

Summary
The HIV-1 core consists of capsid proteins (CA) surrounding viral genomic RNA. After virus-cell fusion, the core enters the cytoplasm and the capsid shell is lost through uncoating. CA loss precedes nuclear import and HIV integration into the host genome, but the timing and location of uncoating remain unclear. By visualizing single HIV-1 infection, we find that CA is required for core docking at the nuclear envelope (NE), whereas early uncoating in the cytoplasm promotes proteasomal degradation of viral complexes. Only docked cores exhibiting accelerated loss of CA at the NE enter the nucleus. Interestingly, a CA mutation (N74D) altering virus engagement of host factors involved in nuclear transport does not alter the uncoating site at the NE, but reduces the nuclear penetration depth. Thus, CA protects HIV-1 complexes from degradation, mediates docking at the nuclear pore before uncoating, and determines the depth of nuclear penetration en route to integration.
Keywords: Single particle tracking, reverse transcription, nuclear pore, HIV uncoating, integration, cyclophilin A, HIV inhibitors, capsid protein, proteasomal degradation, nuclear import
eTOC Blurb
HIV-1 genome is enclosed in a capsid shell composed of CA proteins. By imaging virions labeled with a fluorescent CA marker, Francis and Melikyan reveal that premature CA loss leads to proteasomal degradation of viral complexes, whereas CA release at the nuclear pore results in nuclear entry and productive integration.
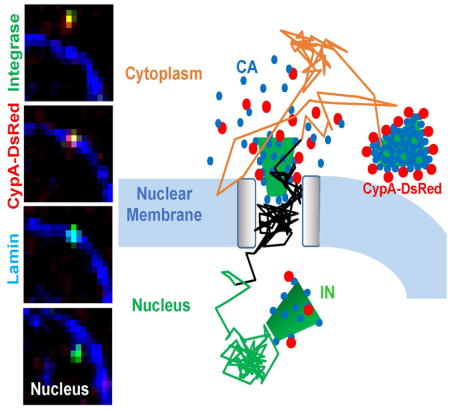
Introduction
Mature HIV-1 core consists of ~1,500 capsid proteins (CA) arranged in a hexagonal cone-shaped lattice surrounding the reverse transcriptase (RT), integrase (IN), nucleocapsid (NC) and two copies of viral genomic RNA (Briggs et al., 2004; Ganser-Pornillos et al., 2008). Following the delivery of the HIV-1 core into the cytoplasm, the viral RNA is reverse transcribed into cDNA and the capsid shell is lost through a process referred to as uncoating (reviewed in (Ambrose and Aiken, 2014; Campbell and Hope, 2015)). Loss of CA is required for penetration of the pre-integration complexes (PICs) through the nuclear pore. After the PIC enters the nucleus, IN catalyzes integration of the viral cDNA into host genome. Knowledge of the spatio-temporal regulation of uncoating is essential for understanding the multiple roles of CA in HIV-1 core trafficking, evasion of host innate immune sensors, nuclear entry of PICs and optimal integration into the host genome (reviewed in (Campbell and Hope, 2015)).
The extent, timing and cellular location of HIV-1 uncoating, which appears to proceed through multiple steps (Xu et al., 2013), remain poorly understood (Arhel et al., 2007; Francis et al., 2016; Hulme et al., 2011; Mamede et al., 2017; Xu et al., 2013). This largely stems from the use of indirect bulk assays reporting different aspects of uncoating and a lack of techniques capable of monitoring this process in a spatially and temporally resolved manner, in the context of productive infection. Live cell single-virus imaging provides a powerful means to elucidate dynamics of virus entry and uncoating, but direct CA labeling has proven difficult. To circumvent this problem, we have introduced an alternative approach to label the HIV-1 capsid and visualize its loss from single cores in live cells through incorporation of a tetrameric cyclophilin A-DsRed (CypA-DsRed) marker (Francis et al., 2016). Incorporation of CypA-DsRed through high-avidity binding to HIV-1 CA/Gag does not affect infectivity in TZM-bl cells and functionally compensates for lack of CypA in target Jurkat CypA-null cells. We have observed CypA-DsRed loss from post-fusion cores both at early and late times post-infection (Francis et al., 2016), but the functional relevance of these uncoating phenotypes remained unclear.
Here, we imaged the productive HIV-1 uncoating pathway by visualizing the release of CypA-DsRed/CA from single IN complexes that entered the nucleus and established infection. Virtually all cores that docked at the nuclear membrane contained CypA-DsRed and only those that subsequently lost CypA-DsRed/CA entered the nucleus and established infection. In contrast, early uncoating in the cytoplasm led to virus degradation in proteasomes. These results provide conclusive evidence that cores that retain a significant portion of CA are protected from degradation in the cytoplasm and are thus able to dock at the nuclear pore, where they undergo uncoating, prior to nuclear import and integration in the host genome.
Results
Sub-stoichiometric incorporation of CypA-DsRed does not affect the HIV-1 capsid’s interactions with host factors
We have previously shown that CypA-DsRed incorporation into HIV-1 particles through the high-avidity binding to CA/Gag does not significantly affect infectivity (Francis et al., 2016). Nearly 90% of IN-superfolderGFP (INsfGFP) labeled HIV-1 cores incorporated detectable amounts of CypA-DsRed (Fig. S1A, B). In spite of its efficient incorporation into virions (Fig. S1A, B), CypA-DsRed occupied only approximately half of the CypA binding sites on the mature HIV-1 capsid/core (Fig. S1C, D). Sub-stoichiometric amounts of incorporated CypA-DsRed do not alter the activity of CA-interacting restriction factors, TRIMCyp (which binds to the CypA-binding loop of CA) and the cytoplasm-resident CPSF6 fragment, CPSF6-358 (Lee et al., 2010) (Fig. S1E), or the kinetics of HIV-1 reverse transcription and integration (Fig. S1F). These results further support the utility of CypA-DsRed as a non-invasive marker for HIV-1 CA. We used HeLa-derived TZM-bl cells in this study as a model for HIV-1 infection, because these cells (i) enable single particle tracking and (ii) show similar host factor requirement for HIV-1 nuclear import and integration site selection to primary cells (reviewed in (Ambrose and Aiken, 2014; Campbell and Hope, 2015)). In addition, the ability of HIV-1 to infect these cells irrespective of endogenous CypA expression (Hatziioannou et al., 2005; Sokolskaja et al., 2004) should minimize possible unaccounted effects of CypA-DsRed on virus entry.
HIV-1 uncoating in the cytoplasm results in a gradual loss of IN signal
We have previously observed abrupt loss of CypA-DsRed from the overwhelming majority of post-fusion cores within the first hour of TZM-bl cell infection (Francis et al., 2016). The remaining (≤5%) post-fusion cores retained CypA-DsRed for several hours and tended to slowly release this marker (half-time ~1 h), implying that uncoating could proceed gradually, perhaps through multiple steps. We thus operationally define uncoating as a loss of the single core-associated CypA-DsRed/CA down to the background level or to a low level that remains constant throughout a long-term imaging experiment. Hereafter, post-uncoating HIV-1 structures will be referred to as IN complexes.
To determine which of the previously observed distinct uncoating phenotypes is relevant to infection, we followed the fate of INsfGFP-labeled HIV-1 particles pseudotyped with VSV-G after the release of CypA-DsRed and found that the INsfGFP signal tended to decay after uncoating (Fig. 1A, B). Although a few INsfGFP puncta remained visible beyond 1 h after loss of CypA-DsRed (Fig. 1A, B), on average, the IN signal disappeared within ~25 min (Fig. 1C). The relatively short lifetime of the cytoplasmic INsfGFP spots after uncoating should preclude the visualization of their entry into the nucleus, which takes several hours in TZM-bl cells (Albanese et al., 2008; Burdick et al., 2017).
Fig. 1. HIV-1 uncoating in the cytoplasm results in proteasomal degradation of RTC/PICs.

TZM-bl cells were infected with VSV-G pseudotyped HIV-1 labeled with INsfGFP/CypA-DsRed and imaged 0–2 h.p.i. (A) Examples of single uncoating events exhibiting loss of IN signal (1, 2) or relatively stable signal (3). (B) CypA-DsRed and INsfGFP fluorescence intensity traces for particles 2 and 3 in panel A. (C) The rate of disappearance of post-uncoating INsfGFP spots in cells pretreated with 10 μM MG132 or left untreated (DMSO). Stable INsfGFP spots (e.g., the bottom panel in (A)) were not included in this analysis. (D) Maximum intensity projection images showing fluorescent HIV-1 complexes in cells treated with DMSO or MG132 at 4 h.p.i and immunostained for p24. (E) The number of INsfGFP complexes positive and negative for CypA-DsRed or CA/p24 at 4 h.p.i. determined from 100 cells in 4 independent experiments. (F and G) Effect of MG132 on the nuclear import and infectivity of INsfGFP/CypA-DsRed labeled VSV-G pseudoviruses encoding for RFP. Error bars in (E–G) represent SEM from 4 independent experiments. (H) Images representative of data in F and G showing nuclear import (top) and mRFP expression in infected cells (bottom). Scale bars 5 μm in (D) and (H, top) and 50 μm (H, bottom). Cells infected at MOI 0.008 (A–C) or 0.2 (D–H). See related Figure S3.
The relatively rapid loss of INsfGFP after uncoating was not due to CypA-DsRed interference with binding of target cell CypA to the core. Similar levels of infection were observed for control and CypA-DsRed labeled pseudoviruses, both in parental TZM-bl cells and in cells lacking endogenous CypA (referred to as PPIA−/−, Fig. S2A, B). CypA knockout did not affect the kinetics of early particle uncoating (Fig. S2C and (Francis et al., 2016)), reverse transcription and integration (Figs. S1F and S2D) or the nuclear import of INsfGFP complexes (Fig. S2E), while the efficiency of nuclear import of INsfGFP complexes slightly increased in PPIA−/− cells (Fig. S2F, G).
The viral capsid protects RTC/PICs from proteasomal degradation
Decay of INsfGFP signal following the loss of CypA-DsRed (Fig. 1A–C) could occur due to destabilization/degradation of post-uncoating complexes or to shedding of INsfGFP. To investigate the cause of INsfGFP loss, we imaged virus entry into PPIA−/− cells treated with a proteasome inhibitor MG132 that delays the loss of YFP-Vpr puncta after viral fusion (Sood et al., 2017). This inhibitor affected neither the extent nor the kinetic of early uncoating (Fig. S2C), but markedly delayed post-uncoating loss of INsfGFP (Fig. 1C). The latter effect resulted in accumulation of cytoplasmic (Fig. 1D, E) and intra-nuclear (Fig. 1F, H) IN complexes at late times post-infection and enhanced infection (Fig. 1G). The preferential enrichment in cellular IN-only complexes vs. nuclear complexes and infection (5-fold vs. ~2.5-fold, Fig. 1D–H) suggests that nuclear entry of prematurely uncoated cores is disfavored, even in the absence of proteasomal degradation. Accumulation of cytoplasmic IN complexes was also detected in PPIA−/− cells pretreated with another proteasome inhibitor lactacystin (Fig. S3A, B) and in TZM-bl cells pretreated with either proteasome inhibitor (Fig. S3C–F), regardless of whether or not the viruses were labeled with CypA-DsRed. These results imply that post-uncoating complexes are degraded by proteasomes.
Nuclear entry of INsfGFP complexes and their subsequent disappearance correlate with productive infection
Correlation between the number of nuclear IN complexes and infection (Fig. 1E–H) prompted further examination of this relationship by long-term imaging of cells infected with INsfGFP/CypA-DsRed labeled pseudoviruses encoding for eGFP (Fig. 2A and movie S1). These experiments confirmed the marked loss of fluorescent puncta over the first few hours of infection. Approximately 2% of cell-bound particles entered the nucleus, reaching a plateau by 8 h.p.i. (Fig. 2B and Fig. S2E) prior to manifestation of infection by eGFP expression (Fig. 2A, last panel). In agreement with the previous studies (Burdick et al., 2013; Peng et al., 2014), a CA-binding inhibitor, PF-3450074 (PF74, 2 μM), but not the reverse transcription inhibitor, Nevirapine (NVP, 10 μM), blocked the nuclear entry of IN complexes (Fig. S4A, B). These results support the essential role of CA in nuclear import of PICs, whereas non-infectious complexes can enter the nucleus in the absence of reverse transcription.
Fig. 2. Nuclear INsfGFP complexes are likely to establish infection.

(A) EBFP2-Lamin expressing TZM-bl cells infected with INsfGFP/CypA-DsRed labeled pseudoviruses encoding for eGFP were continuously imaged, starting at 0 h.p.i. An arrow marks a single particle docking at the NE, losing CypA-DsRed and entering the nucleus. An arrowhead shows subsequent disappearance of the nuclear INsfGFP signal prior to eGFP reporter expression. Scale bar 5 μm. (B) Kinetics of live-cell nuclear import of INsfGFP complexes in TZM-bl and PPIA−/− cells infected with INsfGFP/CypA-DsRed labeled viruses (≥5 independent experiments). (C and D). Analysis of the number of nuclear IN complexes in infected (eGFP+) vs. uninfected (eGFP−) cells and correlation between the nuclear complexes and the probability of eGFP expression after inoculation with pseudoviruses at MOI 0.2. (E) Correlation between the number of INsfGFP puncta associated with each cell after incubation for 10 min at 37°C and infection. (F) The nuclear INsfGFP complex disappearance events were detected and correlated with the infection outcome. Inset: A fraction of IN-spots that disappeared in infected vs. uninfected cells. Total cells analyzed from ≥3 independent experiments in panels C–F: GFP+ (PPIA−/−, n=97; TZM-bl, n=74) and GFP- (PPIA−/−, n=154; TZM-bl, n=130). (G) Time course of nuclear detection of INsfGFP spots in TZM-bl cells after infection with pR9ΔE/VSV-G pseudovirus labeled with INsfGFP and CypA-DsRed in the presence of a fully inhibitory concentration of Raltegravir (10 μM) or DMSO (control). Infection was carried out in the presence of aphidicolin (5 μM). Cells were fixed at indicated time points and the number of nuclear IN complexes measured. Error bars are SEM from 4 fields of view, from a representative of 2 independent experiments. An additional plot (right axis, green circles) shows the kinetics of nuclear INsfGFP puncta disappearance measured in live TZM-bl cells in parallel experiments (n=97; cumulative of 2 independent experiments). See related Figure S4 and movie S1.
The probability with which nuclear IN complexes productively integrate into the host genome was assessed from the above experiments (Fig. 2A) by relating the number of the nuclear INsfGFP spots (if any, MOI 0.2) to subsequent expression of eGFP in the same cell. Cells without detectable nuclear IN spots were rarely (3.1%) infected, whereas all cells that contained >7 nuclear INsfGFP complexes expressed eGFP (Fig. 2C, D). Based on the slope of the probability of productive integration vs. the number of nuclear IN spots (0.12), on average, 1 out of 8 nuclear IN complexes establishes infection in PPIA−/− and TZM-bl cells (Fig. 2D). The very low probability of infection in cells with no detectable nuclear INsfGFP spots (Fig. 2C, D) implies that we visualize virtually all relevant IN complexes in the nucleus, consistent with the high efficiency (~90%) of virus labeling with INsfGFP (Fig. S1A, B). In contrast, only 1 in 200–300 pseudoviruses bound to cells establishes infection (Fig. 2E). The above results show that the nuclear INsfGFP complexes are better predictors of productive infection than unstable post-uncoating complexes in the cytoplasm.
Interestingly, a fraction of IN complexes disappeared after varied times following the nuclear entry (Fig. 2A, arrowhead) and this loss of signal correlated very well with the subsequent expression of eGFP (Fig. 2F and movie S1). More than 80% of cells, in which loss of a single nuclear IN complex was detected, expressed eGFP. The relationship between loss of nuclear INsfGFP puncta and integration into the host genome is further supported by the time course of single IN complex accumulation in the nuclei (Fig. 2G). Whereas the number of INsfGFP spots peaked at ~6 h.p.i. and decreased, this number steadily increased up to 24 h.p.i. in the presence of Raltegravir. This result, along with the overlapping kinetics of disappearance of the nuclear INsfGFP complexes without a drug measured in parallel live cell experiments (Fig. 2G, green circles), strongly support the notion that loss of the nuclear IN complexes corresponds to HIV-1 integration into the host genome. Moreover, the time course of disappearance overlapped with the time course of integration measured by adding a fully inhibitory concentration of Raltegravir at varied time points (Figs. S1F and S2D). Interestingly, the probability of INsfGFP disappearance in the nuclei of cells prior to manifestation of infection was ~10-fold greater than in cells that failed to express eGFP within that time frame (Fig. 2F, Inset), likely due to different permissiveness of these cells to infection.
The strikingly strong correlation between disappearance of INsfGFP foci within the nucleus and eGFP expression (n=97) suggests that these events correspond to productive HIV-1 integration into the host genome, in agreement with the previous reports (Arhel et al., 2006; Borrenberghs et al., 2016). Importantly, the examples shown in Fig. 2A and movie S1 are representative of 37 out of 97 events, in which docking at the NE and loss of the CA marker could be resolved prior to nuclear entry, disappearance of INsfGFP particles and subsequent expression of eGFP. The loss of CypA-DsRed could not be resolved for majority of infectious entry events owing to the limited temporal resolution employed for the long-term imaging experiments. These results indicate that productive uncoating may occur at the nuclear pore and that systematic analysis of single HIV-1 uncoating that culminates in nuclear entry will define a productive entry pathway en route to infection.
CA mediates HIV-1 docking at the nuclear envelope
Tracking of INsfGFP/CypA-DsRed puncta at late times post-infection revealed stable association of a small fraction of HIV-1 cores with the nuclear envelope (NE) (Fig. 3 and (Francis et al., 2016)). We define core docking as stable (≥15 min) co-localization with the NE labeled with eBFP-LaminB1 associated with highly confined motion. The nature of interactions responsible for HIV-1 docking at the NE was explored using PF74; this compound binds to the same pocket, formed by the NTD-CTD interface of the CA hexamer, as the cellular factors, CPSF6 and Nup153 (Bhattacharya et al., 2014; Blair et al., 2010; Matreyek et al., 2013; Price et al., 2014). Since CPSF6 and Nup153 are involved in transport of HIV-1 complexes across the nuclear pore, the observed block of nuclear entry by PF74 (Fig. S4A, B) could be due to inhibition of CA-dependent binding to these host factors.
Fig. 3. Docking at the nuclear pore is mediated by CA.

FAP-Lamin expressing PPIA−/− cells infected with INsfGFP/CypA-DsRed labeled viruses were imaged for 1 hour between 4 and 8 h.p.i. PF74 or CsA (10μM) was added to cells after ~30 min of imaging. Docked cores were identified based upon colocalization with lamin and restricted motion (see STAR Methods). (A to C) Images, fluorescent intensity traces and trajectories corresponding to a single core displacement from the NE by PF74. (D to F) Images, fluorescent intensity traces and trajectories showing CypA-DsRed dissociation from a core that remains docked after CsA addition. Scale bar 2 μm in (A) and (D). (G) Analysis of the number of stably (>15 min) docked cores before and after PF74 or CsA addition. Error bars are SEM from 5 independent experiments. (H) PPIA−/− cells infected with INsfGFP/CypA-DsRed labeled viruses (MOI 0.5) containing wild-type (WT) CA or the E45A or K203A mutants for 4 h, were fixed and immunostained for lamin. Scale bar 10 μm. (I) The average number of INsfGFP spots per nucleus for WT and CA mutants. Error bars = SEM from 4 fields of view containing ~90 cells. (J and K) Fluorescent intensity traces and trajectory of a representative E45A CA mutant-containing core. See related Figure S4 and movie S2.
To test this notion, we assessed the ability of PF74 to displace single INsfGFP/CypA-DsRed cores docked at the NE by live cell imaging. Addition of 10 μM PF74 displaced docked HIV-1 cores from the NE (Fig. 3A–C and movie S2), whereas addition of CsA in control experiments displaced CypA-DsRed, while the cores remained docked (Fig. 3D–F and movie S2). Multiple experiments confirmed a strong core displacing effect of PF74, but not CsA (Fig. 3G). A lower concentration of PF74 (2 μM) also displaced docked cores (Fig. S4C–E). The PF74 effect suggests that docking is mediated by CA-interacting host factors involved in nuclear import (perhaps CPSF6 and Nup153) and that, therefore, functional cores should retain a certain amount of hexameric CA to stably attach to the NE.
To further assess the role of CA in core docking, we examined CA mutants with altered core stability. At 5 h.p.i., the double-labeled (presumably intact, Fig. 3H) cores made of the hyper-stable E45A CA mutant accumulated in the cytoplasm to a greater extent than WT/CA cores, whereas there were rare surviving IN complexes after the quick uncoating of the unstable K203A CA mutant (Figs. 3H and (Francis et al., 2016)). In spite of its marked accumulation around the NE, nuclear entry of the E45A mutant was dramatically reduced compared to WT cores (Fig. 3H, I). Accordingly, and in agreement with a recent study (Burdick et al., 2017), the hyperstable mutant cores did not exhibit stable docking and tended to alternate between confined and less restricted motion along the nuclear envelope (Fig. 3J, K). These results support the essential role of CA in protecting viral complexes from proteasomal degradation and in docking at the nuclear pore complex.
Accelerated loss of CA at the nuclear envelope is a prerequisite for nuclear import
In order to identify productive uncoating events, we visualized loss of CypA-DsRed from single HIV-1 cores that entered the nucleus. Almost without an exception, INsfGFP complexes that entered the nucleus of TZM-bl cells were positive for CypA-DsRed at the time of arrival at the NE and lost a large portion of this marker prior to nuclear import (Fig. 4A–F and movies S3 and S4). Importantly, a slow release of the CA marker from perinuclear cores was greatly (~20-fold) accelerated after docking at the NE (Fig. 4G), likely due to capsid interactions with the nuclear pore complex. Shortly after shedding CypA-DsRed at the nuclear membrane, IN complexes quickly moved into the nucleoplasm and exhibited a slower, more restricted motion, while remaining within a few microns from the NE (Fig. 4C, F, Fig. S5 and movies S3 and S4). The same progression through docking, uncoating, nuclear entry and intra-nuclear motion was observed for PPIA−/− cells (Fig. 4 and Fig. S5A–C) and TZM-bl cells (Fig. S5D–F and movie S1).
Fig. 4. Uncoating at the nuclear pore is a pre-requisite for HIV-1 nuclear entry.

EBFP2-Lamin expressing PPIA−/− cells were infected (MOI 0.2) with INsfGFP/CypA-DsRed labeled pseudoviruses. (A to F). Time-lapse images, fluorescent intensity traces and trajectories obtained using slow (every 3–5 min, A–C) and fast (every 20 sec, D–F) acquisition are shown. Increase and decrease in the lamin signal (blue) in B and E mark docking and nuclear import, respectively. Analysis of the core docking time and post-uncoating lag before nuclear import is illustrated by arrows in B. Scale bar 2 μm in (A) and (D). (G) Analysis of uncoating at the NE by ensemble averaging multiple events. Individual tracks were aligned by the time of arrival at the NE (defined as time = 0), and the average CypA-DsRed intensity was calculated separately for the cytoplasmic cores (negative time scale) and for docked particles that entered the nucleus (blue). Error bars are not shown for clarity. Similar analysis was applied to stably docked cores that fail to or enter the nucleus (black; time = 0 corresponds to the onset of imaging). Error bars represent SEM. Fluorescence intensities were normalized to the initial values at the onset of imaging. (H) Docking time distributions for single- (INsfGFP) and double- (INsfGFP/CypA-DsRed) labeled viruses in TZM-bl and PPIA−/− cells. Note that the difference between single- vs. double-labeled viruses in TZM-bl cells barely reached statistical significance. (I) Post-uncoating lag time distributions for double-labeled viruses in TZM-bl and PPIA−/− cells. Only single particle tracks from experiments with relatively frequent (20 sec to 5min) image acquisition are analyzed in H and I. (J and K) Fluorescence intensity traces and trajectory of a single docked particle that did not enter the nucleus during the observation window. (L) The fraction of the CypA-DsRed signal remaining by the end of observation (Docked) or after nuclear entry (Imported). See related Figure S5 and movies S3–5.
As mentioned above, INsfGFP spots lacking detectable CypA-DsRed rarely docked or entered the nucleus (n=3). These single-labeled particles most likely represent INsfGFP cores that failed to incorporate detectable amounts of CypA-DsRed upon assembly (Fig. S1A), for which the uncoating status could not be assessed. The rarity of docking or nuclear import of post-uncoating INsfGFP complexes, which was abrogated by the destabilizing K203A mutation (Fig. 3H), further supports the notion that loss of CypA-DsRed/CA in the cytoplasm precludes nuclear entry.
Kinetic analysis revealed that, on average, HIV-1 cores remained docked for ~35 min prior to nuclear import and this distribution was not considerably affected by CypA knockdown in target cells or by the virus-incorporated CypA-DsRed (Fig. 4H). In addition, “post-uncoating lag” was measured as an interval between loss of CypA-DsRed and nuclear entry of INsfGFP puncta (illustrated in Fig. 4B). After loss of CypA-DsRed, IN complexes resided at the NE for ~17 min and this timing was not modulated by endogenous CypA (Fig. 4I), suggesting the existence of an additional kinetic barrier for nuclear import of IN complexes.
Importantly, docked cores that gradually lost up to 70% of the initial CypA-DsRed signal during a 2-hour imaging window failed to enter the nucleus (Fig. 4J–L and movie S5). On average, these cores lost CypA-DsRed with half-time of 77 min (Fig. 4G). In a few instances, cores retaining a large portion of the initial CypA-DsRed signal after several hours of docking detached from the NE, suggesting that failure to uncoat could lead to undocking (data not shown). In contrast, imported IN complexes exhibited a profound and rather abrupt loss of the CypA-DsRed signal at the NE (Fig. 4A–G, L and movie S5). A clear segregation of the extents of CypA-DsRed loss from the docked/not imported vs. imported complexes, suggests an operational definition for functionally relevant HIV-1 uncoating at the NE as loss of the CypA-DsRed/CA to nearly a background level. Experiments described below provide evidence that loss of CypA-DsRed at the NE reflects uncoating and not displacement of this marker from capsid.
Nuclear IN complexes are stable and can contain trace amounts of CypA-DsRed/CA
To determine whether IN complexes retain trace amounts of CA/CypA-DsRed upon nuclear import, we fixed the cells at varied times after infection, permeabilized and immunostained for p24. Imaging was performed under more stringent conditions than live cell imaging (see STAR Methods) to achieve higher signal-to-background ratio and detect minute amounts of CypA-DsRed and CA. In agreement with previous studies (Burdick et al., 2017; Chin et al., 2015; Hulme et al., 2015; Peng et al., 2014), we found that approximately a third of the nuclear INsfGFP spots were positive for CypA-DsRed and/or p24, and that the extent of IN/p24 co-localization was not affected by CypA-DsRed labeling (Fig. 5A, B). Nuclear IN complexes contained lower amounts of p24 and CypA-DsRed compared to the cytoplasmic complexes (Fig. 5C). The INsfGFP signal itself was also diminished after nuclear import, in line with previous reports (Borrenberghs et al., 2016; Francis et al., 2014).
Fig. 5. Nuclear IN complexes are stable and can contain trace amounts of CA/CypA-DsRed.

TZM-bl cells were infected with INsfGFP/CypA-DsRed labeled pseudoviruses for 4 h, fixed and immunolabeled for CA/p24. (A) Nuclear INsfGFP complexes containing or lacking detectable CypA-DsRed and CA/p24 signal are shown. Scale bar 5 μm. (B) The fractions of intra-nuclear INsfGFP complexes that colocalized with CA/p24 or CypA-DsRed for single- and double-labeled viruses was measured for ~120 nuclei. (C) Distributions of sum fluorescence intensities for INsfGFP, CypA-DsRed and CA/p24 puncta in the cytoplasm and nucleus. The numbers above the graph represent mean fluorescence values for the respective box-plots below. (D) Decrease in the mean fluorescence of the cytosolic CypA-DsRed and p24 signals upon nuclear import calculated from panel C. (E) Decrease in the mean fluorescence of cytosolic CypA-DsRed and p24 signals upon nuclear import in the presence of 10 μM NVP. (F and G) Images and fluorescence intensity information of a nuclear INsfGFP complex containing CypA-DsRed that was tracked for several hours. (H) Fraction of nuclear IN containing or lacking detectable CypA-DsRed signal that disappeared prior to eGFP expression was determined by live-cell imaging. Analysis is based on 13 infected cells and 50 PICs, of which 22 contained CypA-DsRed. Cells were infected with MOI 1.5 in (A–E) and 0.2 in (F–H). Data are means and SEM from 4 independent experiments in (B), from 3 independent experiments in (C and D), from a representative of 2 experiments (4 fields of view) in (E), and from 13 infected cells in (H). Scale 2 μm in F. See related Figure S6 and movie S6.
Importantly, an identical, ~2.5-fold drop in the amount of core-associated CA/p24 and CypA-DsRed for the nuclear vs. the cytosolic IN complexes (Fig. 5D) strongly implies that loss of CypA-DsRed reflects uncoating and is not caused by CypA-DsRed displacement from the cores upon CA-nucleoporin binding. The loss of the CypA-DsRed/CA or INsfGFP signals upon nuclear import was not affected by inhibition of reverse transcription, which delays single virus cytoplasmic uncoating (Fig. 5E) (Francis et al., 2016; Mamede et al., 2017). Of note, incorporation of CypA-DsRed into virions did not affect the reduction of CA/p24 signal associated with the nuclear entry (data not shown). This result, along with the ability of CypA-DsRed to remain associated with residual CA on the nuclear PICs, further supports its use as a reliable marker for HIV-1 uncoating.
In sharp contrast to post-uncoating HIV-1 complexes that were degraded in the cytoplasm, most nuclear INsfGFP spots containing or lacking detectable CypA-DsRed could be continuously tracked for several hours (Fig. 5F, G, Fig. S6 and movie S6). Thus, unlike the cytoplasm, the nucleoplasm is a safe environment for IN complexes and loss of INsfGFP spots in the nucleus that is blocked by Raltegravir (Fig. 2G) appears to reflect productive integration. Importantly, the stable CypA-DsRed signal for nuclear complexes (e.g., Fig. 5G) implies that the releasable CA/CypA-DsRed pool is lost at the NE and that the remaining CA molecules are tightly associated with PICs.
As shown in Figure 2F, disappearance of a fraction of nuclear INsfGFP complexes strongly correlates with productive integration. Live cell imaging revealed that the nuclear complexes lacking or containing detectable amounts of CypA-DsRed disappeared with similar probability (Fig. 5H). Thus, based upon the loss of the nuclear INsfGFP signal as a proxy for integration, it appears that the nuclear IN complexes can be functional irrespective of the presence or the relative level of the CA marker associated with them.
HIV-1 CA/host factor interactions determine the depth of nuclear penetration
We next ask if the N74D CA mutant, which utilizes an alternative, CPSF6- and NUP153-independent nuclear entry pathway (Ambrose et al., 2012; Lee et al., 2010; Matreyek and Engelman, 2011; Schaller et al., 2011), uncoats at the nuclear pore. The N74D mutation did not affect HIV-1 infectivity (Fig. S7A) or the core stability, as measured by the number of post-fusion core at 3 h.p.i. (Fig. S7B), in TZM-bl cells. Also similar to WT viruses, N74D cores co-labeled with INsfGFP and CypA-DsRed docked at the NE and lost the CA marker (n=45, Fig. 6A, B), whereas post-uncoating IN complexes were not observed to dock (data not shown). However, after uncoating at the NE, N74D IN complexes typically remained colocalized with lamin (Fig. 6A, B and Fig. S7C–E).
Fig. 6. HIV-1 CA-host factor interactions determine the nuclear penetration of IN complex.

EBFP2-Lamin expressing TZM-bl cells were infected with INsfGFP/CypA-DsRed HIV-1 pseudoviruses bearing the N74D CA mutation. (A, B). Fluorescent intensity traces and trajectory of a single N74D CA particle docked at the NE. Decrease in the CypA-DsRed intensity after association with lamin (blue) in panel A marks uncoating after particle docking. The relatively constant lamin signal after uncoating shows the lack of significant penetration into the nucleoplasm. (C, D) Images and analysis of nuclear penetration in fixed TZM-bl cells at 4 h.p.i. with WT and N74D pseudoviruses labeled with INsfGFP. Cells were immunolabeled for NUP358. Analysis included only imported N74D complexes that clearly separated from the nuclear membrane, whereas a significant fraction of membrane-colocalized post-uncoating complexes were not considered. Scale bar 5 μm. (E) EBFP2-Lamin expressing TZM-bl cells infected with INsfGFP/CypA-DsRed labeled pseudoviruses containing the N74D CA mutant and encoding for eGFP were continuously imaged, starting at 1 h.p.i. White arrows show 2 docked particles at the NE that loose CypA-DsRed without detectable nuclear penetration. Arrowheads show INsfGFP disappearance prior to eGFP reporter expression. Scale bar 10 μm. (F) Correlation between the number of N74D IN complexes that remained associated with the NE after uncoating and infection measured by eGFP expression. N=68 infected cells and n=157 for uninfected cells (>3 independent experiments). (G) Correlation between the INsfGFP disappearance after uncoating at the NE and infection (n=91 in infected and n=6 in uninfected cells). (H) Waiting time for uncoating (defined as terminal loss of CypA-DsRed) after arrival at the NE for single WT and N74D cores. MOI 0.2 was used in panels A–I. Only single particle tracks from experiments with relatively frequent (20 sec to 5 min) image acquisition are analyzed (n=51 for N74D and n=84 for WT). See related Figure S7 and movie S7.
To assess whether the mutant virus penetrated into the nucleoplasm, we fixed the cells, immuno-stained the nuclei for NUP358 and performed high signal-to-background confocal imaging (Fig. 6C), as well as super-resolution STED imaging (Fig. S7C). In agreement with the previous reports (Albanese et al., 2008; Burdick et al., 2013), we found that imported WT PICs tended to reside at the nuclear periphery (on average, 1.8 μm from the NE). In contrast, post-uncoating N74D complexes remained very close to the NE, even when imaged by super-resolution microscopy. The fraction of cores that separated from the NE traveled on average only 0.5 μm (Fig. 6D).
In spite of poor apparent nuclear penetration, a fraction of N74D uncoating events culminated in disappearance of the INsfGFP signal at the NE followed by eGFP expression (Fig. 6E and movie S7), implying that the observed uncoating led to nuclear import and productive integration.
Moreover, disappearance of post-uncoating N74D IN complexes at the NE resulted in eGFP expression with a probability similar to the intra-nuclear WT IN complexes (compare Figs. 6F and 2D). Also, disappearance of post-uncoating N74D IN complexes at the NE showed a high degree of correlation with productive integration that was virtually identical to correlation observed for disappearing intra-nuclear WT IN complexes (compare Figs. 6G and 2F). Together, these results imply that uncoating of N74D cores at the nuclear membrane leads to nuclear import and productive integration in TZM-bl cells.
Analysis of single N74D core docking and uncoating revealed that, interestingly, the loss of CypA-DsRed from after docking was significantly (>3-fold) slower than for WT cores (Fig. 6H and Fig. S7). The above findings support the notion that uncoating at the nuclear pore is mediated by CA-host factor interactions and that these interactions determine the depth of nuclear penetration of IN complexes and the sites of integration.
Discussion
Here, we visualized the single HIV-1 entry process in its entirety, from the cytoplasmic transport to uncoating, nuclear import and infection. This unique strategy revealed that: (1) uncoating (loss of protective capsid) in the cytoplasm exposes the IN complexes to host degradation machinery; (2) a large part of CA lattice must be preserved for HIV-1 docking at the nuclear pore; (3) accelerated loss of nearly all CA molecules at the nuclear pore is a pre-requisite for nuclear import of IN complexes that are likely to establish infection; (4) nuclear IN complexes are long-lived and loss of the INsfGFP signal correlates with productive integration; and (5) CA-host factor interactions define the HIV-1 nuclear penetration depth, which likely contributes to the integration site preference. Our results thus show that uncoating at the nuclear pore represents an infectious pathway in HeLa-based cell lines. Similar models for the protective role of CA and uncoating at the nuclear pore have been proposed for primary cells (Hilditch and Towers, 2014; Rasaiyaah et al., 2013).
Extensive validation of the CypA-DsRed marker did not reveal any considerable effects on major steps of HIV-1 entry, including the ability of cores to dock at the NE and the docking time distribution prior to nuclear entry. These results, along with the minor effects of endogenous CypA knockout on HIV-1 entry and uncoating observed in our experiments, are consistent with CypA-independence of HeLa cell infection (Hatziioannou et al., 2005; Sokolskaja et al., 2004). Our approach is compatible with imaging of HIV-1 uncoating and nuclear import in CD4 T cells and primary human macrophages, in which the infectivity of CypA-DsRed labeled viruses remains comparable to unlabelled viruses. We were able to visualize single virus uncoating in these cells, however, further optimization of imaging conditions is required to demonstrate uncoating at the nuclear membrane. It would also be interesting to explore possible effects of envelope glycoprotein (VSV-G vs. HIV-1 Env) on post-fusion steps of HIV-1 entry.
A recent indirect approach to visualize single HIV-1 uncoating in living cells took advantage of a small amount of free GFP trapped in an intact mature core (Mamede et al., 2017). Release of this GFP pool within 30 min of virus entry correlated with infection, suggesting that productive uncoating occurs in the cytoplasm. We surmise that the release of trapped GFP at early times post-infection reflects the initiation of uncoating (compromised core integrity), whereas terminal loss of CA occurs at the nuclear pore and is a prerequisite for nuclear import and infection. Furthermore, by monitoring loss of CA/CypA-DsRed, we show that complete uncoating in the cytoplasm triggers proteasomal degradation of HIV-1 complexes, although partial loss of CA in the cytoplasm appears to be tolerated (Figs. 4D, E and S5D, E). It should be noted, however, that the CypA-DsRed-based uncoating assay is not sensitive to loss of a minor fraction of CA molecules. The model that can reconcile the above results is that productive uncoating proceeds through multiple steps, including loss of capsid integrity at early times post-infection and a subsequent terminal release of CA at the nuclear pore.
Protection of RTC/PICs from proteasomal degradation by capsid observed in our experiments is in line with the enhancement of HIV-1 infection by proteasome inhibitors (Schwartz et al., 1998; Wei et al., 2005). Exposure of IN on post-uncoating complexes could provide a signal for degradation (e.g., (Devroe et al., 2003; Mulder and Muesing, 2000)). On the other hand, disfavored stable docking and nuclear import of hyper-stable E45A HIV-1 cores (Fig. 3K and (Burdick et al., 2017)) is in line with the notion that partial CA release may be required for functional binding to the nuclear pore (Mamede et al., 2017; Matreyek et al., 2013). However, the infectivity defect conferred by the E45A CA mutation appears complex and may be unrelated to the increased core stability (Yang et al., 2012). Further studies are needed to elucidate the mechanism by which post-uncoating cytoplasmic complexes are recognized and targeted for degradation.
While our results are in general agreement with a recent study by Burdick and co-authors (Burdick et al., 2017), shorter core docking times were observed under our conditions (~30 min vs. 90 min). The reasons for this discrepancy are unclear. It is conceivable that the choice of a fluorescent NE marker could affect the kinetics of nuclear import, since POM121 used in (Burdick et al., 2017) is at the center of the nuclear pore, whereas Lamin-B1 is at the inner nuclear membrane.
The use of an alternative nuclear import pathway by the N74D CA mutant has been shown to alter the HIV-1 integration sites (Ambrose et al., 2012; Matreyek and Engelman, 2011; Schaller et al., 2011). Our live cell imaging experiments revealed the requirement for nuclear pore-associated uncoating of N74D complexes. The delayed completion of uncoating of docked N74D mutant cores at the NE compared to WT cores (Fig. 6H) further supports the essential role of CA interactions with the nuclear pore-resident factors in uncoating and nuclear import. Importantly, consistent with the nuclear penetration distances of the viral DNA for WT and N74D CA (Albanese et al., 2008; Chin et al., 2015; Di Primio et al., 2013), the N74D mutant complexes failed to undergo significant transport into the nucleoplasm after uncoating at the NE (Figs. 6A–E and S7). This result suggests that the difference in the integration site preference for the N74D and WT CA might be determined by the CA/host factor-dependent nuclear penetration of PICs.
In summary, the current study shows the critical role of CA in protecting the viral replication complexes from host surveillance and ensuring their successful docking at the nuclear pore. We provide conclusive evidence that uncoating, as defined by the terminal loss of CA at the nuclear pore, represents the productive pathway for HIV-1 nuclear entry and infection.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Gregory Melikyan (gmeliki@emory.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Plasmids
The pCypA-DsRed and Vpr-INsfGFP plasmids have been described previously (Francis et al., 2014; Francis et al., 2016). The Vpr-INmNeonGreen was constructed by swapping sfGFP with mNeonGeen using BamHI and NotI enzymes (NEB, New England Biolabs Inc., MA). The pR9ΔEnv vector containing the WT CA or CA mutations E45A, and K203A was described previously (Forshey et al., 2002). pMD2.G expressing VSV-G glycoprotein and the psPAX2 vectors were obtained from Addgene (Cat# 1259 and 12260) were a gift from Didier Trono. The pFUPI-TrimCyp126, pFUPI-TrimCyp322 and the lentiviral vector encoding for huTRIMCyp were gifts from Dr. Jeremy Luban (University of Massachusetts). The pHIV-eGFP (NL4.3 R-E- eGFP) and N74D-eGFP plasmids were a gift from Dr. Christopher Aiken (Vanderbilt University). The pHIV-RFP vector was a gift from Dr. Vineet KewalRamani (NCI). The NL4.3-Nef-HA plasmid was provided by Dr. Massimo Pizzato (University of Trento). The pLenti-CRISPR.V2.sgPPIA plasmid, as validated in GECKO library, was obtained from GenScript. The pLenti.EBFP2-LaminB1-10 construct was made by excising the EBFP2-Lamin encoding fragment from EBFP2-LaminB1-10 (Addgene#55244), using AgeI and BamHI, and cloning into pLentiCRISPR.V2 plasmid by replacing Cas9. The pLenti.FAP-Lamin construct was prepared by replacing EBFP2 with the Fluorogen Activating Peptide (FAP, (Szent-Gyorgyi et al., 2008)) using AgeI and XhoI.
Cell lines and reagents
HEK293T/17 cells (from ATCC, Manassas, VA) and TZM-bl cells derived from human cervical carcinoma HeLa cells (from NIH AIDS Reference and Reagent Program) were grown in high-glucose Dulbecco’s Modified Eagle Medium (DMEM, Mediatech, Manassas VA) supplemented with 10% Fetal Bovine Serum (FBS, Sigma, St. Louis, MO) and 100 U/ml penicillin-streptomycin (Gemini Bio-Products, Sacramento, CA). The growth medium for HEK 293T/17 was supplemented with 0.5 mg/ml G418 sulfate (Mediatech). Cyclosporin A (CsA) was obtained from Calbiochem (Burlington, MA), dissolved in DMSO at 50 mM and stored in aliquots at −20°C. TZM-bl PPIA−/− cells (referred to as PPIA−/− cells) were generated by lentivirus-mediated transduction of TZM-bl cells with pLentiCRISPR.V2.sgPPIA (MOI 0.2) followed by puromycin selection. EBFP2-LaminB1-10 expressing TZM-bl and PPIA−/− cells were generated using a lentiviral vector Lenti.EBFP2-LaminB1-10 encoding for LaminB1. Following puromycin selection, cells were subjected to limited dilution and clones expressing optimal levels of EBFP2-Lamin were selected. TZM-bl expressing the restriction- competent CPSF6-358 fragment and control cells were a kind gift from Drs. Vineet KewalRamani (NCI) and Zandrea Ambrose (University of Pittsburgh). TZM-bl cells stably expressing huTRIMCyp isoforms were prepared by transduction with pFupi-Empty, pFupi-TRIMCyp322 WT or TRIMCyp126 expressing vectors (InvivoGen, San Diego) followed by puromycin selection. All cell lines were cultured in a 37°C incubator supplemented with 5% CO2, and were passaged in regular intervals.
The following reagents were obtained from the NIH AIDS Reference and Reagent Program, Division of AIDS, NIAID, NIH: pNL4-3.Luc.R-E- from Dr. Nathaniel Landau (He et al., 1995), TZM-bl cells expressing CD4, CXCR4 and CCR5 from Drs. J.C. Kappes and X. Wu (Platt et al., 1998); anti-p24 antibody AG3.0 donated by Dr. J. Alan (Simm et al., 1995); RT inhibitor Nevirapine and IN inhibitor Raltegravir (Merck & Company, Inc.).
The HIV-1 CA binding inhibitors, PF74 (#PF-3450074), Aphidicolin (#A0781) and mouse anti-tubulin antibody (#T6074) was purchased from Sigma-Aldrich. Bright-Glo luciferase assay kit was from Promega (Madison, WI). Puromycin was obtained from InvivoGen. Antibodies to Lamin-B (#ab16048), CypA (#ab3563) and donkey anti-rabbit AF405 antibody (#ab175651) were purchased from Abcam (San Francisco, CA). Anti-mouse HRP antibodies were from SantaCruz (#sc-2005, Dallas, TX), anti-rabbit-HRP from Millipore (#AP188P, Burlington, MA), Cy5 conjugated anti-mouse antibody was from SouthernBiotech (Birmingham, AL). Rabbit-polyclonal antibody against NUP358/RanBP2 was purchased from Abcam (#ab64276). The FAP binding fluorescent probe Se-Red-S was obtained from SharpEdgeLabs (Pittsburgh, PA). Phosphate buffered saline containing Mg2+/Ca2+ (dPBS) and Mg/Ca-free (PBS) was purchased from Corning (MediaTech.Inc., Manassas, VA).
Pseudovirus production and characterization
Fluorescently labeled pseudoviruses were produced and characterized, as described previously (Francis et al., 2016). Briefly, HEK293T/17 cells grown in 6-well culture plates were transfected with the following plasmids: HIV-1 pR9ΔEnv (1.8 μg), VSV-G (0.2 μg), Vpr-INsfGFP (0.4 μg) and, where indicated, CypA-DsRed (0.3 μg), using the JetPrime Transfection reagent (VWR, Radnor, PA). Alternatively, HIV-1 NL4-3-based (pHIVeGFP/RFP) pseudoviruses were produced by co-transfecting VSV-G (0.2 μg), pHIVeGFP/RFP (1.8 μg), Nef-HA (0.4 μg) and CypA-DsRed (0.3 μg). For generation of EBFP2-Lamin expressing cells and for PPIA−/− (CypA knockout) cells, lentivirus encoding for pLenti.EBFP2-LaminB1-10 or pLenti-CRISPR.V2.sgPPIA (2 μg), respectively, was co-transfected with the Gag/Gag-Pol expressing psPAX2 (1 μg) and VSV-G (0.2 μg) vectors. Six hours after transfection, the medium was replaced with 2 ml of fresh DMEM/10% FBS without phenol red, and the sample incubated for additional 36 h at 37°C, 5% CO2. Viral supernatant was collected, filtered through a 0.45 μm filter and the RT activity measured using the PERT protocol (Pizzato et al., 2009). For live-cell imaging of uncoating/nuclear import, fluorescent viruses were purified through a 20% sucrose cushion and re-suspended in FluoroBrite medium containing 10% FBS. Viruses were aliquoted and stored at −80°C.
Virus quantification for RT activity (PERT Assay)
Viral RT activity was measured using the qPCR based Product Enhanced Reverse Transcriptase (PERT) assay (Pizzato et al., 2009). The RT activity of virus preparations was determined by comparing to a standard curve obtained for the recombinant M-MLV RT enzyme (ThermoFisher; #28025013) activity. Briefly, 5 μl of viral supernatant was lysed for 10 min in a 2x lysis buffer (0.25% Triton X-100, 50 mM KCl, 100 mM TrisHCl pH 7.4, 40% glycerol). The RNase inhibitor Ribolock (Fermentas, # E00381) was included in the 2x virus lysis buffer to minimize RNA degradation. The lysate was diluted 1:200 with ddH20. The qPCR reaction was carried out using 2.5 μl virus lysate. The reaction mixture consisted of 5 μl of 2x iTaq™ Universal SYBR® green Supermix (BIORAD; #172-5121), 2.5 μl of 1:200 diluted viral lysate, 2.5 μl solution of MS2 RNA template (7 picomoles/ml, Roche #10165948001), and 10 μM primers (see Key Resources) to amplify MS2 sequences. The reaction was performed in a 96-well transparent qPCR plate (GeneMate; # 490003-822) which was loaded onto a qPCR-machine (Applied Biosystem®7500; #4351104). The PERT reaction consisted of 4 steps. Step-1: 42°C (20 min); Step-2: 95°C (5 min); Step-3: (40-cycles)/95°C (20 sec) and 60°C (1min); Step-4: melting curve. The Auto-Ct values were determined and the RT-units (RTU)/ml of viral supernatants were later calculated using the M-MLV based standard curve.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-LaminB1 antibody | Abcam | #ab16048; RRID:AB_443298 |
| Anti-Cyclophilin A | Abcam | #ab3563; RRID:AB_303910 |
| Anti-RanBP2 antibody | Abcam | #ab64276; RRID:AB_1142517 |
| Donkey Anti-Rabbit IgG H&L (Alexa Fluor® 405) | AbCam | #ab175651 |
| anti-p24 antibody AG3.0 | NIH AIDS Reagent Program, NIAID, NIH; (Simm et al., 1995) | #120227 |
| Goat Anti-mouse HRP | Santa Cruz Biotechnology | #sc-2005; RRID:AB_631736 |
| Goat anti-mouse Cy5 | SouthernBiotech | #1030-15 |
| Anti-rabbit-HRP | Millipore | #AP188P; RRID:AB_92637 |
| Monoclonal Anti-α-Tubulin | SIGMA-Aldrich | #T 6074; RRID:AB_477582 |
| Bacterial and Virus Strains | ||
| NEB® 5-alpha Competent E.coli (High Efficiency) | New England Biolabs | #C2987I |
| One Shot™ Stbl3™ Chemically Competent E.coli | Invitrogen, ThermoFisher | #C737303 |
| pR9ΔEnv | Dr. Christopher Aiken, Vanderbilt University (Forshey et al., 2002) | N/A |
| pR9ΔEnv-CA E45A mutant | Dr. Christopher Aiken, Vanderbilt University (Forshey et al., 2002) | N/A |
| pR9ΔEnv-CA K203A mutant | Dr. Christopher Aiken, Vanderbilt University (Forshey et al., 2002) | N/A |
| pR9ΔEnv-CA N74D mutant | Dr. Christopher Aiken, Vanderbilt University (Lee et al., 2010) | N/A |
| pHIV-eGFP | NIH AIDS Reagent program, NIAID NIH (Zhang et al., 2004) | N/A |
| pHIV-N74D/CA-eGFP | Dr. Christopher Aiken, Vanderbilt University (Shah et al., 2013) | N/A |
| pHIV-mRFP | Dr. Vineet KewalRamani, NCI (Ambrose et al., 2012) | N/A |
| Biological Samples | ||
| N/A | N/A | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Aphidicolin | Sigma Aldrich | #A0781 |
| CA binding inhibitor PF74 (PF-3450074) | Sigma Aldrich | #SML0835 |
| high-glucose Dulbecco’s Modified Eagle Medium | Mediatech | #10-013-CV |
| Fluorobrite medium | Gibco | #A18967-01 |
| Live cell Imaging buffer | Invitrogen | #A14291DJ |
| Phosphate buffered Saline Ca-/Mg- | Mediatech | #21-040-CV |
| Dulbecco Phosphate buffered Saline Mg2+/Ca2+ (dPBS) | Mediatech | #17-513Q |
| Puromycin | Invivogen | #ant-pr-1 |
| Gentamycin G418 | Mediatech | #30-234-CR |
| Cyclosporine A (CsA) | Calbiochem | #239835 |
| Penicillin - streptomycin | Mediatech | #30-002-CI |
| RT inhibitor Nevirapine | NIH AIDS Reagent Program; NIAID, NIH | #4666 |
| Hoechst-33342 | ThermoFisher | #H3570 |
| Integrase Inhibitor Raltegravir (ISENTRESS/MK-0518) | NIH AIDS Reagent Program, NIAID, NIH; Merck & Company, Inc. | #11680 |
| TritonX-100 | SIGMA-ALDRICH | #234729 |
| PFA | Electron Microscopy Sciences | #1570-S |
| MG132 | SIGMA-ALDRICH | #474791 |
| Lactacystin, Synthetic | SIGMA-ALDRICH | #426100 |
| M-MLV Reverse Transcriptase | ThermoFisher | #28025013 |
| MS2 RNA | Roche | #10165948001 |
| Ribolock RNase Inhibitor | Fermentas | # E00381 |
| iTaq™ Universal SYBR® green Supermix | BIORAD | #172-5121 |
| Digitonin | SIGMA-ALDRICH | #D141 SIGMA |
| FAP binding fluorescent probe Se-Red-S | SharpEdgeLabs | Se-Red-S |
| Critical Commercial Assays | ||
| Bright-Glo™ Luciferase Assay System | Promega | # E2620 |
| Jet Prime®transfection reagent | Polyplus | #712-60 and #114-07 |
| Deposited Data | ||
| N/A | N/A | N/A |
| Experimental Models: Cell Lines | ||
| TZM-bl cells expressing CD4, CXCR4 and CCR5 | NIH AIDS Reagent Program, NIAID, NIH (Platt et al., 1998) | #8129 |
| Endogenous CypA KO TZM-bl PPIA−/− cells expressing CD4, CXCR4 and CCR5 | This paper | N/A |
| TZM-bl expressing the restriction- competent CPSF6-358 fragment | Drs. Vineet KewalRamani (NCI) and Zandrea Ambrose (University of Pittsburgh); (Lee et al., 2010) | N/A |
| HEK293T/17 cells | ATCC | #CRL11268 |
| Experimental Models: Organisms/Strains | ||
| N/A | N/A | N/A |
| Oligonucleotides | ||
| crRNA-PPIA: ATGGACCAACCTGCTGTCTT | GenScript®; (Sanjana et al., 2014) | GEKCO Library |
| PERT Fwd: 5′-tcctgctcaacttcctgtcgag-3′ | Invitrogen; (Pizzato et al., 2009) | N/A |
| PERT Rev: 5′-CACAGGTCAAACCTCCTAGGAATG-3′ | Invitrogen; (Pizzato et al., 2009) | N/A |
| Recombinant DNA | ||
| pFupi-Empty, | Dr. Jeremy Luban. University of Massachusetts (Neagu et al., 2009) | N/A |
| pFupi-TRIMCyp322 | Dr. Jeremy Luban. University of Massachusetts (Neagu et al., 2009) | N/A |
| pFupi-TRIMCyp126 | Dr. Jeremy Luban. University of Massachusetts (Neagu et al., 2009) | N/A |
| pLentiCRISPR.V2.sgPPIA | Dr. Feng Zhang, Broad institute MIT. (Sanjana et al., 2014) | Addgene Plasmid #52961 |
| pLentiCRISPR.V2.sgPPIA | GenScript®; (Sanjana et al., 2014) | GEKCO Library |
| pLentiCRISPR.V2.EBFP-LaminB1 | This paper | N/A |
| pLentiCRISPR.V2.FAP-LaminB1 | This Paper and (Szent-Gyorgyi et al., 2008) | N/A |
| pCypA-DsRed | (Francis et al., 2016) | N/A |
| pVpr-IN-superfolderGFP (IN-sfGFP) | (Francis et al., 2016) | N/A |
| Vpr-INmNeonGreen | This paper | N/A |
| pMD2.G expressing VSV-G envelope | Dr. Didier Trono, École Polytechnique Fédérale de Lausanne | Addgene Plasmid #1259 |
| psPax2 | Dr. Didier Trono, École Polytechnique Fédérale de Lausanne | Addgene Plasmid #12260 |
| EBFP2-LaminB1-10 | Dr. Michael Davidson, Florida State University | Addgene Plasmid #55244 |
| Software and Algorithms | ||
| ICY Image analysis software | icy.bioimageanalysis.org (de Chaumont et al., 2012) | N/A |
| Huygens essential deconvolution software | Scientific Volume Imaging B.V., Netherlands; | N/A |
| ImageJ | NIH.GOV | N/A |
| Other | ||
| 35 mm glass bottom dishes | MatTek corp., Ashland, MA | #P35GCOL-1.5-10.C |
| 96-well plate | Corning | # 3904 |
METHOD DETAILS
Single-cycle infection assays
TZM-bl or PPIA−/− cells were plated onto a 96-well plate (Corning, Corning, Kennebunk ME) at 104 cells/well. Serial dilutions of virus supernatants containing equal RT activity were used to infect the cells in triplicates. Virus binding to cells was enhanced by a 30 min spin at 1,200×g at 4°C. Cells were cultured at 37°C for 48 h, lysed and the luciferase activity measured using the bright glow luciferase substrate (Promega). Where mentioned, MG132, Raltegravir, nevirapine or PF74 were used at indicated concentrations and times after infection. Percent infected cells was determined by counting the number of eGFP or mRFP reporter expressing cells at 24 h.p.i. (4 random fields of view) and normalizing to the total number of cells as identified by the Hoechst nuclear stain.
Live-cell nuclear import and infectivity assays
TZM-bl or PPIA−/− cells (5·105) stably expressing EBFP2-Lamin or FAP-Lamin fusion proteins were grown on 35 mm glass bottom dishes (MatTek corp., Ashland, MA). VSV-G pseudotyped pHIV-eGFPΔEnv particles containing WT/CA or the N74D/CA mutant were co-labeled with INsfGFP and CypA-DsRed and used to infect the cells at MOI of 0.2. Viruses were bound to cells by spinoculation at 1200×g for 30 min at 4°C, and cells were either incubated for additional 10min in a CO2 incubator at 37°C or the sample was moved to a Zeiss LSM880 confocal microscope stage maintained at 37°C with 5% CO2. Multiple (4–25) adjacent fields of view, was imaged at high spatial resolution with multiple line averaging and Z-stacks from 0–10 min to 16–24 h.p.i., in the multiple experiments and the data was pooled. The time and number of intra-nuclear PICs entry and disappearance was manually determined for each nuclei that either became eGFP+ (infected) or remained eGFP- (un-infected cells). For the N74D/CA mutant, IN complexes that remained associated with the NE after uncoating and subsequent disappearance of a fraction of these complexes were quantified. Where indicated, Aphidicolin (5μM) was added immediately post spinoculation and maintained until 24 hours of imaging. Cells that underwent mitosis or those that moved outside the field of view were not included in the analysis.
Live-cell uncoating and nuclear entry of HIV-1
Single HIV-1 uncoating in live cells was visualized, as previously described (Francis et al., 2016). In brief, VSV-G pseudotyped pR9ΔEnv particles co-labeled with INsfGFP and CypA-DsRed were bound to 5·105 TZM-bl or PPIA−/− cells (10 pg of p24, MOI 0.008) by spinoculation at 1500×g, 4°C for 30 min. Prior to virus binding, cell nuclei were stained for 10 min with 2 μg/ml Hoechst-33342. The cells were washed twice, and virus entry was synchronously initiated on a temperature- and CO2-controlled microscope stage by adding pre-warmed live-cell imaging buffer (Invitrogen, Grand Island, NY). When indicated, DMSO, CsA (10 μM), PF74 (10 μM) or nevirapine (10 μM) were added to cells decorated with viruses before beginning the image acquisition and maintained throughout the experiment. MG132 treatment was carried out by pre-incubating TZM-bl or PPIA−/− cells with 5 μM MG132 for 30 min prior to virus binding, and imaging in the presence of MG132. Where indicated, CsA (10 μM) was added at 75–80 min after beginning of image acquisition to allow quantification of the number of post-fusion cores that did not uncoat.
To visualize uncoating, docking and nuclear entry, FAP or EBFP2-LaminB1-10 expressing TZM-bl or PPIA−/− cells were incubated 30–90 min at 37°C in a CO2 incubator with pR9ΔEnv or HIV-eGFPΔEnv pseudoviruses co-labeled with INsfGFP and CypA-DsRed. Excess virus was washed off and the sample was either further incubated in FluoroBrite/10% FBS in a CO2 incubator or moved to a temperature- and CO2-controlled microscope stage for imaging.
Immunofluorescence assay
TZM-bl or PPIA−/− cells (105) were infected at MOI 0.5–3 with fluorescent viruses by spinoculation, as above, and washed prior to incubation at 37°C in a CO2. Drugs were added at indicated concentrations immediately after virus binding, except for MG132 that was added 30 min prior to infection (see above). Cells were fixed with 2% PFA (Electron Microscopy Sciences, #1570-S) for 7 min at indicated time points. Fixed cells were permeabilized with 0.1% TX-100 for 5 min at room temperature, washed and blocked in 3% BSA with 0.1% Tween-20 in dPBS. The mouse monoclonal anti-CA/p24 AG3.0 antibody (1:100) and polyclonal rabbit anti-LaminB1 antibody (1:1000) diluted in a blocking solution were allowed to bind for 1 h at room temperature or overnight at 4°C. Cells were washed with dPBS supplemented with 0.1% Tween-20, incubated with secondary goat anti-mouse Cy5 antibody and, after 3x washes, with goat anti-rabbit-AlexaFluor405 antibody, each for 1 h at room temperature. As controls for p24 background staining, cells were incubated only with secondary antibodies.
Image acquisition
3D time-lapse live cell imaging was carried out on a Zeiss LSM780 or LSM880 laser scanning confocal microscopes using a C-Apo 40x/1.2NA water-immersion objective or 63x/1.4NA oil-immersion objective. For live-cell uncoating experiments, a field of view was selected, and full cell volume was imaged by acquiring 8–12 Z-stacks spaced by 1 μm every 30 sec. Acquired image series were converted to maximum intensity projections and analyzed, as previously described (Francis et al., 2016). The loss of CypA-DsRed signal from INsfGFP labeled viral particles was blindly annotated by two trained students. When imaging fixed cells, greater laser powers and line averaging were used, along with more stringent axial sampling (~45 Z-stacks spaced by 0.3 μm) in order to improve signal-to-background ratio. STED images were acquired using a Leica-SP8 3X STED microscope (Leica Microsystems Inc. IL). The NUP358-AF647 fluorescence was depleted using lasers (715 nm) and INmNeonGreen was depleted using lasers (592 nm).
To visualize uncoating and nuclear entry, multiple fields of view were imaged between 0.5 and 24 h.p.i. by acquiring 11–15 Z-stacks spaced by 1 or 0.7 μm every 3–5 min (slow acquisition; low-temporal resolution) or every 20–60 sec (fast acquisition; high-temporal resolution). Cells were imaged at 37°C using the Zeiss environmental chamber maintained at 5% CO2. The DefiniteFocus module (Carl Zeiss) was utilized to correct for axial drift. EBFP2-Lamin, INsfGFP, CypA-DsRed and FAP-lamin fluorescence was excited using highly attenuated 405, 488, 561 and 633 nm laser lines, respectively. 3D-image series were processed off-line using ICY image analysis software (icy.bioimageanalysis.org) (de Chaumont et al., 2012). Docked viral cores that were co-localized with the nuclear lamin were imaged at high temporal resolution for 2hr-windows, starting at 4 h.p.i. To detect core-displacement from the nuclear lamin by PF74, cells were imaged for 30 min before and 30 min after drug addition, images were acquired every 20–40 sec. Live-cell nuclear import and infection correlation assays were visualized by imaging infection starting at 0–10 min through 16–24 h.p.i. (long-term imaging). For these experiments, high signal/noise ratio images were acquired every 10–30 min, using multiple line averaging and Z-stacks spaced by 0.5–0.7 μm.
QUANTIFICATION AND STATISTICAL ANALYSIS
Single-particle tracking and image analysis
Initial annotation of uncoating cores and INsfGFP spots that entered the nucleus was done manually. Single particle tracking was performed using the ICY image analysis platform. 2D or 3D INsfGFP objects were detected using the wavelet spot detection plugin and tracked using the spot-tracking plugin. The coordinates for nucleus and single particle movements were visualized with Track Manager. Cellular motion artifacts were corrected by tracking single nuclear volume, using the Active-Cells plugin and subtracting the nucleus trajectory from the single IN-complex trajectory. Single particle intensity and trajectory were obtained using the fluorescence intensity and coordinate plugins. The background signal for CypA-DsRed marker was determined by imaging single-labeled INsfGFP trafficking and nuclear import.
Single particle intensity traces were normalized to initial fluorescence intensity of INsfGFP/CypA-DsRed after background subtraction. The time of IN survival after uncoating or the time of completion of CypA-DsRed release was determined by the signal drop below <5% of initial intensity. The nuclear FAP or EBFP2-Lamin signals were normalized by subtracting the background signal and setting the peak intensity at the mid lamin section as 100%.
Single docked cores at the nuclear membrane were identified manually based upon the apparent co-localization to the nuclear membrane and tracked using ICY, as described above. After trajectory analysis, cores were considered docked if they exhibited restricted movement not exceeding 1 μm in three dimensions, while remaining co-localized with the nuclear lamin. Particle departure from the initial localization with the nuclear membrane was interpreted as undocking. After visual inspection, the intensity and trajectory information was used to localize single particles to cytoplasm, the time of arrival (docking) at nuclear membrane and time of penetration/import into the nucleus. The time of residence at the nuclear membrane (docking time) was calculated as an interval between particle arrival/docking at the nuclear membrane and nuclear entry. The lag between uncoating and nuclear entry (post-uncoating lag) was defined as an interval between loss of CypA-DsRed and nuclear entry. For N74D/CA mutant, the time of uncoating at the NE was measured as the interval between single particle arrival/docking at the nuclear membrane and loss of CypA-DsRed. The docking, uncoating and nuclear entry times were determined manually for those events that were not amenable to single-particle tracking. The extent of CypA-DsRed loss for docked cores that did not enter the nucleus or for IN complexes that entered the nucleus was calculated by averaging the CypA-DsRed fluorescence intensity from the last 5 time frames of docked cores and the first 5 time frames after nuclear entry of the IN complex.
For assessing the CA/p24 and CypA-DsRed signals in the cytoplasm and nucleus, stringent imaging conditions were used (2x laser power, 4-line averaging, with 0.12 μm/pixel in X-Y and 0.3 μm/pixel in Z). In order to separate the cytoplasmic INsfGFP from the nuclear spots an in-house protocol was created using the ICY protocols module. Briefly, the nuclear volume in three dimensions was detected using the EBFP-lamin intensity, which was identified by the HK-means and connected components plugins in ICY. To avoid detection of nuclear membrane-associated INsfGFP spots, the obtained 3-dimensional ROI corresponding to the nuclear volume was shrunk by 0.5 μm in X-Y-Z using the ROI-erosion plugin. The IN complexes within the eroded ROI were verified and considered as nuclear spots, all remaining spots were deemed to reside in the cytoplasm. The sum intensity of the CA/p24 and CypA-DsRed signals within each cell-associated INsfGFP spot was measured. The respective background fluorescence intensities were determined using a secondary antibody only control for CA/p24 or single-labeled INsfGFP for CypA-DsRed. Outliers (likely viral aggregates) with the INsfGFP, p24 or CypA-DsRed intensities falling outside of a standard deviation range were excluded from analysis. The fraction of intra-nuclear INsfGFP spots that contained above-background levels of CA/p24 or CypA-DsRed were considered co-localized with these markers.
Spatial distribution of nuclear IN complexes was measured, as described previously (Albanese et al., 2008; Di Primio et al., 2013). Briefly, infected cells were immunostained for nucleoporin NUP358, and images were acquired and deconvolved using Huygens essential deconvolution software (Scientific Volume Imaging B.V., Netherlands). Confocal slices (spaced by 4 μm) of the central nuclear volume were used for this analysis. The shortest distance between the center of a single intra-nuclear IN complex and the center of the NE was determined manually in 2D using ImageJ (NIH).
Statistical analysis
Statistical significance was determined by the Mann-Whitney rank-sum test or the Student t-Test, as indicated. p<0.05 (*) was considered significant; ** and *** denote p<0.01 and p<0.001, respectively. The number of experiments and error bars are indicated in the figure legends.
Supplementary Material
Example-1: TZM-bl cells expressing EBFP2-lamin were infected with VSV-G pseudotyped HIV-1 labeled with INsfGFP (green) and CypA-DsRed (red) and imaged from 30 min to 21 h.p.i. Time-lapse Z-stack images (shown as maximum intensity projections through a 4 μm central section) were acquired every 10 min. A cell showing single particle uncoating, nuclear entry and INsfGFP disappearance prior to infection (Related to Fig. 2A). Example-2: PPIA−/− cells expressing EBFP2-Lamin were infected and imaged (30 min/frame) from 30 min post-infection, as above. A cell showing single particle uncoating, nuclear entry and disappearance prior to infection is shown. Scale bar 5 μm.
Left: A single core co-labeled with CypA-DsRed/INsfGFP docked at the nuclear lamin is displaced after addition of 10 μM PF-74. Scale bar 2 μm. Right: Control experiments showing, a single core co-labeled with CypA-DsRed/INsfGFP docked at the nuclear lamin. CsA (10 μM) is added at 15 min of the video. A single Z-plane is shown. Imaging was performed between 4 and 5 h.p.i. Scale bar 1 μm.
Arrival of a single HIV-1 pseudovirus co-labeled with CypA-DsRed/INsfGFP at the nuclear envelope is followed by uncoating and nuclear import. Time-lapse Z-stack images (shown as maximum intensity projections through a 1 μm central section) were acquired every 3 min, starting at 2.5 h.p.i. Scale bar 5 μm.
Arrival of a single HIV-1 pseudovirus co-labeled with CypA-DsRed/INsfGFP at the nuclear envelope is followed by uncoating and nuclear import. Time-lapse Z-stack images (shown as maximum intensity projections through a 1 μm central section) were acquired every 20 sec, starting after 1 h.p.i. Images were temporal averaged using a 3 frame window. Scale bar 5 μm.
Top: A single HIV-1 pseudovirus co-labeled with CypA-DsRed/INsfGFP docked at the nuclear envelope, loses CypA-DsRed (red) and enters the nucleus. Bottom: A single HIV-1 pseudovirus docked at the nuclear envelope does not lose a significant portion of CypA-DsRed and does not enter the nucleus. Time-lapse Z-stack images (shown as maximum intensity projections through a 1 μm central section) were acquired every 20 sec, starting at ~5 h.p.i. Images were temporally averaged, using a 3-frame window. Scale bar 2 μm.
Arrival of a single HIV-1 pseudovirus co-labeled with CypA-DsRed/INsfGFP at the nuclear envelope (top Z slice) is followed by uncoating and nuclear import (middle Z slice) was visualized in TZM-bl cells every 5 min. Virus transport is shown from 10 min p.i. through 14 h.p.i. Maximum intensity projections of 1 μm top- and central nuclear volume are shown. Scale bar 5 μm.
TZM-bl cells expressing EBFP2-Lamin were infected with VSV-G pseudotyped HIV-CA/N74D mutant labeled with INsfGFP (green) and CypA-DsRed (red) and imaged from 1 to 18 h.p.i. Time-lapse Z-stack images (shown as maximum intensity projections through a 2 μm central section) were acquired every 15 min. Example-1: A cell with 2 docked particles that uncoat without an apparent separation from lamin and disappeared prior to infection (expression of eGFP). Example-2: A cell with 3 docked particles that uncoat and remain at the nuclear membrane for the duration of the experiment without losing the IN signal or establishing infection. Scale bar 5 μm.
Highlights.
Loss of CA in the cytoplasm promotes proteasomal degradation of HIV-1 complexes
HIV-1 docking at the nuclear pore is mediated by CA
Docking at the nuclear pore results in accelerated uncoating
Loss of CA at the nuclear pore precedes nuclear import and productive integration
Acknowledgments
The authors thank the NIH AIDS Reagent Program and Drs. C. Aiken, V. KewalRamani, J. Luban and M. Pizzato for providing reagents, Caleb Mason for technical assistance, and the team at ICY for help with image analysis. Critical reading of the manuscript by Dr. C. Aiken and the members of Melikyan laboratory is gratefully acknowledged. We also thank Riya Mathew for the artwork in Figure 7. This work was supported by the NIH R01 GM054787 and AI129862 grants to GBM.
Fig. 7. A model for HIV-1 uncoating and nuclear entry.

Functional HIV-1 uncoating occurs through multiple steps involving early core opening and subsequent terminal loss of CA at the NE, prior to intra-nuclear transport to sites of integration. Terminal uncoating in the cytoplasm leads to proteasomal targeting of the IN complexes, while the failure to uncoat after docking at the NE results in block to nuclear import. The N74D/CA mutant that uses an alternative nuclear import pathway also uncoats at the NE, but fails to move away from the NE, leading to peripheral integration.
Footnotes
Author contributions. ACF and GBM conceived the study, ACF performed the experiments, ACF and GBM analyzed the results and co-wrote the manuscript.
Declaration of Interests. The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Albanese A, Arosio D, Terreni M, Cereseto A. HIV-1 pre-integration complexes selectively target decondensed chromatin in the nuclear periphery. PloS one. 2008;3:e2413. doi: 10.1371/journal.pone.0002413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrose Z, Aiken C. HIV-1 uncoating: connection to nuclear entry and regulation by host proteins. Virology. 2014;454–455:371–379. doi: 10.1016/j.virol.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrose Z, Lee K, Ndjomou J, Xu H, Oztop I, Matous J, Takemura T, Unutmaz D, Engelman A, Hughes SH, et al. Human immunodeficiency virus type 1 capsid mutation N74D alters cyclophilin A dependence and impairs macrophage infection. J Virol. 2012;86:4708–4714. doi: 10.1128/JVI.05887-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arhel N, Genovesio A, Kim KA, Miko S, Perret E, Olivo-Marin JC, Shorte S, Charneau P. Quantitative four-dimensional tracking of cytoplasmic and nuclear HIV-1 complexes. Nat Methods. 2006;3:817–824. doi: 10.1038/nmeth928. [DOI] [PubMed] [Google Scholar]
- Arhel NJ, Souquere-Besse S, Munier S, Souque P, Guadagnini S, Rutherford S, Prevost MC, Allen TD, Charneau P. HIV-1 DNA Flap formation promotes uncoating of the pre-integration complex at the nuclear pore. The EMBO journal. 2007;26:3025–3037. doi: 10.1038/sj.emboj.7601740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya A, Alam SL, Fricke T, Zadrozny K, Sedzicki J, Taylor AB, Demeler B, Pornillos O, Ganser-Pornillos BK, Diaz-Griffero F, et al. Structural basis of HIV-1 capsid recognition by PF74 and CPSF6. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:18625–18630. doi: 10.1073/pnas.1419945112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair WS, Pickford C, Irving SL, Brown DG, Anderson M, Bazin R, Cao J, Ciaramella G, Isaacson J, Jackson L, et al. HIV capsid is a tractable target for small molecule therapeutic intervention. PLoS Pathog. 2010;6:e1001220. doi: 10.1371/journal.ppat.1001220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrenberghs D, Dirix L, De Wit F, Rocha S, Blokken J, De Houwer S, Gijsbers R, Christ F, Hofkens J, Hendrix J, et al. Dynamic Oligomerization of Integrase Orchestrates HIV Nuclear Entry. Scientific reports. 2016;6:36485. doi: 10.1038/srep36485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs JA, Simon MN, Gross I, Krausslich HG, Fuller SD, Vogt VM, Johnson MC. The stoichiometry of Gag protein in HIV-1. Nat Struct Mol Biol. 2004;11:672–675. doi: 10.1038/nsmb785. [DOI] [PubMed] [Google Scholar]
- Burdick RC, Delviks-Frankenberry KA, Chen J, Janaka SK, Sastri J, Hu WS, Pathak VK. Dynamics and regulation of nuclear import and nuclear movements of HIV-1 complexes. PLoS Pathog. 2017;13:e1006570. doi: 10.1371/journal.ppat.1006570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdick RC, Hu WS, Pathak VK. Nuclear import of APOBEC3F-labeled HIV-1 preintegration complexes. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E4780–4789. doi: 10.1073/pnas.1315996110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell EM, Hope TJ. HIV-1 capsid: the multifaceted key player in HIV-1 infection. Nature reviews Microbiology. 2015;13:471–483. doi: 10.1038/nrmicro3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin CR, Perreira JM, Savidis G, Portmann JM, Aker AM, Feeley EM, Smith MC, Brass AL. Direct Visualization of HIV-1 Replication Intermediates Shows that Capsid and CPSF6 Modulate HIV-1 Intra-nuclear Invasion and Integration. Cell reports. 2015;13:1717–1731. doi: 10.1016/j.celrep.2015.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Chaumont F, Dallongeville S, Chenouard N, Herve N, Pop S, Provoost T, Meas-Yedid V, Pankajakshan P, Lecomte T, Le Montagner Y, et al. Icy: an open bioimage informatics platform for extended reproducible research. Nat Methods. 2012;9:690–696. doi: 10.1038/nmeth.2075. [DOI] [PubMed] [Google Scholar]
- Devroe E, Engelman A, Silver PA. Intracellular transport of human immunodeficiency virus type 1 integrase. J Cell Sci. 2003;116:4401–4408. doi: 10.1242/jcs.00747. [DOI] [PubMed] [Google Scholar]
- Di Primio C, Quercioli V, Allouch A, Gijsbers R, Christ F, Debyser Z, Arosio D, Cereseto A. Single-cell imaging of HIV-1 provirus (SCIP) Proceedings of the National Academy of Sciences of the United States of America. 2013;110:5636–5641. doi: 10.1073/pnas.1216254110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forshey BM, von Schwedler U, Sundquist WI, Aiken C. Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J Virol. 2002;76:5667–5677. doi: 10.1128/JVI.76.11.5667-5677.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis AC, Di Primio C, Quercioli V, Valentini P, Boll A, Girelli G, Demichelis F, Arosio D, Cereseto A. Second generation imaging of nuclear/cytoplasmic HIV-1 complexes. AIDS Res Hum Retroviruses. 2014;30:717–726. doi: 10.1089/aid.2013.0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis AC, Marin M, Shi J, Aiken C, Melikyan GB. Time-Resolved Imaging of Single HIV-1 Uncoating In Vitro and in Living Cells. PLoS Pathog. 2016;12:e1005709. doi: 10.1371/journal.ppat.1005709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganser-Pornillos BK, Yeager M, Sundquist WI. The structural biology of HIV assembly. Curr Opin Struct Biol. 2008;18:203–217. doi: 10.1016/j.sbi.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatziioannou T, Perez-Caballero D, Cowan S, Bieniasz PD. Cyclophilin interactions with incoming human immunodeficiency virus type 1 capsids with opposing effects on infectivity in human cells. J Virol. 2005;79:176–183. doi: 10.1128/JVI.79.1.176-183.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Choe S, Walker R, Di Marzio P, Morgan DO, Landau NR. Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J Virol. 1995;69:6705–6711. doi: 10.1128/jvi.69.11.6705-6711.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilditch L, Towers GJ. A model for cofactor use during HIV-1 reverse transcription and nuclear entry. Current opinion in virology. 2014;4:32–36. doi: 10.1016/j.coviro.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme AE, Kelley Z, Foley D, Hope TJ. Complementary assays reveal a low level of CA associated with nuclear HIV-1 viral complexes. J Virol. 2015 doi: 10.1128/JVI.00476-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme AE, Perez O, Hope TJ. Complementary assays reveal a relationship between HIV-1 uncoating and reverse transcription. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:9975–9980. doi: 10.1073/pnas.1014522108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Ambrose Z, Martin TD, Oztop I, Mulky A, Julias JG, Vandegraaff N, Baumann JG, Wang R, Yuen W, et al. Flexible use of nuclear import pathways by HIV-1. Cell host & microbe. 2010;7:221–233. doi: 10.1016/j.chom.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamede JI, Cianci GC, Anderson MR, Hope TJ. Early cytoplasmic uncoating is associated with infectivity of HIV-1. Proceedings of the National Academy of Sciences of the United States of America. 2017;114:E7169–E7178. doi: 10.1073/pnas.1706245114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matreyek KA, Engelman A. The requirement for nucleoporin NUP153 during human immunodeficiency virus type 1 infection is determined by the viral capsid. J Virol. 2011;85:7818–7827. doi: 10.1128/JVI.00325-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matreyek KA, Yucel SS, Li X, Engelman A. Nucleoporin NUP153 phenylalanine-glycine motifs engage a common binding pocket within the HIV-1 capsid protein to mediate lentiviral infectivity. PLoS Pathog. 2013;9:e1003693. doi: 10.1371/journal.ppat.1003693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder LC, Muesing MA. Degradation of HIV-1 integrase by the N-end rule pathway. The Journal of biological chemistry. 2000;275:29749–29753. doi: 10.1074/jbc.M004670200. [DOI] [PubMed] [Google Scholar]
- Neagu MR, Ziegler P, Pertel T, Strambio-De-Castillia C, Grutter C, Martinetti G, Mazzucchelli L, Grutter M, Manz MG, Luban J. Potent inhibition of HIV-1 by TRIM5-cyclophilin fusion proteins engineered from human components. The Journal of clinical investigation. 2009;119:3035–3047. doi: 10.1172/JCI39354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng K, Muranyi W, Glass B, Laketa V, Yant SR, Tsai L, Cihlar T, Muller B, Krausslich HG. Quantitative microscopy of functional HIV post-entry complexes reveals association of replication with the viral capsid. eLife. 2014;3:e04114. doi: 10.7554/eLife.04114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzato M, Erlwein O, Bonsall D, Kaye S, Muir D, McClure MO. A one-step SYBR Green I-based product-enhanced reverse transcriptase assay for the quantitation of retroviruses in cell culture supernatants. J Virol Methods. 2009;156:1–7. doi: 10.1016/j.jviromet.2008.10.012. [DOI] [PubMed] [Google Scholar]
- Platt EJ, Wehrly K, Kuhmann SE, Chesebro B, Kabat D. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J Virol. 1998;72:2855–2864. doi: 10.1128/jvi.72.4.2855-2864.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AJ, Jacques DA, McEwan WA, Fletcher AJ, Essig S, Chin JW, Halambage UD, Aiken C, James LC. Host cofactors and pharmacologic ligands share an essential interface in HIV-1 capsid that is lost upon disassembly. PLoS Pathog. 2014;10:e1004459. doi: 10.1371/journal.ppat.1004459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasaiyaah J, Tan CP, Fletcher AJ, Price AJ, Blondeau C, Hilditch L, Jacques DA, Selwood DL, James LC, Noursadeghi M, et al. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature. 2013;503:402–405. doi: 10.1038/nature12769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller T, Ocwieja KE, Rasaiyaah J, Price AJ, Brady TL, Roth SL, Hue S, Fletcher AJ, Lee K, KewalRamani VN, et al. HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog. 2011;7:e1002439. doi: 10.1371/journal.ppat.1002439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz O, Marechal V, Friguet B, Arenzana-Seisdedos F, Heard JM. Antiviral activity of the proteasome on incoming human immunodeficiency virus type 1. J Virol. 1998;72:3845–3850. doi: 10.1128/jvi.72.5.3845-3850.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah VB, Shi J, Hout DR, Oztop I, Krishnan L, Ahn J, Shotwell MS, Engelman A, Aiken C. The host proteins transportin SR2/TNPO3 and cyclophilin A exert opposing effects on HIV-1 uncoating. J Virol. 2013;87:422–432. doi: 10.1128/JVI.07177-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simm M, Shahabuddin M, Chao W, Allan JS, Volsky DJ. Aberrant Gag protein composition of a human immunodeficiency virus type 1 vif mutant produced in primary lymphocytes. J Virol. 1995;69:4582–4586. doi: 10.1128/jvi.69.7.4582-4586.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolskaja E, Sayah DM, Luban J. Target cell cyclophilin A modulates human immunodeficiency virus type 1 infectivity. J Virol. 2004;78:12800–12808. doi: 10.1128/JVI.78.23.12800-12808.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sood C, Francis AC, Desai TM, Melikyan GB. An improved labeling strategy enables automated detection of single-virus fusion and assessment of HIV-1 protease activity in single virions. The Journal of biological chemistry. 2017 doi: 10.1074/jbc.M117.818088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szent-Gyorgyi C, Schmidt BF, Creeger Y, Fisher GW, Zakel KL, Adler S, Fitzpatrick JA, Woolford CA, Yan Q, Vasilev KV, et al. Fluorogen-activating single-chain antibodies for imaging cell surface proteins. Nat Biotechnol. 2008;26:235–240. doi: 10.1038/nbt1368. [DOI] [PubMed] [Google Scholar]
- Wei BL, Denton PW, O’Neill E, Luo T, Foster JL, Garcia JV. Inhibition of lysosome and proteasome function enhances human immunodeficiency virus type 1 infection. J Virol. 2005;79:5705–5712. doi: 10.1128/JVI.79.9.5705-5712.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Franks T, Gibson G, Huber K, Rahm N, De Castillia CS, Luban J, Aiken C, Watkins S, Sluis-Cremer N, et al. Evidence for biphasic uncoating during HIV-1 infection from a novel imaging assay. Retrovirology. 2013;10:70. doi: 10.1186/1742-4690-10-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang R, Shi J, Byeon IJ, Ahn J, Sheehan JH, Meiler J, Gronenborn AM, Aiken C. Second-site suppressors of HIV-1 capsid mutations: restoration of intracellular activities without correction of intrinsic capsid stability defects. Retrovirology. 2012;9:30. doi: 10.1186/1742-4690-9-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Zhou Y, Alcock C, Kiefer T, Monie D, Siliciano J, Li Q, Pham P, Cofrancesco J, Persaud D, et al. Novel single-cell-level phenotypic assay for residual drug susceptibility and reduced replication capacity of drug-resistant human immunodeficiency virus type 1. J Virol. 2004;78:1718–1729. doi: 10.1128/JVI.78.4.1718-1729.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Example-1: TZM-bl cells expressing EBFP2-lamin were infected with VSV-G pseudotyped HIV-1 labeled with INsfGFP (green) and CypA-DsRed (red) and imaged from 30 min to 21 h.p.i. Time-lapse Z-stack images (shown as maximum intensity projections through a 4 μm central section) were acquired every 10 min. A cell showing single particle uncoating, nuclear entry and INsfGFP disappearance prior to infection (Related to Fig. 2A). Example-2: PPIA−/− cells expressing EBFP2-Lamin were infected and imaged (30 min/frame) from 30 min post-infection, as above. A cell showing single particle uncoating, nuclear entry and disappearance prior to infection is shown. Scale bar 5 μm.
Left: A single core co-labeled with CypA-DsRed/INsfGFP docked at the nuclear lamin is displaced after addition of 10 μM PF-74. Scale bar 2 μm. Right: Control experiments showing, a single core co-labeled with CypA-DsRed/INsfGFP docked at the nuclear lamin. CsA (10 μM) is added at 15 min of the video. A single Z-plane is shown. Imaging was performed between 4 and 5 h.p.i. Scale bar 1 μm.
Arrival of a single HIV-1 pseudovirus co-labeled with CypA-DsRed/INsfGFP at the nuclear envelope is followed by uncoating and nuclear import. Time-lapse Z-stack images (shown as maximum intensity projections through a 1 μm central section) were acquired every 3 min, starting at 2.5 h.p.i. Scale bar 5 μm.
Arrival of a single HIV-1 pseudovirus co-labeled with CypA-DsRed/INsfGFP at the nuclear envelope is followed by uncoating and nuclear import. Time-lapse Z-stack images (shown as maximum intensity projections through a 1 μm central section) were acquired every 20 sec, starting after 1 h.p.i. Images were temporal averaged using a 3 frame window. Scale bar 5 μm.
Top: A single HIV-1 pseudovirus co-labeled with CypA-DsRed/INsfGFP docked at the nuclear envelope, loses CypA-DsRed (red) and enters the nucleus. Bottom: A single HIV-1 pseudovirus docked at the nuclear envelope does not lose a significant portion of CypA-DsRed and does not enter the nucleus. Time-lapse Z-stack images (shown as maximum intensity projections through a 1 μm central section) were acquired every 20 sec, starting at ~5 h.p.i. Images were temporally averaged, using a 3-frame window. Scale bar 2 μm.
Arrival of a single HIV-1 pseudovirus co-labeled with CypA-DsRed/INsfGFP at the nuclear envelope (top Z slice) is followed by uncoating and nuclear import (middle Z slice) was visualized in TZM-bl cells every 5 min. Virus transport is shown from 10 min p.i. through 14 h.p.i. Maximum intensity projections of 1 μm top- and central nuclear volume are shown. Scale bar 5 μm.
TZM-bl cells expressing EBFP2-Lamin were infected with VSV-G pseudotyped HIV-CA/N74D mutant labeled with INsfGFP (green) and CypA-DsRed (red) and imaged from 1 to 18 h.p.i. Time-lapse Z-stack images (shown as maximum intensity projections through a 2 μm central section) were acquired every 15 min. Example-1: A cell with 2 docked particles that uncoat without an apparent separation from lamin and disappeared prior to infection (expression of eGFP). Example-2: A cell with 3 docked particles that uncoat and remain at the nuclear membrane for the duration of the experiment without losing the IN signal or establishing infection. Scale bar 5 μm.