

Abstract
Mitogen-activated protein kinases (MAPKs) regulate diverse cellular processes including proliferation, cell survival, differentiation, and apoptosis. While conventional MAPK constituents have well-defined roles in oncogenesis, the MAPK kinase 5-extracellular signal-regulated kinase 5 (MEK5-ERK5) pathway has only recently emerged in cancer research. In this review, we consider the MEK5 signaling cascade, focusing specifically on its involvement in drug resistance and regulation of aggressive cancer phenotypes. Moreover, we explore the role of MEK5 in tumorigenesis and metastatic progression, discussing the discrepancies in preclinical studies and assessing its viability as a therapeutic target for anti-cancer agents.
Keywords: mitogen-activated protein kinase, MEK5-ERK5, cellular signaling, kinase inhibitors, targeted therapies
1. Introduction
The mitogen-activated protein kinase kinase 5-extracellular signal-regulated kinase 5 (MEK5-ERK5) pathway contains many features that are structurally and functionally distinct from other MAPKs, all of which increase its viability as a novel target for future therapeutics [1–3]. In the MAPK signaling network, MEK5 most resembles MEK1/2 by sequence alignment but remains the only known direct MEK activator of ERK5 [4]. MEK5 protein kinase is encoded by MAP2K5. Alternative splicing results in two isoforms of MEK5 (50 kDa α and 40 kDa β) differing in the N-terminus, which accounts for their relative binding affinities for ERK5 [5]. MEK5α contains a distinct docking site in its N-terminal extension, a phox and Bem1p (PB1) domain, crucial to ERK5 activation and transcriptional induction via myocyte enhancer factor 2C (MEF2C). Accordingly, MEK5α is a stronger activator of ERK5 than MEK5β, which lacks this consensus motif [6]. Moreover, the PB1 domain, present in all three components of this signaling cascade (Figure 1), acts as a scaffold to facilitate and maintain specificity of MEKK2–MEK5–ERK5 interaction and signaling [7].
Figure 1. Structure of MEK5 signaling components.
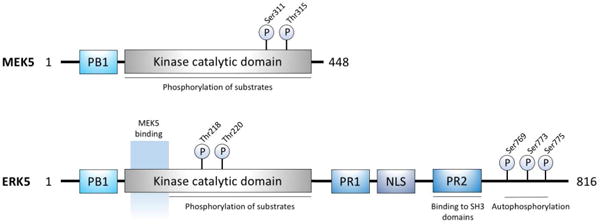
Linear representation of MEK5 and ERK5, PB1 - Phox and Bem1p, PR1 – Proline rich domain 1, NLS – Nuclear localization, PR1 – Proline rich domain 2
Due to its extended C-terminus containing a nuclear localization signal (NLS), two proline-rich regions, and a transcriptional activation domain (TAD), ERK5, or big MAP kinase 1 (BMK1) encoded by the MAPK7 gene, is more than twice the molecular weight of other MAPKs (110 kDa). This structural distinction enables active ERK5 to undergo autophosphorylation of its C-terminal TAD, an ability unique to ERK5, thereby exerting direct control over gene transcription [8]. In the unphosphorylated state, ERK5 presents an inactive conformation, where its N- and C-terminal domains are associated together while in the cytosol. Activation by MEK5 induces an open conformation of ERK5, exposing the NLS, to relieve the autoinhibitory effects and facilitate ERK5 translocation to the nucleus [9–11]. ERK5 activity is also regulated by splice variants (a, b, and c) [12]. While ERK5a is the most highly expressed isoform, ERK5b and c, both deficient in protein kinase activity, can inhibit MEK5-mediated ERK5a stimulation.
Known substrates of ERK5 include transcription factors Sap-1a, c-FOS, c-MYC and MEF2 (A, C, and D) and kinases, such as RSK and serum/glucocorticoid-regulated kinase (SGK) (Figure 2) [13–17]. Similar to other proline (Pro)-directed MAPKs, ERK5 substrate recognition and subsequent phosphorylation occurs on amino acids Ser or Thr adjacent to a Pro residue (-X-Ser/Thr-Pro-X-sequence). Additionally, ERK5 protein kinase activity can be non-Pro-directed, as in the case of ERK5 autophosphorylation and ERK5-mediated MEK5 phosphorylation on Ser/Thr sites not directly preceding Pro residues [18]. These findings further distinguish ERK5 from other conventional MAPK family members.
Figure 2. MEK5 activation and downstream substrates.
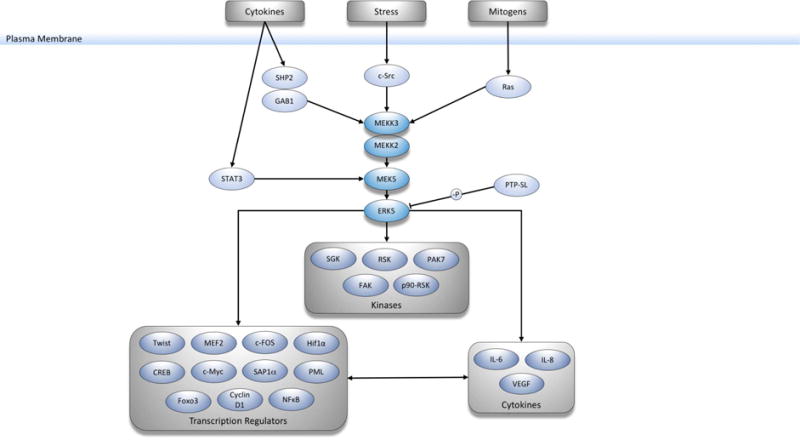
MEK5/ERK5 pathway can be activated by stress, mitogens or cytokines, leading to the regulation of various downstream targets including kinases and transcription factors.
2. Upstream activators of MEK5-ERK5 signaling
MEK5-ERK5 signal transduction can be activated by environmental stress, growth factors, and cytokines [13]. In response to these extracellular stimuli, MEKK2 or MEKK3 binds to the N-terminal domain of MEK5 and phosphorylates Ser311 and Thr315; however, the mechanisms of MEKK2/3 activation by external stimuli have not been fully elucidated [19]. MEKK2 has a higher binding affinity for MEK5 relative to MEKK3, but both MEKKs can also activate other conventional MAPK pathways, including JNK and p38 MAPK, via phosphorylation of their respective upstream MAP2Ks [20, 21]. Overexpression of MEKK2 has been detected in prostate and colorectal cancers, while elevated MEKK3 expression has been identified in breast, cervical, lung, kidney, and esophageal cancers [22–25].
MEKK2 is necessary for epidermal growth factor receptor (EGFR)- and human epidermal growth factor receptor 2 (HER2)-dependent activation of ERK5. Knockdown of MEKK2 inhibited tumor growth of triple-negative MDA-MB-231 and HER2-positive BT474 breast cancer xenografts and diminished metastasis of the TNBC cells [26]. MEKK2 has also been shown to regulate breast cancer cell migration by inducing focal adhesion turnover, specifically ubiquitylation and consequent removal of paxillin from focal adhesion complexes [27, 28]. To date, there are no selective MEKK2 inhibitors, though six compounds with potent in vitro MEKK2 inhibitory activity have recently been reported. Among this list of kinase inhibitors, Ponatinib (AP24534, Iclusig) is an FDA-approved drug indicated for BCR-ABL-targeting in treatment of chronic myeloid leukemia, suggesting its potential both as a preclinical research tool to elucidate the role of MEKK2 in cancer and as a drug repurposed for MEKK2-dependent cancers in the clinical setting [29].
The role of MEKK3 as a regulator of NF-κB signaling is well-documented [30, 31]. Overexpression of MEKK3 in glioma and ovarian cancer cells enhanced NF-κB activation and increased expression of cell survival factors to confer resistance to cytotoxic effects of chemotherapeutic agents [24, 32]. Conversely, silencing of MEKK3 by RNAi sensitized breast cancer cells to tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) cytotoxicity through suppression of nuclear factor κB (NF-κB) transcriptional activity [33]. Furthermore, MEKK3 depletion induced cell death in renal cancer cells and reduced tumor growth of breast cancer cells, but did not significantly affect the frequency of metastasis [23, 26]. Despite their involvement in processes essential to tumorigenesis and malignancy, MEKK2/3 are understudied kinases. Instead, efforts have focused on parallel MEKKs and downstream effectors of MEKK2/3.
3. Pharmacological inhibitors of MEK5 cascade
Interest in the MEK5 pathway has emerged in cancer research partly due to its overlap with the MEK1/2 pathway along with the discovery that first-generation MEK1/2 inhibitors PD98059, U0126, and PD184352 also exhibit activity toward MEK5, providing impetus for the development of MEK5 selective inhibitors to parse the role of these pathways in cancer progression [10, 34]. The indolinone-6-carboxamides BIX02188 and BIX02189 (Boehringer Ingelheim Pharmaceuticals) were the first selective small-molecule ATP-site inhibitors of MEK5 signaling to be described, inhibiting MEK5 catalytic function with IC50 4.3 and 1.5 nM, respectively [35]. BIX02189 also displayed more potent suppression of ERK5 kinase activity with IC50 59 nM compared to that of BIX02188 (810 nM). Both compounds also inhibited transcriptional activity of MEF2, a downstream substrate of the MEK5 signaling cascade, in a dose-dependent manner. These MEK5 inhibitors blocked ERK5 phosphorylation without affecting activation of ERK1/2, p38 MAPK, or JNK [35].
Synthesis of XMD8-92 stemmed incidentally from a screen of analogs of BI-2536, a highly selective, ATP-competitive polo kinase inhibitor [36]. XMD8-92 selectivity for ERK5 was validated through profiling first against a diverse panel of 402 kinases and then against all detectable kinases in HeLa cell lysates, identifying ERK5 as most potently inhibited target with IC50 of 1.5 μM. MEK5 and ERK1/2 were not inhibited by XMD8-92, but the compound did significantly reduce ERK5-dependent MEF2C-driven gene expression. Pharmacokinetics and tolerability of XMD8-92 was also evaluated in Sprague-Dawley rats. A single intravenous or oral dose of XMD8-92 was found to have a 2-hour half-life clearance of 26 mL/min/kg and high oral bioavailability with 69% dose absorption. After a single oral dose of 2 mg/kg, maximal plasma concentrations reached 500 nM within 30 minutes, with 34 nM remaining 8 hours post drug administration. To assess tolerability, plasma concentrations of XMD8-92 were maintained at high levels, 10 μM following IP dosing of 50 mg/kg, for 2 weeks. Animals did not show signs of morbidity or mortality [36].
TG02, an oral pyrimidine-based multi-kinase inhibitor, blocks CDKs 1, 2, 3, 5, and 9 with IC50 values below 10 nM in addition to janus kinase 2 (JAK2), p38δ, and ERK5 with IC50 values of 19, 56, and 43 nM, respectively [37–39]. The pharmacokinetic profile showed drug levels retained in tumors were above the IC50 for 8 and 24 hours after a single oral dose of 30 or 60 mg/kg, respectively [38]. TG02 treatment was well-tolerated in mice, even at maximum oral dosing of 40 mg/kg daily, with no body weight loss at endpoint. This novel anti-cancer agent has recently completed phase I of clinical trials for treatment of leukemia and multiple myeloma patients, the results of which may unveil the potential for MEK5 signaling inhibitors in cancer therapy.
4. Role of MEK5 pathway in drug resistance
Cytotoxic therapy
Drug resistance, both primary (intrinsic) and acquired, is a major obstacle in cancer therapeutics, indicative of more clinically aggressive tumor cells contributing to disease progression. The efficacy of cytotoxic agents used in chemotherapy, the standard-of-care for various cancer types, is mitigated by activation of signaling pathways, such as MEK5, that confer drug resistance [40]. Our lab and others have shown that MEK5 signaling promotes epithelial-to-mesenchymal transition (EMT), cell survival, and evasion of apoptosis – mechanisms linked to adaptive resistance [40–42].
Through expression profiling, we observed MEK5 upregulation in apoptotically resistant (APO−) MCF-7 breast cancer cell variants compared to apoptotically sensitive (APO+) cells. Transfection of dominant-negative (DN) ERK5 plasmid into APO− cells reduced cell viability in a dose-dependent manner versus vector control, and the cytotoxic effects of DN-ERK5 expression were augmented by treatment with apoptotic-inducing agents etoposide, tumor necrosis factor (TNF), or TNF-related apoptosis-inducing ligand (TRAIL). Furthermore, phorbol ester (PMA) stimulation failed to rescue cell viability of DN-ERK5-transfected cells treated with TRAIL [43]. In basal-like breast cancer subtypes, overexpression of MEK5 in conjunction with ERK5 was associated with poor relapse- and metastasis-free survival in patients who received chemotherapy compared to patients not treated with chemotherapy, which suggests that MEK5-ERK5 expression could serve as a predictive marker for patient benefit from systemic treatments in the ER-negative breast cancer setting [44]. Moreover, in MDA-MB-231 cells ERK5 inhibition by TG02 augmented anti-cancer effects of chemotherapeutic agents conventionally used in triple-negative breast cancer (TNBC) treatment, including taxotere, vinorelbine, and cisplatin [45]. These results support the role of MEK5 signaling in regulation of survival and apoptosis and implicate MEK5 pathway involvement in chemoresistance [43].
The pyrimidine analog 5-fluorouracil (5-FU), a widely used chemotherapeutic agent, is the common backbone of all standard polychemotherapy regimens for colorectal cancer [46]. While clinical efficacy of 5-FU exceeds that of other drugs, only 30% of colon cancer patients initially respond to therapy and the majority of which will develop resistance [47]. In vitro treatment of colon cancer cells HCT116 and SW620 with 5-FU reduced activation of both MEK5 and ERK5. Constitutive activation of MEK5 conferred a survival advantage to HCT116 cells exposed to 5-FU compared to empty vector cells, whereas downregulation of MEK5 signaling, either by transfection of dominant-negative ERK5 construct or treatment with a highly-selective ERK5 inhibitor XMD8-92, enhanced sensitivity of HCT116 cells to 5-FU-induced cytotoxicity through stimulation of p53-dependent transcriptional activation of p21 and Puma. The anti-apoptotic effects of 5-FU treatment in conjunction with ERK5 inhibition were recapitulated in vivo using an HCT116 xenograft model. Combination therapy using 5-FU and XMD8-92 significantly increased apoptosis and reduced tumor burden in comparison to monotherapy of each compound [48]. Consistent with this study, ERK5 inhibition via XMD8-92 treatment combined with doxorubicin, another chemotherapeutic agent, demonstrated synergistic induction of p53 and promoted significant tumor regression in both HeLa cervical cancer cells and A549 lung cancer cells [49]. Furthermore, small hairpin RNA (shRNA)-mediated knockdown of ERK5, as a mirror of ERK5 pharmacological inhibition, sensitized HMESO malignant mesothelioma cells to doxorubicin in vitro and synergized with doxorubicin in enhancing anti-tumor activity compared to vector control [40]. These findings provide rationale for the application of MEK5 pathway inhibitors coupled with 5-FU- or doxorubicin-based chemotherapy to enhance therapeutic efficacy and potentially delay the onset of drug resistance.
Targeted therapy
Pursuit of mechanism-based, individualized therapeutics has led to the development of small-molecule inhibitors and monoclonal antibodies targeting key signaling molecules or networks that drive cancer progression. Targeted therapies, though diverse in their mechanisms of action, have not overcome the hurdle of drug resistance. MEK5 signal transduction has been implicated as a critical factor in mediating sensitivity to several targeted therapies.
Endocrine resistance, either de novo or acquired, is evident in up to 50% of patients on an antiestrogen regimen, the mainstay in treatment of estrogen receptor alpha (ER-α)-positive breast cancer. ER-α signaling is an integral component of breast cancer biology as well as an important molecular mechanism perverted in endocrine therapy resistance [50]. Our lab has demonstrated that overexpression of MEK5 in the antiestrogen-sensitive, ER-α-positive (ER+) MCF-7 cell line downregulated ER-α expression and transcriptional activity in an ERK5-dependent manner and increased clonogenic survival following endocrine treatment [51]. These results delineate the role of MEK5-ERK5 signaling in progression to a more malignant estrogen-independent phenotype.
In breast tumors positive for human epidermal growth factor receptor 2 (HER2) expression, anti-HER2 therapy, such as trastuzumab, has demonstrated clinical efficacy in the adjuvant setting, yet approximately 20% of patients experience relapse [52]. High ERK5 expression in patients with HER2-positive breast cancer was associated with worse disease-free survival [53]. HER2-enriched breast cancer cell lines SKBR3 and BT-474 have been shown to express constitutively active ERK5 [54]. Downregulation of ERK5 expression or activation potentiated anti-proliferative effects of trastuzumab in BT-474 cells, indicating that pharmacological inhibition of ERK5 may enhance anti-cancer action of trastuzumab [53]. Moreover, ERK5 inhibitor XMD8-92 synergized with heat shock protein (Hsp90) inhibition, proposed as a therapeutic target in TNBC, to suppress breast tumor formation in vivo [55]. In another cancer model, expression of dominant-negative ERK5 increased sensitivity of myeloma cells to apoptosis induced by the proteasome inhibitor PS341. Furthermore, overexpression of ERK5 in these cells abrogated the effects of PS341 on cell death [56]. Taken together, these studies implicate the MEK5-ERK5 pathway as a fundamental component of drug resistance in cancer therapy. Defining the mechanisms by which MEK5 promotes a therapeutically resistant phenotype may provide insight for the next generation of potent anti-cancer agents.
5. Role of MEK5 pathway in tumorigenesis
The MEK5-ERK5 cascade has been emerging as an important mediator of cell proliferation through induction of cell cycle regulators, including cyclin D1, c-MYC, n-MYC, SGK, RSK2, and NF-κB [15, 57–62]. Through phosphorylation of MEF2 transcription factors, MEK5 has been shown to regulate the expression of c-JUN, a proto-oncogene vital to cell growth [14, 63]; moreover, the ERK5-MEF2 axis has been reported in activation of survival signaling [64]. It has also been demonstrated that ERK5 can phosphorylate S403 and T409 of tumor suppressor promyelocytic leukemia protein (PML) and inhibit its activity, thereby downregulating the induction of p21 expression and enabling cells to overcome the G1-S phase [36, 65]. Constitutive activation of MEK5 in prostate and colon cancer cell lines accelerated cell cycle progression and increased proliferation [48, 66, 67]. Similarly, ERK5 knockdown studies using RNA interference (RNAi) or pharmacological inhibition by XMD8-92 treatment delayed cell cycle progression and decreased proliferation in various cancer types (Table 1). There are, however, conflicting reports showing that in cell lines harboring K-Ras or B-Raf mutations neither MEK5 inhibition, via BIX02189 or dominant-negative (DN) construct, nor siRNA-mediated downregulation of ERK5 affected cell growth, suggesting that in this cell context ERK5 is a dispensable proliferative signal [48, 68]. Interestingly, these results were also shown in ERK5-amplified SNU449 and KYSE30 cells [68], directly contradicting previous research demonstrating that knockdown of ERK5 resulted in cell growth inhibition in the ERK5-dysregulated hepatocellular and esophageal cancer cells, respectively [69, 70]. Recent findings have presented delineations between kinase activity and transcriptional activity of ERK5 that may account for discrepancies in determining ERK5 regulation of cellular proliferative responses [71]. Through a noncanonical mechanism involving Hsp90 dissociation, cell division cycle 37 (Cdc37) overexpression induced nuclear translocation of catalytically inactive but transcriptionally active ERK5 and collaborated with overexpressed ERK5 to promote cell proliferation [72]. Another study showed that XMD8-92 exhibited off-target kinase activity on bromodomain-containing protein 4 (BRD4), and using ERK5-selective derivatives, suggested that inhibition of ERK5 kinase activity was not responsible for XMD8-92-mediated anti-proliferative effects [73]. Further research is needed to elaborate on the nuclear function of ERK5 independent of its catalytic status.
Table 1.
Effects of MEK5/ERK5 signaling on in vitro cancer cell proliferation.
| Disease | Cell line | Targeted Approach | Effects (compared to control) | Mechanism | Ref. |
|---|---|---|---|---|---|
| T cell leukemia | Jurkat T | shRNA | did not affect cell cycle progression, sensitized cells to TNF-α | ERK5 activates NF-κB signaling and promotes nuclear localization and transcriptional activity of p65. | [60] |
| breast carcinoma | BT-549 | shRNA | decreased cell proliferation | TG02 decreased expression of antiapoptotic proteins MCL1 and BCL2 and triggered cell death through caspase-dependent and -independent mechanisms. | [45] |
| Hs-578T | |||||
| HCC1187 | |||||
| MDA-MB-231 | |||||
| MDA-MB-231 | TG02 | delayed cell cycle progression, induced apoptosis | |||
| HCC1187 | |||||
| prostate carcinoma | LNCaP | CA-MEK5 | increased proliferative index | CA-MEK5 expression increased percentage of cell in S phase. | [66] |
| PC3 | overexpression | increased proliferative index | High levels of ERK5 is associated with accelerated cell cycle progression. | [74] | |
| multiple myeloma | MM1S | TG02 | suppressed cell cycle progression and induced apoptosis | TG02 activated apoptosis through intrinsic and extrinsic pathways. | [37] |
| cervical adenocarcinoma | HeLa | DN-ERK5 (Ala-Glu-Phe [AEF]) | inhibited proliferation | ERK5 inhibited PML function and p21 expression to regulate cell proliferation. | [36] |
| XMD8-92 | |||||
| lung carcinoma | A549 | XMD8-92 | |||
| NCI-H1793 | siRNA | did not affect cell proliferation or death | Dysregulated ERK5 signaling drives cell proliferation. | [70] | |
| esophageal carcinoma | KYSE30 | siRNA | decreased proliferation, increased cell death | ||
| hepatocellular carcinoma | SNU449 | siRNA | decreased proliferation | ||
| hepatocellular carcinoma | HepG2 | siRNA | decrease in proliferation | XMD8-92 treatment decreased expression of cyclin D1. | [63] |
| Huh-7 | |||||
| HepG2 | XMD8-92 | decrease in proliferation, reduction of cells in S phase and increased percentage of cells in G0/G1, no indications of apoptosis | |||
| Huh-7 | |||||
| SNU449 | siRNA | inhibited cell growth | RNAi knockdown of ERK5 decreased mitotic index, implicating ERK5 involvement in regulation of mitotic entry. | [69] | |
| SNU449 | siRNA | inhibition of ERK5 expression or activity did not decrease proliferation | K-Ras/B-Raf-mutated or ERK5 amplified cancer cells are not dependent on MEK5 pathway for proliferation. | [68] | |
| BIX02189 | |||||
| colon carcinoma | HCT116 | siRNA | did not affect proliferation | ||
| BIX02189 | inhibited proliferation at high doses (>10 μM) | ||||
| HT29 | BIX02189 | inhibited proliferation at high doses (>10 μM), did not affect proliferation in 3D culture | |||
| HCT116 | DN-MEK5 | did not affect proliferation index (however, ERK5 overactivation by CA-MEK5 increased cell proliferation) | ERK5 inhibition was associated with increased p53 transcriptional activity, upregulating p21 and Puma. | [48] | |
| SW620 | did not affect proliferation index | ||||
| SW620 | DN-MEK5 | accelerated cell cycle progression in CA-MEK5 cells, which was abolished by XMD8-92 treatment | MEK5 signaling promotes cell cycle progression through degradation of IκB leading to NF-κB activation. | [67] | |
| CA-MEK5 |
Note: ERK5 inhibition; MEK5 inhibition
Using a conditional ERK5 knockout mouse model, Hayashi et al. demonstrated that tumor cells inoculated subcutaneously into the right flank region of the animals exhibited impaired vasculature development and reduced tumor growth, suggesting the involvement of ERK5 in the regulation of tumor-associated angiogenesis as well as tumor formation [58]. Studies since then have supported the involvement of the MEK5/ERK5 pathway in cancer progression (Table 2). We showed that hyperactivation of MEK5 in ER+ breast cancer cells enhanced estrogen-independent tumorigenesis [51], while others observed that ERK5 overexpression supported prostate tumor growth [74]. The role of ERK5 in tumor formation was further established as its silencing by shRNA impaired growth of malignant mesothelioma, T cell leukemia, and hepatocellular carcinoma xenografts through regulation of pro-inflammatory cytokines or NF-κB signaling [40, 60, 63]. Moreover, XMD8-92 treatment decreased tumor volume of various cancer types [36, 48, 63]. TG02, a multi-kinase inhibitor that targets ERK5, has also been shown to be an efficacious anti-tumor agent in the multiple myeloma and breast cancer settings [37, 45]. Based on studies demonstrating that shRNA-mediated knockdown of ERK5 did not alter growth dynamics of triple-negative breast cancer xenografts [26, 75], the anti-proliferative effects have been proposed as an artifact of TG02 activity against CDK targets. However, partial silencing of ERK5 may not be sufficient to exert anti-tumor effects in certain cell lines. For example, 70% ERK5 inhibition in SNU449 cells decreased proliferation while not affecting apoptosis, whereas 90% reduction of ERK5 expression in KYSE30 cells resulted in suppression of cell growth and significant induction of cell death [70]. As the Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-associated protein-9 (CRISPR/Cas9) knockout system has been widely adopted for precision genome editing, it would be a beneficial tool in delineating the involvement of MEK5/ERK5 in tumorigenesis. Despite the controversy surrounding this research arena, MEK5 signaling remains a viable therapeutic target and elucidation of this pathway may provide insights to stratify the anti-cancer armamentarium, especially in regard to neoplasms such as hepatocellular carcinoma that require novel molecular therapies.
Table 2.
Effects of MEK5/ERK5 signaling on in vivo tumorigenesis.
| Disease | Cell line | Targeted Approach | Effects (compared to control) | Ref. |
|---|---|---|---|---|
| lung carcinoma | LL/2 | deletion of host gene | delayed tumor development, reduced tumor vasculature | [58] |
| melanoma | B16F10 | |||
| prostate carcinoma | PC3 | overexpression | enhanced tumor formation | [74] |
| breast adenocarcinoma | MCF7 | CA-MEK5 | enhanced tumor growth independent of estrogen, shRNA-downregulation of ERK5 decreased MCF7-MEK5 tumor growth | [51] |
| MDA-MB-231 | TG02 | delayed tumor growth | [45] | |
| MDA-MB-231 | shRNA | did not significantly affect tumor growth | [26] | |
| MDA-MB-23◆ | shRNA | did not significantly affect tumor growth | [75] | |
| multiple myeloma | MM1S | TG02 | inhibited tumor growth | [37] |
| OPM2 | inhibited tumor growth | |||
| malignant mesothelioma | HMESO | shRNA | impaired tumor formation | [40] |
| H2373 | ||||
| T cell leukemia | EL-4 | shRNA | impaired tumor formation | [60] |
| hepatocellular carcinoma | Huh-7 | shRNA | suppressed tumor growth by 100-fold | [63] |
| XMD8-92 | suppressed tumor growth due to reduction in cell proliferation, no change in levels of apoptosis | |||
| colon carcinoma | HCT116 | XMD8-92 | inhibited tumor growth by 46% | [48] |
| pancreatic adenocarcinoma | AsPC-1 | XMD8-92 | inhibited tumor growth and decreased tumor volume | [76] |
| cervical adenocarcinoma | HeLa | XMD8-92 | suppressed tumor growth, blocked tumor cell proliferation | [36] |
| lung carcinoma | LL/2 |
Note: ERK5 inhibition; MEK5 inhibition
(4175 TGL variant)
6. Role of MEK5 pathway in metastatic progression
Dysregulated MEK5 signaling is associated with metastatic risk in prostate, breast, colon, kidney, bone, and oral cancers as well as less favorable survival outcome [51, 67, 77–81]. Molecular inhibition of ERK5 in vitro suppressed cell motility and invasion of liver, breast, and prostate cancer cells [63, 82] and decreased metastasis of breast cancer xenografts in vivo [26, 75]. Conversely, cancer cells overexpressing MEK5 or ERK5 exhibited a migratory and invasive phenotype [66, 74], denoted by an increase in tumor metastases [67, 77, 83].
Metastasis, a complex process in which malignant cells originating from the primary tumor infiltrate and colonize distal organs, is organized into simplified steps: local invasion, intravasation of cells into the circulation, dissemination, extravasation of cells at distant sites, and colonization. Epithelial-to-mesenchymal transition (EMT) is an integral part of metastatic progression whereby cells adopt motile and invasive capabilities through loss of epithelial markers, namely Cadherin 1/E-Cadherin (CDH1), and acquisition of mesenchymal markers, such as vimentin (VIM) and Cadherin 2/N-Cadherin (CDH2). MEK5 signaling has been implicated in the activation of EMT and transcription factors linked to EMT induction, including NF-κB and FOS-Like Antigen 1 (FRA-1) [16, 51, 67]. Furthermore, ERK5 signaling has been shown to regulate the expression of matrix metalloproteinase (MMP) family members (MMP-1, 2, 9, 12, and 16), known for their role in degradation of the extracellular matrix (ECM) to potentiate cancer cell dissemination [40, 66, 77, 84], and other proteins involved in migration and invasion, such as tissue inhibitor of metalloproteinases 2 (TIMP2) and bone morphogenic protein 5 (BMP5) [40, 77]. While many studies have presented a positive correlation between ERK5 expression and EMT induction, dissenting observations have been reported. Inhibition of ERK5 in A549 lung cancer cells by XMD8-92 treatment did not affect transforming growth factor-β1 (TGF-β1)-induced EMT, whereas BIX02189 abrogated the pro-metastatic effects of TGF-β1 surprisingly through suppression of TGF-β type I receptor (TβRI) activation, not MEK5/ERK5 signaling, although the level of MEK5/ERK5 activation was not determined [85]. In another investigation using metastatic A549 cells, knockdown of ERK5 resulted in reduced protein expression of CDH1 and ZO-1, upregulation of snail family zinc finger 1 (SNAI1), CDH2, and VIM, and enhanced cell migration [86]. Contrary to these findings, a recent study utilizing the same lung cancer cell line cited EMT suppressive, or MET inducing, effects of ERK5 depletion, including increased levels of CDH1 and reduction in cell migration, through regulation of SNAI2 with no change observed in SNAI1 levels [75]. These morphogenetic changes were recapitulated in a highly aggressive mesenchymal breast cancer model where suppression of ERK5 induced an epithelial phenotype and decreased intravascular invasion, leading to significantly fewer circulating tumor cells (CTCs) derived from primary orthotopic xenografts and reduction of metastatic lesions [75].
In addition to tumor cell intravasation, EMT has been linked to enrichment of the cancer stem cell (CSC)-like phenotype, further cementing its role in the metastatic cascade [87]. CSCs exhibit tumor-initiating potential, vital for metastatic colonization, attributed to their ability to self-renew and generate differentiated progeny that do not bear CSC cell-surface markers. From the multitude of studies establishing connections between MEK5 signaling and EMT, it follows that ERK5 would be involved in regulation of CSCs. Indeed, ERK5 activation was associated with enhanced CSC tumor sphere formation and tumor-initiating capacity [88]. Inhibition of ERK5 abrogated the effects of MEK5 activity on tumorigenicity of A549 spheres through hypoxia-inducible factor 1α (HIF1α)-mediated upregulation of apoptosis-associated genes BCL2 interacting protein 3 (BNIP3) and BNIP3 like (BNIP3L).
Involvement of the MEK5 pathway has also been described in disruption of actin dynamics leading to alterations in cell migration/invasion potential and metastatic dissemination. For instance, transfection of ERK5 expression construct in prostate cancer cells promoted formation of invadopodia, actin-rich protrusions of the plasma membrane associated with increased invasiveness of cancer cells [77]. Additionally, novel roles of ERK5 have been demonstrated in cytoskeletal remodeling pathways. PMA-stimulated ERK5 activity was implicated in regulation of cell morphology through phosphorylation of focal adhesion kinase (FAK) on S910 [89]. Moreover, integrin-mediated FAK signaling was linked to ERK5 activation in prostate and breast cancer cells, resulting in enhanced cell motility [82]. The ER-α/ERK5/cofilin (CFL1) network is another regulatory pathway of actin organization. While it has previously been shown that MEK5 signaling represses ER-α expression in breast cancer cells thereby promoting a more malignant hormone-independent phenotype, the role of ER-α was recently discovered in nuclear recruitment of ERK5 and CFL1, restricting their colocalization to cytoplasmic regions of actin remodeling, to suppress metastatic capacity [51, 90]. Notably, in ER-negative cell lines introduction of ER-α or ERK5 inhibition using XMD8-92 impaired cell motility and invasiveness [90].
Cell division cycle 42 (Cdc42), a member of the Rho GTPase family was shown to exert breast cancer cell line-specific effects on metastatic potential in part through regulation of the ERK5 pathway. It was reported that knockdown of Cdc42 increased ERK5 phosphorylation and suppressed cell motility and invasion in moderately metastatic Hs-578T breast cancer cells, suggesting that ERK5 signaling negatively correlates with metastatic progression [91, 92]. However, Cdc42 depletion enhanced cell migration and invasion in highly aggressive MDA-MB-231 breast cancer cells [91]. If activation of ERK5 associated with Cdc42 silencing, then ERK5 would exert pro-metastatic effects in these highly invasive cells; yet ERK5 activity was shown to decrease the invasive potential of MDA-MB-231 cells [92]. These conflicting results highlight the nuanced and cellular context-dependence of ERK5 function in modulating the invasive phenotype and further supports continued investigation of MEK-ERK5 signaling in regulation of metastatic progression.
7. Future perspective
Conventional MAPK family members, such as MEK1/2, are currently undergoing clinical trials, evaluated by potential to reduce tumor burden and improve progression-free survival in advanced-stage cancers. Studies have also assessed combinations of MEK1/2 inhibitors and other targeted agents or cytotoxic chemotherapy aimed at mitigating resistance mechanisms and enhancing patient response [45, 93, 94]. In particular, trametinib, a MEK1/2 inhibitor, has exhibited anti-tumor activity in the treatment of BRAF-mutated melanoma and has gained FDA approval as both a stand-alone agent and in combination with BRAF inhibitor dabrafenib [95, 96].
Recent advancements in unravelling the role of MEK5-ERK5 signaling in oncogenesis have shed light on its potential as a target in novel cancer therapeutics. Deregulation of the MEK5 pathway has been implicated in metastatic prostate cancer, colon cancer, and invasive osteosarcoma, demonstrating its broad range of application across various cancer types [66, 67, 97]. Elevated levels of ERK5 expression and activity correlates with worse prognosis in patients with triple-negative breast cancer, an aggressive subtype for which there are currently no targeted therapies available [45, 51]. Preclinical studies have shown that inhibition of the MEK5 cascade decreased intravascular invasion leading to decreased circulating tumor cells and formation of metastatic lesions, implicating its role in tumor progression and metastasis. Moreover, MEK5 signaling is strongly linked to chemoresistance. Overall, our work as well as others highlight the importance of this understudied pathway in cancer biology.
Due to redundancies in the MAPK signal transduction cascade and high degree of overlap in downstream targets of the MEK1/2 and MEK5 pathways, further investigation is warranted in determining potential synergy of combined MEK1/2 and MEK5 inhibition in targeting aggressive cancer types to delay the onset of drug resistance and maximize patient response to therapy. Understanding the MEK5-ERK5 pathway will provide a pivotal stage to expand the current spectrum of MEK inhibitor therapies and lead to wider application of such treatments.
MAPKs regulate diverse cellular processes including proliferation, cell survival, differentiation, and apoptosis
The MEK5-ERK5 cascade has emerged as an important mediator of tumorigenesis and metastatic progression
These studies implicate the MEK5-ERK5 pathway as a fundamental component of drug resistance in cancer therapy
Acknowledgments
This research was supported by National Institutes of Health - CA176496 (JE Cavanaugh) CA125806 (ME Burow), and CA174785 (ME Burow), The Office of Naval Research N00014-16-1-1136 (ME Burow). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest: The authors declare no conflict of interest.
References
- 1.Buschbeck M, et al. Phosphotyrosine-specific phosphatase PTP-SL regulates the ERK5 signaling pathway. J Biol Chem. 2002;277(33):29503–9. doi: 10.1074/jbc.M202149200. [DOI] [PubMed] [Google Scholar]
- 2.Cavanaugh JE, et al. Differential regulation of mitogen-activated protein kinases ERK1/2 and ERK5 by neurotrophins, neuronal activity, and cAMP in neurons. J Neurosci. 2001;21(2):434–43. doi: 10.1523/JNEUROSCI.21-02-00434.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cavanaugh JE, et al. Neuroprotective role of ERK1/2 and ERK5 in a dopaminergic cell line under basal conditions and in response to oxidative stress. J Neurosci Res. 2006;84(6):1367–75. doi: 10.1002/jnr.21024. [DOI] [PubMed] [Google Scholar]
- 4.Pearson G, et al. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22(2):153–83. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 5.English JM, et al. Isolation of MEK5 and differential expression of alternatively spliced forms. J Biol Chem. 1995;270(48):28897–902. doi: 10.1074/jbc.270.48.28897. [DOI] [PubMed] [Google Scholar]
- 6.Seyfried J, et al. A novel mitogen-activated protein kinase docking site in the N terminus of MEK5alpha organizes the components of the extracellular signal-regulated kinase 5 signaling pathway. Mol Cell Biol. 2005;25(22):9820–8. doi: 10.1128/MCB.25.22.9820-9828.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakamura K, Johnson GL. Noncanonical function of MEKK2 and MEK5 PB1 domains for coordinated extracellular signal-regulated kinase 5 and c-Jun N-terminal kinase signaling. Mol Cell Biol. 2007;27(12):4566–77. doi: 10.1128/MCB.00125-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morimoto H, et al. Activation of a C-terminal transcriptional activation domain of ERK5 by autophosphorylation. J Biol Chem. 2007;282(49):35449–56. doi: 10.1074/jbc.M704079200. [DOI] [PubMed] [Google Scholar]
- 9.Buschbeck M, Ullrich A. The unique C-terminal tail of the mitogen-activated protein kinase ERK5 regulates its activation and nuclear shuttling. J Biol Chem. 2005;280(4):2659–67. doi: 10.1074/jbc.M412599200. [DOI] [PubMed] [Google Scholar]
- 10.Nishimoto S, Nishida E. MAPK signalling: ERK5 versus ERK1/2. EMBO Rep. 2006;7(8):782–6. doi: 10.1038/sj.embor.7400755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kondoh K, et al. Regulation of nuclear translocation of extracellular signal-regulated kinase 5 by active nuclear import and export mechanisms. Mol Cell Biol. 2006;26(5):1679–90. doi: 10.1128/MCB.26.5.1679-1690.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yan C, et al. Molecular cloning of mouse ERK5/BMK1 splice variants and characterization of ERK5 functional domains. J Biol Chem. 2001;276(14):10870–8. doi: 10.1074/jbc.M009286200. [DOI] [PubMed] [Google Scholar]
- 13.Kamakura S, Moriguchi T, Nishida E. Activation of the protein kinase ERK5/BMK1 by receptor tyrosine kinases. Identification and characterization of a signaling pathway to the nucleus. J Biol Chem. 1999;274(37):26563–71. doi: 10.1074/jbc.274.37.26563. [DOI] [PubMed] [Google Scholar]
- 14.Kato Y, et al. BMK1/ERK5 regulates serum-induced early gene expression through transcription factor MEF2C. Embo Journal. 1997;16(23):7054–7066. doi: 10.1093/emboj/16.23.7054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hayashi M, et al. BMK1 mediates growth factor-induced cell proliferation through direct cellular activation of serum and glucocorticoid-inducible kinase. J Biol Chem. 2001;276(12):8631–4. doi: 10.1074/jbc.C000838200. [DOI] [PubMed] [Google Scholar]
- 16.Terasawa K, Okazaki K, Nishida E. Regulation of c-Fos and Fra-1 by the MEK5-ERK5 pathway. Genes Cells. 2003;8(3):263–73. doi: 10.1046/j.1365-2443.2003.00631.x. [DOI] [PubMed] [Google Scholar]
- 17.Ranganathan A, et al. The MAP kinase ERK5 binds to and phosphorylates p90 RSK. Arch Biochem Biophys. 2006;449(1–2):8–16. doi: 10.1016/j.abb.2006.02.023. [DOI] [PubMed] [Google Scholar]
- 18.Mody N, et al. An analysis of the phosphorylation and activation of extracellular-signal-regulated protein kinase 5 (ERK5) by mitogen-activated protein kinase kinase 5 (MKK5) in vitro. Biochem J. 2003;372(Pt 2):567–75. doi: 10.1042/BJ20030193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chao TH, et al. MEKK3 Directly Regulates MEK5 Activity as Part of the Big Mitogen-activated Protein Kinase 1 (BMK1) Signaling Pathway. Journal of Biological Chemistry. 1999;274(51):36035–36038. doi: 10.1074/jbc.274.51.36035. [DOI] [PubMed] [Google Scholar]
- 20.Cheng J, et al. Dimerization through the catalytic domain is essential for MEKK2 activation. J Biol Chem. 2005;280(14):13477–82. doi: 10.1074/jbc.M414258200. [DOI] [PubMed] [Google Scholar]
- 21.Sun W, et al. MEKK2 associates with the adapter protein Lad/RIBP and regulates the MEK5-BMK1/ERK5 pathway. J Biol Chem. 2001;276(7):5093–100. doi: 10.1074/jbc.M003719200. [DOI] [PubMed] [Google Scholar]
- 22.Jiang L, et al. Overexpression of MEKK2 is associated with colorectal carcinogenesis. Oncol Lett. 2013;6(5):1333–1337. doi: 10.3892/ol.2013.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu H, et al. The expression and role of MEKK3 in renal clear cell carcinoma. Anat Rec (Hoboken) 2015;298(4):727–34. doi: 10.1002/ar.23093. [DOI] [PubMed] [Google Scholar]
- 24.Samanta AK, et al. Overexpression of MEKK3 confers resistance to apoptosis through activation of NFkappaB. J Biol Chem. 2004;279(9):7576–83. doi: 10.1074/jbc.M311659200. [DOI] [PubMed] [Google Scholar]
- 25.Hasan R, et al. Mitogen activated protein kinase kinase kinase 3 (MAP3K3/MEKK3) overexpression is an early event in esophageal tumorigenesis and is a predictor of poor disease prognosis. BMC Cancer. 2014;14(1):2. doi: 10.1186/1471-2407-14-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cronan MR, et al. Defining MAP3 kinases required for MDA-MB-231 cell tumor growth and metastasis. Oncogene. 2012;31(34):3889–900. doi: 10.1038/onc.2011.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mirza AA, et al. MEKK2 regulates focal adhesion stability and motility in invasive breast cancer cells. Biochim Biophys Acta. 2014;1843(5):945–54. doi: 10.1016/j.bbamcr.2014.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ameka M, et al. MEKK2 regulates paxillin ubiquitylation and localization in MDA-MB 231 breast cancer cells. Biochem J. 2014;464(1):99–108. doi: 10.1042/BJ20140420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahmad S, Johnson GL, Scott JE. Identification of ponatinib and other known kinase inhibitors with potent MEKK2 inhibitory activity. Biochem Biophys Res Commun. 2015;463(4):888–93. doi: 10.1016/j.bbrc.2015.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang J, et al. The essential role of MEKK3 in TNF-induced NF-kappaB activation. Nat Immunol. 2001;2(7):620–4. doi: 10.1038/89769. [DOI] [PubMed] [Google Scholar]
- 31.Huang Q, et al. Differential regulation of interleukin 1 receptor and Toll-like receptor signaling by MEKK3. Nat Immunol. 2004;5(1):98–103. doi: 10.1038/ni1014. [DOI] [PubMed] [Google Scholar]
- 32.Samanta AK, et al. MEKK3 expression correlates with nuclear factor kappa B activity and with expression of antiapoptotic genes in serous ovarian carcinoma. Cancer. 2009;115(17):3897–908. doi: 10.1002/cncr.24445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo SY, et al. RNAi silencing of the MEKK3 gene promotes TRAIL-induced apoptosis in MCF-7 cells and suppresses the transcriptional activity of NF-kappaB. Oncol Rep. 2012;27(2):441–6. doi: 10.3892/or.2011.1509. [DOI] [PubMed] [Google Scholar]
- 34.Squires MS, Nixon PM, Cook SJ. Cell-cycle arrest by PD184352 requires inhibition of extracellular signal-regulated kinases (ERK) 1/2 but not ERK5/BMK1. Biochem J. 2002;366(Pt 2):673–80. doi: 10.1042/BJ20020372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tatake RJ, et al. Identification of pharmacological inhibitors of the MEK5/ERK5 pathway. Biochem Biophys Res Commun. 2008;377(1):120–5. doi: 10.1016/j.bbrc.2008.09.087. [DOI] [PubMed] [Google Scholar]
- 36.Yang Q, et al. Pharmacological inhibition of BMK1 suppresses tumor growth through promyelocytic leukemia protein. Cancer Cell. 2010;18(3):258–67. doi: 10.1016/j.ccr.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alvarez-Fernandez S, et al. Potent antimyeloma activity of a novel ERK5/CDK inhibitor. Clin Cancer Res. 2013;19(10):2677–87. doi: 10.1158/1078-0432.CCR-12-2118. [DOI] [PubMed] [Google Scholar]
- 38.Goh KC, et al. TG02, a novel oral multi-kinase inhibitor of CDKs, JAK2 and FLT3 with potent anti-leukemic properties. Leukemia. 2012;26(2):236–43. doi: 10.1038/leu.2011.218. [DOI] [PubMed] [Google Scholar]
- 39.William AD, et al. Discovery of kinase spectrum selective macrocycle (16E)-14-methyl-20-oxa-5,7,14,26-tetraazatetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8(27),9,11,16,21,23-decaene (SB1317/TG02), a potent inhibitor of cyclin dependent kinases (CDKs), Janus kinase 2 (JAK2), and fms-like tyrosine kinase-3 (FLT3) for the treatment of cancer. J Med Chem. 2012;55(1):169–96. doi: 10.1021/jm201112g. [DOI] [PubMed] [Google Scholar]
- 40.Shukla A, et al. Extracellular signal-regulated kinase 5: a potential therapeutic target for malignant mesotheliomas. Clin Cancer Res. 2013;19(8):2071–83. doi: 10.1158/1078-0432.CCR-12-3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Housman G, et al. Drug resistance in cancer: an overview. Cancers (Basel) 2014;6(3):1769–92. doi: 10.3390/cancers6031769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou C, et al. Proteomic analysis of tumor necrosis factor-alpha resistant human breast cancer cells reveals a MEK5/Erk5-mediated epithelial-mesenchymal transition phenotype. Breast Cancer Res. 2008;10(6):R105. doi: 10.1186/bcr2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weldon CB, et al. Identification of mitogen-activated protein kinase kinase as a chemoresistant pathway in MCF-7 cells by using gene expression microarray. Surgery. 2002;132(2):293–301. doi: 10.1067/msy.2002.125389. [DOI] [PubMed] [Google Scholar]
- 44.Miranda M, et al. MEK5-ERK5 pathway associates with poor survival of breast cancer patients after systemic treatments. Oncoscience. 2015;2(2):99–101. doi: 10.18632/oncoscience.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ortiz-Ruiz MJ, et al. Therapeutic potential of ERK5 targeting in triple negative breast cancer. Oncotarget. 2014;5(22):11308–18. doi: 10.18632/oncotarget.2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wolpin BM, Mayer RJ. Systemic treatment of colorectal cancer. Gastroenterology. 2008;134(5):1296–310. doi: 10.1053/j.gastro.2008.02.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mader RM, Muller M, Steger GG. Resistance to 5-fluorouracil. Gen Pharmacol. 1998;31(5):661–6. doi: 10.1016/s0306-3623(98)00191-8. [DOI] [PubMed] [Google Scholar]
- 48.Pereira DM, et al. MEK5/ERK5 signaling inhibition increases colon cancer cell sensitivity to 5-fluorouracil through a p53-dependent mechanism. Oncotarget. 2016 doi: 10.18632/oncotarget.9107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang Q, et al. BMK1 is involved in the regulation of p53 through disrupting the PML-MDM2 interaction. Oncogene. 2013;32(26):3156–64. doi: 10.1038/onc.2012.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Normanno N, et al. Mechanisms of endocrine resistance and novel therapeutic strategies in breast cancer. Endocr Relat Cancer. 2005;12(4):721–47. doi: 10.1677/erc.1.00857. [DOI] [PubMed] [Google Scholar]
- 51.Antoon JW, et al. MEK5/ERK5 signaling suppresses estrogen receptor expression and promotes hormone-independent tumorigenesis. PLoS One. 2013;8(8):e69291. doi: 10.1371/journal.pone.0069291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.O’Sullivan CC, et al. Efficacy of Adjuvant Trastuzumab for Patients With Human Epidermal Growth Factor Receptor 2-Positive Early Breast Cancer and Tumors </= 2 cm: A Meta-Analysis of the Randomized Trastuzumab Trials. J Clin Oncol. 2015;33(24):2600–8. doi: 10.1200/JCO.2015.60.8620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Montero JC, et al. Expression of Erk5 in early stage breast cancer and association with disease free survival identifies this kinase as a potential therapeutic target. PLoS One. 2009;4(5):e5565. doi: 10.1371/journal.pone.0005565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Esparis-Ogando A, et al. Erk5 Participates in Neuregulin Signal Transduction and Is Constitutively Active in Breast Cancer Cells Overexpressing ErbB2. Molecular and Cellular Biology. 2002;22(1):270–285. doi: 10.1128/MCB.22.1.270-285.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Al-Ejeh F, et al. Kinome profiling reveals breast cancer heterogeneity and identifies targeted therapeutic opportunities for triple negative breast cancer. Oncotarget. 2014;5(10):3145–58. doi: 10.18632/oncotarget.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carvajal-Vergara X, et al. Multifunctional role of Erk5 in multiple myeloma. Blood. 2005;105(11):4492–9. doi: 10.1182/blood-2004-08-2985. [DOI] [PubMed] [Google Scholar]
- 57.Mulloy R, et al. Activation of cyclin D1 expression by the ERK5 cascade. Oncogene. 2003;22(35):5387–98. doi: 10.1038/sj.onc.1206839. [DOI] [PubMed] [Google Scholar]
- 58.Hayashi M, et al. Big mitogen-activated protein kinase 1/extracellular signal-regulated kinase 5 signaling pathway is essential for tumor-associated angiogenesis. Cancer Res. 2005;65(17):7699–706. doi: 10.1158/0008-5472.CAN-04-4540. [DOI] [PubMed] [Google Scholar]
- 59.Cude K, et al. Regulation of the G2–M cell cycle progression by the ERK5–NFκB signaling pathway. The Journal of Cell Biology. 2007;177(2):253–264. doi: 10.1083/jcb.200609166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Garaude J, et al. ERK5 Activates NF-B in Leukemic T Cells and Is Essential for Their Growth In Vivo. The Journal of Immunology. 2006;177(11):7607–7617. doi: 10.4049/jimmunol.177.11.7607. [DOI] [PubMed] [Google Scholar]
- 61.English JM. Identification of Substrates and Regulators of the Mitogen-activated Protein Kinase ERK5 Using Chimeric Protein Kinases. Journal of Biological Chemistry. 1998;273(7):3854–3860. doi: 10.1074/jbc.273.7.3854. [DOI] [PubMed] [Google Scholar]
- 62.Umapathy G, et al. The kinase ALK stimulates the kinase ERK5 to promote the expression of the oncogene MYCN in neuroblastoma. Sci Signal. 2014;7(349):ra102. doi: 10.1126/scisignal.2005470. [DOI] [PubMed] [Google Scholar]
- 63.Rovida E, et al. The mitogen-activated protein kinase ERK5 regulates the development and growth of hepatocellular carcinoma. Gut. 2015;64(9):1454–65. doi: 10.1136/gutjnl-2014-306761. [DOI] [PubMed] [Google Scholar]
- 64.Liu L, et al. ERK5 activation of MEF2-mediated gene expression plays a critical role in BDNF-promoted survival of developing but not mature cortical neurons. Proc Natl Acad Sci U S A. 2003;100(14):8532–7. doi: 10.1073/pnas.1332804100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang Q, Lee JD. Targeting the BMK1 MAP kinase pathway in cancer therapy. Clin Cancer Res. 2011;17(11):3527–32. doi: 10.1158/1078-0432.CCR-10-2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mehta PB, et al. MEK5 overexpression is associated with metastatic prostate cancer, and stimulates proliferation, MMP-9 expression and invasion. Oncogene. 2003;22(9):1381–9. doi: 10.1038/sj.onc.1206154. [DOI] [PubMed] [Google Scholar]
- 67.Simoes AE, et al. Aberrant MEK5/ERK5 signalling contributes to human colon cancer progression via NF-kappaB activation. Cell Death Dis. 2015;6:e1718. doi: 10.1038/cddis.2015.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lochhead PA, et al. Tumor cells with KRAS or BRAF mutations or ERK5/MAPK7 amplification are not addicted to ERK5 activity for cell proliferation. Cell Cycle. 2016;15(4):506–18. doi: 10.1080/15384101.2015.1120915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zen K, et al. ERK5 is a target for gene amplification at 17p11 and promotes cell growth in hepatocellular carcinoma by regulating mitotic entry. Genes Chromosomes Cancer. 2009;48(2):109–20. doi: 10.1002/gcc.20624. [DOI] [PubMed] [Google Scholar]
- 70.Gavine PR, et al. Identification and validation of dysregulated MAPK7 (ERK5) as a novel oncogenic target in squamous cell lung and esophageal carcinoma. BMC Cancer. 2015;15:454. doi: 10.1186/s12885-015-1455-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gomez N, Erazo T, Lizcano JM. ERK5 and Cell Proliferation: Nuclear Localization Is What Matters. Front Cell Dev Biol. 2016;4:105. doi: 10.3389/fcell.2016.00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Erazo T, et al. Canonical and kinase activity-independent mechanisms for extracellular signal-regulated kinase 5 (ERK5) nuclear translocation require dissociation of Hsp90 from the ERK5-Cdc37 complex. Mol Cell Biol. 2013;33(8):1671–86. doi: 10.1128/MCB.01246-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lin EC, et al. ERK5 kinase activity is dispensable for cellular immune response and proliferation. Proc Natl Acad Sci U S A. 2016;113(42):11865–11870. doi: 10.1073/pnas.1609019113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McCracken SR, et al. Aberrant expression of extracellular signal-regulated kinase 5 in human prostate cancer. Oncogene. 2008;27(21):2978–88. doi: 10.1038/sj.onc.1210963. [DOI] [PubMed] [Google Scholar]
- 75.Javaid S, et al. MAPK7 Regulates EMT Features and Modulates the Generation of CTCs. Mol Cancer Res. 2015;13(5):934–43. doi: 10.1158/1541-7786.MCR-14-0604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sureban SM, et al. XMD8-92 inhibits pancreatic tumor xenograft growth via a DCLK1-dependent mechanism. Cancer Lett. 2014;351(1):151–61. doi: 10.1016/j.canlet.2014.05.011. [DOI] [PubMed] [Google Scholar]
- 77.Ramsay AK, et al. ERK5 signalling in prostate cancer promotes an invasive phenotype. Br J Cancer. 2011;104(4):664–72. doi: 10.1038/sj.bjc.6606062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yue B, et al. ERK5 silencing inhibits invasion of human osteosarcoma cell via modulating the Slug/MMP-9 pathway. European Review for Medical and Pharmacological Sciences. 2014;18(18):2640–2647. [PubMed] [Google Scholar]
- 79.Sticht C, et al. Activation of MAP Kinase Signaling Through ERK5 But Not ERK1 Expression Is Associated with Lymph Node Metastases in Oral Squamous Cell Carcinoma (OSCC) Neoplasia. 2008;10(5):462–IN4. doi: 10.1593/neo.08164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hu B, et al. Expression of the phosphorylated MEK5 protein is associated with TNM staging of colorectal cancer. BMC Cancer. 2012;12:127. doi: 10.1186/1471-2407-12-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Arias-González L, et al. ERK5/BMK1 Is a Novel Target of the Tumor Suppressor VHL: Implication in Clear Cell Renal Carcinoma. Neoplasia. 2013;15(6):649–IN17. doi: 10.1593/neo.121896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sawhney RS, Liu W, Brattain MG. A novel role of ERK5 in integrin-mediated cell adhesion and motility in cancer cells via Fak signaling. J Cell Physiol. 2009;219(1):152–61. doi: 10.1002/jcp.21662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liu F, Zhang H, Song H. Upregulation of MEK5 by Stat3 promotes breast cancer cell invasion and metastasis. Oncol Rep. 2017;37(1):83–90. doi: 10.3892/or.2016.5256. [DOI] [PubMed] [Google Scholar]
- 84.Deryugina EI, Quigley JP. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006;25(1):9–34. doi: 10.1007/s10555-006-7886-9. [DOI] [PubMed] [Google Scholar]
- 85.Park SJ, et al. BIX02189 inhibits TGF-beta1-induced lung cancer cell metastasis by directly targeting TGF-beta type I receptor. Cancer Lett. 2016;381(2):314–22. doi: 10.1016/j.canlet.2016.08.010. [DOI] [PubMed] [Google Scholar]
- 86.Chen R, Yang Q, Lee JD. BMK1 kinase suppresses epithelial-mesenchymal transition through the Akt/GSK3beta signaling pathway. Cancer Res. 2012;72(6):1579–87. doi: 10.1158/0008-5472.CAN-11-2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Scheel C, Weinberg RA. Cancer stem cells and epithelial-mesenchymal transition: concepts and molecular links. Semin Cancer Biol. 2012;22(5–6):396–403. doi: 10.1016/j.semcancer.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Song C, et al. Inhibition of BMK1 pathway suppresses cancer stem cells through BNIP3 and BNIP3L. Oncotarget. 2015;6(32):33279–89. doi: 10.18632/oncotarget.5337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Villa-Moruzzi E. Targeting of FAK Ser910 by ERK5 and PP1delta in non-stimulated and phorbol ester-stimulated cells. Biochem J. 2007;408(1):7–18. doi: 10.1042/BJ20070058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Madak-Erdogan Z, et al. Novel roles for ERK5 and cofilin as critical mediators linking ERalpha-driven transcription, actin reorganization, and invasiveness in breast cancer. Mol Cancer Res. 2014;12(5):714–27. doi: 10.1158/1541-7786.MCR-13-0588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zuo Y, Wu Y, Chakraborty C. Cdc42 negatively regulates intrinsic migration of highly aggressive breast cancer cells. J Cell Physiol. 2012;227(4):1399–407. doi: 10.1002/jcp.22853. [DOI] [PubMed] [Google Scholar]
- 92.Zuo Y, et al. Modulation of ERK5 is a novel mechanism by which Cdc42 regulates migration of breast cancer cells. J Cell Biochem. 2015;116(1):124–32. doi: 10.1002/jcb.24950. [DOI] [PubMed] [Google Scholar]
- 93.Flaherty KT, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367(18):1694–703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Janne PA, et al. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol. 2013;14(1):38–47. doi: 10.1016/S1470-2045(12)70489-8. [DOI] [PubMed] [Google Scholar]
- 95.Infante JR, et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur J Cancer. 2014;50(12):2072–81. doi: 10.1016/j.ejca.2014.04.024. [DOI] [PubMed] [Google Scholar]
- 96.Kim KB, et al. Phase II study of the MEK1/MEK2 inhibitor Trametinib in patients with metastatic BRAF-mutant cutaneous melanoma previously treated with or without a BRAF inhibitor. J Clin Oncol. 2013;31(4):482–9. doi: 10.1200/JCO.2012.43.5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tesser-Gamba F, et al. MAPK7 and MAP2K4 as prognostic markers in osteosarcoma. Hum Pathol. 2012;43(7):994–1002. doi: 10.1016/j.humpath.2011.08.003. [DOI] [PubMed] [Google Scholar]