

Summary
Cognitive disabilities that occur with age represent a growing and expensive health problem. Age-associated memory deficits are observed across many species, but the underlying molecular mechanisms remain to be fully identified. Here we report elevations in the levels and activity of the striatal-enriched phosphatase (STEP) in the hippocampus of aged memory-impaired mice and rats, in aged rhesus monkeys, and in people diagnosed with amnestic mild cognitive impairment (aMCI). The accumulation of STEP with aging is related to dysfunction of the ubiquitin-proteasome system that normally leads to the degradation of STEP. Higher level of active STEP is linked to enhanced dephosphorylation of its substrates GluN2B and ERK1/2, CREB inactivation, and a decrease in total levels of GluN2B and BDNF. These molecular events are reversed in aged STEP knock-out and heterozygous mice, which perform similarly to young control mice in the Morris water maze (MWM) and Y-maze tasks. In addition, administration of the STEP inhibitor TC-2153 to old rats significantly improved performance in a delayed alternation T-maze memory task. In contrast, viral-mediated STEP overexpression in the hippocampus is sufficient to induce memory impairment in the MWM and Y-maze tests, and these cognitive deficits are reversed by STEP inhibition. In old LOU/C/Jall rats, a model of healthy aging with preserved memory capacities, levels of STEP and GluN2B are stable, and phosphorylation of GluN2B and ERK1/2 is unaltered. Altogether, these data suggest that elevated levels of STEP that appear with advancing age in several species contribute to the cognitive declines associated with aging.
Keywords: Phosphatase STEP, Memory, Aging, Animal Models, Animal Behaviors, Hippocampus
Graphical abstract
The molecular changes leading to memory deficits during aging remain to be fully identified. Castonguay et al. provide genetic, behavioral and molecular evidence that accumulation of the phosphatase STEP is a common molecular event that oppose synaptic strengthening and contributes to age-related memory deficits in mice, rats, monkeys and humans.
Introduction
Cognitive disabilities that occur with age are a common and expensive health problem. Cellular and molecular alterations that occur during aging can lead to memory deficits, ranging from subjective forgetfulness complaints to amnestic mild cognitive impairment (aMCI), a distinct clinical diagnosis that increases the risk of developing Alzheimer's disease (AD) or other types of dementia [1-4]. Many features of age-related cognitive decline in humans are found across other species, but it remains unclear whether the underlying mechanisms are common or divergent between species.
The striatal-enriched protein tyrosine phosphatase (STEP) was found to be implicated in diverse brain disorders [5], but its impact on memory deficits during normal aging still need to be investigated. STEP is highly expressed in regions where memory consolidation occurs, such as the hippocampus and cortex [6-8]. STEP61 is the only splice isoform expressed in these regions and is localized mainly within postsynaptic compartments [6, 8]. Dephosphorylation of regulatory tyrosine residues by STEP decreases synaptic strengthening by inactivating its substrates (ERK1/2, Fyn, Pyk2, p38), and by promoting internalization of surface glutamate N-methyl-D-aspartate receptors (NMDARs) containing the GluN2B subunit and GluA1/GluA2-containing α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) [9-14].
Several studies have shown that decreased levels of GluN2B-containing NMDARs are associated with memory decline during normal aging [15-18]. Transgenic mice overexpressing GluN2B in the cortex or hippocampus show significant improvement in learning and memory that last into old age [19, 20]. In contrast, GluN2B ablation in the forebrain or hippocampus reduces long-term potentiation (LTP) and impairs spatial memory performance in mice [21]. In non-human primates, a decrease in NMDARs is observed during aging in the prefrontal cortex and hippocampus, two brains regions involved in memory that are vulnerable to aging [22-26]. However, it remains to determine if dysregulation of STEP levels during aging could compromised glutamatergic function by decreasing cell surface expression of GluN2B-containing NMDARs at the synapse.
STEP also dephosphorylates and inactivates ERK1/2 [27], which is involved in LTP as well as learning and memory [28]. Decreased tyrosine phosphorylation and lower activity of ERK1/2 was observed in aged (24 months old) rats compared to 12 and 6 months old rat [29]. ERK activates the signaling pathway of the cAMP response element-binding protein (CREB) and brain-derived neurotrophic factor (BDNF), two main molecules implicated in memory consolidation [30, 31]. Overactivation of STEP also directly leads to decrease BDNF expression [32]. Interestingly, ERK and CREB were found to be more activated in aged memory-impaired rats [33].
Here we provide genetic, behavioral and molecular evidence that excessive accumulation of the tyrosine phosphatase STEP induces alteration in NMDAR and ERK signaling pathways that contributes to age-related memory deficits in mice, rats, rhesus monkeys, and humans.
Results
Reduction of STEP alleviates age-related memory deficits
The spatial memory abilities of aged C57BL/6J mice were evaluated in the reference memory version of the Morris water maze (MWM) [34], a hippocampus-dependent behavioral task widely used to detect aged memory deficits in animal models [33, 35-37]. We tested 22-24 months old male mice (n=34) and characterized them as aged memory-impaired (AI) or aged memory-unimpaired (AU) based on their performance during the acquisition phase and probe trial, and on their search strategy in the MWM test. As described previously [33, 35, 36], aged mice were categorized as AI (n=9) if their escape latencies on testing days 3 and 4 were greater than 1 or 2 standard deviations (SD), respectively, from the mean of the young (Y) adult group (6 months old; n=34) (Figure 1A). Animals were classified as AU (n=9) if their escape latencies on days 3 and 4 were less than 0.5 SD from the Y group. The distance to reach the hidden escape platform, a measurement independent of the swimming speed of the mice, was also longer for AI compared to AU and Y mice (Figure 1B).
Figure 1. Aged memory impairment is associated in mice with higher STEP levels and is attenuated by genetic reduction of STEP.

Aged (22-24 mo old) memory-impaired (AI; n=9), aged memory-unimpaired (AU; n=9), aged STEP knock-out (KO; n=9) and heterozygous (Het; n=9), and young (Y; n=34) adult (6 mo old) mice were tested in the Morris water maze (MWM) (three-way ANOVA). During the acquisition phase of the MWM task (3 trials/day; 60 s; 30 m inter-trial interval), escape latency (A) and distance swam (B) before finding the hidden platform was longer for AI than AU and Y animals. * or + indicates significant differences compared to Y or AU mice, respectively. Performance of STEP KO mice were similar to Y mice, and lower than AI mice on days 3 and 4 ($ P<0.05, $$ P<0.01). STEP Het mice had longer latency than Y animal on day 4 (& P<0.05) but performed better than AI mice on days 3 and 4 (# P<0.05). (C) Swim speed was not significantly (n.s.) different between all aged groups but was decreased compared to Y mice (* P<0.05, two-way ANOVA, post-hoc Tukey's HSD test). During the probe trial performed 24 h after the last acquisition day, the AI mice crossed the platform location less frequently than all the other groups (two-way ANOVA, Tukey's HSD test) (D), and showed no preference for the target quadrant compared to the opposite one (three-way ANOVA) (E). # represents significant variations compared to AI for the target quadrant, and & indicates time differences for the target and opposite quadrants within each group. (F) During the Y-maze, all groups, except the AI mice, spent more time in the new available arm than in the other arm previously explored during the training phase (three-way ANOVA). (G) Representative immunoblots of non-phosphorylated STEP (non pSTEP) at S221, STEP, pGluN2B Y1472, GluN2B, PSD-95, GAPDH, pERK1/2 Y204/187, ERK1/2 in synaptosomal (P2) fractions prepared from the dorsal hippocampus. The levels of pCREB S133 and CREB were measured in the nuclear fraction and BDNF levels in the cytosolic fraction. (H) Densitometric quantification of changes expressed as the mean ratio of proteins (n=9 in each group, two independent experiments). * or + indicates significant variations compared to Y or AI mice, respectively. (I) Representative immunoblots of polyubiquitinated STEP isolated from the P2 hippocampal fraction and probed with an anti-STEP antibody. (J) The phosphatase activity of STEP immunoprecipitated from the P2 hippocampal fraction was assayed by using Fyn-phosphorylated GST-GluN2B as substrate. Dephosphorylation of GluN2B Y1472 was normalized to the initial level of Fyn-phosphorylated GST-GluN2B (input) not treated with immunoprecipitated STEP. * or + indicates significant differences compared to Y or AU mice, respectively (n=9 in each group, two independent experiments. All histograms are presented as mean values ± s.e.m. *,+,$,&,# P<0.05, **,++,$$,## P<0.01, ***,+++,###,&&& P<0.001. See also Figures S1, S2 and S3.
To evaluate the impact of STEP in age-related memory impairment, we tested aged (22-24 mo old) STEP knock-out (KO; n=9) and heterozygote (Het; n=9) mice in the MWM task under the same behavioral conditions. A three-way ANOVA revealed a significant genotype × age × acquisition day interaction (P<0.05). We found that aged STEP KO and Het mice performed similarly to young WT and AU mice (P>0.05) during the acquisition phase of the MWM task (Figures 1A and 1B).
The search strategy of each mouse was also analyzed and two mice out of eleven were excluded from the AI group before behavioral analysis because of thigmotaxis, a non-specific memory search strategy that consists of swimming close to the wall without paying attention to the spatial cues to locate the platform. Young mice swam faster than aged mice, but no significant difference was observed in swimming speed between all aged groups (Figure 1C). We also found that young 6 mo old STEP KO and Het mice performed as well as young WT mice in the MWM (Supplemental Figure S1).
To confirm memory status, the number of platform crossings (Figure 1D) and the time spent in the target and opposite quadrants (Figure 1E) were evaluated during a probe trial 24 h after the last acquisition day. All the groups (Y, AU, STEP Het and KO mice) crossed the previous platform location more frequently than AI mice (P<0.01; two-way ANOVA) during the probe trial (Figure 1D). A three-way ANOVA analysis revealed a significant genotype × age × quadrant interaction (P<0.01; Figure 1E). The STEP Het and KO mice spent as much time as Y and AU mice in the target zone, whereas AI mice showed no preference for the target zone. Similar escape latencies were found among all the groups during the cued trial when the platform was visible (Y, 7.6±1.0 s; AI, 7.3±1.2 s; AU, 8.4±1.6 s; STEP Het, 7.8±1.4 s; STEP KO, 7.9±1.5 s; mean ± s.e.m.; P>0.05; two-way ANOVA), which indicated no motivational or sensorimotor differences between all groups to escape from water.
The effect of STEP reduction in aged mice was also tested in another hippocampus-dependent memory task, the Y-maze task Figure 1F). During this test, mice needed to remember which two arms were visited during the training phase before recognizing the new arm and spending more time exploring it during the testing phase. A three-way ANOVA revealed a significant genotype × age × arm interaction (P<0.05). We found that STEP Het and KO mice spent as much time as Y and AU mice exploring the new arm, whereas AI mice showed no preference for the new arm. Taken together, these results indicate that lower STEP levels have beneficial effects on memory functioning in aged mice.
Higher level of STEP is observed in AI mice
The synaptosomal (P2) membrane fraction prepared from hippocampus of each mouse (n=9 per group) was extracted 6 h after the last behavioral task (Y-maze test) and analyzed by western blots (Figures 1G and 1H). This 6 h time point was selected to determine the impact of STEP alteration on the phosphorylation of its substrates but also on more downstream events such as CREB phosphorylation and BDNF levels. The enrichment of synaptic proteins in the P2 fraction was first confirmed by western blots (Supplemental Figure S2). A two-way ANOVA followed by a post-hoc Tukey's HSD test revealed two main findings. First, total STEP levels were increased in older animals (AI and AU compare to Y mice), as measured by using an antibody that recognizes total levels of STEP (both its phosphorylated and non-phosphorylated forms) (Figure 1G). Since STEP is activated when its epitope S221 is not phosphorylated, we used an antibody that recognized active STEP when non-phosphorylated at S221 (non pSTEP S221). Elevation of total STEP level was accompanied by a proportional elevation of non-phosphorylated active STEP.
Second, STEP expression was significantly higher in AI compared to AU mice, and this enhancement was associated with more pronounced dephosphorylation of the STEP substrates GluN2B and ERK1/2. No changes in STEP levels and phosphorylation of GluN2B and ERK1/2 were found in the occipital cortex, a control brain region not involved in hippocampus-dependent memory process (Supplemental Figure S3). Because the hippocampus is especially vulnerable to aging, one could predict that the involvement of STEP on age-related memory deficits is more pronounced for tasks involving the hippocampus as shown here, although it cannot be excluded that other brain regions implicated in memory could also be affected by a decrease in STEP levels during aging. We also observed that the total level of GluN2B decreased with age (AI and AU compare to Y mice), and that reduced STEP levels in STEP KO and Het mice counteracted this phenomenon. The phosphorylation of GluN2B (pGluN2B Y1472) relative to total GluN2B level after normalization with GAPDH was also diminished in AI when compared to all the other groups (Figure 1H).
Although no significant cell death is apparent during normal aging, synapse and spine losses were shown to be associated with aging [15, 38]. To account for spine loss, we measured the level of the postsynaptic density protein-95 (PSD-95), an anchoring protein that tethers NMDARs at the synapse. We found that this protein decreased with age as previously reported [39]. The total levels of GluN2B were significantly lower in AI compared to AU and Y mice after normalization to PSD-95. Higher levels of GluN2B were also observed in STEP KO and Het mice after normalization to PSD-95.
We then analyzed the phosphorylation state of CREB in the nuclear fraction and BDNF expression in the cytosolic fraction. Both proteins are downstream targets of ERK1/2 and have been shown to be involved in memory consolidation [30, 31]. When compared to young mice the phosphorylation of CREB and total BDNF levels were significantly decreased in AI mice but not in aged STEP KO and Het mice. Taken together, these results suggest that old mice with memory impairments have higher STEP levels, which reduces the phosphorylation and activity of its primary substrates GluN2B and ERK1/2 and consequently CREB and BDNF. We also found that lower STEP level in mice improved age-related memory deficits in two hippocampus-dependent behavioral tasks, the MWM and Y-maze tests.
STEP accumulation involves the ubiquitin-proteasome system
STEP is normally ubiquitinated and degraded by the proteasome after synaptic activation [10]. To determine whether STEP elevation in aged animals resulted from a disruption of the ubiquitin-proteasome system (UPS), we analyzed the accumulation of ubiquitin-conjugated STEP in the hippocampus from these mice (Figure 1I). There was a significant increase of polyubiquitinated STEP in AI compared to AU and Y animals (P<0.05; two-way ANOVA, Tukey's HSD test).
To confirm that accumulated STEP was in its active form, we performed a phosphatase assay with a GST-GluN2B construct phosphorylated at Y1472, the residue dephosphorylated by STEP (Figure 1J). STEP immunoprecipitation followed by a tyrosine phosphatase assay showed a significant increase in STEP activity in AI mice compared to AU and Y mice (P<0.05; two-way ANOVA, Tukey's HSD test). These results suggest that decreased degradation of STEP leads to an accumulation of active STEP levels in aged-memory impaired animals.
Overexpression of STEP in the hippocampus of young mice induces memory impairment
To address more directly the functional role of higher STEP levels in memory deficits, STEP was overexpressed in the CA1 region of dorsal hippocampus of young (6 months old) C57BL/6J male mice. Marked expression of STEP, or EGFP as control, was achieved by stereotaxic microinjection of AAV-STEP and AAV-EGFP vectors into the CA1 region of the hippocampus. Fluorescence microscopy of EGFP and STEP showed pronounced levels of both proteins 4 weeks after microinjections (Figure 2A). The immunostaining was predominant in cell bodies and dendrites of pyramidal neurons of the CA1 region of the dorsal hippocampus. STEP overexpression in the dorsal hippocampus was also confirmed by real-time qRT-PCR in another group of six mice (Figure 2A). The mRNA level of STEP was significantly higher in the CA1 area of the hippocampus microinjected with AAV-STEP when compared to the control contralateral hippocampus injected with AAV-EGFP (P<0.01, paired t test).
Figure 2. Hippocampal expression of STEP and EGFP in mice following microinjection of AAV vectors containing the transgenes.
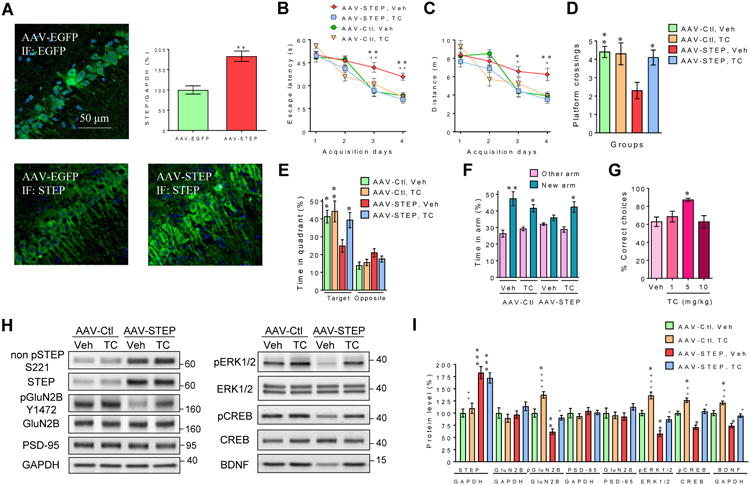
(A) Microinjection of AAV-EGFP (upper left) into the CA1 region of dorsal hippocampus induced marked green immunofluorescence (IF) using an EGFP antibody, indicating efficient AAV vector transfection and expression of the EGFP reporter gene. STEP immunostaining in the CA1 region was more intense following AAV-STEP injection compared to control AAV-EGFP injection (lower panel). Real-time qRT-PCR analysis (upper right) of STEP mRNA expression normalized to GAPDH mRNA 4 weeks following microinjection of control AAV-EGFP or AAV-STEP in the CA1 region in the contralateral dorsal hippocampus (n=6; ** P<0.01, paired t test). Reported values are log10 transforms of the STEP/GAPDH ratio. Escape latency (B) and distance swam (C) to reach the hidden platform was longer for AAV-STEP mice (n=10) injected with vehicle (Veh; 0.1% DMSO, i.p., v/v) when compared to AAV-STEP mice (n=10) treated with TC-2153 (i.p., 10mg/kg, 3h prior daily trials) or AAV-EGFP treated with Veh (n=10) or TC-2153 (n=10) (three-way ANOVA). * and + represents statistical significant variations between AAV-EGFP + Veh and AAV-STEP + TC-2153, respectively. Contrary to all the other groups, AAV-STEP + Veh mice crossed the previous platform location less often (two-way ANOVA, Tukey's HSD test) (D), and showed no preference for the target quadrant during the probe trial 24 h after the last acquisition day (E) (three-way ANOVA). * represents a statistical significant variation between AAV-STEP + Veh and other groups for platform crossings or the target quadrant. (F) During the Y-maze, the AAV-STEP + Veh was the only group showing no preference for the new available arm (three-way ANOVA). (G) The effects of TC-2153 (1, 5 and 10 mg/kg, i.p.) in aged rats performing the delayed alternation memory task. TC-2153 improved performance in the aged rats at the dose of 5mg/kg (one-way ANOVA, Tukey's HSD test). (H) Representative immunoblots of total and phosphorylated levels of proteins in synaptosomal (P2) fractions prepared from the dorsal hippocampus. CREB and pCREB S133 levels were evaluated in the nuclear fraction and BDNF levels in the cytosolic fraction (I) Densitometric quantification of changes between groups expressed as the mean ratio of proteins (n=10 in each group, two independent experiments). * or + indicates significant variations compared to AAV-Ctl + Veh or AAV-STEP + Veh mice, respectively. All histograms are presented as mean values ± s.e.m. *,+ P<0.05, **,++ P<0.01, ***,+++ P<0.001. See also Figure S4.
Mice microinjected bilaterally in the dorsal hippocampus with either AAV-STEP or AAV-EGFP vectors were tested in the MWM task. A three-way ANOVA revealed a significant vector × treatment × acquisition day interaction (P<0.05). Higher hippocampal levels of STEP were associated with memory deficits during the acquisition phase of the MWM relative to control mice microinjected with AAV-EGFP (Figures 2B and 2C).
The STEP inhibitor TC-2153 [40] was then administered to determine if this would reverse the cognitive deficits induced by higher STEP expression. Using the same TC-2153 treatment (i.p., 10 mg/Kg, 3 h before MWM testing) that we performed previously to reverse memory deficits in a transgenic AD mouse model [40], we found that TC-2153 alleviated memory impairment in AAV-STEP mice on acquisition days 3 and 4 (Figures 2B and 2C). The STEP inhibitor had no significant effect on the cognitive performance of mice injected with AAV-EGFP. No difference was detected in the swimming speed across all the groups (AAV-STEP + Veh, 21.0±0.8 cm/s; AAV-STEP + TC, 19.8±0.5 cm/s; AAV-EGFP + Veh, 18.8±0.7 cm/s; AAV-EGFP + TC, 20.2±0.7 cm/s; mean ± s.e.m.; P>0.05; two-way ANOVA). Similar escape latencies were found between groups during the cued trial (AAV-STEP + Veh, 8.4±1.6 s; AAV-STEP + TC, 8.2±1.8 s; AAV-EGFP + Veh, 7.9±1.8 s; AAV-EGFP + TC, 8.3±1.7 s; mean ± s.e.m.; P>0.05; two-way ANOVA).
Memory dysfunction in mice overexpressing STEP was also observed during the probe trial 24 h after the last acquisition day of the MWM test (Figures 2D and 2E). Compared to AAV-EGFP + Veh mice, AAV-STEP + Veh mice crossed the previous platform location less frequently (P<0.01; two-way ANOVA, Tukey's HSD test), and spent less time in the target quadrant (P<0.01; three-way ANOVA,). These cognitive deficits were reversed by administration of TC-2153 (Figures 2D and 2E).
To determine the functional specificity of the AAV-STEP microinjection in affecting only the hippocampus-dependent task, mice were also tested in auditory-cued fear conditioning, a behavioral paradigm known to predominantly involve the amygdala instead of the hippocampus [41, 42]. Mice overexpressing STEP in the hippocampus demonstrated intact auditory-cued fear memory acquisition (P>0.05; three-way ANOVA; Supplemental Figure S4). The hippocampus-dependent memory impairment of AAV-STEP mice was also confirmed in the Y-maze task (Figure 2F). A three-way ANOVA analysis revealed a significant vector × treatment × arm interaction (P<0.05). During the testing phase, control AAV-EGFP mice spent more time exploring the new arm, whereas AAV-STEP + Veh mice showed no preference for the new arm. Injecting TC-2153 3h prior to the Y-maze test prevented memory deficits in AAV-STEP mice (Figure 2F).
The effect of TC-2153 on age-related memory deficits was evaluated in another series of experiments using old Sprague-Dawley male rats (20-22 mo old) trained in the delayed alternation T-maze memory task (Figure 2G). TC-2153 significantly improved delayed alternation performance in rats when compared with vehicle (P<0.05, one-way ANOVA, Tukey's HSD test). No toxicity or side effects were observed with TC-2153 at 10 mg/kg in aged rats, which indicates that aged rats required a lower dose of TC-2153 compared to mice overexpressing STEP in the hippocampus to induce beneficial effects in this memory task.
The hippocampus of each mouse was extracted 6 h after the last behavioral task (Y-maze task) for western blot analysis (Figures 2H and 2I). AAV-STEP mice showed higher levels of total STEP compared to AAV-EGFP mice (P<0.001; two-way ANOVA, Tukey's HSD test). The level of non pSTEP S221 was proportional to total STEP accumulation, and treatment with TC-2153 did not affect the level of STEP in either AAV-EGFP or AAV-STEP mice. Overexpression of STEP resulted in marked dephosphorylation of its substrates GluN2B and ERK1/2, an effect that was reversed by TC-2153. Levels of pCREB and BDNF were also decreased in AAV-STEP mice relative to AAV-EGFP mice and returned to their basal levels after TC-2153 treatment.
Higher STEP levels in AI rats but not in a rat model of healthy aging
To determine if STEP elevation is found in other species with cognitive decline, we performed a series of experiments with aged (22-24 mo old) Long-Evans rats (Figure 3). A large cohort of 24 mo old Long-Evans rats (n=108) were previously tested in the reference memory version of the MWM task as reported earlier [33]. Here are shown the cognitive performances of the rats used for immunoblotting analysis in this study (n=8 in each group). In line with the results we obtained with aged mice, rats characterized as AI in the MWM task showed increased STEP expression that was associated with enhanced dephosphorylation of GluN2B and ERK1/2.
Figure 3. STEP is elevated in aged memory-impaired Long-Evans rats.
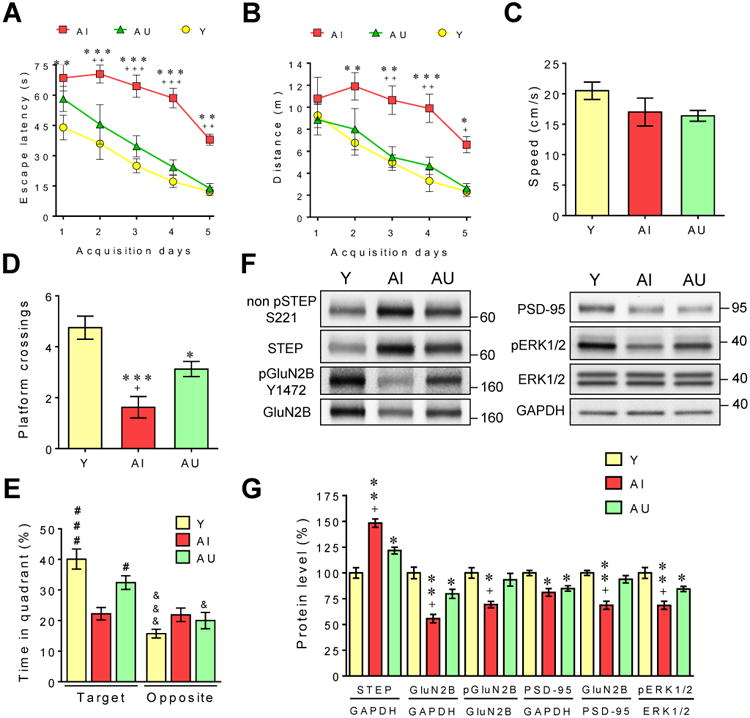
A large cohort of 24mo old Long-Evans rats (n=108) were previously tested in the reference memory version of the MWM task as reported earlier [33]. Here are shown the cognitive performances of the rats used for immunoblotting analysis in this study (n=8 in each group). During the acquisition phase of the MWM task, escape latency (A) and distance swam (B) to reach the hidden platform was longer for AI than AU and Y animals (two-way ANOVA, Tukey's HSD test). (C) No significant difference was observed between the speed of Y, AI and AU rats (P>0.05; one-way ANOVA). During the probe trial, AU and Y mice crossed the platform location more frequently (one-way ANOVA) (D) and spent more time in the target quadrant (two-way ANOVA) (E) than AI mice. # represents variations compared to AI for the target quadrant, and & indicates differences for the target and opposite quadrants within each group. (F) Representative immunoblots of total and phosphorylated levels of proteins in synaptosomal (P2) fractions prepared from the hippocampus. (G) Densitometric quantification of changes between Y, AI and AU mice expressed as the mean ratio of proteins (n=8 in each group, two independent experiments). * or + indicates significant variations compared to Y or AU mice, respectively. All histograms are presented as mean values ± s.e.m. *,+,&,# P<0.05, **,++ P<0.01, ***,+++,###,&&& P<0.001.
We next tested whether STEP level was affected in aging LOU/C/Jall rats, a model of healthy aging [43] with preserved recognition memory [44, 45] (Fig. 4). There was no significant spatial memory impairment in 24 mo old LOU/C/Jall rats compared to mature (12 mo) and young adult (6 mo) animals during the acquisition phase and probe trial in the MWM (Figures 4A and 4C). There were no changes in STEP levels or in the Tyr phosphorylation of its substrates GluN2B or ERK1/2 in these aged rats with preserved spatial memory (Figures 4D and 4E). Taken together, these data indicate that increase STEP level is associated with age-related memory impairment not only in mice but also in rats.
Figure 4. Stable STEP expression is associated with preserved memory capacity in aged LOU/C/Jall rats.
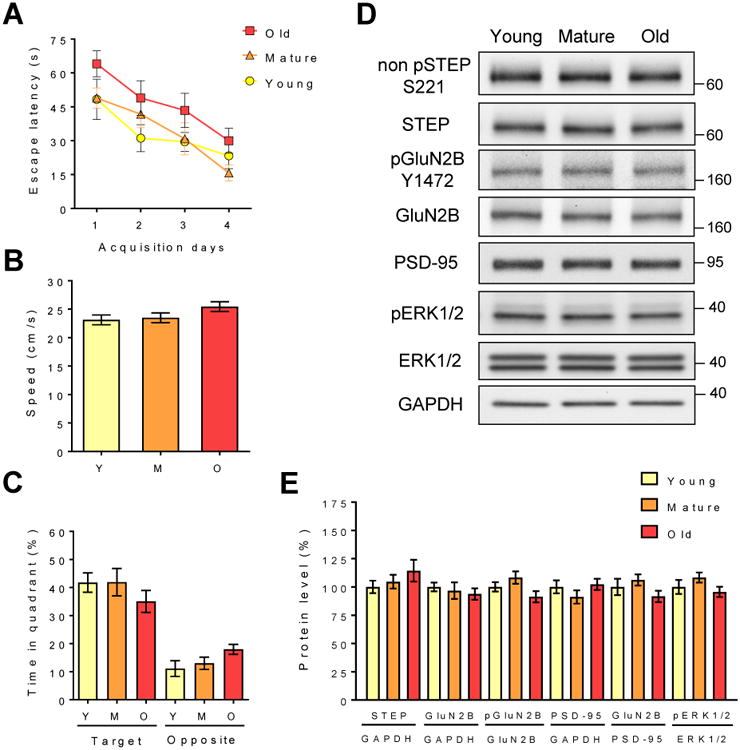
During the MWM task, escape latency (P>0.05; two-way ANOVA, Tukey's HSD test) (A), swim speed (P>0.05; one-way ANOVA) (B), and percent time spent in the target and opposite quadrants (P>0.05; two-way ANOVA, Tukey's HSD test) (C) were not significantly different between all groups (P>0.05). (D) Representative immunoblots of total and phosphorylated levels of proteins in hippocampal P2 fractions. (E) Densitometric quantification of changes expressed as the mean ratio of proteins. Error bars indicate ± s.e.m. (n=8, two independent experiments).
STEP level is increased in aged monkeys and human aMCI individuals
To further investigate if STEP is found in age-related memory decline in other species, we analyzed its level in rhesus monkeys and aged humans with aMCI. Rhesus monkeys (macaca mulatta) are frequently used as non-human primates to study cognitive decline during normal aging. Macaques of 6 to 8, 10 to 12, and 24 to 26 years of age were tested in two behavioral paradigms: the paired associate learning (PAL), and delayed matching to sample (DMTS) tasks. Results of these tests were reported in a previous study [46], and both paradigms demonstrated cognitive deficits in aged compared to mature primates. As with our observations in mice and Long-Evans rats, decreases in memory capacities of aged macaques were associated with increases in total STEP levels in the synaptosomal fraction of the hippocampus, and proportionally of non-phosphorylated active STEP (one-way ANOVA; Figures 5A and 5B). The phosphorylation of GluN2B and ERK1/2, as well as total level of GluN2B before or after normalization to PSD-95, was decreased in aged impaired monkeys.
Figure 5. STEP level is increased in aged rhesus monkey and human with aMCI.

(A and B) Representative immunoblots and densitometric quantification of total and phosphorylated proteins obtained from synaptosomal fraction of the hippocampus of young adult (6–8 y), mature (10–12 y), and old (24–26 y) rhesus macaques found to have cognitive deficits through previous testing using the paired associate learning (PAL) and delayed matching to sample (DMTS) behavioral paradigms (n=3 in each group) [46]. * or + indicates significant variations compared to young or mature macaques, respectively (one-way ANOVA). (C and D) Changes of protein levels in the synaptosomal fraction of the hippocampus of aMCI (n=11) and control (Ctl) aged non-demented subjects (n=12). # represents differences between Ctl and MCI subjects (unpaired t test). Error bars indicate ± s.e.m. *,+,# P<0.05, **,## P<0.01, ### P<0.001. Correlations between the level of STEP normalized to GAPDH with the amount of soluble amyloid-beta (Aβ) and neurofibrillary tangles (NFTs) found in the hippocampus (E and F), and the MMSE scores (G) of aMCI and control individuals. See also Figure S5.
Lastly, we tested if STEP level was increased in human hippocampus of aMCI individuals (n=11) with no other diagnosed brain disease when compared to age-matched control subjects (n=12) with a clinical diagnosis of no cognitive impairment (Table 1). The mean post-mortem interval in both groups was short (3.7±0.7 h for aMCI vs 3.1±0.1 h for Ctl; Table 1), and not significantly different between the two groups (P>0.05; unpaired t test). Compared to control individuals, aMCI individuals had higher levels of soluble amyloid-beta (Aβ) and neurofibrillary tangles (NFTs) in the hippocampus (P<0.05; unpaired t test; Table 1). Individuals with aMCI also had lower scores in the mini-mental state examination (MMSE) but not in the other cognitive tests (Table 1), suggesting that the MMSE conducted closest to the date of death was the most sensitive test to detect cognitive alterations in this cohort. In these aMCI individuals, the level of STEP and non pSTEP S221 was proportionally higher relative to control, and these events were accompanied with lower levels of pGluN2B, pERK1/2 and PSD-95 (unpaired t test; Figures 5C and 5D).
Table 1. Characteristic of human brain samples.
The diagnosis was performed by pathologists and clinicians at the Alzheimer's Disease Center at the University of Kentucky. Control and aMCI cases were determined based on both clinical and neuropathological evaluations as previously described [2, 64]. Neurofibrillary tangles (NFTs) and total amount of soluble amyloid-beta (Aβ total) were determined in the hippocampus by enzyme-linked immunosorbent assay (ELISA) as previously described [64].
| Characteristic | Units | Control | aMCI |
|---|---|---|---|
| Age | years | 86.9 ± 1.7 | 89.5 ± 1.6 |
|
| |||
| Gender | ♀ | Female n = 7 | Female n = 5 |
| ♂ | Male n = 5 | Male n = 6 | |
|
| |||
| PMI | hours | 3.1 ± 0.1 | 3.7 ± 0.7 |
|
| |||
| Education | years | 16.2 ± 0.7 | 16.6 ± 0.6 |
|
| |||
| NFTs | #/mm2 | 2.5 ± 1.4 | * 12.2 ± 4.0 |
|
| |||
| Aβ total | ρmol/g | 522.0 ± 135.1 | * 2006.8 ± 733.4 |
|
| |||
| MMSE | score | 27.8 ± 0.7 | * 24.4 ± 1.0 |
|
| |||
| ANIMALS | score | 13.1 ± 1.5 | 13.7 ± 1.7 |
|
| |||
| BNT | score | 14.1 ± 0.3 | 13.9 ± 0.4 |
|
| |||
| Cowa | score | 38.3 ± 3.7 | 35.0 ± 4.1 |
|
| |||
| TRAILA | score | 71.3 ± 10.5 | 78.0 ± 12.2 |
|
| |||
| LogMemI | score | 13.2 ± 1.2 | 12.4 ± 1.5 |
PMI, post-mortem interval; MMSE, mini-mental state examination; ANIMALS, animal naming test; BNT, Boston naming test; Cowa, controlled oral word association test; TRAILA = trail making test A; LogMemI, logical memory story A immediate recall.
P < 0.05,
P < 0.01, ± s.e.m.
These results are in line with studies showing a decrease in pGluN2B and PSD-95 [47, 48], whereas other have reported no changes in ERK levels [49]. No changes in protein levels were found in the occipital cortex, a control brain region for aMCI individuals (Supplemental Figure S5). The clinical report associated with each human tissue always contained the MMSE score of the patient (n=23), and in some cases the level of NFTs (n=22) and soluble Aβ (n=13) in the hippocampus. When combining both groups, the total level of STEP was correlated with the amount of soluble Aβ and NFTs detected in the hippocampus (Figures 5E and 5F), and inversely correlated with MMSE scores (Figure 5G). These results indicate that age-related modification in STEP levels parallel cognitive disabilities in non-human primates and humans.
Discussion
The results reported here indicate that STEP levels are increased during aging. Moreover, memory performances were impaired when the expression and activity of STEP are elevated in mice, rats, monkeys, and individuals with aMCI. Genetic reduction of STEP in aged STEP KO and Het mice alleviated age-related memory decline in the MWM and Y-maze tasks. In contrast, overexpression of STEP specifically in the CA1 region of dorsal hippocampus was sufficient to induce memory impairment in hippocampus-dependent behavioral tests (MWM and Y-maze), but not in an amygdala-dependent task (auditory-cued fear conditioning). The STEP inhibitor TC-2153 reversed the cognitive deficits and the tyrosine dephosphorylation of GluN2B and ERK1/2 in mice overexpressing STEP. TC-2153 also alleviated age-related memory deficits in the T-maze task. Based on this functional inhibition of STEP with TC-2153, it would be interesting to study the precise kinetic of TC-2153 biodistribution from the periphery to the central nervous following its systemic injection. In the LOU/C/Jall rat, a model of healthy aging, levels of STEP, GluN2B, pGluN2B and pERK1/2 were stable in young, mature and old animals. In aMCI individuals, higher STEP levels correlated with poorer cognitive scores in the MMSE, and with increased levels of soluble Aβ and NFTs in the hippocampus.
Increased levels of active STEP in aged animals with memory deficits resulted in a significant dephosphorylation and loss of GluN2B-containing NMDARs at the synapse and inactivation of ERK1/2. Previous studies indicate that decreased hippocampal GluN2B levels are associated with memory decline during aging [15-18]. Although many studies have looked at the implication of the GluN2B subunit on memory process, it is still unclear how exactly GluN2B-containing NMDA receptors are related to learning and memory (for review see [50]). Our data suggest that the elevation of STEP activity in aged brains may contribute to the cognitive deficits by decreasing the total level of GluN2B-containing NMDARs at the synapse. These results do not exclude the possibility that other substrates of STEP could also be affected in the context of age-related memory decline.
The direct functional role of increased STEP expression on the dephosphorylation of GluN2B and ERK1/2 was confirmed by overexpression of STEP activity in young mice, while STEP inhibition with TC-2153 compound restored phosphorylation of both proteins to control basal level. During aging, a subpopulation of some rat strains develops memory impairments in the MWM task, such as Long-Evans, Sprague-Dawley and Fisher 344 rats. In contrast, aged LOU/C/Jall rats showed fewer cognitive deficits with aging and perform similarly to young rats in various memory tasks as reported here and previously [44, 45]. There were stable levels of STEP and unaltered phosphorylation of STEP substrates GluN2B and ERK1/2 in hippocampus from aged LOU/C/Jall rats. These results are in line with previous reports showing no alteration of total GluN2B levels in the hippocampus of 24mo old LOU/C/Jall rats [44, 45].
The UPS regulates a wide variety of cellular processes, including synaptic plasticity and memory formation [51, 52]. Compelling evidences have pinpointed the vulnerability of the UPS in the hippocampus during aging [53-57]. A previous study demonstrated that synaptic NMDAR activation promotes ubiquitination and degradation of STEP in normal condition [10]. Conversely, the level of polyubiquitinated STEP was increased in 12 mo old AD mice (Tg2576) compared to age-matched WT mice due to a disruption of the UPS [58].
In line with these data, the current study suggests that disruption of the UPS lowers STEP degradation, leading to an accumulation of active STEP levels in aged-memory impaired animals. Since UPS dysfunctions can also impact the accumulation of other proteins, it cannot be excluded that decrease degradation of additional proteins might also participate in age-related cognitive impairment. This increased level of STEP during aging might also favor its dimerization and inactivation as observed in a previous study [59]. Since the functional role of these dimers is still unclear, one could argue that STEP dimerization is a protective mechanism to attenuate the deleterious effect of STEP when it accumulates during aging. Because these dimers are not found in the synaptosomal (P2) membrane fraction used here [59], further studies will be necessary to uncover the impact of STEP dimerization on memory.
One aspect emerging from research performed on STEP is that its level is up-regulated in some neuropsychiatric diseases (AD, schizophrenia, Fragile × syndrome) or down-regulated in others (Huntington's disease, stroke/ischemia, pain) [5]. Thus, some diseases could benefit from an increase in STEP activity, whereas others might benefit from STEP inhibition. One could also expect that large reductions in STEP activity in brain regions where STEP is highly expressed such as the striatum, could have adverse effects in illnesses affecting these specific brain regions.
This is the first study showing an involvement of the phosphatase STEP in age-associated memory deficits. MCI is a diagnostic entity distinct from any other dementia that might, in some cases, be a precursor of AD or other neurodegenerative disorders [1-4, 60]. Thus, our findings might be relevant to on-going AD research where many of the clinical trials underway are recruiting aMCI after unsuccessful trials in mild-to-moderate AD patients. Our results suggest that STEP elevation occurs at early stages of cognitive deficits in aMCI individuals, instead of being an outcome of more advanced AD pathology as shown previously [58]. Since synapse dysfunction is an early event in AD and the best correlate of cognitive decline associated with the illness [61-63], drugs targeting molecules that oppose synaptic strengthening such as STEP might lead to new disease modifying therapies effective at the preclinical stages of AD, before neurodegeneration occurs and produces irreversible damage to the brain. Thus, in future clinical trials it could be of interest to determine if treatment with STEP inhibitors would have beneficial effects in aMCI individuals.
In summary, STEP activity rises with aging and increased STEP expression could be viewed as a molecular mechanism that participates in at least some aspects of the cognitive declines that can occur during aging. Since this novel role of STEP applies to rodents, non-human primates and humans, it justifies the need of additional studies to validate if STEP is a promising target for drug development to enhance cognitive performances in older adults and MCI individuals.
Contact For Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jonathan Brouillette (jonathan.brouillette@umontreal.ca).
Experimental Model and Subject Details
Animal models
Aged (22-24 mo old) and young adult (6 mo old) wild-type (WT) C57BL/6J male mice were purchased from Jackson Laboratory (Bar Harbor, ME). STEP KO, STEP Het and WT male littermate mice were maintained on a C57BL/6J background and generated at Yale University from heterozygous animals backcrossed for at least nine generations. Mice were group housed (3-5 per cage). Long-Evans male rats were purchased from Charles River Laboratories (St-Constant, QC). Young (6 mo), mature (12 mo), and old (24 mo) LOU/C/Jall rats were obtained from the Quebec Network for Research on Aging. Rats were group housed (2-3 per cage). Aging animals were monitored on a weekly basis by a veterinarian and/or qualified technicians for weight loss, obesity, abscesses, cataracts, sebaceous cysts, tumours, and malocclusions. Young adult (6-8 y), mature (10-12 y), and old (24-26 y) rhesus macaques (macaca mulatta) were housed in individual primate cages in an AAALAC-accredited facility. Rhesus macaques care was supervised by veterinarians skilled in the healthcare and maintenance of non-human primates. The experimenter was blind to monkey ages when performing western blots for the present study. Animals were kept in a climate-controlled facility under a 12 h light-dark cycle with ad libitum access to food and water. Each animal model used here were tested during the light phase of the cycle, in the same animal facility, and under the same behavioral conditions. The experimenter was blind to treatment or animal genotype for all behavioral testing.
All procedures were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at Yale University. All procedures conducted at Research Center of the Douglas Mental Health University Institute, Centre Hospitalier de l'Université de Montréal, or Hôpital du Sacré-Coeur de Montréal were approved by their respective Animal Care Committee in compliance with the Canadian Council for Animal Care.
Human brain tissues
Brain specimens from aMCI and age-matched control non-demented subjects were generously provided by the Alzheimer's Disease Center at the University of Kentucky. The diagnosis for aMCI and control subjects were performed as previously described [64]. Briefly, control and aMCI cases were determined based on both clinical and neuropathological evaluations [2]. Standardized mental status testing, neurologic and physical examinations were performed annually or biannually. Each participant was followed for at least 2 years (many for more than10 years) before death, and had no history of substance abuse, head injury, encephalitis, meningitis, epilepsy, stroke/transient ischemic attack, chronic infectious disease, or any other major psychiatric illness that limited cognitive performance or influenced affect. The MMSE conducted closest to the date of death was the most consistently available cognitive measure across all patients and was used as an indicator of overall cognitive status. All control subjects were at Braak stage lower than three and typically scored 28 or higher on the MMSE. Control and aMCI subjects were primarily selected based on the shortest PMIs that were available at the brain bank of the Alzheimer's Disease Center at the University of Kentucky to avoid as much as possible protein degradation that might occur during PMI. Further details are provided in Table 1.
Method Details
Morris water maze
The Morris water maze (MWM) was conducted as previously described [35, 36]. Mice and Long-Evans rats were trained to swim in a 1.4 m diameter pool to find a 14 cm diameter submerged platform located 1 cm below the surface of water (24°C), rendered opaque by the addition of non-allergenic white gouache paint. Rodents were pseudo-randomly started from a different position at each trial and used distal visuo-spatial cues to find the hidden escape platform that remained in the center of the same quadrant throughout all acquisition days (3 trials per day). Acquisition measures included escape latency and path length to reach the platform. When animals failed to find the platform, they were guided to it and remained there for 10 s before removal. Twenty-four hours after the acquisition phase, the platform was removed, and a probe trial of 90 s was given to evaluate swim speed, platform location crossings, and time spent in the target quadrant (previously containing the escape platform) and the opposite quadrant. To assess visual deficits and motivation to escape from water, the probe test was followed by a cued task (60 s; three trials per animal) during which the platform was visible. The visible platform was moved to different locations between each trial. After each trial, animals were immediately placed under a warming lamp to dry and prevent hypothermia. Aged animals falling between 0.5 and 2.0 standard deviations of the performance of young rodents were not used in any further tests in this study. Behavioral data from acquisition trials, probe and cued tests were acquired and analyzed using the AnyMaze automated tracking system (Stoelting, IL, USA).
Y-maze
Mice were tested for short-term, hippocampus-dependent spatial memory using a two trials Y-maze task as described [65]. Briefly, experiments were conducted with an ambient light level of 6 lux. To eliminate olfactory cues, the floor of the maze was covered with bedding that was mixed after each trial and the maze was wipe off with 70% alcohol between each trial. Various extra-maze cues were placed on the surrounding walls. During the training phase, mice had 5 min to freely explore two arms (the “start arm” and the “other arm”), without access to the third one (the “new arm”) closed by an opaque door. Mice were then removed from the maze and returned to their home cage for 5 min. During the testing phase, mice could freely explore all three arms for 5 min. The trials were started when the mouse reached the center of the maze and the time spent in each arm was recorded and analyzed using the AnyMaze automated tracking system. Discriminating the new (previously blocked) arm from the two familiar arms was considered as an index of spatial recognition memory performance.
T-maze
The T-maze tests were conducted as previously described [66]. Briefly, animals were fully habituated to the T-maze and to eating food rewards in the maze (criterion of eating 10 rewards in 8 min for two consecutive days). Subsequently, rats were habituated to handling procedures on the maze (criterion of eating 10 food rewards in 5 min while being picked up five times for two consecutive days). The animal was picked up immediately after eating the reward (or picked up with no reward if the incorrect arm is chosen) and placed in the start box between trials. The choice point of the maze was wiped with alcohol to prevent olfactory cues from guiding behavior. Each daily test session consisted of 10 trials. For the assessment of the effects of TC-2153 on delayed alternation performance, delays were adjusted between 0 and 15 s to maintain performance between 60-70% of correct choices, thus allowing room for either improvement or impairment with drug treatment. The delay was increased in 5 s intervals if a rat performed at ∼90% for two consecutive testing sessions or ∼80% for three consecutive testing sessions. Thus, the delay needed to maintain baseline performance at 60-70% correct can be used as a measure of general performance on the task. The animals were also given a week of wash out between various drug dosages, which also accounts for the time they need to achieve their baseline for the next drug injection.
Auditory-cued fear conditioning
AAV-STEP and AAV-GFP mice were tested for amygdala-dependent auditory-cued fear conditioning Experiments were conducted with an ambient 6 lux light. During the training phase, mice were placed in a chamber (30cm × 30cm × 30cm) with white walls and stainless-steel grid floor (context 1). After freely exploring the chamber for 3 min, a tone (Conditioned stimulus (CS), 30s, 10kHz, 75dB SPL, pulsed 5Hz) immediately followed by a footshock (Unconditioned stimulus (US), 0.7 mA, 2 s, constant current) was delivered through the grid floor. The fear conditioning chamber was thoroughly cleaned with 70% ethanol between each mouse. Twenty-four hours following the training phase, each mouse was placed in a novel context (same size chamber with black walls and a plain black floor cleaned with 1% acetic acid). Mice were exposed to this novel context for 3 min (pre-CS phase) followed by a 3 min period of auditory stimulation (CS phase). Freezing and mean activity was recorded using Smart 3.0 software (Harvard Apparatus, MA, USA).
Subcellular fractionation and western blot analysis
Dorsal hippocampi dissected from all species (mouse, rat, monkey, human) were homogenized in a solution containing 0.32 M sucrose, 20 mM HEPES (pH 7.4), 1 mM EDTA, 5 mM NaF, 1 mM sodium vanadate, and protease inhibitor cocktail (Roche, Nutley, NJ), and processed as described [67]. In brief, homogenates were centrifuged at 2800 rpm for 10 min at 4°C. The pellet (nuclear fraction) contains nuclei and large cell debris. The supernatant was centrifuged at 12000 rpm for 10 min at 4°C. The cytosolic fraction (supernatant) was removed and the crude synaptosomal (P2) fraction (pellet) was resuspended and sonicated in protein RIPA lysis and extraction buffer (Pierce, Thermofisher, IL) containing 5 mM NaF, 1 mM sodium vanadate, and protease inhibitor cocktail (Roche). The enrichment of synaptic proteins in the P2 fraction was confirmed by western blots (Supplemental Fig. S2). The total protein content was determined using a BCA protein assay kit (Thermo Scientific). The levels of each protein were tested in the fraction where it is the most concentrate to obtain a more sensible signal. Eight different antibodies were tested in the synaptosomal (P2) fraction: STEP, non-phosphorylated STEP S221, GluN2B, pGluN2B Y1472, ERK1/2, pERK1/2 Y204/187, PSD-95, GAPDH. Three antibodies were tested in the nucleus fraction (CREB, pCREB S133, GAPDH), and two in the cytosolic fraction (BDNF, GAPDH). Western blots were performed as described [65], and membranes were quantified with Image J 1.33 (National Institutes of Health).
Ubiquitinated protein pull-down
Polyubiquitinated STEP was isolated from P2 hippocampal fractions using Agarose-TUBE2 (Tandem Ubiquitin Binding Entity, LifeSensors, PA) affinity chromatography according to the manufacturer's protocol. Eluted polyubiquitinated proteins were subjected to western blots probed with anti-STEP antibody (23E5).
STEP immunoprecipitation and activity
STEP was immunoprecipitated from P2 hippocampal fractions with anti-STEP antibody (23E5; 10 μL) and A/G PLUS-Agarose beads according to the manufacturer's protocol (Santa Cruz, CA). The STEP-bead complex was resuspended in the phosphatase assay buffer (in mM): 25 HEPES pH 7.3, 5 EDTA, 10 DTT. STEP activity was measured with a GST-GluN2B construct containing the amino acid sequence 1361-1482 phosphorylated at Y1472 by Fyn (Upstate Biotechnology, NY) as described [58].
Immunofluorescence
Immunofluorescence was performed on coronal brain section (16 μm thick) fixed with 4% formaldehyde and mounted on Fisherbrand superfrost plus slides (Fisher Scientific) according to the protocol established by Cell Signaling Technology for STEP (23E5: 1:100) and GFP (D5.1: 1:100) antibodies using Alexa Fluor 488 conjugated secondary antibodies (1:1000) and prolong gold antifade reagent with DAPI (Cell Signaling). Immunofluorescence was visualized using a Zeiss Axio Observer.Z1 microscope.
Adeno-associated virus vector constructs and microinjections
Recombinant adeno-associated virus (AAV) serotype 1 vectors were constructed and amplified by the Penn Vector Core (Philadelphia, PA). The transgene for mouse STEP and the reporter gene, enhanced green florescent protein (EGFP), were expressed into an AAV backbone containing the cytomegalovirus enhancer/chicken β-actin promoter, the woodchuck post-transcriptional regulatory element (WPRE), and the recombinant bovine growth hormone (AAV-STEP and AAV-EGFP). Titer of AAV-STEP was 5.12e12 genome copies per mL (GC/mL) and titer of AAV-EGFP was 7.36e12 GC/ml. For AAV vector microinjections, mice were anaesthetized with ketamine/xylazine (75/10 mg/kg, i.p.) and maintained with 2% isoflurane. To limit vector diffusion, microinjections were performed at a slow rate of 20 nL/min for 50 min using a 10 μl Hamilton syringe with a 30 gauge blunted-tip needle (Hamilton Company) and a dual-piston pump (CMA 402, Harvard Apparatus, MA). Bilateral viral infusion (1 μL) were performed in the CA1 region of the dorsal hippocampus (-2.3 mm AP, ±1.5 mm ML, -1.4 mm DV from bregma according to Paxinos and Franklin mouse brain atlas). Behavioral analysis was started 4 weeks after vector infusion when transgene protein expression had peaked to remain at stable levels [68]. Transgene expression was observed at least 2 mm on either side of the injection site in the dorsal hippocampus as previously reported [69]. Gene transfer and expression of the vectors into the CA1 region in dorsal hippocampus was confirmed by immunofluorescence, real-time quantitative RT-PCR, and western blots (Figure 2).
Real-time quantitative RT-PCR
Real-time quantitative RT-PCR (qRT-PCR) was performed with TaqMan reagents using the Applied Biosystems Real-Time PCR System according to the manufacturer's protocol (Applied Biosystems, Foster City, CA) using STEP gene-specific primers (ThermoFisher Scientific). Conditions of the PCR reaction: 48°C for 30 min, 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60° C for 1 min. All the reactions were performed in triplicate.
Quantification and Statistical Analysis
Statistical analysis of multiple samples was carried out using one-, two-, or three-way analysis of variance (ANOVA) as indicated in the text and figure legends. Simple main effects were analyzed using the F-test when the interaction was significant. Tukey's Honestly Significant Difference (HSD) test was used for post-hoc pair-wise comparisons when necessary. The statistical significance between two groups was analyzed using two-tailed Student's t test. The alpha level used was P<0.05.
Supplementary Material
Highlights.
- Increased STEP levels in aged memory-impaired mice, rats, monkeys and aMCI people.
- Genetic and pharmacological reduction of STEP improved memory performance in rodents.
- Viral-mediated STEP overexpression in the hippocampus induced memory impairments.
Acknowledgments
We thank laboratory members for helpful discussions and critical reading of the manuscript. We thank Jian Xu for providing the GST-GluN2B construct and Mitchell Powell for technical assistance. We thank Dr. Marilee Ogren for critical reading of the manuscript. Thank you also to Dr. Erin Abner at the Sanders-Brown Center on Aging for assistance with human case histories. This work was funded by FRQS and Hôpital du Sacré-Coeur de Montréal (HSCM) grant (J.B), NIH grants MH091037 and MH052711 (P.J.L.), AG047270 (A.C.N.), and AG024190, AG027297, AG028383, and Center's ADC grant P30AG028383 (C.M.N.). This research was supported by an unrestricted grant from Motac Holding, Inc. (J.S.S. and E.B), and a CNRS and ANR-10-MALZ-002 grants (B.B.). This project was funded by grants from the Canadian Institutes of Health Research (CIHR) (R.Q.), and the Quebec Network for Research on Aging (a network funded by Fonds de Recherche du Québec – Santé) (P.G. and G.F.). C.M. holds a postdoctoral fellowship from CIHR.
Footnotes
Author Contributions: J.B. designed the study, carried out the experiments, interpreted results and wrote the manuscript. D.C. and J.D.G performed immunofluorescence, western blots, and behavioral testing. C.M. carried out the MWM and the behavioral analysis for the Long-Evans and LOU/C/Jall rats. M.C. and J.B. performed the T-maze task. R.Q. provided the Long-Evans rats, and P.G. and G. F. the LOU/C/Jall rats. E.B., J.S.S. and B.B. provided the rhesus monkey tissues. C.M.N. provided the post-mortem MCI and control samples obtained from the neuropathology core of the Alzheimer's Disease Center (ADC) at the University of Kentucky. P.J.L., P.G., E.B., G.F., C.M.N., J.S.S., B.B., R.Q., A.C.N., and A.F.T.A. supervised the experiments and participated in study conception and manuscript writing. All authors discussed the results and commented on the manuscript.
Declaration of Interest: The authors declare no competing interests.
The authors declare no biomedical financial interests or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mufson EJ, Binder L, Counts SE, DeKosky ST, de Toledo-Morrell L, Ginsberg SD, Ikonomovic MD, Perez SE, Scheff SW. Mild cognitive impairment: pathology and mechanisms. Acta Neuropathol. 2012;123:13–30. doi: 10.1007/s00401-011-0884-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petersen RC, Parisi JE, Dickson DW, Johnson KA, Knopman DS, Boeve BF, Jicha GA, Ivnik RJ, Smith GE, Tangalos EG, et al. Neuropathologic features of amnestic mild cognitive impairment. Arch Neurol. 2006;63:665–672. doi: 10.1001/archneur.63.5.665. [DOI] [PubMed] [Google Scholar]
- 3.Cloutier S, Chertkow H, Kergoat MJ, Gauthier S, Belleville S. Patterns of Cognitive Decline Prior to Dementia in Persons with Mild Cognitive Impairment. J Alzheimers Dis. 2015;47:901–913. doi: 10.3233/JAD-142910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grundman M, Petersen RC, Ferris SH, Thomas RG, Aisen PS, Bennett DA, Foster NL, Jack CR, Jr, Galasko DR, Doody R, et al. Mild cognitive impairment can be distinguished from Alzheimer disease and normal aging for clinical trials. Arch Neurol. 2004;61:59–66. doi: 10.1001/archneur.61.1.59. [DOI] [PubMed] [Google Scholar]
- 5.Goebel-Goody SM, Baum M, Paspalas CD, Fernandez SM, Carty NC, Kurup P, Lombroso PJ. Therapeutic implications for striatal-enriched protein tyrosine phosphatase (STEP) in neuropsychiatric disorders. Pharmacol Rev. 2012;64:65–87. doi: 10.1124/pr.110.003053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Braithwaite SP, Paul S, Nairn AC, Lombroso PJ. Synaptic plasticity: one STEP at a time. Trends Neurosci. 2006;29:452–458. doi: 10.1016/j.tins.2006.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lombroso PJ, Murdoch G, Lerner M. Molecular characterization of a protein-tyrosine-phosphatase enriched in striatum. Proc Natl Acad Sci USA. 1991;88:7242–7246. doi: 10.1073/pnas.88.16.7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boulanger LM, Lombroso PJ, Raghunathan A, During MJ, Wahle P, Naegele JR. Cellular and molecular characterization of a brain-enriched protein tyrosine phosphatase. J Neurosci. 1995;15:1532–1544. doi: 10.1523/JNEUROSCI.15-02-01532.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- 10.Xu J, Kurup P, Zhang Y, Goebel-Goody SM, Wu PH, Hawasli AH, Baum ML, Bibb JA, Lombroso PJ. Extrasynaptic NMDA receptors couple preferentially to excitotoxicity via calpain-mediated cleavage of STEP. J Neurosci. 2009;29:9330–9343. doi: 10.1523/JNEUROSCI.2212-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y, Venkitaramani DV, Gladding CM, Zhang Y, Kurup P, Molnar E, Collingridge GL, Lombroso PJ. The tyrosine phosphatase STEP mediates AMPA receptor endocytosis after metabotropic glutamate receptor stimulation. J Neurosci. 2008;28:10561–10566. doi: 10.1523/JNEUROSCI.2666-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nguyen TH, Liu J, Lombroso PJ. Striatal enriched phosphatase 61 dephosphorylates Fyn at phosphotyrosine 420. J Biol Chem. 2002;277:24274–24279. doi: 10.1074/jbc.M111683200. [DOI] [PubMed] [Google Scholar]
- 13.Xu J, Kurup P, Bartos JA, Patriarchi T, Hell JW, Lombroso PJ. Striatal-enriched protein-tyrosine phosphatase (STEP) regulates Pyk2 kinase activity. J Biol Chem. 2012;287:20942–20956. doi: 10.1074/jbc.M112.368654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paul S, Nairn AC, Wang P, Lombroso PJ. NMDA-mediated activation of the tyrosine phosphatase STEP regulates the duration of ERK signaling. Nat Neurosci. 2003;6:34–42. doi: 10.1038/nn989. [DOI] [PubMed] [Google Scholar]
- 15.Burke SN, Barnes CA. Neural plasticity in the ageing brain. Nat Rev Neurosci. 2006;7:30–40. doi: 10.1038/nrn1809. [DOI] [PubMed] [Google Scholar]
- 16.Clayton DA, Browning MD. Deficits in the expression of the NR2B subunit in the hippocampus of aged Fisher 344 rats. Neurobiol Aging. 2001;22:165–168. doi: 10.1016/s0197-4580(00)00196-2. [DOI] [PubMed] [Google Scholar]
- 17.Clayton DA, Mesches MH, Alvarez E, Bickford PC, Browning MD. A hippocampal NR2B deficit can mimic age-related changes in long-term potentiation and spatial learning in the Fischer 344 rat. J Neurosci. 2002;22:3628–3637. doi: 10.1523/JNEUROSCI.22-09-03628.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang Z, Krause M, Rao G, McNaughton BL, Barnes CA. Synaptic commitment: developmentally regulated reciprocal changes in hippocampal granule cell NMDA and AMPA receptors over the lifespan. J Neurophysiol. 2008;99:2760–2768. doi: 10.1152/jn.01276.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cao X, Cui Z, Feng R, Tang YP, Qin Z, Mei B, Tsien JZ. Maintenance of superior learning and memory function in NR2B transgenic mice during ageing. Eur J Neurosci. 2007;25:1815–1822. doi: 10.1111/j.1460-9568.2007.05431.x. [DOI] [PubMed] [Google Scholar]
- 20.Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M, Liu G, Tsien JZ. Genetic enhancement of learning and memory in mice. Nature. 1999;401:63–69. doi: 10.1038/43432. [DOI] [PubMed] [Google Scholar]
- 21.von Engelhardt J, Doganci B, Jensen V, Hvalby O, Gongrich C, Taylor A, Barkus C, Sanderson DJ, Rawlins JN, Seeburg PH, et al. Contribution of hippocampal and extra-hippocampal NR2B-containing NMDA receptors to performance on spatial learning tasks. Neuron. 2008;60:846–860. doi: 10.1016/j.neuron.2008.09.039. [DOI] [PubMed] [Google Scholar]
- 22.Gazzaley AH, Siegel SJ, Kordower JH, Mufson EJ, Morrison JH. Circuit-specific alterations of N-methyl-D-aspartate receptor subunit 1 in the dentate gyrus of aged monkeys. Proc Natl Acad Sci USA. 1996;93:3121–3125. doi: 10.1073/pnas.93.7.3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang M, Yang Y, Wang CJ, Gamo NJ, Jin LE, Mazer JA, Morrison JH, Wang XJ, Arnsten AF. NMDA receptors subserve persistent neuronal firing during working memory in dorsolateral prefrontal cortex. Neuron. 2013;77:736–749. doi: 10.1016/j.neuron.2012.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morrison JH, Baxter MG. The ageing cortical synapse: hallmarks and implications for cognitive decline. Nat Rev Neurosci. 2012;13:240–250. doi: 10.1038/nrn3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang M, Gamo NJ, Yang Y, Jin LE, Wang XJ, Laubach M, Mazer JA, Lee D, Arnsten AF. Neuronal basis of age-related working memory decline. Nature. 2011;476:210–213. doi: 10.1038/nature10243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rapp PR, Amaral DG. Evidence for task-dependent memory dysfunction in the aged monkey. J Neurosci. 1989;9:3568–3576. doi: 10.1523/JNEUROSCI.09-10-03568.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paul S, Nairn AC, Wang P, Lombroso PJ. NMDA-mediated activation of the tyrosine phosphatase STEP regulates the duration of ERK signaling. Nat Neurosci. 2003;6:34–42. doi: 10.1038/nn989. [DOI] [PubMed] [Google Scholar]
- 28.Thomas GM, Huganir RL. MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci. 2004;5:173–183. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- 29.Zhen X, Uryu K, Cai G, Johnson GP, Friedman E. Age-associated impairment in brain MAPK signal pathways and the effect of caloric restriction in Fischer 344 rats. J Gerontol A Biol Sci Med Sci. 1999;54:B539–548. doi: 10.1093/gerona/54.12.b539. [DOI] [PubMed] [Google Scholar]
- 30.Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell. 1994;79:59–68. doi: 10.1016/0092-8674(94)90400-6. [DOI] [PubMed] [Google Scholar]
- 31.Bramham CR, Messaoudi E. BDNF function in adult synaptic plasticity: the synaptic consolidation hypothesis. Prog Neurobiol. 2005;76:99–125. doi: 10.1016/j.pneurobio.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 32.Xu J, Kurup P, Baguley TD, Foscue E, Ellman JA, Nairn AC, Lombroso PJ. Inhibition of the tyrosine phosphatase STEP61 restores BDNF expression and reverses motor and cognitive deficits in phencyclidine-treated mice. Cell Mol Life Sci. 2016;73:1503–1514. doi: 10.1007/s00018-015-2057-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Menard C, Quirion R. Successful cognitive aging in rats: a role for mGluR5 glutamate receptors, homer 1 proteins and downstream signaling pathways. PloS one. 2012;7:e28666. doi: 10.1371/journal.pone.0028666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 35.Brouillette J, Quirion R. Transthyretin: a key gene involved in the maintenance of memory capacities during aging. Neurobiol Aging. 2008;29:1721–1732. doi: 10.1016/j.neurobiolaging.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 36.Quirion R, Wilson A, Rowe W, Aubert I, Richard J, Doods H, Parent A, White N, Meaney MJ. Facilitation of acetylcholine release and cognitive performance by an M(2)-muscarinic receptor antagonist in aged memory-impaired. J Neurosci. 1995;15:1455–1462. doi: 10.1523/JNEUROSCI.15-02-01455.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aubert I, Rowe W, Meaney MJ, Gauthier S, Quirion R. Cholinergic markers in aged cognitively impaired Long-Evans rats. Neuroscience. 1995;67:277–292. doi: 10.1016/0306-4522(95)00056-o. [DOI] [PubMed] [Google Scholar]
- 38.Morrison JH, Hof PR. Life and death of neurons in the aging brain. Science. 1997;278:412–419. doi: 10.1126/science.278.5337.412. [DOI] [PubMed] [Google Scholar]
- 39.VanGuilder HD, Yan H, Farley JA, Sonntag WE, Freeman WM. Aging alters the expression of neurotransmission-regulating proteins in the hippocampal synaptoproteome. J Neurochem. 2010;113:1577–1588. doi: 10.1111/j.1471-4159.2010.06719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu J, Chatterjee M, Baguley TD, Brouillette J, Kurup P, Ghosh D, Kanyo J, Zhang Y, Seyb K, Ononenyi C, et al. Inhibitor of the tyrosine phosphatase STEP reverses cognitive deficits in a mouse model of Alzheimer's disease. PLoS Biol. 2014;12:e1001923. doi: 10.1371/journal.pbio.1001923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim JJ, Fanselow MS. Modality-specific retrograde amnesia of fear. Science. 1992;256:675–677. doi: 10.1126/science.1585183. [DOI] [PubMed] [Google Scholar]
- 42.Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci. 1992;106:274–285. doi: 10.1037//0735-7044.106.2.274. [DOI] [PubMed] [Google Scholar]
- 43.Alliot J, Boghossian S, Jourdan D, Veyrat-Durebex C, Pickering G, Meynial-Denis D, Gaumet N. The LOU/c/jall rat as an animal model of healthy aging? J Gerontol A Biol Sci Med Sci. 2002;57:B312–320. doi: 10.1093/gerona/57.8.b312. [DOI] [PubMed] [Google Scholar]
- 44.Kollen M, Stephan A, Faivre-Bauman A, Loudes C, Sinet PM, Alliot J, Billard JM, Epelbaum J, Dutar P, Jouvenceau A. Preserved memory capacities in aged Lou/C/Jall rats. Neurobiol Aging. 2010;31:129–142. doi: 10.1016/j.neurobiolaging.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 45.Menard C, Quirion R, Bouchard S, Ferland G, Gaudreau P. Glutamatergic signaling and low prodynorphin expression are associated with intact memory and reduced anxiety in rat models of healthy aging. Front Aging Neurosci. 2014;6:81. doi: 10.3389/fnagi.2014.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schneider JS, Pioli EY, Jianzhong Y, Li Q, Bezard E. Effects of memantine and galantamine on cognitive performance in aged rhesus macaques. Neurobiol Aging. 2013;34:1126–1132. doi: 10.1016/j.neurobiolaging.2012.10.020. [DOI] [PubMed] [Google Scholar]
- 47.Mohmmad Abdul H, Baig I, Levine H, 3rd, Guttmann RP, Norris CM. Proteolysis of calcineurin is increased in human hippocampus during mild cognitive impairment and is stimulated by oligomeric Abeta in primary cell culture. Aging Cell. 2011;10:103–113. doi: 10.1111/j.1474-9726.2010.00645.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sultana R, Banks WA, Butterfield DA. Decreased levels of PSD95 and two associated proteins and increased levels of BCl2 and caspase 3 in hippocampus from subjects with amnestic mild cognitive impairment: Insights into their potential roles for loss of synapses and memory, accumulation of Abeta, and neurodegeneration in a prodromal stage of Alzheimer's disease. J Neurosci Res. 2010;88:469–477. doi: 10.1002/jnr.22227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mufson EJ, He B, Nadeem M, Perez SE, Counts SE, Leurgans S, Fritz J, Lah J, Ginsberg SD, Wuu J, et al. Hippocampal proNGF signaling pathways and beta-amyloid levels in mild cognitive impairment and Alzheimer disease. J Neuropathol Exp Neurol. 2012;71:1018–1029. doi: 10.1097/NEN.0b013e318272caab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shipton OA, Paulsen O. GluN2A and GluN2B subunit-containing NMDA receptors in hippocampal plasticity. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130163. doi: 10.1098/rstb.2013.0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yi JJ, Ehlers MD. Emerging roles for ubiquitin and protein degradation in neuronal function. Pharmacol Rev. 2007;59:14–39. doi: 10.1124/pr.59.1.4. [DOI] [PubMed] [Google Scholar]
- 52.Lip PZ, Demasi M, Bonatto D. The role of the ubiquitin proteasome system in the memory process. Neurochem Int. 2017;102:57–65. doi: 10.1016/j.neuint.2016.11.013. [DOI] [PubMed] [Google Scholar]
- 53.Paz Gavilan M, Vela J, Castano A, Ramos B, del Rio JC, Vitorica J, Ruano D. Cellular environment facilitates protein accumulation in aged rat hippocampus. Neurobiol Aging. 2006;27:973–982. doi: 10.1016/j.neurobiolaging.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 54.Figueiredo LS, de Freitas BS, Garcia VA, Dargel VA, Kobe LM, Kist LW, Bogo MR, Schroder N. Iron Loading Selectively Increases Hippocampal Levels of Ubiquitinated Proteins and Impairs Hippocampus-Dependent Memory. Mol Neurobiol. 2016;53:6228–6239. doi: 10.1007/s12035-015-9514-6. [DOI] [PubMed] [Google Scholar]
- 55.Keller JN, Hanni KB, Markesbery WR. Possible involvement of proteasome inhibition in aging: implications for oxidative stress. Mech Ageing Dev. 2000;113:61–70. doi: 10.1016/s0047-6374(99)00101-3. [DOI] [PubMed] [Google Scholar]
- 56.Keller JN, Gee J, Ding Q. The proteasome in brain aging. Ageing Res Rev. 2002;1:279–293. doi: 10.1016/s1568-1637(01)00006-x. [DOI] [PubMed] [Google Scholar]
- 57.Ding Q, Cecarini V, Keller JN. Interplay between protein synthesis and degradation in the CNS: physiological and pathological implications. Trends Neurosci. 2007;30:31–36. doi: 10.1016/j.tins.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 58.Kurup P, Zhang Y, Xu J, Venkitaramani DV, Haroutunian V, Greengard P, Nairn AC, Lombroso PJ. Abeta-mediated NMDA receptor endocytosis in Alzheimer's disease involves ubiquitination of the tyrosine phosphatase STEP61. J Neurosci. 2010;30:5948–5957. doi: 10.1523/JNEUROSCI.0157-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rajagopal S, Deb I, Poddar R, Paul S. Aging is associated with dimerization and inactivation of the brain-enriched tyrosine phosphatase STEP. Neurobiol Aging. 2016;41:25–38. doi: 10.1016/j.neurobiolaging.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Petersen RC. Mild cognitive impairment as a diagnostic entity. J Int Med. 2004;256:183–194. doi: 10.1111/j.1365-2796.2004.01388.x. [DOI] [PubMed] [Google Scholar]
- 61.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 62.Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 63.Coleman PD, Yao PJ. Synaptic slaughter in Alzheimer's disease. Neurobiol Aging. 2003;24:1023–1027. doi: 10.1016/j.neurobiolaging.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 64.Abdul HM, Sama MA, Furman JL, Mathis DM, Beckett TL, Weidner AM, Patel ES, Baig I, Murphy MP, LeVine H, 3rd, et al. Cognitive decline in Alzheimer's disease is associated with selective changes in calcineurin/NFAT signaling. J Neurosci. 2009;29:12957–12969. doi: 10.1523/JNEUROSCI.1064-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brouillette J, Caillierez R, Zommer N, Alves-Pires C, Benilova I, Blum D, De Strooper B, Buee L. Neurotoxicity and memory deficits induced by soluble low-molecular-weight amyloid-beta1-42 oligomers are revealed in vivo by using a novel animal model. J Neurosci. 2012;32:7852–7861. doi: 10.1523/JNEUROSCI.5901-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Franowicz JS, Kessler LE, Borja CM, Kobilka BK, Limbird LE, Arnsten AF. Mutation of the alpha2A-adrenoceptor impairs working memory performance and annuls cognitive enhancement by guanfacine. J Neurosci. 2002;22:8771–8777. doi: 10.1523/JNEUROSCI.22-19-08771.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, Li XY, Aghajanian G, Duman RS. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329:959–964. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.During MJ, Young D, Baer K, Lawlor P, Klugmann M. Development and optimization of adeno-associated virus vector transfer into the central nervous system. Methods Mol Med. 2003;76:221–236. doi: 10.1385/1-59259-304-6:221. [DOI] [PubMed] [Google Scholar]
- 69.Klugmann M, Wymond SC, Leichtlein CB, Klaussner BK, Dunning J, Fong D, Young D, During MJ. AAV-mediated hippocampal expression of short and long Homer 1 proteins differentially affect cognition and seizure activity in adult rats. Mol Cell Neurosci. 2005;28:347–360. doi: 10.1016/j.mcn.2004.10.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.