

Abstract
Prostate cells are hormonally driven to grow and divide. Typical treatments for prostate cancer involve blocking activation of the androgen receptor by androgens. Androgen deprivation therapy can lead to the selection of cancer cells that grow and divide independently of androgen receptor activation. Prostate cancer cells that are insensitive to androgens commonly display metastatic phenotypes and reduced long-term survival of patients. In this study we provide evidence that androgen-insensitive prostate cancer cells have elevated PLD activity relative to the androgen-sensitive prostate cancer cells. PLD activity has been linked with promoting survival in many human cancer cell lines; and consistent with the previous studies, suppression of PLD activity in the prostate cancer cells resulted in apoptotic cell death. Of significance, suppressing the elevated PLD activity in androgen resistant prostate cancer lines also blocked the ability of these cells to migrate and invade Matrigel™. Since survival signals are generally an early event in tumorigenesis, the apparent coupling of survival and metastatic phenotypes implies that metastasis is an earlier event in malignant prostate cancer than generally thought. This finding has implications for screening strategies designed to identify prostate cancers before dissemination.
1. Introduction
Most prostate cancers require androgens for proliferation and survival – and as a consequence are sensitive to androgen deprivation therapy [1]. However, patients develop resistance to androgen deprivation and it is amongst these androgen resistant cancers that the most aggressive cancers emerge [2–4]. A common mechanism for promoting resistance to androgen deprivation is persistent androgen receptor signaling [1]. Genetic alterations at the androgen receptor locus such as mutations in the ligand binding domain or amplification of the androgen receptor gene have been suggested to promote androgen receptor signals under conditions of low serum testosterone [5].
Another route to androgen independence is activation of the phosphatidylinositol-3-kinase-AKT-mTOR (mammalian target of rapamycin) signaling pathway [5]. This pathway is clearly emerging as an important signaling node that promotes androgen resistance and stimulates tumor growth in the setting of reduced levels of androgens. This pathway appears to be altered at the genomic and transcriptional level in most metastatic prostate cancers [6–8]. There are several points of therapeutic intervention for prostate cancers where the phosphatidylinositol-3-kinase -AKT-mTOR signaling pathway is promoting survival and metastasis [5].
An under-appreciated component of the intra-cellular signals leading to the activation of mTOR is phospholipase D (PLD) [9]. PLD generates a metabolite phosphatidic acid (PA) [10] that is required for the stability of the mTOR complexes – mTORC1 and mTORC2 [11]. We previously reported that elevated PLD activity provided an mTOR-dependent survival signal in the absence of estrogen in estrogen receptor positive breast cancer cells [12, 13]. We also reported that PLD activity was elevated in estrogen receptor negative breast cancer cell lines deprived of serum and provided an mTOR dependent survival signal [14]. In addition to providing a survival signal, the elevated PLD activity in the estrogen receptor negative cells enhanced cell migration and invasion of Matrigel™, linking survival with metastatic phenotypes [14]. Since survival signals are of necessity an early event in tumorigenesis to suppress default apoptotic programs, we proposed that the coupling of survival and migration signals in hormone independent breast cancer cells promoted early metastasis [14].
We report here that there is elevated PLD activity in androgen-insensitive prostate cancer cell lines and that elevated PLD activity promotes both survival and migration signals. The study links survival and migration in hormone independent prostate cancer cells and provides a rationale for metastasis occurring early in androgen-resistant prostate cancers.
2. Materials and methods
2-1. Cells and cell culture conditions
The human cancer cell line lines DU145, PC-3 and LNCaP were obtained from the American Tissue Type Culture Collection (ATCC). The DU145 cancer cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Sigma D6429) and supplemented with 10% Fetal Bovine Serum (FBS) (Sigma F4135). The human cancer cell line PC-3 was cultured in Roswell Park Memorial Institute medium (RPMI-1640) (Sigma R8758) and supplemented with 10% FBS. The human cancer cell line LNCaP was cultured in RPMI-1640 supplemented with 10% FBS and 10 nM testosterone (Sigma T1500).
2-2. Materials
Reagents were obtained from the following sources: Antibodies against cleaved PARP (9541), HA-Tag (2367), PLD1 (3832), PLD2 (13904) and prostate specific antigen (PSA) (2475) were obtained from Cell Signaling; β-actin (60008) was obtained from ProteinTech; anti-mouse and anti-rabbit HRP conjugated secondary antibodies were obtained from Promega. Negative control scrambled siRNA (D-001810) and siRNAs targeted against PLD1 (L-009413), PLD2 (L-005064) were obtained from Dharmacon. Lipofectamine RNAiMax (Invitrogen, 56532) was used for siRNA transfection. Plasmids with hemagglutinin (HA)-tagged PLD1 (pCMV3-HA-PLD1) (Sino Biological HG13850) and HA-tagged PLD2 (pcDNA3.1-HA-PLD2) (gift from M. Frohman, SUNY Stony Brook) were transfected with Lipofectamine 3000 Transfection Kit (Invitrogen, L3000015). PLD inhibitors VU0359595 (PLD1) and VU02855655-A (PLD2) and phosphatidyl butanol (258637) were obtained from Avanti Polar Lipids. [3H]-myristate (NET830005MC) was obtained from PerkinElmer.
2-3. Cell viability
Cell viability was determined by trypan blue exclusion. After various treatments, cells were harvested, washed and treated with trypan blue at a concentration of 0.4% (w/v). After 20 minutes, trypan blue uptake was scored using a hemacytometer.
2-4. Phospholipase D assay
Cells were plated in 6-well plates at 2 × 105 cells per well. Cells were then prelabeled for 4 hours with of [3H]-myristate (3 μCi; 60 Ci/mmol) in 2 ml of medium. PLD catalyzed transphosphatidylation for 20 minutes in the presence of 0.8% 1-butanol. Labeled media was removed and the cells were washed and then lysed of with 500 μl of acidified methanol (methanol: 6N HCl (50:2)). The cells were then added to the first extraction tube (155 μl 1N NaCl and 500 μl CHCl3), vortexed for 30 seconds and centrifuged for 3 minutes at 16,100 × g. The organic phase was then transferred to the second extraction tube (350 μl H20, 115 μl CH3OH, and 115 μl NaCl), vortexed for 15 seconds and centrifuged for 3 minutes at 16,100 × g. The organic phase (350 ul), spiked with 10 μl of 12 μM unlabeled phosphatidylbutanol to visualize, was loaded onto a silica gel thin layer chromatography plate and the mobile phase (the organic phase of the mixture of ethyl acetate, isooctane, glacial acetic acid, and water at a 77:35:14:70 ratio) was allowed to proceed across the length of the thin layer chromatography plate. The phosphatidylbutanol band was visualized with iodine vapor and marked. The band was scraped from the plate and a scintillation count was performed.
2-5. Transient transfections
Cells were plated in 6-well plates in medium containing 10% FBS. The next day transfections with plasmid (1 μg/ml) in Lipofectamine 3000 or siRNAs (50 nM) in Lipofectamine RNAiMax were performed in the absence of serum. After 6 hours reagents were replaced with fresh media with 10% FBS and cells were allowed to incubate for an additional 48 (plasmid) to 96 hours (siRNA).
2-6. Transwell migration and invasion assays
The assays were carried out using BIOCOAT™ cell culture inserts that have polyethylene terephthalate filters (8 um pore size on the bottom). For migration assays, inserts were used directly without coating (BD Bioscience 354578); for invasion assays, the inserts were pre-coated with Matrigel™ purified from the Engelbreth-Holm-Swarm mouse sarcoma, a tumor rich in extracellular matrix proteins, which closely mimics the basement membrane in vivo (BD Bioscience 354480). Five hundred microliters of single cell suspensions with a concentration of 5 × 104 cells/ml were added into the inserts. The inserts were set into 24-well plates that held 0.75 ml/well of growth media and incubated under normal growth condition for 24 hours. Cells that had not penetrated the filters were wiped out with cotton swabs and cells that had migrated or invaded to the lower surface of the filters were fixed in methanol and then stained with a 0.2% (v/v) solution of crystal violet in 2% (v/v) ethanol. The number of migrated or invaded cells was counted under the microscope. The mean of five individual fields in the center of the filter where migration and invasion was the highest was obtained for each well.
2-7. Western blot analysis
Proteins were extracted from cultured cells in M-PER (Thermo Scientific 78501) Equal amounts of proteins were subjected to SDS-PAGE on polyacrylamide separating gels. Electrophoresed proteins were then transferred to nitrocellulose membrane. After transfer, membranes were blocked in an isotonic solution containing 5% non-fat dry milk in phosphate buffered saline. Membranes were then incubated with primary antibodies as described in the text. The dilutions were used as per vendor’s instructions. Depending on the origin of the primary antibody, either anti-mouse or anti-rabbit HRP conjugated IgG was used for detection using ECL system (Thermo Scientific 34080).
2-8. Wound healing assay
Cells were plated in 24-well plates in medium containing 10% FBS at 90% confluency. The next day a path was cleared of cells and the medium was replaced with fresh medium. Closure was then observed after 24 hours. The area of closure was quantified using the MRI Wound Healing Tool macro for ImageJ.
2-9. Statistical Analysis
Statistical data are presented as the mean ± standard error of mean of three individual experiments preformed in triplicate. Statistical analysis was carried out using the one-way analysis of variance or the unpaired Student’s t-test using GraphPad Prism software. The level of significance was set at a P-value of <0.10.
3. Results
3-1. Androgen-insensitive prostate cancer cells have elevated PLD activity relative to androgen-sensitive prostate cancer cells
We previously demonstrated that there is elevated PLD activity in estrogen receptor negative breast cancer cells [13, 14]. The elevated PLD activity was largely restricted to cells deprived of serum [14] – leading us to speculate that the elevated PLD activity was a stress response to the absence of serum. Since prostate cancer, like breast cancer, is largely a hormonally driven cancer [2], we wanted to evaluate the level of PLD activity in androgen responsive and androgen non-responsive prostate cancer cells. We looked at three prostate cancer cell lines – DU145 and PC-3, which are androgen non-responsive; and LNCaP, which are androgen responsive [15]. We first examined the responsiveness of the three prostate cancer cell lines to androgen as observed by the ability to induce expression of PSA, which is induced by androgen [16]. As expected, androgen treatment caused an increase in the levels of PSA in the androgen-responsive LNCaP cells, but not in the androgen non-responsive PC-3 and DU145 cells (Fig. 1A). We next evaluated the level of PLD activity in all three cell lines in both the presence and absence of serum. As shown in Fig. 1B, there was elevated PLD activity in the androgen refractory DU145 and PC-3 cells relative to the androgen responsive LNCaP cells. Unlike what was observed with breast and other cancer cells [14], the level of PLD was elevated in both the presence and absence of serum – although the level in the absence of serum was slightly elevated relative to the cells in the presence of serum. We also examined the levels of PLD1 and PLD2 protein in the three prostate cancer cell lines. As shown in Fig. 1C, there was clearly less PLD1 protein in the LNCaP cells relative to the DU145 and PC-3 cells. However, there was no clear correlation between PLD protein and activity levels between the DU145 and PC-3 cells. Rumsby and colleagues recently reported significantly higher levels of PLD1 in PC-3 cells relative to LNCaP cells [17].
Fig. 1.
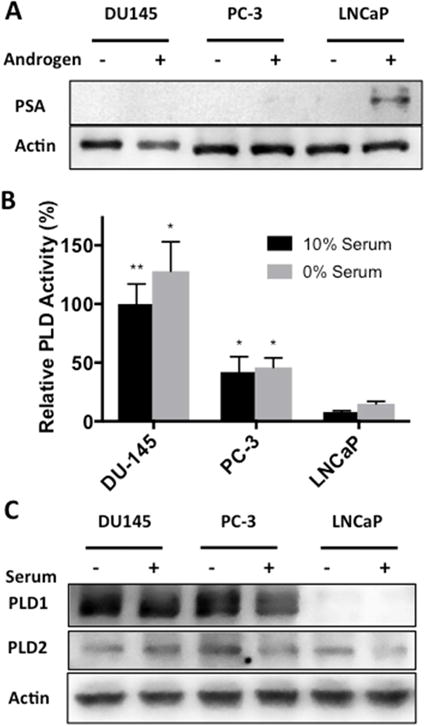
Phospholipase D activity is elevated in androgen insensitive prostate cancer cell lines. (A) DU145, PC-3 and LNCaP cells were plated at density of 200,000 cells/35 mm dish in complete media containing 10% charcoal stripped serum or 10% charcoal stripped serum supplemented with 10 nM testosterone. Cells were harvested and the levels of PSA were determined by Western blot analysis. (B) DU145, PC-3 and LNCaP cells were plated as in Fig 1A in complete media containing 10% serum or 0% serum overnight. Cells were then pre-labeled for 4 hr with [3H]-myristate followed by the addition of 0.8% 1-butanol for 20 min. PLD activity was determined by measuring the levels of the transphosphatidylation product phosphatidylbutanol as described in Materials and Methods. (C) DU145, PC-3 and LNCaP cells were plated as in Fig 1A in complete media containing 10% serum or 0% serum overnight. Cells were harvested and the levels of PLD1 and PLD2 were determined by Western blot analysis. No significance was determined for the difference in serum conditions within each cell line (p > 0.10). (*, p < 0.10; **, p < 0.05; ***, p < 0.01)
3-2. Suppression of PLD activity in prostate cancer cells leads to apoptotic cell death
We have reported that elevated PLD activity in a variety of cancer cells provides an mTOR-dependent survival signal that suppresses apoptotic cell death [13, 14, 18, 19]. We therefore investigated the effect of suppressing PLD activity in the prostate cancer cells. We previously used inhibitors of PLD1 and PLD2 [20, 21] to suppress the effects of PLD in the nutrient induction of mTOR [22, 23]. We found that inhibitors of both PLD1 and PLD2 were most effective when used together at 10 μM each [22]. Suppression of PLD activity in the androgen insensitive DU145 and PC-3 cells with the PLD1 and PLD2 inhibitors led to a loss of cell viability as determined by the uptake of trypan blue (Fig. 2). Surprisingly, the androgen-responsive LNCaP cells were killed more efficiently than the DU145 and PC-3 cells by the PLD inhibitors, which is why we examined cell viability 24 hr earlier than in the DU145 and PC-3 cells. The relatively low levels of PLD activity in the LNCaP cells may have contributed to the more rapid onset of cell death. The PLD inhibitors also induced cleavage of the caspase 3 substrate PARP – indicating apoptotic cell death. As observed with cell viability, PARP cleavage was observed earlier in the LNCaP cells – 24 hr for the LNCaP versus 48 hr for the DU145 and PC-3 cells. Thus, PLD activity is providing a survival signal in the prostate cancer cells. While the PLD activity was substantially lower in the LNCaP cells relative to the DU145 cells, the LNCaP cells apparently need a basal level of PLD activity for survival. Similar results were obtained recently with PC-3 and LNCaP cells using PLD inhibitors at similar concentrations that we used here and previously [22] – including the same PLD1 inhibitor used in this study [17]. The kinetics for cell death were also similar. This group used a different cell viability assay to measure cell viability. These data further support a role for PLD in promoting the survival of prostate cancer cells.
Fig. 2.
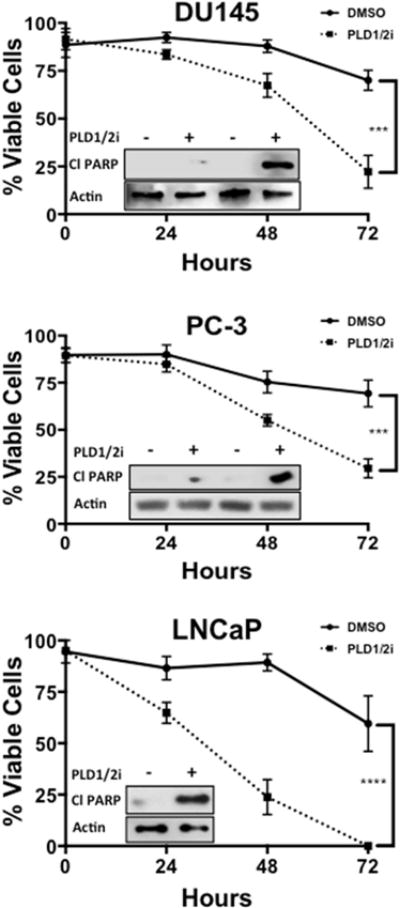
Suppression of PLD activity in prostate cancer cell lines leads to apoptotic cell death. DU145, PC-3 and LNCaP cells were plated at a density of 50,000 cells/35 mm dish in complete media containing 10% serum. Cells were treated with PLD1 and PLD2 inhibitors (10 μM) for the indicated times. Cells were then harvested and cell viability was measured through trypan blue exclusion as described in Materials and Methods. Cells were harvested and the levels of cleaved PARP were determined by Western blot analysis (DU145 and PC-3 at 24 and 48 hr, LNCaP at 24 hr). (*, p < 0.10; **, p < 0.05, ***, p < 0.01, ****, p < 0.001)
3-3. DU145 and PC-3 cells have higher migration and invasion than LNCaP cells
In addition to promoting survival of breast cancer cells, elevated PLD activity also promoted cell migration and invasion of Matrigel™ [14]. We therefore examined the ability of androgen refractory cancer cells, which have high levels of PLD activity, to migrate and invade Matrigel™ relative to the androgen responsive LNCaP cells, which have very low levels of PLD activity. As shown in Fig. 3A, both the DU145 and PC-3 cells migrated and invaded Matrigel™ in transwell chamber assays. In contrast, the ability of LNCaP cells to migrate and invade Matrigel™ was negligible (Fig. 3A). We also employed a wound healing assay to evaluate the ability of the DU145, PC-3 and LNCaP cells to migrate in culture. As shown in Fig. 3B, the DU145 and PC-3 cells significantly migrated into the wound created by scraping the center of the culture dish. These data reveal a correlation between the level of PLD activity in prostate cancer cells and the ability to migrate and invade Matrigel™.
Fig. 3.
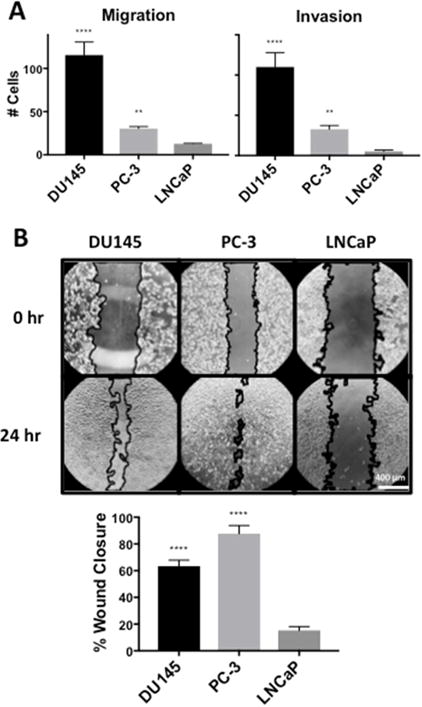
DU145 and PC-3 cells have higher migration and invasion than LNCaP cells. (A) DU145, PC-3 and LNCaP cells were placed in the upper chamber for a transwell migration assay at a density of 25,000 cells/6.5 mm transwell chamber in complete media containing 10% serum. Cells were allowed to migrate through the pores of the membrane to the lower chamber for 24 hr and were fixed, stained and scored under a microscope. Chambers with Matrigel™ indicated invasion while chambers without Matrigel™ indicated migration. (B) DU145, PC-3 and LNCaP cells were plated at a density of 100,000 cells/16 mm dish in complete media containing 10% serum. Cells were allowed to reach full confluence and a “wound gap” was scratched through the monolayer of cells. The media was changed to fresh media containing 10% serum and the “wound gap” was allowed to close for 24 hr. The area of closure was quantified using the MRI Wound Healing Tool macro for ImageJ. (**, p < 0.05, ****, p < 0.001)
3-4. Inhibition of PLD in DU145 and PC-3 cells decreases migration and invasion
The data presented in Fig. 3 reveal a correlation between the level of PLD activity in prostate cancer cells and the ability to migrate in culture and invade Matrigel™. We therefore examined the ability of DU145 and PC-3 cells to migrate and invade Matrigel™ in the presence of PLD inhibitors or PLD knockdown. As shown in Fig. 4A, the ability of both DU145 and PC-3 cells to migrate and invade Matrigel™ in transwell chamber assays was significantly suppressed by PLD inhibitors – especially the ability to invade Matrigel™. Similarly, the ability of the DU145 and PC-3 cells to migrate in the wound-healing assay was suppressed by the PLD inhibitors (Fig. 4B). We next examined the effect of the PLD inhibitors individually and in combination for the wound-healing assay. We also examined the impact of siRNA knockdown of both PLD1 and PLD2 in the wound-healing assay; and as shown in Fig. 4C, the knockdown of PLD1 and PLD2 similarly suppressed migration of DU145 and PC-3 cells. The ability of the PLD inhibitors to suppress wound healing in the DU145 and PC-3 cells required inhibitors of both PLD1 and PLD2 (Fig. 4D). This is consistent with our previous study where we observed that suppressing mTOR kinase activity required inhibitors for both PLD isoforms [22]. Thus, the ability of the DU145 and PC-3 cells to migrate and invade Matrigel™ is dependent on the elevated PLD activity in these androgen insensitive prostate cancer cells. Although the ability of the DU145 and PC-3 cells to migrate/invade did not correlate exactly with the level of PLD activity in these cells, the DU-145 and PC-3 cells are genetically distinct with the PC-3 cells harboring a mutation in PTEN and the DU145 cells harboring mutations in the CDKN2A, RB, and LKB1 genes [24]. While there are many possible explanations why there is not an exact correlation between PLD activity and the ability to migrate and invade, the dependence of migration and invasion on PLD is clear.
Fig. 4.
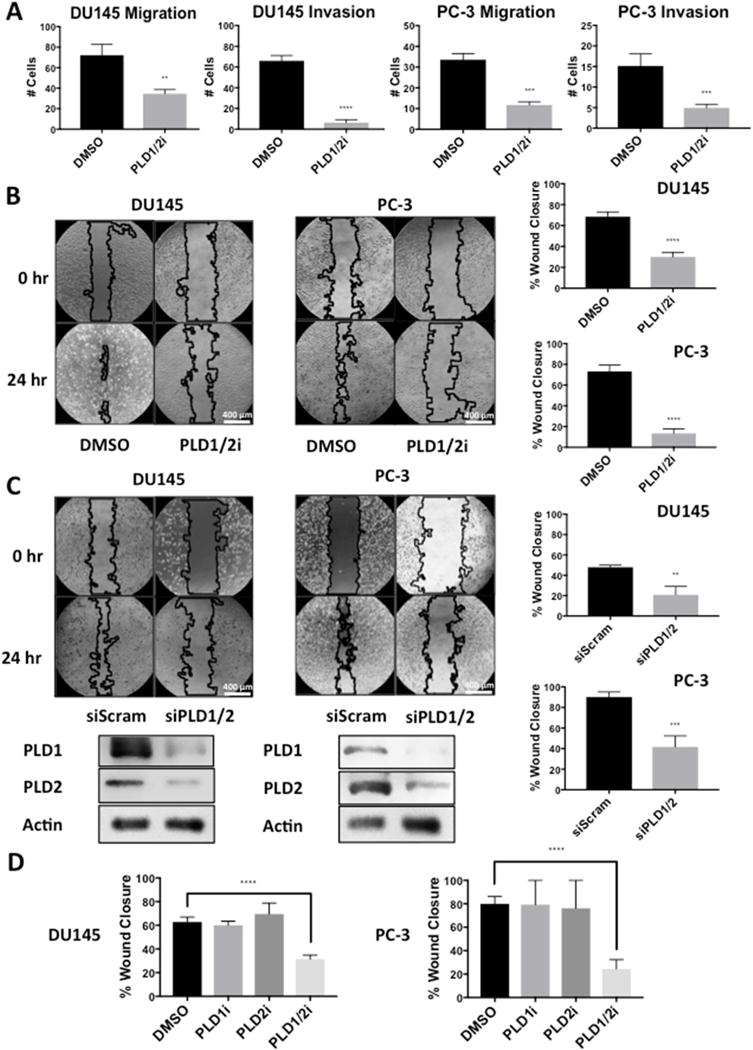
Inhibition of PLD in DU145 and PC-3 cells decreases migration and invasion. (A) DU145 cells were plated for migration and invasion transwell assays as in Fig. 3A and treated with PLD1 and PLD2 inhibitors (10 μM). After 24 hr cells that migrated through the pores of the membrane were scored as in Fig. 3A. (B) DU145 and PC-3 cells were plated for wound healing assay and treated with PLD1 and PLD2 inhibitors (10 μM). A wound was scratched and allowed to fill in for 24 hr at which time the degree of closure was determined as in Fig. 3B. (C) DU145 and PC-3 cells were plated at a density of 200,000 cells/35 mm dish in media containing 10% serum and no antibiotic overnight. Cells were transfected with negative control scrambled siRNA or siRNAs for PLD1 and PLD2 as indicated. Five hours later the cells were shifted to complete media containing antibiotic. Seventy-two hours after transfection, cells were replated for wound healing assay and scored for wound closures as in Fig. 3B, after which they were harvested and the levels of PLD1 and PLD2 were determined by Western blot analysis. (D) DU145 and PC-3 cells were plated for wound healing assay as in (B). A wound was scratched and allowed to fill in for 24 hr in the presence the PLD1 and PLD2 inhibitors (10 μM) individually or together. The degree of closure was then determined as in (B). (**, p < 0.05, ***, p < 0.01, ****, p < 0.001)
3-5. Overexpression of PLD2 in LNCaP cells increases migration and invasion
We next asked whether elevated PLD activity in LNCaP cells would lead to increased migration and invasion of Matrigel™. The LNCaP cells were transfected with plasmids expressing HA-tagged PLD1 and PLD2. The level of PLD activity was elevated very slightly by PLD1, but PLD activity was substantially more elevated in cells transfected with PLD2 (Fig. 5A). If PLD1 and PLD2 were introduced together into the LNCaP cells, the level of PLD activity was less than that observed with PLD2 alone (Fig. 5A) – suggesting that elevated PLD1 expression might be inhibitory. As shown, in Fig 5B, the HA-tagged PLD1 and PLD2 were both expressed in the LNCaP cells. The ability to migrate and invade Matrigel™ in transwell chambers was then examined; and as shown in Fig. 5C, LNCaP cells with elevated PLD2 expression and elevated PLD activity displayed an increased ability to migrate and invade Matrigel™. In cells transfected with either PLD1 or both PLD1 and PLD2 did not significantly elevate migration and invasion. Similarly, LNCaP cells transfected with PLD2 also displayed increased migration in the wound-healing assay (Fig. 5D). These data indicate that elevated PLD activity in prostate cancer cells confers an increased ability to migrate and invade Matrigel™. The reason for the apparent inhibitory effect of PLD1 on migration/invasion in LNCaP cells is not clear, but may be related to the observation in Fig. 1C that PLD1 expression is highly suppressed.
Fig. 5.
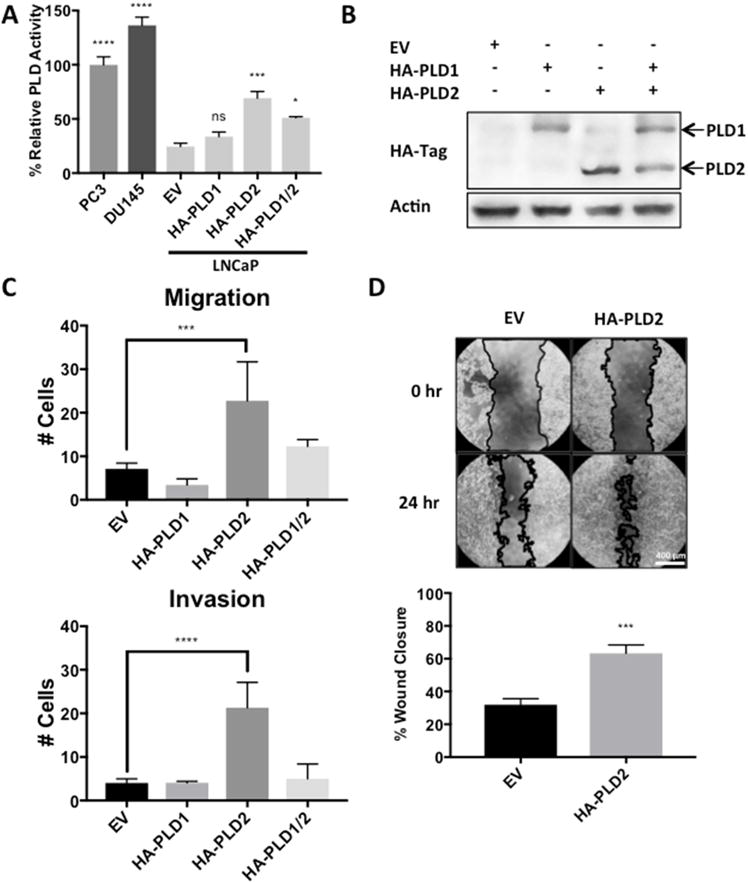
Overexpression of PLD2 in LNCaP cells increases migration and invasion. LNCaP cells were plated for transfection as in Fig. 4C with plasmids expressing empty vector or HA-PLD1 and/or HA-PLD2 as indicated. After 24 hours, cells were replated for PLD assay as in Fig. 1B (A), for Western blot analysis to determine levels of HA-tagged PLD1 and PLD2 (B), for migration and invasion transwell assay as in Fig. 3A (C), and for wound healing assay as in Fig. 3B (D). (ns, p > 0.10; *, p < 0.10, ***, p < 0.01, ****, p < 0.001)
4. Discussion
In this report, we have demonstrated that there are substantially higher levels of PLD activity in the androgen refractory DU145 and PC-3 prostate cancer cell lines relative to androgen responsive LNCaP prostate cancer cells. While the observation reported here with just two androgen-insensitive and one androgen responsive cell line do not establish a precedent they are consistent with a similar correlation with elevated PLD activity in hormone insensitive breast cancer cells and increased migration and invasion [14]. We have also reported previously that elevated PLD activity in many human cancer cells promotes both survival signals [9, 12, 13, 25–27] and metastatic phenotypes [14, 28]. In this study with androgen refractory and androgen responsive prostate cancer cells, we find that the elevated PLD activity detected in the androgen refractory DU145 and PC-3 correlates with the ability to migrate and invade Matrigel™. Surprisingly, inhibition of PLD killed all three cell lines – including the LNCaP cells which displayed low levels of PLD activity. Although the LNCaP cells are androgen responsive, these cells are not dependent on the presence of androgens for survival. They were however, dependent upon PLD for survival. This differs from what we observed with hormone responsive breast cancer cells in that these cells are dependent on estrogen for survival [12, 29]. Thus, the LNCaP cells have apparently evolved to a state of androgen independence, while retaining the ability to respond androgens. The LNCaP cells are however dependent on the low levels of PLD activity for survival, but have not attained the levels of PLD activity necessary for the metastatic phenotypes observed in the PC-3 and DU145 cells.
We previously proposed that elevated PLD activity in breast cancer cells represented a shift from hormone-dependent to hormone-independent cancer – where survival was originally dependent on estrogen, but then shifted to a dependence on PLD generated PA for survival [14]. This shift had the added consequence of enhancing cell migration and invasion that was dependent on PLD activity [14]. The coupling of survival and migration signals has important implications for early detection and treatment. Signals that suppress default apoptotic programs (survival signals) are generally early events in cancer [30, 31]. This is because driver mutations such as activating KRas mutations, in the absence of a survival signal, will lead to either apoptosis or senescence [31, 32]. The observation that PLD is able to provide both survival and migration signals suggests that metastasis could be a much earlier event in breast and prostate cancers. The coupling of survival and migration could explain in part why PSA tests and mammograms do not result in significant reductions in mortality [33]. This point was made recently where breast cancer cells disseminated very early in mice [34–36].
We proposed previously that the coupling of survival and migration signals represented a stress response where the cells elevated their PLD activity in response to the stress of serum withdrawal [14]. The rationale for this proposal was that during early stages of tumorigenesis prior to vascularization, emerging tumor would respond to the lack of growth factors and nutrients with a stress program that would suppress default apoptotic programs and promote migration to blood vessels where nutrients could be obtained. In this report, the PLD activity in prostate cancer cells was not dramatically elevated in response to serum withdrawal as was observed with breast cancer cells. However, suppressing the PLD activity reduced migration and stimulated apoptosis – indicating that PLD was critical for both survival and migration. Of interest, was a very recent report that androgen receptor inhibition results in a shift in castration-resistant prostate cancer to cancers that are androgen receptor-null and dependent elevated fibroblast growth factor and MAP kinase signals [37, 38]. Significantly, fibroblast growth factor has been reported to activate MAP kinase via the PA- and PLD-dependent recruitment of Raf to the plasma membrane [39]. Thus, the shift from androgen receptor-dependence to fibroblast growth factor-dependence could also be accomplished by elevated PLD activity.
Highlights.
A key hallmark of cancer is the suppression of default apoptotic programs that likely represent the first line of defense against cancer.
Signals that suppress apoptosis are known as survival signals and occur early in tumorigenesis.
A common survival signal activated in human cancer cells is elevated phospholipase D activity that results in the production of phosphatidic acid – a key regulator of mTOR.
We find that elevated phospholipase D activity promotes both survival and metastatic phenotypes in androgen-independent prostate cancer cells.
Since survival signals are by necessity an early event in tumorigenesis, the implication is that metastasis is an earlier event in prostate cancer than generally appreciated.
Acknowledgments
This study was supported by National Institute of Health grants R01-CA046677 and R01-CA179542 to DAF. A pilot grant from Clinical Translational Science Center at Weill Cornell Medicine (14091346-03) (DAF and YSZ) and a pilot project award (to DAF) from the Research Centers in Minority Institutions award RP-03037 from the National Center for Research Resources of the National Institute of Health are also acknowledged. Support from the New York State Department of Health Prostate Cancer Initiative (C30330GG) is acknowledged. We thank Mike Frohman (Stony Brook University) for PLD expression vectors.
Abbreviations
- FBS
fetal bovine serum
- mTOR, HA
hemagglutinin; mammalian target of rapamycin
- PA
phosphatidic acid
- PARP
poly-ADP-ribose polymerase
- PLD
phospholipase D
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors declare no potential conflict of interest.
References
- 1.Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer. 2015;15:701–711. doi: 10.1038/nrc4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kirby M, Hirst C, Crawford ED. Characterising the castration-resistant prostate cancer population: a systematic review. Int J Clin Pract. 2011;65:1180–1192. doi: 10.1111/j.1742-1241.2011.02799.x. [DOI] [PubMed] [Google Scholar]
- 3.Gingrich JR, Barrios RJ, Kattan MW, Nahm HS, Finegold MJ, Greenberg NM. Androgen-independent prostate cancer progression in the TRAMP model. Cancer Res. 1997;57:4687–4691. [PubMed] [Google Scholar]
- 4.Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. 2001;1:34–45. doi: 10.1038/35094009. [DOI] [PubMed] [Google Scholar]
- 5.Edlind MP, Hsieh AC. PI3K-AKT-mTOR signaling in prostate cancer progression and androgen deprivation therapy resistance. Asian J Androl. 2014;16:378–386. doi: 10.4103/1008-682X.122876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wise HM, Hermida MA, Leslie NR. Prostate cancer, PI3K, PTEN and prognosis. Clin Sci (Lond) 2017;131:197–210. doi: 10.1042/CS20160026. [DOI] [PubMed] [Google Scholar]
- 8.Chen H, Zhou L, Wu X, Li R, Wen J, Sha J, Wen X. The PI3K/AKT pathway in the pathogenesis of prostate cancer. Frontiers in bioscience. 2016;21:1084–1091. doi: 10.2741/4443. [DOI] [PubMed] [Google Scholar]
- 9.Foster DA. Phosphatidic acid signaling to mTOR: signals for the survival of human cancer cells. Biochim Biophys acta. 2009;1791:949–955. doi: 10.1016/j.bbalip.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Foster DA, Salloum D, Menon D, Frias MA. Phospholipase D and the maintenance of phosphatidic acid levels for regulation of mammalian target of rapamycin (mTOR) J Biol Chem. 2014;289:22583–22588. doi: 10.1074/jbc.R114.566091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Toschi A, Lee E, Xu L, Garcia A, Gadir N, Foster DA. Regulation of mTORC1 and mTORC2 complex assembly by phosphatidic acid: competition with rapamycin. Mol Cell Biol. 2009;29:1411–1420. doi: 10.1128/MCB.00782-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rodrik V, Zheng Y, Harrow F, Chen Y, Foster DA. Survival signals generated by estrogen and phospholipase D in MCF-7 breast cancer cells are dependent on Myc. Mol Cell Biol. 2005;25:7917–7925. doi: 10.1128/MCB.25.17.7917-7925.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhong M, Shen Y, Zheng Y, Joseph T, Jackson D, Foster DA. Phospholipase D prevents apoptosis in v-Src-transformed rat fibroblasts and MDA-MB-231 breast cancer cells. Biochem Biophys Res Commun. 2003;302:615–619. doi: 10.1016/s0006-291x(03)00229-8. [DOI] [PubMed] [Google Scholar]
- 14.Zheng Y, Rodrik V, Toschi A, Shi M, Hui L, Shen Y, Foster DA. Phospholipase D couples survival and migration signals in stress response of human cancer cells. The J Biol Chem. 2006;281:15862–15868. doi: 10.1074/jbc.M600660200. [DOI] [PubMed] [Google Scholar]
- 15.Vue B, Zhang X, Lee T, Nair N, Zhang S, Chen G, et al. 5- or/and 20-O-alkyl-2,3-dehydrosilybins: Synthesis and biological profiles on prostate cancer cell models. Bioorg Med Chem. 2017;25:4845–4854. doi: 10.1016/j.bmc.2017.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cleutjens KB, van Eekelen CC, van der Korput HA, Brinkmann AO, Trapman J. Two androgen response regions cooperate in steroid hormone regulated activity of the prostate-specific antigen promoter. J Biol Chem. 1996;271:6379–6388. doi: 10.1074/jbc.271.11.6379. [DOI] [PubMed] [Google Scholar]
- 17.Noble AR, Maitland NJ, Berney DM, Rumsby MG. Phospholipase D inhibitors reduce human prostate cancer cell proliferation and colony formation. Br J Cancer. 2017 doi: 10.1038/bjc.2017.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Y, Rodrik V, Foster DA. Alternative phospholipase D/mTOR survival signal in human breast cancer cells. Oncogene. 2005;24:672–679. doi: 10.1038/sj.onc.1208099. [DOI] [PubMed] [Google Scholar]
- 19.Joseph T, Bryant A, Frankel P, Wooden R, Kerkhoff E, Rapp UR, et al. Phospholipase D overcomes cell cycle arrest induced by high-intensity Raf signaling. Oncogene. 2002;21:3651–3658. doi: 10.1038/sj.onc.1205380. [DOI] [PubMed] [Google Scholar]
- 20.Lewis JA, Scott SA, Lavieri R, Buck JR, Selvy PE, Stoops SL, et al. Design and synthesis of isoform-selective phospholipase D (PLD) inhibitors. Part I: Impact of alternative halogenated privileged structures for PLD1 specificity. Bioorg Med Chem Lett. 2009;19:1916–1920. doi: 10.1016/j.bmcl.2009.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown HA, Thomas PG, Lindsley CW. Targeting phospholipase D in cancer, infection and neurodegenerative disorders. Nat Rev Drug Discov. 2017;16:351–367. doi: 10.1038/nrd.2016.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu L, Salloum D, Medlin PS, Saqcena M, Yellen P, Perrella B, et al. Phospholipase D mediates nutrient input to mammalian target of rapamycin complex 1 (mTORC1) J Biol Chem. 2011;286:25477–25486. doi: 10.1074/jbc.M111.249631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mukhopadhyay S, Saqcena M, Chatterjee A, Garcia A, Frias MA, Foster DA. Reciprocal regulation of AMP-activated protein kinase and phospholipase D. J Biol Chem. 2015;290:6986–6993. doi: 10.1074/jbc.M114.622571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ikediobi ON, Davies H, Bignell G, Edkins S, Stevens C, O’Meara S, et al. Mutation analysis of 24 known cancer genes in the NCI-60 cell line set. Mol Cancer Ther. 2006;5:2606–2612. doi: 10.1158/1535-7163.MCT-06-0433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Foster DA. Targeting mTOR-mediated survival signals in anticancer therapeutic strategies. Expert Rev Anticancer Ther. 2004;4:691–701. doi: 10.1586/14737140.4.4.691. [DOI] [PubMed] [Google Scholar]
- 26.Hui L, Rodrik V, Pielak RM, Knirr S, Zheng Y, Foster DA. mTOR-dependent suppression of protein phosphatase 2A is critical for phospholipase D survival signals in human breast cancer cells. The Journal of biological chemistry. 2005;280:35829–35835. doi: 10.1074/jbc.M504192200. [DOI] [PubMed] [Google Scholar]
- 27.Shi M, Zheng Y, Garcia A, Xu L, Foster DA. Phospholipase D provides a survival signal in human cancer cells with activated H-Ras or K-Ras. Cancer letters. 2007;258:268–275. doi: 10.1016/j.canlet.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aguirre-Ghiso JA, Frankel P, Farias EF, Lu Z, Jiang H, Olsen A, et al. RalA requirement for v-Src- and v-Ras-induced tumorigenicity and overproduction of urokinase-type plasminogen activator: involvement of metalloproteases. Oncogene. 1999;18:4718–4725. doi: 10.1038/sj.onc.1202850. [DOI] [PubMed] [Google Scholar]
- 29.Rodrik V, Gomes E, Hui L, Rockwell P, Foster DA. Myc stabilization in response to estrogen and phospholipase D in MCF-7 breast cancer cells. FEBS Letters. 2006;580:5647–5652. doi: 10.1016/j.febslet.2006.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kauffmann-Zeh A, Rodriguez-Viciana P, Ulrich E, Gilbert C, Coffer P, Downward J, et al. Suppression of c-Myc-induced apoptosis by Ras signalling through PI(3)K and PKB. Nature. 1997;385:544–548. doi: 10.1038/385544a0. [DOI] [PubMed] [Google Scholar]
- 31.Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411:342–348. doi: 10.1038/35077213. [DOI] [PubMed] [Google Scholar]
- 32.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 33.Gotzsche PC, Olsen O. Is screening for breast cancer with mammography justifiable? Lancet. 2000;355:129–134. doi: 10.1016/S0140-6736(99)06065-1. [DOI] [PubMed] [Google Scholar]
- 34.Ghajar CM, Bissell MJ. Metastasis: Pathways of parallel progression. Nature. 2016 doi: 10.1038/nature21104. [DOI] [PubMed] [Google Scholar]
- 35.Hosseini H, Obradovic MM, Hoffmann M, Harper KL, Sosa MS, Werner-Klein M, et al. Early dissemination seeds metastasis in breast cancer. Nature. 2016 doi: 10.1038/nature20785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harper KL, Sosa MS, Entenberg D, Hosseini H, Cheung JF, Nobre R, et al. Mechanism of early dissemination and metastasis in Her2+ mammary cancer. Nature. 2016 doi: 10.1038/nature20609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brennen WN, Isaacs JT. Cellular Origin of Androgen Receptor Pathway-Independent Prostate Cancer and Implications for Therapy. Cancer Cell. 2017;32:399–401. doi: 10.1016/j.ccell.2017.09.011. [DOI] [PubMed] [Google Scholar]
- 38.Bluemn EG, Coleman IM, Lucas JM, Coleman RT, Hernandez-Lopez S, Tharakan R, et al. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell. 2017;32:474–489. doi: 10.1016/j.ccell.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rizzo MA, Shome K, Vasudevan C, Stolz DB, Sung TC, Frohman MA, S C, et al. Phospholipase D and its product, phosphatidic acid, mediate agonist-dependent raf-1 translocation to the plasma membrane and the activation of the mitogen-activated protein kinase pathway. J Biol Chem. 1999;274:1131–1139. doi: 10.1074/jbc.274.2.1131. [DOI] [PubMed] [Google Scholar]