.11.13. [DOI] [PubMed] [Google Scholar]
- 12.Gallez B, De Meester C, Debuyst R, Dejehet F, Dumont P. Mutagenicity of nitroxyl compounds: structure–activity relationships. Toxicol Lett. 1992;63:35–45. doi: 10.1016/0378-4274(92)90105-s. [DOI] [PubMed] [Google Scholar]
- 13.Sies H, Mehlhorn R. Mutagenicity of nitroxide-free radicals. Arch Biochem Biophys. 1986;251:393–396. doi: 10.1016/0003-9861(86)90087-1. [DOI] [PubMed] [Google Scholar]
- 14.Suy S, Mitchell JB, Ehleiter D, Haimovitz-Friedman A, Kasid U. Nitroxides tempol and tempo induce divergent signal transduction pathways in MDA-MB 231 breast cancer cells. J Biol Chem. 1998;273:17871–17878. doi: 10.1074/jbc.273.28.17871. [DOI] [PubMed] [Google Scholar]
- 15.Guo X, Mittelstaedt RA, Guo L, Shaddock JG, Heflich RH, Bigger AH, Moore MM, Mei N. Nitroxide TEMPO: a genotoxic and oxidative stress inducer in cultured cells. Toxicol In Vitro. 2013;27:1496–1502. doi: 10.1016/j.tiv.2013.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jacob KD, Hooten N Noren, Trzeciak AR, Evans MK. Markers of oxidant stress that are clinically relevant in aging and age-related disease. Mech Ageing Dev. 2013;134:139–157. doi: 10.1016/j.mad.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simon HU, Haj-Yehia A, Levi-Schaffer F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis. 2000;5:415–418. doi: 10.1023/a:1009616228304. [DOI] [PubMed] [Google Scholar]
- 18.Chao DT, Korsmeyer SJ. BCL-2 family: regulators of cell death. Annu Rev Immunol. 1998;16:395–419. doi: 10.1146/annurev.immunol.16.1.395. [DOI] [PubMed] [Google Scholar]
- 19.Tsujimoto Y. Role of Bcl-2 family proteins in apoptosis: apoptosomes or mitochondria? Genes to cells: devoted to molecular & cellular mechanisms. 1998;3:697–707. doi: 10.1046/j.1365-2443.1998.00223.x. [DOI] [PubMed] [Google Scholar]
- 20.Raisova M, Hossini AM, Eberle J, Riebeling C, Wieder T, Sturm I, Daniel PT, Orfanos CE, Geilen CC. The Bax/Bcl-2 ratio determines the susceptibility of human melanoma cells to CD95/Fas-mediated apoptosis. J Invest Dermatol. 2001;117:333–340. doi: 10.1046/j.0022-202x.2001.01409.x. [DOI] [PubMed] [Google Scholar]
- 21.Han LL, Xie LP, Li LH, Zhang XW, Zhang RQ, Wang HZ. Reactive oxygen species production and Bax/Bcl-2 regulation in honokiol-induced apoptosis in human hepatocellular carcinoma SMMC-7721 cells, Environ. Toxicol Pharmacol. 2009;28:97–103. doi: 10.1016/j.etap.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 22.Hildeman DA, Mitchell T, Aronow B, Wojciechowski S, Kappler J, Marrack P. Control of Bcl-2 expression by reactive oxygen species. Proc Natl Acad Sci USA. 2003;100:15035–15040. doi: 10.1073/pnas.1936213100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hsieh CJ, Kuo PL, Hsu YC, Huang YF, Tsai EM, Hsu YL. Arctigenin, a dietary phytoestrogen, induces apoptosis of estrogen receptor-negative breast cancer cells through the ROS/p38 MAPK pathway and epigenetic regulation. Free Radical Biol Med. 2014;67:159–170. doi: 10.1016/j.freeradbiomed.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 24.Lee JH, Won YS, Park KH, Lee MK, Tachibana H, Yamada K, Seo KI. Celastrol inhibits growth and induces apoptotic cell death in melanoma cells via the activation ROS-dependent mitochondrial pathway and the suppression of PI3K/AKT signaling. Apoptosis. 2012;17:1275–1286. doi: 10.1007/s10495-012-0767-5. [DOI] [PubMed] [Google Scholar]
- 25.Chen S, Xuan J, Wan L, Lin H, Couch L, Mei N, Dobrovolsky VN, Guo L. Sertraline, an antidepressant, induces apoptosis in hepatic cells through the mitogen-activated protein kinase pathway. Toxicol Sci. 2014;137:404–415. doi: 10.1093/toxsci/kft254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li ZJ, Li XM, Piao YJ, Choi DK, Kim SJ, Kim JW, Sohn KC, Kim CD, Lee JH. Genkwadaphnin induces reactive oxygen species (ROS)-mediated apoptosis of squamous cell carcinoma (SCC) cells. Biochem Biophys Res Commun. 2014;450:1115–1119. doi: 10.1016/j.bbrc.2014.06.118. [DOI] [PubMed] [Google Scholar]
- 27.Son Y, Cheong YK, Kim NH, Chung HT, Kang DG, Pae HO. Mitogen-activated protein kinases and reactive oxygen species: how can ROS activate MAPK pathways? J Signal Transduction. 2011;2011:792639. doi: 10.1155/2011/792639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23:2838–2849. doi: 10.1038/sj.onc.1207556. [DOI] [PubMed] [Google Scholar]
- 29.Picco V, Pages G. Linking JNK activity to the DNA damage response. Genes Cancer. 2013;4:360–368. doi: 10.1177/1947601913486347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rogalska A, Koceva-Chyla A, Jozwiak Z. Aclarubicin-induced ROS generation and collapse of mitochondrial membrane potential in human cancer cell lines. Chem Biol Interact. 2008;176:58–70. doi: 10.1016/j.cbi.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 31.Sinha K, Das J, Pal PB, Sil PC. Oxidative stress: the mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch Toxicol. 2013;87:1157–1180. doi: 10.1007/s00204-013-1034-4. [DOI] [PubMed] [Google Scholar]
- 32.Park GB, Choi Y, Kim YS, Lee HK, Kim D, Hur DY. ROS-mediated JNK/p38-MAPK activation regulates Bax translocation in Sorafenib-induced apoptosis of EBV-transformed B cells. Int J Oncol. 2014;44:977–985. doi: 10.3892/ijo.2014.2252. [DOI] [PubMed] [Google Scholar]
- 33.Tang Y, Jacobi A, Vater C, Zou X, Stiehler M. Salvianolic acid B protects human endothelial progenitor cells against oxidative stress-mediated dysfunction by modulating Akt/mTOR/4EBP1, p38 MAPK/ATF2, and ERK1/2 signaling pathways. Biochem Pharmacol. 2014;90:34–49. doi: 10.1016/j.bcp.2014.04.008. [DOI] [PubMed] [Google Scholar]
- 34.Dikalova AE, Bikineyeva AT, Budzyn K, Nazarewicz RR, McCann L, Lewis W, Harrison DG, Dikalov SI. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ Res. 2010;107:106–116. doi: 10.1161/CIRCRESAHA.109.214601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gariboldi MB, Ravizza R, Petterino C, Castagnaro M, Finocchiaro G, Monti E. Study of in vitro and in vivo effects of the piperidine nitroxide Tempol – a potential new therapeutic agent for gliomas. Eur J Cancer. 2003;39:829–837. doi: 10.1016/s0959-8049(02)00742-6. [DOI] [PubMed] [Google Scholar]
- 36.Soule BP, Hyodo F, Matsumoto K, Simone NL, Cook JA, Krishna MC, Mitchell JB. Therapeutic and clinical applications of nitroxide compounds. Antioxid Redox Signal. 2007;9:1731–1743. doi: 10.1089/ars.2007.1722. [DOI] [PubMed] [Google Scholar]
- 37.Balcerczyk A, Grzelak A, Soszynski M, Bartosz G. Pro-oxidative effects of Tempo in systems containing oxidants. Redox Rep. 2004;9:153–159. doi: 10.1179/135100004225004805. [DOI] [PubMed] [Google Scholar]
- 38.Balcerczyk A, Luczak K, Soszynski M, Bartosz G. Prooxidative effects of TEMPO on human erythrocytes. Cell Biol Int. 2004;28:585–591. doi: 10.1016/j.cellbi.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 39.Zhao QL, Fujiwara Y, Kondo T. Mechanism of cell death induction by nitroxide and hyperthermia. Free Radical Biol Med. 2006;40:1131–1143. doi: 10.1016/j.freeradbiomed.2005.10.064. [DOI] [PubMed] [Google Scholar]
- 40.Guha P, Dey A, Sen R, Chatterjee M, Chattopadhyay S, Bandyopadhyay SK. Intracellular GSH depletion triggered mitochondrial Bax translocation to accomplish resveratrol-induced apoptosis in the U937 cell line. J Pharmacol Exp Ther. 2011;336:206–214. doi: 10.1124/jpet.110.171983. [DOI] [PubMed] [Google Scholar]
- 41.Lee JE, Park JH, Shin IC, Koh HC. Reactive oxygen species regulated mitochondria-mediated apoptosis in PC12 cells exposed to chlorpyrifos. Toxicol Appl Pharmacol. 2012;263:148–162. doi: 10.1016/j.taap.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 42.Moungjaroen J, Nimmannit U, Callery PS, Wang L, Azad N, Lipipun V, Chanvorachote P, Rojanasakul Y. Reactive oxygen species mediate caspase activation and apoptosis induced by lipoic acid in human lung epithelial cancer cells through Bcl-2 down-regulation. J Pharmacol Exp Ther. 2006;319:1062–1069. doi: 10.1124/jpet.106.110965. [DOI] [PubMed] [Google Scholar]
- 43.Shen HM, Zhang Z, Zhang QF, Ong CN. Reactive oxygen species and caspase activation mediate silica-induced apoptosis in alveolar macrophages. Am J Physiol Lung Cell Mol Physiol. 2001;280:L10–17. doi: 10.1152/ajplung.2001.280.1.L10. [DOI] [PubMed] [Google Scholar]
- 44.Kitajima S, Kurioka M, Yoshimoto T, Shindo M, Kanaori K, Tajima K, Oda K. A cysteine residue near the propionate side chain of heme is the radical site in ascorbate peroxidase. FEBS J. 2008;275:470–480. doi: 10.1111/j.1742-4658.2007.06214.x. [DOI] [PubMed] [Google Scholar]
- 45.Renault TT, Teijido O, Antonsson B, Dejean LM, Manon S. Regulation of Bax mitochondrial localization by Bcl-2 and Bcl-x(L): keep your friends close but your enemies closer. Int J Biochem Cell Biol. 2013;45:64–67. doi: 10.1016/j.biocel.2012.09.022. [DOI] [PubMed] [Google Scholar]
- 46.Swanton E, Savory P, Cosulich S, Clarke P, Woodman P. Bcl-2 regulates a caspase-3/caspase-2 apoptotic cascade in cytosolic extracts. Oncogene. 1999;18:1781–1787. doi: 10.1038/sj.onc.1202490. [DOI] [PubMed] [Google Scholar]
- 47.Luanpitpong S, Chanvorachote P, Stehlik C, Tse W, Callery PS, Wang L, Rojanasakul Y. Regulation of apoptosis by Bcl-2 cysteine oxidation in human lung epithelial cells. Mol Biol Cell. 2013;24:858–869. doi: 10.1091/mbc.E12-10-0747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen S, Dobrovolsky VN, Liu F, Wu Y, Zhang Z, Mei N, Guo L. The role of autophagy in usnic acid-induced toxicity in hepatic cells. Toxicol Sci. 2014;142:33–44. doi: 10.1093/toxsci/kfu154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen S, Xuan J, Couch L, Iyer A, Wu Y, Li QZ, Guo L. Sertraline induces endoplasmic reticulum stress in hepatic cells. Toxicology. 2014;322:78–88. doi: 10.1016/j.tox.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dong J, Ramachandiran S, Tikoo K, Jia Z, Lau SS, Monks TJ. EGFR-independent activation of p38 MAPK and EGFR-dependent activation of ERK1/2 are required for ROS-induced renal cell death. Am J Physiol Renal Physiol. 2004;287:F1049–1058. doi: 10.1152/ajprenal.00132.2004. [DOI] [PubMed] [Google Scholar]
- 51.Lu H, Tian A, Wu J, Yang C, Xing R, Jia P, Yang L, Zhang Y, Zheng X, Li Z. Danshensu inhibits beta-adrenergic receptors-mediated cardiac fibrosis by ROS/p38 MAPK axis. Biol Pharm Bullet. 2014;37:961–967. doi: 10.1248/bpb.b13-00921. [DOI] [PubMed] [Google Scholar]
- 52.Mao X, Yu CR, Li WH, Li WX. Induction of apoptosis by shikonin through a ROS/JNK-mediated process in Bcr/Abl-positive chronic myelogenous leukemia (CML) cells. Cell Res. 2008;18:879–888. doi: 10.1038/cr.2008.86. [DOI] [PubMed] [Google Scholar]
- 53.Son Y, Kim S, Chung HT, Pae HO. Reactive oxygen species in the activation of MAP kinases. Methods Enzymol. 2013;528:27–48. doi: 10.1016/B978-0-12-405881-1.00002-1. [DOI] [PubMed] [Google Scholar]
- 54.Wu Y, Beland FA, Chen S, Fang JL. Extracellular signal-regulated kinases 1/2 and Akt contribute to triclosan-stimulated proliferation of JB6 Cl 41–5a cells. Arch Toxicol. 2014 doi: 10.1007/s00204-014-1308-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shan R, Price JO, Gaarde WA, Monia BP, Krantz SB, Zhao ZJ. Distinct roles of JNKs/p38 MAP kinase and ERKs in apoptosis and survival of HCD-57 cells induced by withdrawal or addition of erythropoietin. Blood. 1999;94:4067–4076. [PubMed] [Google Scholar]
- 56.Wang X, Martindale JL, Liu Y, Holbrook NJ. The cellular response to oxidative stress: influences of mitogen-activated protein kinase signalling pathways on cell survival. Biochem J. 1998;333(Pt 2):291–300. doi: 10.1042/bj3330291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 58.Fan M, Goodwin M, Vu T, Brantley-Finley C, Gaarde WA, Chambers TC. Vinblastine-induced phosphorylation of Bcl-2 and Bcl-XL is mediated by JNK and occurs in parallel with inactivation of the Raf-1/MEK/ERK cascade. J Biol Chem. 2000;275:29980–29985. doi: 10.1074/jbc.M003776200. [DOI] [PubMed] [Google Scholar]
- 59.Inoshita S, Takeda K, Hatai T, Terada Y, Sano M, Hata J, Umezawa A, Ichijo H. Phosphorylation and inactivation of myeloid cell leukemia 1 by JNK in response to oxidative stress. J Biol Chem. 2002;277:43730–43734. doi: 10.1074/jbc.M207951200. [DOI] [PubMed] [Google Scholar]
- 60.Suy S, Mitchell JB, Samuni A, Mueller S, Kasid U. Nitroxide tempo, a small molecule, induces apoptosis in prostate carcinoma cells and suppresses tumor growth in athymic mice. Cancer. 103(2005):1302–1313. doi: 10.1002/cncr.20898. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.