

Abstract
Methylphosphonate synthase (MPnS) produces methylphosphonate, a metabolic precursor to methane in the upper ocean. Here we determine a 2.35-Å resolution structure of MPnS and discover that it has an unusual 2-histidine-1-glutamine iron-coordinating triad. We further solve the structure of a related enzyme, hydroxyethylphosphonate dioxygenase from Streptomyces albus (SaHEPD), and find that it displays the same motif. SaHEPD can be converted into an MPnS by mutation of glutamine-adjacent residues, identifying the molecular requirements for methylphosphonate synthesis. Using these sequence markers, we find numerous putative MPnSs in marine microbiomes, and confirm that MPnS is present in the abundant Pelagibacter ubique. The ubiquity of MPnS-containing microbes supports the proposal that methylphosphonate is a source of methane in the upper, aerobic ocean, where phosphorus-starved microbes catabolize methylphosphonate for its phosphorus.
Methane concentrations in oxygenated surface ocean waters are super-saturated relative to the atmosphere (1). This methane has been shown to derive from a biological source (2), however, canonical methanogenesis only occurs in obligate anaerobic archaea (3). Although anoxic microenvironments could provide habitats for methanogenic archaea (4), in situ methanogenesis has not been directly observed. Thus, the source of biological methane from the aerobic upper ocean is unknown, a conundrum referred to as the “oceanic methane paradox” (5). Recently, the C-P lyase pathway was identified as a biogenic source of methane in aerobic organisms prevalent in surface ocean waters (6, 7). Under phosphorus stress, these organisms use the C-P lyase pathway to cleave the C-P bond in methylphosphonate (MPn), releasing methane and sequestering the phosphorus in the form of phosphate (6, 7). The release of methane through metabolism of MPn has been proposed as a resolution to the oceanic methane paradox (8). Indeed, MPn was recently shown to be ubiquitous in dissolved organic matter from the ocean (9), and an MPn biosynthetic enzyme was recently identified but thus far only shown to be present in one marine microbe, the abundant archaeon Nitrosopumilus maritimus (10). The key enzyme discovered, MPn synthase (MPnS), uses molecular oxygen and a non-heme mononuclear Fe(II) center to convert 2-hydroxyethylphosphonate (2-HEP) into MPn and CO2 (10) (Fig. 1). MPnS is similar in sequence to hydroxyethylphosphonate dioxygenase (HEPD) (11) and hydroxypropylphosphonate epoxidase (HppE) (10, 12), which catalyze the formation of hydroxymethylphosphonate (HMP) from 2-HEP (Fig. 1) (11) and the formation of the antibiotic fosfomycin from 2-S-hydroxypropylphosphonate (13), respectively.
Figure 1.
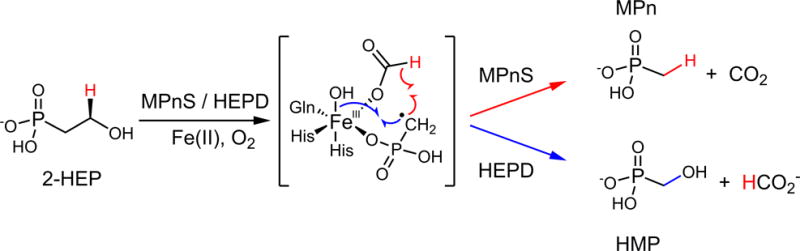
The reactions of MPnS and HEPD have been proposed to proceed through a common radical intermediate. The hydrogen derived from the C2 pro-R position in 2-HEP is highlighted red throughout. The radical recombination in MPnS or HEPD (17) is colored red or blue, respectively. For an alternative mechanism involving a cationic intermediate, see Fig. S9.
To establish the molecular determinants of MPnS activity and thereby establish sequence markers to use in the search for additional MPnSs, we determined X-ray structures of MPnS from N. maritimus in both the substrate-free and the Fe(II)- and substrate-bound states to 2.37 and 2.35 Å resolution, respectively (Table S1). As expected based on sequence (Fig. S1), the overall structure of MPnS is highly similar to the HEPD from Streptomyces viridochromogenes (SvHEPD) (Fig. S2) (11). Both enzymes are dimeric and contain the same domain structure: two β-sheet domains (β1 and β2) which together comprise a bicupin fold and two entirely α-helical domains (α1 and α2) (Fig. S2A). The active site is situated in the first of the two cupin folds and completed by contributions from the N-terminal α-helical domain and the C-terminal tail of the second protomer. As noted previously (11), the dimeric architectures of SvHEPD, and now MPnS, are analogous to the tetramer structure of HppE from Streptomyces wedmorensis (12) with each protomer of HppE corresponding to a single α-helical domain and a single β cupin fold.
Mononuclear non-heme iron enzymes are traditionally characterized by a three-residue iron-binding motif composed of two His residues and one carboxylate-bearing residue (Asp or Glu), termed the facial triad (14). In the active site of MPnS, a Gln residue (Q152) coordinates the Fe(II) along with two His residues (H148 and H190), forming an unusual 2-His-1-Gln facial triad (Figs. 2A and S3A). The position of Gln152 in MPnS is distinct from that of the iron coordinating Glu (E176) in SvHEPD (11), originating from an adjacent β-strand at the analogous position of the iron-coordinating Glu in HppE (12) (Fig. S2B). The substrate 2-HEP coordinates Fe(II) in the active site of MPnS through its hydroxyl oxygen and one phosphonate oxygen (Figs. 2A, S3A, and S4A). The phosphonate moiety is further stabilized by interaction with Arg102, Tyr110, and Asn145 from the cupin barrel; Lys28´ from the α-helical domain of neighboring protomer; and Trp449´ from the C-terminal tail of the neighboring protomer. Interestingly, the Trp449-containing C-terminal tail in MPnS is mobile; in the crystal structure of substrate-free MPnS, residues Ala442 to Ser457 have an alternative position where Trp449 is no longer in the active site and a channel is open from the active site to bulk solvent (Fig. S5, A and B). Thus, movement of the C-terminal tail and Trp449 appears to open and close the active site in MPnS. Although Trp449 is conserved between MPnS and SvHEPD, the residue has not been visualized in the SvHEPD structures (11). Three other residues of the cupin fold in MPnS (Y108, I126, F192) further contribute to substrate binding through hydrophobic interactions (Fig. 2A, Fig. S3A).
Figure 2.
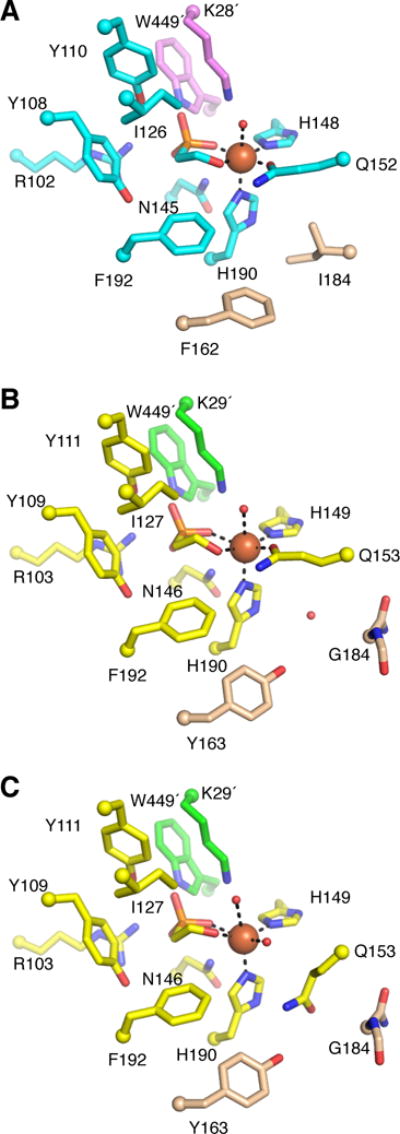
Active site structure of NmMPnS and SaHEPD. (A) The NmMPnS active site is composed of residues from the β1 domain (cyan) and the α1′ domain (pink), with two key residues discussed in the text in light brown. Fe(II) (orange sphere) is ligated by substrate, Gln152, His148, and His190. Although electron density cannot be used to distinguish oxygen from nitrogen, we are showing the oxygen of the Gln side chain coordinating Fe(II). (B) SaHEPD active site composed of residues from the β1 domain (yellow) and the α1′ domain (green) for two protomers of the asymmetric unit in which Gln153 is in the iron-binding conformation. (C) Alternative conformation of SaHEPD, where Gln153 is displaced from the Fe(II).
To determine whether the presence of a 2-His-1-Gln motif is predictive of MPnS activity, we selected to clone, express, and characterize a gene product from Streptomyces albus (strain NRRL B-16041) that displays a 2-His-1-Gln motif and 34% sequence identity to MPnS. Using a previously described phosphorus nuclear magnetic resonance (NMR) spectroscopy assay (11), this enzyme was shown to produce only HMP when 2-HEP is provided as substrate (Fig. S6), classifying this enzyme as an HEPD. HEPDs containing a 2-His-1-Gln facial triad will hereafter be referred to as class II whereas the previously characterized 2-His-1-Glu HEPDs will be called class I. Identification of class II HEPDs that share the same iron-ligating ligand set as MPnS demonstrates that the 2-His-1-Gln motif is not diagnostic of enzyme function, raising the question of what sequence motifs distinguish an MPnS from a class II HEPD.
To investigate the structural differences between MPnS and a class II HEPD, we determined a crystal structure of HEPD from S. albus (SaHEPD) to 1.8-Å resolution with 2-HEP and Fe(II) bound (Table S1). SaHEPD has the same fold and oligomeric state as MPnS and is highly similar [root mean square deviation of 1.9 Å over 427 out of 450 Cα atoms as calculated with PyMOL (15)] (Fig. S2D). Consistent with the sequence alignment (Fig. S1), all three iron-coordinating residues (H149, Q153, and H190 in SaHEPD) and all residues forming the substrate-binding pocket (R103, Y109, Y111, I127, N146, F192, K29´, and W449´ in SaHEPD) are conserved between SaHEPD and MPnS (Fig. 2, A and B, Fig. S3, A and B, and Fig. S4B). The crystal lattice affords four independent views of this active site, and in two of the four active sites in the asymmetric unit, the electron density for Gln153 is consistent with an iron-coordinating conformation. However, in the other two active sites, the electron density indicates that a water molecule has replaced Gln153 as the ligand to Fe(II) and that Gln153 is now pointing away from the iron (Fig. S4, D and E). This Gln is positioned such that it can hydrogen bond to Tyr163 that packs against Fe(II) ligand His190 (Fig. 2C, Fig. S4E). Interestingly, Tyr163 is one of two residues, Gly184 is the other, that are close to the ligand triad and not identical between SaHEPD (Y163/G184) and MPnS (F162/I184). In MPnS, Phe162 and Ile184 are in van der Waals contact with each other and fill in the area under Gln152 such that movement of Gln away from the Fe(II) does not appear possible (Fig. 2A). In contrast, the two residues (Y163 and G184) in SaHEPD form a cavity that can accommodate a water molecule when Gln153 is a ligand to Fe(II) (Fig. 2B) and can also accommodate an alternative (non-coordinating) conformation of Gln153 (Fig. 2C).
To investigate the importance of the alternative conformation of Gln153 in SaHEPD, we mutated both Tyr163 and Gly184 in SaHEPD to the corresponding residues in MPnS, Phe and Ile, respectively. Our structures suggest that in this double mutant the solvent cavity beneath Gln153 would be filled, restricting Gln153 to an iron-binding configuration as observed in MPnS. Indeed, the purified SaHEPD-Y163F/G184I variant catalyzes the conversion of 2-HEP to MPn with no detectable side products and with kinetic parameters [turnover number (kcat), 1.2 ± 0.1 s−1; and Michaelis constant for HEP (Km,HEP), 15 ± 2 μM] very similar to those of purified wild-type SaHEPD (Fig. S6 and S7, Table S2). The catalytic efficiency is in fact slightly better than for N. maritimus MPnS (NmMPnS) (kcat, 0.25 s−1; Km,HEP, 11 μM) (16). These results demonstrate that switching the identity of only two non-coordinating active site residues is sufficient to transform an HEPD into an MPnS. We then investigated which of these two residues is most important; SaHEPD-Y163F produced only HMP, whereas SaHEPD-G184I produced only MPn (Fig. S8). Thus, disruption of hydrogen bonding between Tyr163 and Gln153 is insufficient to alter the reaction outcome, but preventing Gln153 from occupying the alternative conformation does result in the switch in product formation. We also replaced the unusual Gln iron-coordinating ligand with Glu or Ala in both SaHEPD and NmMPnS. SaHEPD-Q153E was inactive, whereas the Q153A mutant still produced HMP, albeit with reduced efficiency (Fig. S8). NmMPnS-Q152A produced HMP with a very low level of activity, whereas NmMPnS-Q152E was inactive under the same condition (Fig. S8). Thus, removal of the Gln ligand is strongly deleterious for both MPnS and class II HEPD enzymes (for discussion, see Fig. S8). Replacing the Glu ligand in class I HEPD with Gln had no effect on activity (17).
We next considered why restricting Gln movement would alter product formation. An iron-bound MPn-based radical and an iron-bound formate are the proposed reaction intermediates for both enzymes (Fig. 1 and Fig. S9) (18); in MPnS, the MPn-based radical abstracts the formate hydrogen atom to yield MPn and CO2, and in HEPD the MPn-based radical reacts with the iron-bound hydroxyl yielding HMP and HCO2− (16, 17). Therefore, the fate of the intermediate appears to depend directly on the geometric arrangement of formate relative to the MPn-based radical. MPnS requires the formate hydrogen atom to be in close proximity to the radical (Fig. 1). Inspection of the substrate-bound MPnS active site reveals that the positioning of an iron-bound formate intermediate would be most directly affected by the side chain of Gln152 (Fig. 2A and Fig. S3A). With the side-chain oxygen of the Gln coordinating the iron, the side-chain amide nitrogen would be positioned appropriately to interact with the iron-bound formate, securing its position near the iron-bound MPn-radical (Fig. 3A). Thus, residues that fix the position of Gln, like a juxtaposed Ile184, favor MPn production. In contrast, residues that permit Gln to adopt an alternative (non-iron-ligating) position, like a juxtaposed Gly184, should support HMP production; Gln movement allows formate movement, potentially increasing the distance between the formyl hydrogen atom and the iron-bound MPn radical, disfavoring hydrogen-atom abstraction (Fig. 3B). The same arguments can be made if the reaction were to proceed via an ionic mechanism (Fig. S9), except that formate would be appropriately positioned by the Gln for hydride rather than hydrogen atom transfer in MPnS. This model also explains the outcome of all mutant enzymes.
Figure 3.
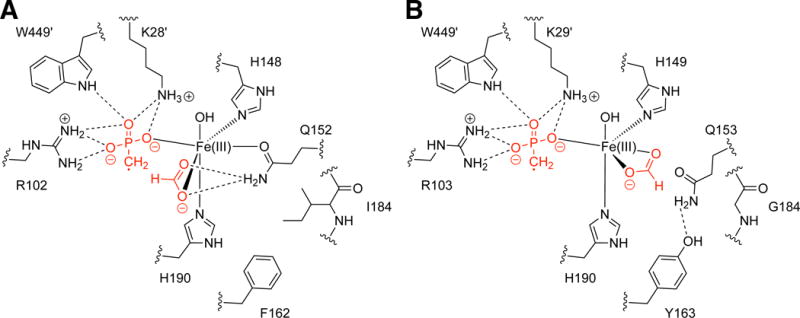
Proposed intermediates for MPnS and SaHEPD. (A) Gln152 in MPnS coordinates the formate intermediate, enabling H-atom abstraction by the MPn radical. (B) Formate displaces Gln153 from its iron-coordinating position in SaHEPD, preventing H-atom abstraction by the MPn-based radical and promoting reaction with the iron-bound hydroxide.
Next, we searched genomes for additional MPnSs. Two homologs that contain Ile at position 184 were identified in the National Center for Biotechnology Information nonredundant protein sequences database. One of these enzymes, a hypothetical protein from Candidatus Pelagibacter ubique strain HTCC7217 (WP_029455269.1), was expressed as a maltose-binding protein fusion enzyme in Escherichia coli. Indeed, this enzyme converts 2-HEP to MPn without detectable side products, consistent with its being an MPnS (Fig. S6). Altogether, these results establish a sequence signature that defines MPnS enzymes: a 2-His-1-Gln facial triad with an Ile at position 184 to pack against the Gln. Phylogenetic analysis suggests that Val at position 184 may also play the same role as Ile (Fig. 4).
Figure 4.
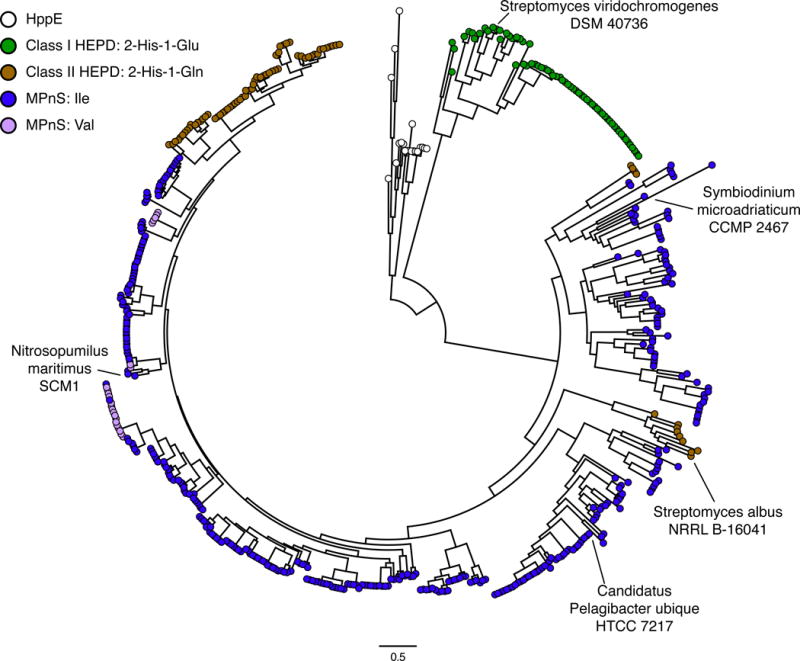
Phylogeny of MPnS, HEPD, and HppE-like proteins. HEPD proteins are colored based on the iron-ligating facial triad: class I HEPD, 2-His-1-Glu (green); or class II HEPD, 2-His-1-Gln (brown). MPnS proteins are colored based on the amino acid at position 184, either Ile (blue) or Val (magenta). Complete labeling of sequence names is provided in Fig. S10.
Extending the MPnS sequence signature to a phylogenetic analysis of genomes, metagenomes, and transcriptome shotgun assemblies reveals additional candidate MPnS-coding genes in uncultivated marine microbes and even eukaryotic marine dinoflagellates, including dinoflagellates of the genera Symbiodinium (Fig. 4). In contrast to HEPD-coding genes, which are present in microbial strains and metagenomes of both marine and terrestrial origin, all candidate MPnS-coding genes identified to date derive from marine environments, suggesting a relevant role for MPn in marine ecosystems. Importantly, P. ubique belongs to the SAR11 clade of α-proteobacteria, and the SAR11 clade, along with thaumarchaeota (e.g. N. maritimus), are two of the most abundant marine microorganisms (19, 20). Thus, the identification of MPnS enzymes in P. ubique and N. maritimus strongly supports a widespread role of MPn in marine carbon and phosphorus cycling, further supporting the MPn hypothesis for oceanic methane production and potentially resolving the oceanic methane paradox.
Supplementary Material
Acknowledgments
We thank the Giovannoni laboratory for genomic DNA from P. ubique HTCC7217 and H. Cooke for the plasmids for NmMPnS-Q152A and Q152E. This work was supported by the National Institutes of Health (GM 077596 to W.A.V). An NIH Molecular Biophysics Training Grant T32 GM008313 supported D.A.B. C.L.D. and W.A.V. are Howard Hughes Medical Institute Investigators. 31P NMR data were collected on a 600 MHz NMR spectrometer funded by NIH (S10-RR028833). This work used Northeastern Collaborative Access Team (NE-CAT) beamlines (GM103403) and a Pilatus detector (RR029205) at the Advanced Photon Source (DE-AC02-06CH11357). Structures of 2-HEP-bound Fe(II)-SaHEPD, substrate-free MPnS, and 2-HEP-bound Fe(II)-MPnS are available through the Protein Data Bank under accession codes 6B9R, 6B9S, and 6B9T, respectively.
References
- 1.Scranton MI, Brewer PG. Occurrence of methane in the near-surface waters of the western subtropical North-Atlantic. Deep Sea Research. 1977;24:127–138. [Google Scholar]
- 2.Holmes ME. Methane production, consumption, and air-sea exchange in the open ocean: An evaluation based on carbon isotopic ratios M. Elizabeth Holmes, 1 Francis J. Sansone, Terri M. Rust, and Brian N. Popp. Global Biogeochemical Cycles. 2000;14:1–10. [Google Scholar]
- 3.Liu Y, Whitman WB. Metabolic, phylogenetic, and ecological diversity of the methanogenic archaea. Annals of the New York Academy of Sciences. 2008;1125:171–189. doi: 10.1196/annals.1419.019. [DOI] [PubMed] [Google Scholar]
- 4.Karl DM, Tilbrook BD. Production and transport of methane in oceanic particulate organic matter. Nature. 1994;368:732–734. [Google Scholar]
- 5.Rogers JE, Whitman WB. Microbial production and consumption of greenhouse gases: methane, nitrogen oxides, and halomethanes. 1991 [Google Scholar]
- 6.Daughton C, Cook A, Alexander M. Biodegradation of phosphonate toxicants yields methane or ethane on cleavage of the CP bond. FEMS Microbiology Letters. 1979;5:91–93. [Google Scholar]
- 7.Carini P, White AE, Campbell EO, Giovannoni SJ. Methane production by phosphate-starved SAR11 chemoheterotrophic marine bacteria. Nature communications. 2014;5 doi: 10.1038/ncomms5346. [DOI] [PubMed] [Google Scholar]
- 8.Karl DM, et al. Aerobic production of methane in the sea. Nature Geoscience. 2008;1:473–478. [Google Scholar]
- 9.Repeta DJ, et al. Marine methane paradox explained by bacterial degradation of dissolved organic matter. Nature Geoscience. 2016;9:884–887. [Google Scholar]
- 10.Metcalf WW, et al. Synthesis of methylphosphonic acid by marine microbes: a source for methane in the aerobic ocean. Science. 2012;337:1104–1107. doi: 10.1126/science.1219875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cicchillo RM, et al. An unusual carbon–carbon bond cleavage reaction during phosphinothricin biosynthesis. Nature. 2009;459:871–874. doi: 10.1038/nature07972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Higgins LJ, Yan F, Liu P, Liu H-w, Drennan CL. Structural insight into antibiotic fosfomycin biosynthesis by a mononuclear iron enzyme. Nature. 2005;437:838–844. doi: 10.1038/nature03924. [DOI] [PubMed] [Google Scholar]
- 13.Liu P, et al. Protein purification and function assignment of the epoxidase catalyzing the formation of fosfomycin. Journal of the American Chemical Society. 2001;123:4619–4620. doi: 10.1021/ja004153y. [DOI] [PubMed] [Google Scholar]
- 14.Straganz G, Nidetzky B. Variations of the 2-His-1-carboxylate Theme in Mononuclear Non-Heme FeII Oxygenases. ChemBioChem. 2006;7:1536–1548. doi: 10.1002/cbic.200600152. [DOI] [PubMed] [Google Scholar]
- 15.DeLano WL. The PyMOL molecular graphics system. 2002 [Google Scholar]
- 16.Cooke HA, Peck SC, Evans BS, van der Donk WA. Mechanistic investigation of methylphosphonate synthase, a non-heme iron-dependent oxygenase. Journal of the American Chemical Society. 2012;134:15660–15663. doi: 10.1021/ja306777w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peck SC, Chekan JR, Ulrich EC, Nair SK, van der Donk WA. A common late-stage intermediate in catalysis by 2-hydroxyethyl-phosphonate dioxygenase and methylphosphonate synthase. Journal of the American Chemical Society. 2015;137:3217. doi: 10.1021/jacs.5b00282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Whitteck JT, et al. On the stereochemistry of 2-hydroxyethylphosphonate dioxygenase. Journal of the American Chemical Society. 2011;133:4236–4239. doi: 10.1021/ja1113326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morris RM, et al. SAR11 clade dominates ocean surface bacterioplankton communities. Nature. 2002;420:806–810. doi: 10.1038/nature01240. [DOI] [PubMed] [Google Scholar]
- 20.Karner MB, DeLong EF, Karl DM. Archaeal dominance in the mesopelagic zone of the Pacific Ocean. Nature. 2001;409:507–510. doi: 10.1038/35054051. [DOI] [PubMed] [Google Scholar]
- 21.Grant SG, Jessee J, Bloom FR, Hanahan D. Differential plasmid rescue from transgenic mouse DNAs into Escherichia coli methylation-restriction mutants. Proceedings of the National Academy of Sciences. 1990;87:4645–4649. doi: 10.1073/pnas.87.12.4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gibson DG, et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 23.Blodgett JA, et al. Unusual transformations in the biosynthesis of the antibiotic phosphinothricin tripeptide. Nature chemical biology. 2007;3:480–485. doi: 10.1038/nchembio.2007.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods in enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [20] [DOI] [PubMed] [Google Scholar]
- 25.Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta Crystallographica Section D: Biological Crystallography. 2002;58:1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- 26.Abrahams J, Leslie A. Methods used in the structure determination of bovine mitochondrial F1 ATPase. Acta Crystallographica Section D: Biological Crystallography. 1996;52:30–42. doi: 10.1107/S0907444995008754. [DOI] [PubMed] [Google Scholar]
- 27.Vonrhein C, Blanc E, Roversi P, Bricogne G. Automated structure solution with autoSHARP. Macromolecular Crystallography Protocols: Volume 2: Structure Determination. 2007:215–230. doi: 10.1385/1-59745-266-1:215. [DOI] [PubMed] [Google Scholar]
- 28.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallographica Section D: Biological Crystallography. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 29.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallographica Section D: Biological Crystallography. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCoy AJ, et al. Phaser crystallographic software. Journal of applied crystallography. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta Crystallographica Section D: Biological Crystallography. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kabsch W. Acta Crystallographica Section D: Biological Crystallography. 2010;66:125–132. doi: 10.1107/S0907444909047337. XDs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bunkóczi G, Read RJ. Improvement of molecular-replacement models with Sculptor. Acta Crystallographica Section D: biological Crystallography. 2011;67:303–312. doi: 10.1107/S0907444910051218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morin A, et al. Collaboration gets the most out of software. elife. 2013;2:e01456. doi: 10.7554/eLife.01456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gasteiger E, et al. ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic acids research. 2003;31:3784–3788. doi: 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ju KS, et al. Discovery of phosphonic acid natural products by mining the genomes of 10,000 actinomycetes. Proceedings of the National Academy of Sciences. 2015;112:12175–12180. doi: 10.1073/pnas.1500873112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Katoh K, Misawa K, Kuma Ki, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic acids research. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Price MN, Dehal PS, Arkin AP. FastTree 2–approximately maximum-likelihood trees for large alignments. PloS one. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. Journal of molecular biology. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 40.Peck SC, et al. O–H Activation by an Unexpected Ferryl Intermediate during Catalysis by 2-Hydroxyethylphosphonate Dioxygenase. Journal of the American Chemical Society. 2017;139:2045–2052. doi: 10.1021/jacs.6b12147. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.