

Abstract
The Hippo signaling pathway and the Notch signaling pathway are evolutionary conserved signaling cascades that have important roles in embryonic development of many organs. In murine liver, disruption of either pathway impairs intrahepatic bile duct development. Recent studies suggested that the Notch signaling receptor Notch2 is a direct transcriptional target of the Hippo signaling pathway effector YAP, and the Notch signaling is a major mediator of the Hippo signaling in maintaining biliary cell characteristics in adult mice. However, it remains to be determined whether the Hippo signaling pathway functions through the Notch signaling in intrahepatic bile duct development. We found that loss of the Hippo signaling pathway tumor suppressor Nf2 resulted in increased expression levels of the Notch signaling pathway receptor Notch2 in cholangiocytes but not in hepatocytes. When knocking down Notch2 on the background of Nf2 deficiency in mouse livers, the excessive bile duct development induced by Nf2 deficiency was suppressed by heterozygous and homozygous deletion of Notch2 in a dose-dependent manner. These results implicated that Notch signaling is one of the downstream effectors of the Hippo signaling pathway in regulating intrahepatic bile duct development.
A major function of the mammalian liver is to produce bile, which is essential for the digestion of lipids in the small intestine. Bile is secreted by hepatocytes, then collected by cholangiocyte-lined intrahepatic bile ducts, and then transported to the duodenum through the extrahepatic bile ducts. Defects in bile duct development and homeostasis impair the flow of bile, which will damage the liver parenchyma and result in a group of both genetic and acquired biliary tract disorders termed cholangiopathies.1,2
Intrahepatic bile duct development (IHBD) starts around E15.5 in the mouse liver when hepatoblasts start to differentiate into biliary precursor cells that form a single cell layer surrounding the portal vein, termed the ductal plate.3,4 Before birth, primitive ductal structures form at discrete points along the ductal plates. During the first 2 weeks of life, the primitive ductal structures incorporate into portal mesenchyme to become mature bile ducts as biliary precursor cells differentiate into mature cholangiocytes correspondingly, while the remaining ductal plates regress and/or differentiate into periportal hepatocytes.5 Several signaling pathways including the Notch signaling, the Hippo signaling, and the ONECUT1/HNF1β transcription cascade have been shown to be critical for bile duct development.6–9
The Hippo signaling pathway is a highly conserved signaling module.10,11 The core Hippo signaling pathway is a kinase cascade. The apical membrane-associated FERM domain (band Four-point-one Ezrin Radixin Moesin homology domain) protein NF2 directly binds and recruits the Nuclear Dbf2-related family kinase, LATS1/2 to the plasma membrane. Membrane recruitment promotes LATS1/2 phosphorylation by the Ste-20 family protein kinase, MST1/2. In turn, LATS1/2 phosphorylates transcription coactivator YAP. Phosphorylated YAP is sequestered in cytoplasm, thus stops YAP's transcriptional outputs.12,13 During intrahepatic bile duct development, YAP activity positively correlates with biliary epithelial cell differentiation and primitive ducts formation.9 Loss of Nf2 (Alb-Cre;Nf2fl/fl), which leads to increased Yap activity, results in increased numbers of biliary epithelial cells and primitive ducts surrounding each portal vein in perinatal mouse livers, while liver-specific Yap deficiency (Alb-Cre;Yapfl/fl) significantly compromises biliary epithelial cell differentiation and bile duct tubulogenesis.9 Nevertheless, the molecular mechanisms by which YAP regulates bile duct development are unclear.
The Notch signaling pathway is an evolutionarily conserved intercellular signaling pathway and has an important role in cell-fate determination. Four Notch receptors (NOTCH1–4) with five canonical ligands (Delta-like ligand1 (DLL1), DLL3, DLL4, JAGGED1, and JAGGED2) are expressed in mammals with tissue-specific patterns. On ligand binding, proteolytic cleavage releases the Notch receptor's intracellular domain (NICD), which translocates to the nucleus and forms a transcriptionally active complex with the DNA-binding protein RBPJ (Recombination signaling-binding protein for immunoglobulin kappa J region). This transcriptionally active complex will promote the expression of Notch target genes such as Hes1 and Sox9.14 Liver-specific deletion of Notch2 (Alb-Cre;Notch2fl/fl) shows similar phenotypes as Yap-deficient mice with defects in bile duct morphogenesis, which led to bile duct paucity.15,16 On the contrary, overexpression of Notch2 intracellular domain Notch2IC in hepatoblasts leads to increased biliary specification of embryonic hepatoblasts,17 a phenotype similar to Nf2-deficient mice.9
The phenotypic similarity between Notch2- and Yap- deficient livers, as well as between Notch2IC overexpression and Nf2-deficient livers suggests that there is a relationship between the Notch signaling pathway and the Hippo signaling pathway in regulating intrahepatic bile duct development. Interestingly, a recent study showed that increased YAP activity induced adult hepatocytes to transfer into a ductal/ progenitor-like fate,18 a similar phenotype to Notch2IC overexpression mice.17 More importantly, it is through direct promotion of Notch2 transcription that YAP induces adult hepatocytes transferring into ductal/progenitor-like cells.18 Based on these studies in the adult mice, it seems possible that the Hippo signaling pathway also functions through Notch2 during the intrahepatic bile duct development. In this study, we tested this hypothesis with mouse genetics. We found that liver-specific Nf2 deficiency significantly increased the Notch signaling activity in cholangiocytes. Reducing the Notch signaling activity with heterozygous and homozygous deletion of the Notch2 gene suppress the Nf2-deficient phenotypes in a dose-dependent manner. Our studies demonstrated the genetic link between the Hippo signaling and the Notch signaling in regulating intrahepatic bile duct development.
Materials and Methods
Mice
All mice were housed at Baylor Scott & White Health animal facility, and handled according to NIH guidelines. The animal studies were approved by the Institutional Animal Care and Use Committee of Baylor Scott & White Health. Liver-specific Nf2 conditional knockout mice (Alb-Cre;Nf2fl/fl) have been described previously.9 Mice carrying conditional knockout alleles for Notch2 (Notch2fl/fl) were obtained from the Jackson laboratory (stock #010525). After multiple rounds of crossing, we obtained the following genotypes that were used in this study: Alb-Cre;Nf2fl/fl;Notch2fl/+; and Alb-Cre;Nf2fl/fl;Notch2fl/fl. All the strains were maintained on a C57Bl6 background. In all the experiments, Alb-Cre-negative littermates served as control. To avoid potential variation related to gender, all the experiments were performed in male mice with paternal inheritance of Alb-Cre.
Histological analysis and Immunostaining
Paraffin-embedded liver tissue was cut into 5 μm sections, which were stained with Sirius Red to visualize fibrosis or performed immunohistochemical staining according to the protocols provided by manufacturers of the respective antibodies. Snap-frozen tissues embedded in O.C.T. were cut into 8 μm sections, which were used for immunofluorescent staining according to the protocols provided by manufacturers of the respective antibodies. The following primary antibodies were used: CK19 for immunohistochemical staining (AbCam; EP1580Y, 1/200), CK19 for immunofluorescent staining (DSHB; TROMA-III; 1:50), Ki67 (AbCAM; Ab15580; 1:100), YAP (Cell Signaling;14074; 1:100). The secondary antibodies used were: Envision anti-rabbit (DAKO; K4002) for immunohistochemical staining, Alexa488-conjugated rabbit secondary antibody and Alexa568-conjugated rat secondary antibody for immuno-fluorescent staining. The immunofluorescent images were visualized using Leica AF 6000 Modular 146 Systems (Leica Biosystems Newcastle). The presented images are representative of at least three independent experiments.
Cholangiocytes and bile duct quantification
To quantify the CK19-positive cholangiocytes and bile ducts in the P0 and P7 livers, we evaluated five randomly chosen × 20 fields per section for each P0 mouse and 10 randomly chosen × 20 fields per section for each P7 mouse with Image Pro software. The numbers of CK19-positive cells and bile ducts were quantified by adding the respective numbers for each × 20 field. To measure cholangiocyte proliferation at P0, the liver sections were co-stained with Ki67 and CK19 and the number of Ki67+/CK19+ cells was calculated as a percentage of the total CK19+ cells counted. A total 100 to 200 CK19+ cells were randomly chosen per liver. n = 3 to 4 for each genotype.
Serum Total Bile Acids, ALT and Total Bilirubin Measurements
Serum levels of total bile acids (#08470; Crystal Chem), total alanine aminotransferase (ALT) (#MAK052; Sigma-Aldrich), and total bilirubin (#MBS756027; My Bio Source) were measured using commercially available kits according to the manufacturer's protocol for 8-week-old animals. Serum ALT and total bilirubin levels in 6-month-old animals were assessed using the Catalyst One chemistry analyzer (IDEXX Laboratories, Westbrook, ME, USA).
Primary Cholangiocyte and Hepatocyte Isolation
The livers of 8-week-old mice were perfused by two-step collagenase perfusion to isolate hepatocytes and cholangiocytes.19 Virtually pure (∼98% by histochemistry for γ-glutamyltransferase, γ-GT)20 mouse cholangiocytes were obtained with immunoaffinity separation21–23 by using a monoclonal antibody, rat IgG2a (a gift from Dr R Faris, Brown University, Providence, RI, USA), against surface EpCAM antigen expressed by all mouse cholangiocytes.22 The isolation of hepatocytes was performed as previously described.24
Quantitative Real-Time PCR
RNA was extracted from isolated cholangiocytes, hepatocytes or whole-liver lysate using PureLink RNA extraction kit (Thermo Fisher) and then was reverse-transcribed with random primers using iScript cDNA synthesis kit (Bio-Rad). Real-time quantitative PCR (qPCR) was performed using the Quantitect SYBR Green PCR kit (Thermo Fisher) on a ViiA7 Realtime PCR System (Applied Biosystems). The qPCR was done in triplicate, using histone H2A.Z and GAPDH as housekeeping controls. Relative differences in the expression of the candidate genes in control and mutant animals were determined using the 2−ΔΔCt method. The primer sequences used are available on request.
Statistical Methods
Unpaired Student's t-tests were used to determine whether there were significant differences between the means of two independent groups. One-way ANOVA and Fisher's SLD were used to determine whether there were significant differences between the means of three or more independent groups.
Results
Ablating Nf2 Promotes the Notch Signaling Activity in Cholangiocytes
In one of our previous studies, we observed no changes in the expression levels of the Notch signaling ligand Jagged1, receptor Notch2, and their target Hes1 when disrupting the Hippo signaling pathway in mouse livers, which suggested that the Notch signaling is not a downstream mediator of the Hippo signaling pathway in regulating intrahepatic bile duct development.9 However, it was later revealed that NF2 and YAP are highly expressed in cholangiocytes and weakly expressed in hepatocytes (www.proteinatlas.org).25 Immuno-histochemical staining of liver sections from P0, P7, and 8-week-old wild-type mice confirmed that YAP mainly locates in cholangiocytes (Supplementary Figure 1). Therefore, ablating Nf2 or Yap may influence the expression levels of the Notch signaling components mainly in the cholangiocytes so that these changes were not detected in the whole-liver lysate as cholangiocytes only account for 3–5% of hepatic cell population. To test this hypothesis, we separately isolated cholangiocytes and hepatocytes from wild-type control and Nf2-deficient mice. Successful cholangiocyte and hepatocyte enrichments were confirmed with high expression levels of the cholangiocyte marker CK19 and low expression levels of the hepatocyte marker Hnf4α in cholangiocyte preparation and vice versa in hepatocyte preparation (Figures 1a and b). The expression levels of CTGF, a demonstrated YAP transcriptional target,26 were significantly upregulated in cholangiocytes but not in hepatocytes of Nf2 livers, when compared with control livers (Figure 1c). These results indicated that NF2 functions to restrict YAP activity mainly in cholangiocytes, which is consistent with its protein location and the pronounced biliary phenotypes of Nf2 mutant livers. In agreement with our hypothesis, increased mRNA levels of Notch2 and Notch2 targets Hes1 and Sox9 were observed in cholangiocytes but not in hepatocytes of Nf2 mutant livers (Figures 1d–f). Thus, NF2 functions to restrict the Notch signaling activity mainly in cholangiocytes.
Figure 1.
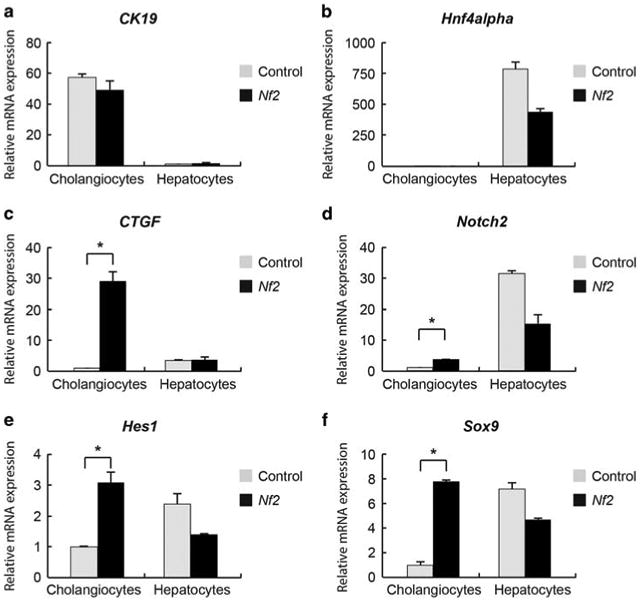
Ablating Nf2 increases the expression levels of Notch2 and Notch2 targets in cholangiocytes. (a–f) Quantitative real-time PCR analysis of selective genes from fresh isolated cholangiocytes and hepatocytes from control and Nf2 mice at the age of 8 weeks. Bars represent mean±s.e.m. (n = 3 mice from each genotype). *P<0.05, compared with control, unpaired t-test.
Suppression of Nf2 Mutant Intrahepatic Bile Duct Development Defects by Loss of Notch2
To determine whether the increased Notch2 expression levels in cholangiocytes account for the biliary phenotypes in Nf2 mutant animals, we combined liver-specific deletions of Nf2 and Notch2 to generate Alb-Cre;Nf2fl/fl;Notch2fl/+ (Nf2; Notch2+/−) and Alb-Cre;Nf2fl/fl;Notch2fl/fl (Nf2;Notch2) mice. Because Nf2- and Notch2-deficient animals displayed contrasting phenotypes for intrahepatic bile duct development,9,15,16 if Notch2 is a downstream effector of the Hippo signaling pathway, which means Nf2-deficient pheno-types are due to increased Notch2 activity, reducing Notch2 activity with heterozygous and homozygous deletion of the Notch2 gene should suppress the Nf2-deficient phenotypes in a dose-dependent manner.
Successful ablation of Nf2 and Notch2 in the liver was confirmed by quantitative RT-PCR analysis of Nf2 and Notch2 mRNA levels in P0, P7, and 8-week-old mutant mice (Supplementary Figure 2). Nf2;Notch2+/− and Nf2;Notch2 mice were born with expected Mendelian frequencies and no overt abnormalities. Because both NF2 and Notch2 mainly influence the primitive bile duct formation, which becomes obvious at perinatal stage (E18.5 – P1), and the bile duct development completes during the first 2 weeks of life, P0 and P7 time points were chosen to analyze the intrahepatic bile duct development. In control mice, at P0, the ductal plates displayed typical appearance with both primitive ducts and nontubular structures formed around large portal veins (Figures 2a and b, Control). As shown previously, all CK19-positive epithelial cells became primitive ducts but not nontubular structures in Nf2 mutant livers (Figures 2a and b, Nf2).9 In contrast to Nf2 mutant livers, the ductal plates of Nf2;Notch2+/− and Nf2;Notch2 livers contain both primitive ducts and nontubular structures (Figures 2a and b, Nf2; Notch2+/− and Nf2;Notch2). Quantification of CK19-positive epithelial cells and primitive ducts numbers indicated that Nf2 deficiency dramatically increased numbers of biliary precursors and primitive ducts, whereas these increases were significantly suppressed by loss of Notch2 in a dose-dependent manner (Figures 1c and d). However, compared with control livers, loss of Notch2 did not completely block the Nf2-deficient phenotype as both Nf2;Notch2+/− and Nf2;Notch2 livers displayed more biliary lineage differentiation and primitive duct formation.
Figure 2.
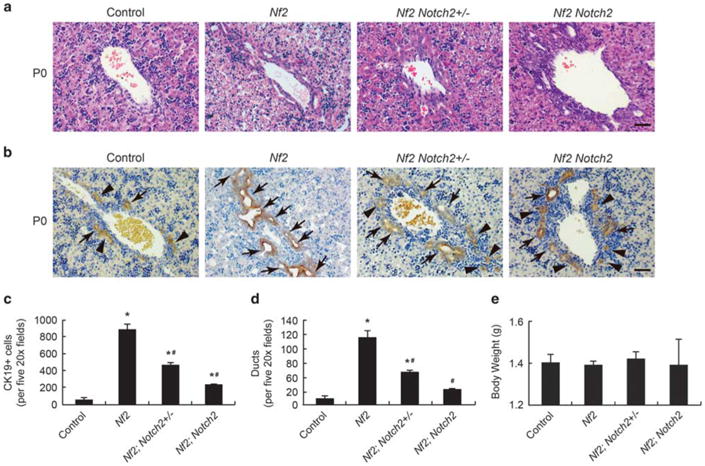
Suppression of Nf2 mutant intrahepatic bile duct development phenotypes by loss of Notch2 (P0). (a and b) H&E staining (a) and CK19 staining (b) of large portal vein area in P0 Control, Nf2, Nf2;Notch2+/− and Nf2;Notch2 livers. Arrows, primitive ducts. Arrowheads, nontubular ductal plate structures. Scale bars =50 μm. (c) Quantification of CK19-positive cholangiocytes for P0 livers. (d) Quantification of primitive ducts for P0 livers. (e) Comparison of body weights for P0 mice. Bars represent mean ±s.e.m. (n= 3 mice from each genotype). *P<0.05, compared with control, #P<0.05, compared with Nf2, one-way ANOVA and Fisher's LSD
At P7, the primitive ducts incorporated into portal mesenchyme and became mature bile ducts in control and all of mutant livers. However, while the ductal plates remnants regressed in control livers, these nontubular structures were still obvious in Nf2;Notch2+/− and Nf2; Notch2 livers (Figures 3a and b). Quantification of CK19-positive cholangiocytes and primitive ducts in control and mutant livers showed similar trends to those in P0 livers. The elevated cholangiocyte numbers and bile duct numbers in Nf2 livers were significantly suppressed by heterozygous and homozygous deletion of Notch2. Nf2;Notch2 livers displayed similar amounts of bile ducts to those of control livers (Figures 3c and d).
Figure 3.
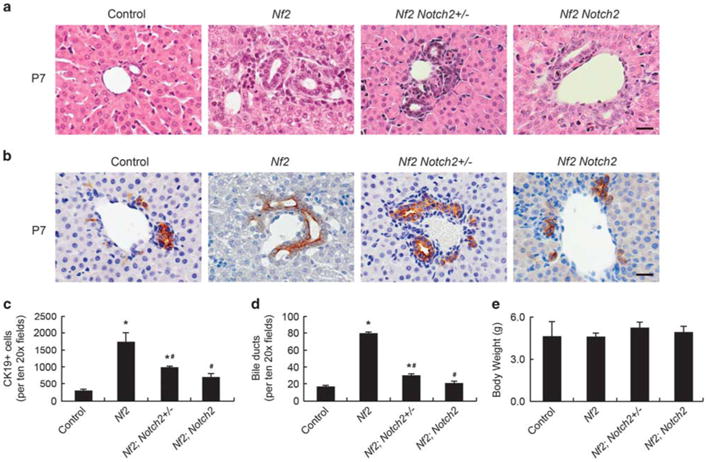
Suppression of Nf2 mutant intrahepatic bile duct development phenotypes by loss of Notch2 (P7). (a and b) H&E staining (a) and CK19 staining (b) of small portal vein area in P7 Control, Nf2, Nf2;Notch2+/− and Nf2;Notch2 livers. Arrows, primitive ducts. Arrowheads, nontubular ductal plate structures. Scale bars =20 μm. (c) Quantification of CK19-positive cholangiocytes for P7 livers. (d) Quantification of primitive ducts for P7 livers. (e) Comparison of body weights for P7 mice. Bars represent mean ±s.e.m. (n= 3 mice from each genotype). *P<0.05, compared with control, #P<0.05, compared with Nf2, one-way ANOVA and Fisher's LSD.
It has been previously reported that ablating Notch2 with Alb-Cre-mediated DNA recombination resulted in jaundice and reduced body weights in postnatal mice.13 These defects were not observed in Nf2;Notch2 mice. All the mutant mice showed similar body weights to those of control mice at P0 and P7 (Figures 2e and 3e) and no signs of jaundice. Taken together, loss of Notch2 suppressed Nf2-deficient intrahepatic bile duct development defects in a dose-dependent manner, indicating that NF2 functions through Notch2 in regulating intrahepatic bile duct development. Nf2;Notch2 mutant livers did not phenocopy Notch2 mutant livers, suggesting the existence of other mediators downstream of the Hippo signaling pathway in regulating intrahepatic bile duct development.
Loss of Notch2 Suppresses Cholangiocyte Overproliferation Induced by Nf2 Deficiency During Intrahepatic Bile Duct Development
A major function of the Hippo signaling pathway is to inhibit cell proliferation10 and loss of Nf2 promoted cholangiocyte proliferation at P0.9 To determine whether the increased Notch signaling activity accounts for the elevated cholangiocyte proliferation in Nf2-deficient livers, we assessed the cholangiocyte proliferation in P0 Nf2;Notch2+/− and Nf2; Notch2 livers. Because in developing mouse livers, hepatocytes undergo substantial proliferation,27 we performed double staining of proliferation marker Ki67 and cholangiocyte marker CK19 to identify proliferating cholangiocytes. In agreement with the previous study,9 Nf2 mutant livers showed enhanced proliferation of the CK19-positive cholangiocytes compared with the livers from wild-type mice (Figures 4a and b). This cholangiocyte overproliferation induced by loss of Nf2 was greatly suppressed by heterozygous and homozygous deletion of Notch2 in a dose-dependent manner (Figures 4a and b). Thus, Notch2 is an effector of the Hippo signaling pathway in mediating cholangiocyte proliferation during intrahepatic bile duct development.
Figure 4.
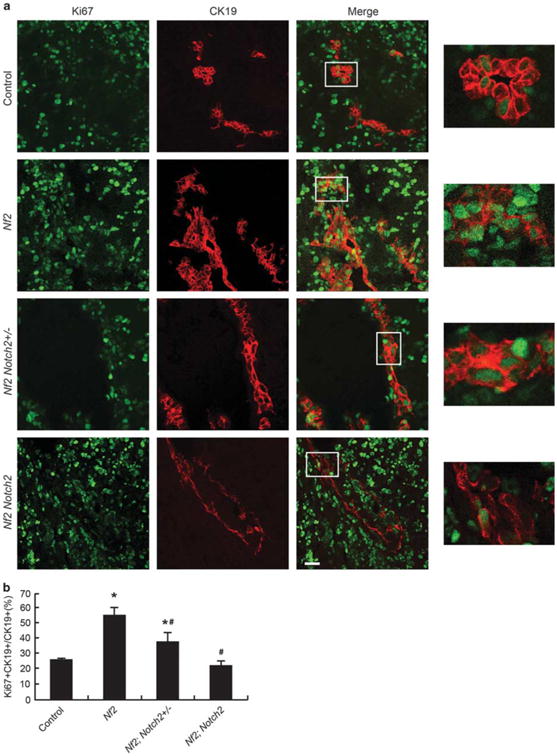
Loss of Notch2 suppresses cholangiocyte overproliferation induced by Nf2 deficiency during IHBD development. (a) Control, Nf2, Nf2;Notch2 +/− and Nf2;Notch2 livers from P0 animals were analyzed for Ki67 staining (green) and counterstained for the cholangiocyte marker CK19 (red). Scale bar= 25 μm. Representative areas are shown with higher magnification. (b) Quantification of Ki67-positive cholangiocytes. Bars represent mean±s.e.m. (n= 3 mice from each genotype). *P<0.05, compared with control, #P<0.05, compared with Nf2, one-way ANOVA and Fisher's LSD.
Cholangiocyte Proliferation Continues in Adult Nf2; Notch2+/− And Nf2;Notch2 Mice
Consistent with the previous study,9 the cholangiocyte compartment of Nf2 livers continued to expand in adult life as revealed by CK19-positive ductular reactions extending from portal area to deep parenchyma in 8-week-old mice (Figures 5b and c, Nf2). The ductular reactions were more severely developed at the liver edge and formed bile duct hamartomas (Figure 5a, Nf2). Similar to what was observed in the Nf2 livers, ductular reaction and hamartoma were also present in Nf2;Notch2+/− and Nf2;Notch2 livers (Figures 5a–c). Owing to the increased cholangiocyte population, the liver/body ratio was significantly increased in Nf2, Nf2;Notch2 +/− and Nf2;Notch2 mice, when compared with their wild-type littermates (Figure 5d). The cholangiocytes continued to proliferate in deep liver parenchymal area in all groups of mutant mice as they grew older (Supplementary Figure 3b). However, the hamartoma region at the edge of liver significantly reduced in 6-month-old Nf2, Nf2;Notch2+/− and Nf2;Notch2 mice, when compared with those at 8 weeks of age (Supplementary Figure 3a). The liver/body ratio of 6-month-old Nf2, Nf2;Notch2+/− and Nf2;Notch2 mice was also significantly higher than their wild-type littermates (Supplementary Figure 3c).
Figure 5.
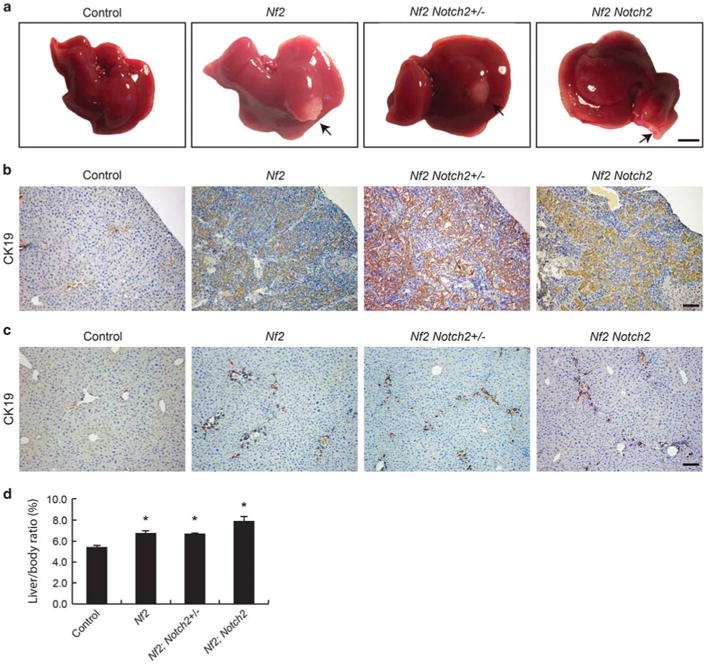
Cholangiocyte proliferation continues in adult Nf2;Notch2+/− and Nf2;Notch2 mice. (a) Gross liver images from 8-week-old animals. Note the formation of biliary hamartoma in Nf2, Nf2;Notch2+/− and Nf2;Notch2 livers (indicated by arrows and confirmed by CK19 staining in b). Scale bar= 0.5 cm. (b) CK19 staining of the biliary hamartoma shown in a, Scale bar= 100 μm. (c) CK19 staining of deep parenchymal area of control, Nf2, Nf2; Notch2+/− and Nf2;Notch2 livers from 8-week-old animals. Scale bar = 100 μm. (d) Quantification of liver-to-body ratio of 8-week-old animals. Bars represent mean± s.e.m. (n = 3 mice from each genotype). *P<0.05, compared with wild-type control, one-way ANOVA and Fisher's LSD.
Nf2;Notch2+/− And Nf2;Notch2 Mice Do Not Suffer Severe Liver Damage
Owing to bile duct paucity caused by loss of Notch2, the adult Alb-Cre; Notch2fl/fl mice suffered jaundice and cholestasis-related liver damage including necrosis and fibrosis.12,13 In contrast to Notch2 liver phenotypes, no signs of jaundice were observed in 8-week-old or 6-month-old Nf2;Notch2+/− and Nf2;Notch2 mice. Consistently, the serum total bilirubin levels were normal in these mice (Figure 6a and Supplementary Figure 3d). Histological analysis of Nf2;Notch2+/ − and Nf2; Notch2 livers revealed absence of necrosis, which was supported by normal serum alanine aminotransferase (ALT) activity (Figure 6b and Supplementary Figure 3e). Interestingly, 8-week-old Nf2 mice showed increased serum total bile acids (TBA) levels (Figure 6c), suggesting hepatic dysfunction caused by Nf2 deficiency.28 The increased TBA levels were rescued by heterozygous and homozygous deletion of Notch2. Nf2;Notch2+/− and Nf2;Notch2 mice displayed comparable serum TBA levels to wild-type controls (Figure 6c). Sirius red staining of liver sections indicated collagen deposition accompanying bile duct hamartoma at the edge of Nf2, Nf2;Notch2+/− and Nf2;Notch2 livers but with little fibrosis in deep parenchymal region (Figures 6d and e). Thus, consistent with mild bile duct development defects during embryonic development, Nf2;Notch2+/− and Nf2; Notch2 displayed normal liver functions in the adulthood.
Figure 6.
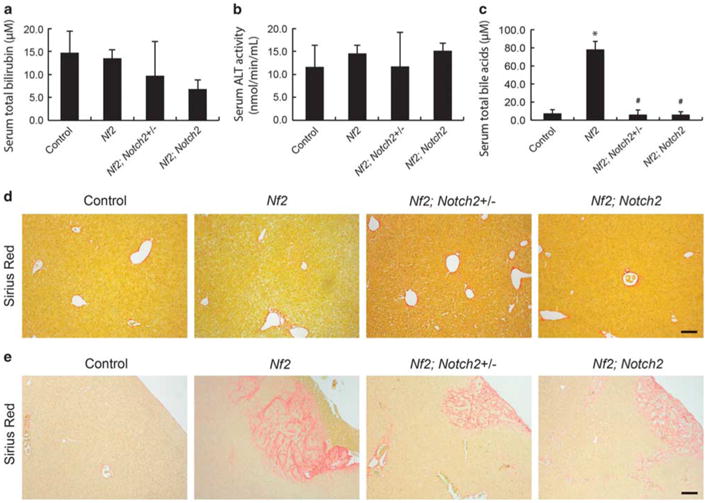
Nf2;Notch2+/− and Nf2;Notch2 mice do not suffer severe liver damage. (a) Serum total bilirubin levels, (b) serum alanine aminotransferase (ALT) activity, (c) serum total bile acids levels in 8-week-old control, Nf2, Nf2;Notch2+/− and Nf2;Notch2 animals. Bars represent mean ± s.e.m. (n =3 mice from each genotype). *P<0.05, compared with control, #P<0.05, compared with Nf2, one-way ANOVA and Fisher's LSD. (d and e) Liver fibrosis evaluation of 8-week-old control, Nf2, Nf2;Notch2+/−, and Nf2;Notch2 animals with Sirius Red staining, a method detecting collagen deposition.
Discussion
The Hippo signaling pathway has recently emerged as a major mechanism that regulates intrahepatic bile duct development.2 The upstream tumor suppressor NF2 and the downstream effector YAP function antagonistically to regulate bile duct formation. However, the mechanisms that link defective Hippo signaling and impaired IHBD development remain elusive. The possibility of the Notch signaling pathway as a mechanistic candidate was initially dismissed because disrupting the Hippo signaling did not affect the expression levels of various Notch signaling components.9 As later studies revealed that both NF2 and YAP mainly locate in the cholangiocytes of liver (www.proteinatlas.org),25 changes in the Notch signaling activity induced by the Hippo signaling disruption may be restricted in the cholangiocytes and not detectable in whole-liver lysate. In this study, we separately isolated the cholangiocytes and the hepatocytes from control and Nf2 KO mice. Indeed, ablating Nf2 in both cholangiocytes and hepatocytes with Alb-Cre-induced DNA recombination increased the expression levels of the Notch signaling receptor Notch2 and the downstream transcriptional targets Hes1 and Sox9 in cholangiocytes. In hepatocytes, Nf2 deficiency resulted in no increases of Notch2, Hes1, and Sox9 expression. The elevated expression levels of Notch2 and the Notch signaling transcriptional targets Hes1 and Sox9 in cholangiocytes of Nf2 deficiency livers suggested that the Notch signaling is a downstream effector of the Hippo signaling in regulating IHBD development. The comparable expression levels of Notch2, Hes1 and Sox9 in hepatocytes of wild-type and Nf2 deficiency mice explained the similar results observed in the previous study using whole-liver lysate.9 We then confirmed that the Notch signaling pathway functions downstream of the Hippo signaling pathway with mouse genetic studies. Heterozygous and homozygous deletion of Notch2 suppressed Nf2-deficient intrahepatic bile duct phenotypes in a dosedependent manner. Our study demonstrated that the Notch signaling pathway is an effector pathway of the Hippo signaling pathway in intrahepatic bile duct development.
However, the Notch signaling pathway does not seem to be the only effector pathway of the Hippo signaling in regulating intrahepatic bile duct development. In the previous study that demonstrated Yap is genetically epistatic to Nf2 in intrahepatic bile duct development, Alb-Cre;Nf2fl/fl;Yapfl/fl mice displayed similar bile duct paucity phenotypes as Alb-Cre;Yapfl/fl animals.9 These results indicated that YAP dictates the signaling flow from NF2 to bile duct formation. In the current study, Alb-Cre; Nf2fl/fl;Notch2fl/fl mice displayed similar bile duct numbers at P0 when compared with those of wild-type animals, which were different from the bile duct paucity phenotypes of Alb-Cre; Notch2fl/fl mice.15,16 These results suggested that Notch2 is not the only effector of the Hippo signaling pathway accounting for its function in bile duct development. Other effector pathways functioning downstream of the Hippo signaling may exist and need further investigation.
It is worthwhile to note that the Notch signaling pathway was proposed as an effector of YAP in mouse intestine. YAPS127A overexpression in murine intestine led to progenitor cell expansion, which was partially due to activation of Notch signaling evidenced by the upregulation of Hes1 expression and the suppression of the phenotypes with Notch inhibitor.29 Ablation of the Hippo signaling pathway tumor suppressors Mst1/Mst2 in mouse intestine also led to expansion of the progenitor cell compartment and activation of the Notch signaling with upregulated Jagged1 expression.30 However, because the Jagged1 is not a known YAP direct target but a known direct target of the Wnt signaling, the authors proposed that YAP indirectly induced Notch activation through directly activating the Wnt signaling.30 Interestingly, during Drosophila oogenesis, the YAP homology, Yki functions as a repressor of Notch activity for both polar cell specification and follicle cell proliferation.31,32 Therefore, the relationship between the Notch signaling pathway and the Hippo signaling pathway may have organism- and tissue-specific nature and needs to be carefully investigated and explained with respect to different contexts.
As defective Notch signaling and Hippo signaling are both linked to human biliary diseases, our study provided a mechanistic basis in searching for potential treatment strategies for these diseases. Alagille syndrome, caused by mutations and the resultant haploinsufficiency of the Notch ligand Jagged1, is a pleiotropic developmental disorder affecting multiple organs including liver, heart, eye, and skeleton.33,34 Bile duct paucity and the resultant severe jaundice are major characteristic features in patients with Alagille syndrome. Liver-specific ablation of Notch2 (Alb-Cre;Notch2fl/fl) exhibited the clinically relevant features of Alagille syndrome and served as the mouse model for Alagille syndrome.15,16 In this study, we showed that ablating Nf2 was able to rescue the phenotypes of Alb-Cre; Notch2fl/fl animals. In contrast to Alb-Cre;Notch2fl/fl animals,15,16 Alb-Cre;Nf2fl/fl;Notch2fl/fl mice displayed normal bile duct numbers during bile duct development, normal body weight range from birth to adulthood, absence of jaundice, and liver injury. Thus, inhibiting the Hippo signaling activity or increasing YAP activity represents a potential treatment strategy for Alagille syndrome. On the other hand, increased YAP expression levels were frequently observed in human cholangiocarcinoma and overexpression of YAP promoted cholangiocarcinoma development in vivo and in vitro.25,35,36 Our study demonstrated that loss of Notch2 suppresses Nf2 deficiency-induced cholangiocyte proliferation, suggesting that YAP-driven cholangiocarcinoma may benefit from treatment with Notch inhibitors.
Supplementary Material
Acknowledgments
This work was supported by the Dr. Nicholas C. Hightower Centennial Chair of Gastroenterology from Scott & White, a VA Research Career Scientist Award, a VA Merit award to Dr. Alpini (5I01BX000574), a VA Merit award to Dr. Meng (5I01 BX001724), a VA Merit award to Dr. Glaser (I01BX002192), a VA Merit award to Dr. DeMorrow (I01BX002638), the NIH grants DK58411, DK07698, DK095291, and DK062975 to Drs Alpini and Meng, the NIH grant DK082435 to Dr DeMorrow, and the Baylor Scott & White Health operational funds to Dr Bai. This material is the result of work supported by resources at the Central Texas Veterans Health Care System. The views expressed in this article are those of the authors and do not necessarily represent the views of the Department of Veterans Affairs.
Footnotes
Disclosure/Conflict of Interest: The authors declare no conflict of interest.
Supplementary Information accompanies the paper on the Laboratory Investigation website (http://www.laboratoryinvestigation.org)
References
- 1.Maroni L, Haibo B, Ray D, et al. Functional and structural features of cholangiocytes in health and disease. Cell Mol Gastroenterol Hepatol. 2015;1:368–380. doi: 10.1016/j.jcmgh.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nguyen Q, Anders RA, Alpini G, et al. Yes-associated protein in the liver: regulation of hepatic development, repair, cell fate determination and tumorigenesis. Dig Liver Dis. 2015;47:826–835. doi: 10.1016/j.dld.2015.05.011. [DOI] [PubMed] [Google Scholar]
- 3.Raynaud P, Carpentier R, Antoniou A, et al. Biliary differentiation and bile duct morphogenesis in development and disease. Int J Biochem Cell Biol. 2011;43:245–256. doi: 10.1016/j.biocel.2009.07.020. [DOI] [PubMed] [Google Scholar]
- 4.Zong Y, Stanger BZ. Molecular mechanisms of liver and bile duct development. Wiley Interdiscip Rev Dev Biol. 2012;1:643–655. doi: 10.1002/wdev.47. [DOI] [PubMed] [Google Scholar]
- 5.Carpentier R, Suner RE, van Hul N, et al. Embryonic ductal plate cells give rise to cholangiocytes, periportal hepatocytes, and adult liver progenitor cells. Gastroenterology. 2011;141:1432–1438. 1438 e1431–1434. doi: 10.1053/j.gastro.2011.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clotman F, Lannoy VJ, Reber M, et al. The onecut transcription factor HNF6 is required for normal development of the biliary tract. Development. 2002;129:1819–1828. doi: 10.1242/dev.129.8.1819. [DOI] [PubMed] [Google Scholar]
- 7.Coffinier C, Gresh L, Fiette L, et al. Bile system morphogenesis defects and liver dysfunction upon targeted deletion of HNF1beta. Development. 2002;129:1829–1838. doi: 10.1242/dev.129.8.1829. [DOI] [PubMed] [Google Scholar]
- 8.Kodama Y, Hijikata M, Kageyama R, et al. The role of notch signaling in the development of intrahepatic bile ducts. Gastroenterology. 2004;127:1775–1786. doi: 10.1053/j.gastro.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 9.Zhang N, Bai H, David KK, et al. The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. Dev Cell. 2010;19:27–38. doi: 10.1016/j.devcel.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pan D. Hippo signaling in organ size control. Genes Dev. 2007;21:886–897. doi: 10.1101/gad.1536007. [DOI] [PubMed] [Google Scholar]
- 11.Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19:491–505. doi: 10.1016/j.devcel.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong J, Feldmann G, Huang J, et al. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130:1120–1133. doi: 10.1016/j.cell.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yin F, Yu J, Zheng Y, et al. Spatial organization of Hippo signaling at the plasma membrane mediated by the tumor suppressor Merlin/NF2. Cell. 2013;154:1342–1355. doi: 10.1016/j.cell.2013.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang H, Zang C, Liu XS, et al. The role of Notch receptors in transcriptional regulation. J Cell Physiol. 2015;230:982–988. doi: 10.1002/jcp.24872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Geisler F, Nagl F, Mazur PK, et al. Liver-specific inactivation of Notch2, but not Notch1, compromises intrahepatic bile duct development in mice. Hepatology. 2008;48:607–616. doi: 10.1002/hep.22381. [DOI] [PubMed] [Google Scholar]
- 16.Lozier J, McCright B, Gridley T. Notch signaling regulates bile duct morphogenesis in mice. PLoS ONE. 2008;3:e1851. doi: 10.1371/journal.pone.0001851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeliazkova P, Jors S, Lee M, et al. Canonical Notch2 signaling determines biliary cell fates of embryonic hepatoblasts and adult hepatocytes independent of Hes1. Hepatology. 2013;57:2469–2479. doi: 10.1002/hep.26254. [DOI] [PubMed] [Google Scholar]
- 18.Yimlamai D, Christodoulou C, Galli GG, et al. Hippo pathway activity influences liver cell fate. Cell. 2014;157:1324–1338. doi: 10.1016/j.cell.2014.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kreamer BL, Staecker JL, Sawada N, et al. Use of a low-speed, iso-density percoll centrifugation method to increase the viability of isolated rat hepatocyte preparations. In Vitro Cell Dev Biol. 1986;22:201–211. doi: 10.1007/BF02623304. [DOI] [PubMed] [Google Scholar]
- 20.Rutenburg AM, Kim H, Fischbein JW, et al. Histochemical and ultrastructural demonstration of gamma-glutamyl transpeptidase activity. J Histochem Cytochem. 1969;17:517–526. doi: 10.1177/17.8.517. [DOI] [PubMed] [Google Scholar]
- 21.Alpini G, Roberts S, Kuntz SM, et al. Morphological, molecular, and functional heterogeneity of cholangiocytes from normal rat liver. Gastroenterology. 1996;110:1636–1643. doi: 10.1053/gast.1996.v110.pm8613073. [DOI] [PubMed] [Google Scholar]
- 22.Ishii M, Vroman B, LaRusso NF. Isolation and morphologic characterization of bile duct epithelial cells from normal rat liver. Gastroenterology. 1989;97:1236–1247. doi: 10.1016/0016-5085(89)91695-8. [DOI] [PubMed] [Google Scholar]
- 23.Renzi A, DeMorrow S, Onori P, et al. Modulation of the biliary expression of arylalkylamine N-acetyltransferase alters the autocrine proliferative responses of cholangiocytes in rats. Hepatology. 2013;57:1130–1141. doi: 10.1002/hep.26105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alpini G, Garrick RA, Jones MJ, et al. Water and nonelectrolyte permeability of isolated rat hepatocytes. Am J Physiol. 1986;251(6 Pt 1):C872–C882. doi: 10.1152/ajpcell.1986.251.6.C872. [DOI] [PubMed] [Google Scholar]
- 25.Bai H, Gayyed MF, Lam-Himlin DM, et al. Expression of Yes-associated protein modulates Survivin expression in primary liver malignancies. Hum Pathol. 2012;43:1376–1385. doi: 10.1016/j.humpath.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao B, Ye X, Yu J, et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008;22:1962–1971. doi: 10.1101/gad.1664408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Timchenko NA, Harris TE, Wilde M, et al. CCAAT/enhancer binding protein alpha regulates p21 protein and hepatocyte proliferation in newborn mice. Mol Cell Biol. 1997;17:7353–7361. doi: 10.1128/mcb.17.12.7353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rudman D, Kendall FE. Bile acid content of human serum. I Serum bile acids in patients with hepatic disease. J Clin Invest. 1957;36:530–537. doi: 10.1172/JCI103450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Camargo FD, Gokhale S, Johnnidis JB, et al. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr Biol. 2007;17:2054–2060. doi: 10.1016/j.cub.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 30.Zhou D, Zhang Y, Wu H, et al. Mst1 and Mst2 protein kinases restrain intestinal stem cell proliferation and colonic tumorigenesis by inhibition of Yes-associated protein (Yap) overabundance. Proc Natl Acad Sci USA. 2011;108:E1312–E1320. doi: 10.1073/pnas.1110428108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen HJ, Wang CM, Wang TW, et al. The Hippo pathway controls polar cell fate through Notch signaling during Drosophila oogenesis. Dev Biol. 2011;357:370–379. doi: 10.1016/j.ydbio.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 32.Yu J, Poulton J, Huang YC, et al. The hippo pathway promotes Notch signaling in regulation of cell differentiation, proliferation, and oocyte polarity. PLoS ONE. 2008;3:e1761. doi: 10.1371/journal.pone.0001761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alagille D, Estrada A, Hadchouel M, et al. Syndromic paucity of interlobular bile ducts (Alagille syndrome or arteriohepatic dysplasia): review of 80 cases. J Pediatr. 1987;110:195–200. doi: 10.1016/s0022-3476(87)80153-1. [DOI] [PubMed] [Google Scholar]
- 34.Emerick KM, Rand EB, Goldmuntz E, et al. Features of Alagille syndrome in 92 patients: frequency and relation to prognosis. Hepatology. 1999;29:822–829. doi: 10.1002/hep.510290331. [DOI] [PubMed] [Google Scholar]
- 35.Marti P, Stein C, Blumer T, et al. YAP promotes proliferation, chemoresistance, and angiogenesis in human cholangiocarcinoma through TEAD transcription factors. Hepatology. 2015;62:1497–1510. doi: 10.1002/hep.27992. [DOI] [PubMed] [Google Scholar]
- 36.Pei T, Li Y, Wang J, et al. YAP is a critical oncogene in human cholangiocarcinoma. Oncotarget. 2015;6:17206–17220. doi: 10.18632/oncotarget.4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.