

Synopsis
Pulmonary alveolar proteinosis (PAP) is a rare syndrome characterized by the accumulation of surfactant in alveoli and terminal airways resulting in respiratory failure. PAP comprises part of a spectrum of disorders of surfactant homeostasis (clearance and production). The surfactant clearance disorders are caused by disruption of GM-CSF signaling (primary PAP) or by an underlying disease that impairs alveolar macrophage functions including surfactant catabolism (secondary PAP). Primary PAP is related to alveolar macrophage dysfunction due to the disruption of GM-CSF signaling caused by a high serum level of anti-GM-CSF autoantibody (autoimmune PAP) (∼90%) or by the mutations in the GM-CSF receptor genes (hereditary PAP). The surfactant production disorders are caused by mutations in genes required for normal surfactant production. The PAP syndrome can be identified based on history, radiologic and bronchoalveolar lavage and/or histopathological findings. The diagnosis of PAP-causing diseases in secondary PAP requires further studies. Whole lung lavage is the current standard therapy and promising new pharmacologic therapies are currently in development.
Keywords: Pulmonary surfactant, GM-CSF, Alveolar macrophages, GM-CSF autoantibody, CSF2RA, CSF2RB, Whole lung lavage, GM-CSF inhalation therapy
Introduction
Pulmonary alveolar proteinosis (PAP) is a rare syndrome characterized by the accumulation of surfactant in alveolar macrophages and alveoli resulting in hypoxemic respiratory failure. In 1958, Rosen et al. first reported PAP as a disorder consisting of filling of alveoli by a PAS-positive proteinaceous material, rich in lipid1. The accumulated material is now known to be comprised primarily of pulmonary surfactant and smaller amounts of cell debris. PAP is often reported in the medical literature as a disease rather than as a syndrome and by the use of various terms, e.g., as pulmonary alveolar lipoproteinosis, idiopathic PAP, acquired PAP and congenital PAP 2. However, it should be noted that PAP is not a single disease. These disorders of surfactant homeostasis can be defined in the context of abnormalities of surfactant production or surfactant clearance. Surfactant production disorders are less common, typically occur in neonates and children, and are associated with alveolar wall distortion and varying degrees of accumulation of dysfunctional surfactant. They are caused by genetic mutations in genes that encode surfactant proteins or proteins involved in surfactant lipid metabolism, e.g., mutation in the SFTPB, SFTPC, ABCA3 or Nkx2.1 genes. Disorders of surfactant clearance are caused by disruption of GM-CSF signaling (primary PAP) or by an underlying disease that impairs alveolar macrophage number or functions including surfactant catabolism (secondary PAP) (Table 1). Among these PAP-causing diseases, autoimmune PAP is most common and will be a focus of this review2-4.
Table 1. Classification of Diseases and Risk Factors Reported in Association with PAP Syndrome1.
| PAP Classification | Risk factor |
|---|---|
| Primary PAP2 | GM-CSF Autoantibodies3 CSF2RA mutations4 CSF2RB mutations4 |
| Secondary PAP5 | Hematologic diseases6 Non-hematologic malignancies Immune deficiency/disruption diseases7 Chronic inflammatory/infectious diseases Toxic inhalation exposures8 SLC7A7 mutations MARS mutations9 |
| Surfactant Production Disorders10 |
SFTPB mutations SFTPC mutations ABCA3 mutations TTF1 (NKX2.1) mutations |
Abridged list. The strength of association of risk factors with the occurrence PAP varies widely.
Defined as PAP associated with a primary disruption of GM-CSF signaling.
Neutralizing GM-CSF autoantibodies mediate disease pathogenesis of what is now called autoimmune PAP (previously idiopathic, acquired, other names), which represents ∼85-90% of all patients with PAP syndrome. The presence of serum GM-CSF autoantibodies is 100% sensitive and specific for this disease.
The associated disease is commonly referred to as hereditary or familial PAP.
Defined as PAP associated with an underlying clinical condition secondarily affecting alveolar macrophage
Myelodysplastic syndrome (MDS) is the most common current disease in this category although many hematologic diseases have been reported in association with PAP.
Includes acquired immunodeficiency syndrome, agammaglobulinemia, amyloidosis, Bechet's disease, juvenile dematomyositis, and Fanconi's syndrome.
Includes inorganic dusts (aluminum, cement, silica, titanium, indium), organic dusts (agricultural, bakery flour, fertilizer, sawdust), and fumes (chlorine, cleaning products, gasoline/petroleum, nitrogen dioxide, paint, synthetic plastic fumes, varnish).
Biallelic MARS mutations cause a lung and liver phenotype and are prevalent in a population on Reunion Island.
Defined as PAP associated with genetic mutations resulting in abnormal surfactant protein or lipid production.
Pathogenesis
Pulmonary surfactant maintains lung function by creating an air-liquid interface on the alveolar surface, reducing surface tension and preventing alveolar collapse. It is comprised of 90% lipid (primary phospholipids) and 10% proteins (SP-A, -B, -C and -D). It is also important for host defense against microbial pathogens 4. Surfactant is synthesized and secreted by alveolar type II epithelial cells, removed by uptake, recycling and catabolism in these epithelial cells, and by uptake and catabolism in alveolar macrophages. The surfactant pool size is tightly regulated by the balance of secretion and removal by these cells (Figure 1). Studies in Csf2-/- (GM-CSF gene deficient) mice study revealed the role of GM-CSF in surfactant homeostasis and suggested the pathogenesis of PAP in 19945,6. This finding was confirmed by an identical pulmonary phenotype in Csf2rb-/- (GM-CSF receptor beta chain gene deficient) mice7,8. These results showed that the catabolism of surfactant in alveolar macrophages requires the presence of GM-CSF in the lungs. Based on these mechanisms, disorders of surfactant clearance are classified as primary PAP, which is caused by disruption of GM-CSF signaling, and secondary PAP, which is associated with an underlying disease that impairs alveolar macrophage functions and/or numbers (Table 1). Although GM-CSF gene deficiency has never been identified in human patients, neutralizing autoantibodies against GM-CSF in BALF from “idiopathic” PAP patients were first reported in 19999. This form of PAP is now classified as autoimmune PAP, which is the most common clinical form of PAP and accounts for 90% of cases. Autoimmune PAP is related to alveolar macrophage dysfunction due to the disruption of GM-CSF signaling, which is in turn caused by a high level of anti GM-CSF autoantibody in the lungs. The pathogenesis of autoimmune PAP was confirmed in a 2009 study where the adoptive transfer of PAP patient-derived autoantibodies into nonhuman primates resulted in PAP10. The incidence and prevalence of autoimmune PAP is 0.49 and 6.2 per million, respectively, in the general population according to a Japanese cohort study11. Autoimmune PAP is twice as common in males, typically presenting in the third through sixth decades, and is rare in children younger than 10 years of age. Among GM-CSF autoantibody-negative PAP patients, gene mutations of GM-CSF receptor alpha and beta genes (CSF2RA and CSF2RB, respectively) were identified. Disruption of GM-CSF signaling by recessive mutations in CSF2RA or CSF2RB causes a hereditary form of PAP that is clinically, physiologically, and histologically indistinguishable from autoimmune PAP12-16. These genetic forms of PAP are now classified as hereditary PAP. Hereditary PAP probably account for <1% of cases. Secondary PAP is associated with various underlying diseases such as hematological diseases (myelodysplastic syndrome [MDS), acute myelogenous leukemia, acute lymphoblastic leukemia, chronic myelocytic leukemia, chronic lymphocytic leukemia, aplastic anemia, multiple myeloma, lymphoma, Waldenstrom's macroglobulinemia), nonhematologic malignancies (lung adenocarcinoma, glioblastoma, melanoma),infectious diseases (Cytomegalovirus, Mycobacterium tuberculosis, Nocardia, Pneumocystis jirovecii), immune deficiency/disruption syndromes (acquired immunodeficiency syndrome, amyloidosis, Fanconi's syndrome, agammaglobulinemia, Bechet disease, juvenile dermatomyositis, renal tubular acidosis, severe combined immunodeficiency disease), and toxic inhalation exposures (Table 1). Inorganic dust (aluminum, cement, silica, titanium, indium), organic dust (agricultural, bakery flour, fertilizer, sawdust), and fumes (chlorine, cleaning products, gasoline/petroleum, nitrogen dioxide, paint, synthetic plastic fumes, varnish) have also been reported to be associated with secondary PAP. These underlying diseases or conditions are thought to impair alveolar macrophage number or functions including surfactant catabolism. Hematological disorders (especially MDS) are the major underlying disease in >75% of cases with adult-onset secondary PAP 17. Secondary PAP accounts for 8-9% of all PAP cases, with an incidence and prevalence of approximately 0.05 and 0.5 per million, respectively. Surfactant production disorders are less common and are caused by genetic mutations in genes that encode surfactant proteins or proteins involved in surfactant lipid metabolism, e.g., mutations in the SFTPB, SFTPC, ABCA3 or Nkx2.1 genes. Other genetic defects such as y+LAT1 (SLC7A7) gene mutations which causes lysinuric protein intolerance 18 and Methionyl-tRNA synthetase (MARS) gene mutations19 were reported to be associated with PAP. The precise mechanisms of PAP pathogenesis induced by these gene mutations have not been elucidated.
Figure 1.
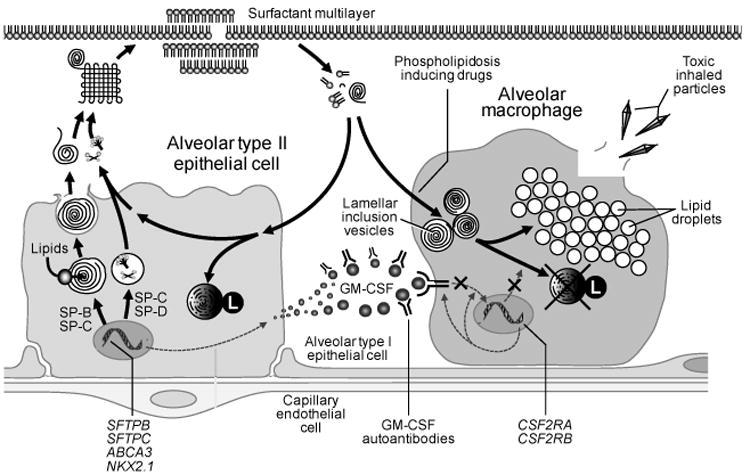
Regulation of surfactant homeostasis. Surfactant lipids and proteins (SP-A, -B, -C and –D) are synthesized in alveolar type II epithelial cell, secreted into the intra-alveolar space, and adsorb to surfactant layer at the air-liquid-tissue interface. The primary function of surfactant is to reduce surface tension within the alveolus to limit collapse of small airspaces. Surfactant lipids and proteins are removed by uptake and recycling in type II epithelial cells or by uptake and catabolism in alveolar macrophages. GM-CSF, a critical regulator of surfactant catabolism in alveolar macrophages, functions by binding to heterooligomeric receptors on alveolar macrophages and stimulating surfactant catabolism. Impaired GM-CSF signaling by GM-CSF autoantibodies or gene mutations in the GM-CSF receptor (CSF2RA, CSF2RB) cause alveolar macrophage dysfunction, which leads to impaired clearance and accumulation of surfactant in the lung (primary PAP).
Clinical Presentation
Common symptoms of PAP include exertional dyspnea, cough, fatigue and weight loss (Table 2). Fever (1% in Japanese cohort, 11% in Italian cohort and 15% in Chinese cohort) and sputum production (4% in Japanese cohort and 1% in Italian cohort) are less common. In 20 patients with hereditary PAP caused by CSF2RA mutations and forms of secondary PAP, similar symptoms have been reported20,17. In secondary PAP with MDS, the most common symptoms were fever (45%), dyspnea on exertion (42%), and cough (42%)21. In autoimmune PAP, a smoking history is commonly present (57% in Japanese cohort, 79% in German cohort, and 64% in Italian cohort), and dust or fume exposure have been reported (26% in Japanese cohort, 54% in German cohort, and 32% in Italian cohort). In several studies, two-thirds of the patients were men (male: female ratio is 2.2)2,11,22-24. The physical examination is generally unremarkable, but crackles, clubbing and cyanosis have been reported in some patients.
Table 2. Clinical Presentation in Patients with PAP Syndrome1.
| Study | Inoue | Bonella | Campo |
|---|---|---|---|
| Geographic location of patients | Japan | Germany | Italy |
| Number of patients | 223 | 70 | 73 |
| Disease / disease category | |||
| Autoimmune PAP (%) | 100 | 91 | 100 |
| Secondary PAP (%) | 0 | 9 | 0 |
| Symptom / Sign | Frequency (% of patients) | ||
|
|
|||
| Dyspnea | 52 | 94 | 67 |
| Cough | 23 | 66 | 31 |
| Fatigue | 0 | 49 | 0 |
| Weight loss | 0.4 | 43 | 0 |
| Fever | 1 | 0 | 11 |
| Sputum | 4 | 0 | 1 |
| None | 31 | 0 | 5 |
| Reference | [11] | [22] | [23] |
Only reports with a comprehensive symptom inventory are shown here. Studies with incomplete symptom assessment and symptom frequency in other PAP-causing diseases can be found in the text.
Diagnosis
PAP syndrome can be identified based on a compatible history, typical radiologic findings, bronchoalveolar lavage cytology and/or lung biopsy findings, and compatible biomarkers. However, diagnosis of the specific PAP-causing disease in secondary PAP syndromes requires further studies. If a patient is suspected to have PAP based on history, radiology studies and other findings, early anti GM-CSF autoantibody testing is useful to make the diagnosis of autoimmune PAP, the most common etiology, and can minimize the use of more invasive procedures (see below). A diagnostic algorithm is shown in Figure 2.
Figure 2.
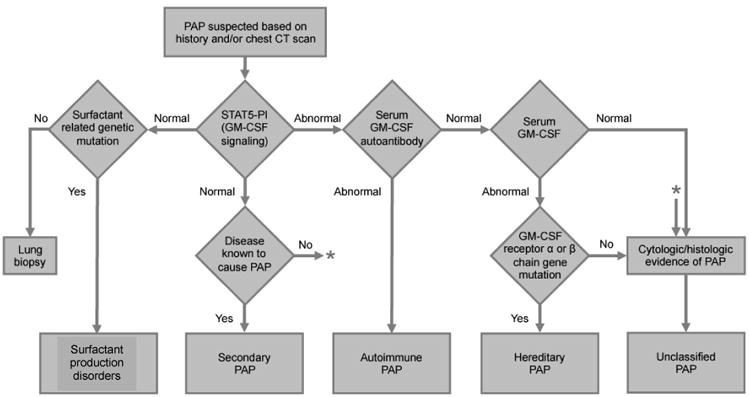
Algorithm used for the differential diagnosis of PAP syndrome. The diagnosis of PAP is made based on a compatible history, typical radiologic findings, compatible biomarkers, and characteristic lung biopsy or bronchoalveolar lavage cytology findings. Patients with high serum GM-CSF autoantibodies but without underlying diseases known to be associated with PAP are diagnosed with auto immune PAP. Patients with negative serum GM-CSF autoantibodies and with underlying diseases known to cause PAP are diagnosed with secondary PAP. Patients with negative serum GM-CSF autoantibodies and elevated levels of serum GM-CSF without apparent underlying diseases known to be associated with PAP need further evaluation for hereditary PAP by analyzing GM-CSF receptor gene (CSF2RA or CSF2RB) mutations. Whole blood GM-CSF signaling tests such as the GM-CSF induced STAT5 phosphorylation index test (STAT5-PI) may support the diagnosis of primary PAP. Surfactant production disorders are diseases due to the gene mutations of SFTPB, SFTPC, ABCA3 and NKX2.1. * Indicates that patients with normal STAT5-PI and no known disease to cause PAP need to be evaluated for cytologic/ histologic evidence of PAP. (From Trapnell BC, Luisetti M. Pulmonary alveolar proteinosis syndrome. In; Broaddus VC, Mason RJ, Ernst JD, et al, eds. Murray and Nadel's textbook of pulmonary medicine, 6th edition. Philadelphia: Elsevier Saunders; 2015; with permission.)
Radiographic Findings
A Chest radiograph may be a useful screening test for THE diagnosis of PAP. The distribution of infiltrates in autoimmune PAP and hereditary PAP is typically bilateral symmetrical opacities in the mid- and lower- lung fields (Figure 3A). High-resolution computed tomography (HRCT) shows ground-glass opacities (GGO) in autoimmune, hereditary and secondary PAP (Figure 3B). GGO presents as a patchy geographic pattern that is distributed in the lower lung fields in autoimmune PAP, whereas GGO typically presents as a diffuse pattern in secondary PAP. A crazing paving appearance (GGO superimposed on septal thickening) and subpleural sparing are frequently seen in autoimmune PAP, but are less frequently apparent in secondary PAP comprising 12% of 27 cases17 in one study.
Figure 3.
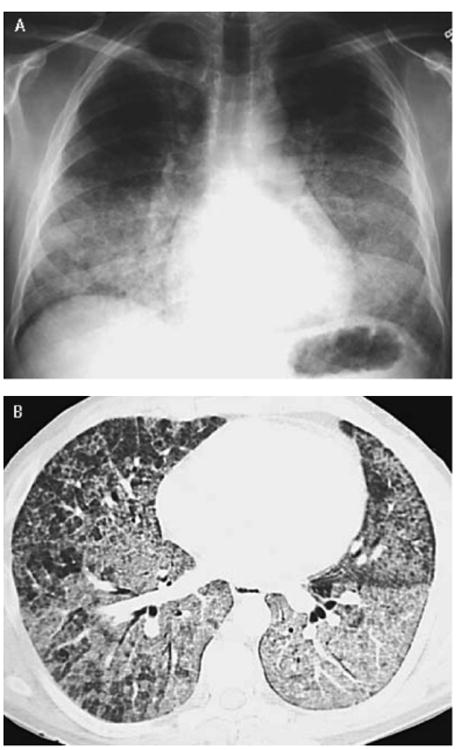
Chest radiograph and chest computed tomography (CT) in autoimmune PAP. (A) Chest radiograph shows diffuse, bilateral infiltrates suggestive of pulmonary edema but without findings suggestive of cardiovascular disease. (B) Chest CT shows a geographic pattern of ground-glass opacification superimposed on thickened interlobular septa commonly referred to as ‘crazy paving’.(From Trapnell BC, Whitsett JA, Nakata K. Pulmonary alveolar proteinosis. NEngl JMed 2003;349:2528, with permission.)
Laboratory Findings
Most of routine laboratory tests are normal in PAP. The serum level of LDH is typically elevated, although this finding is nonspecific. LDH and PaO2 are moderately correlated and LDH and A-aDO2 are more significantly correlated2. Levels of other biomarkers including SP-A, SP-B, SP-C, KL-6, MCP-1, CEA, cytokeratin 19, Cyfra 21-1 and NSE are increased and some of them correlate with disease severity (see below), although none of these findings are specific or diagnostic for PAP.
Pulmonary Function Testing
Pulmonary function testing is of limited usefulness in diagnosing the severity of PAP lung disease. Increased alveolar-arterial oxygen gradient (A-aDO2) correlates better with disease severity. FVC and FEV1 are generally within normal limits. Some patients show decreased forced vital capacity (FVC), consistent with restrictive physiology25. The diffusion capacity of the lung for carbon monoxide (DLCO) is frequently reduced and correlates with disease severity11.
Bronchoscopy, Bronchoalveolar Lavage, Microbiologic Culture
Bronchoscopic examination of the airways is unremarkable in PAP but bronchoalveolar lavage (BAL) fluid shows a milky, turbid appearance and thick sediment. BAL is emerging as a useful diagnostic tool for PAP (rising from 4% of 410 published cases in 2002 to 83% in German series in 2011)2,22. In a Japanese cohort, the diagnosis of PAP was based on HRCT and BAL analysis in 58.7% of patients; HRCT, BALF and transbronchial lung biopsy (TBLB) in 34.1% of patients; BAL analysis or TBLB and video-assisted thoracoscopic surgery (VATS) in 7.2% of patients11. Cytology of PAP BAL fluid reveals Periodic acid Schiff (PAS) stain and oil-red-O stain positive large foamy macrophages and dirty appearing sediment. Fungal, mycobacterial and other infectious etiologies should be ruled out by appropriate staining and culture.
Histopathology
The histopathology of PAP reveals well-preserved alveoli filled with eosinophilic, granular and PAS positive material and foamy alveolar macrophages (Figure 4). It is associated with minimal interstitial inflammation or fibrosis. The combined use of BAL analysis and measurement of serum GM-CSF autoantibody obviates the need for histologic confirmation in many cases of suspected PAP.
Figure 4.
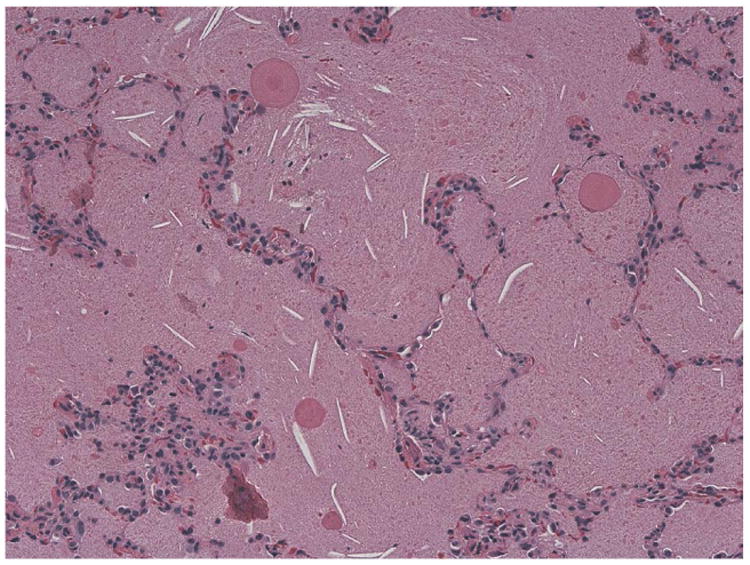
Lung histopathology in auto immune PAP. The alveoli are completely filled with amorphous, acellular eosinophilic material. The alveolar architecture is well preserved, and the alveolar walls are essentially unremarkable. (Periodic acid-Schiff (PAS) stain, original magnification ×20).
Biomarkers
In autoimmune PAPA, only disease-specific biomarker that is known to be elevated in serum is the GM-CSF autoantibody (Figure 5). This test is simple, standardized, and can be used to identify the disease in a large number of patients. The reported sensitivity and specificity for autoimmune PAP approaches 100% 26. GM-CSF autoantibody levels in serum do not correlate with duration of disease, disease severity, pulmonary function (FVC%, VC%, FEV1%,DLCO%), or serum biomarkers (LDH, SP-A, SP-D, CEA, KL-6) 11. The serum GM-CSF autoantibody also does not correlate with age, gender, smoking status, a history of environmental or occupational dust inhalation exposure, or duration of disease in a Japanese series 11, but shows a weak correlation with age and the duration of disease 22. GM-CSF autoantibody-negative PAP patients with an underling disease known to cause PAP are diagnosed with secondary PAP. Serum GM-CSF is elevated in hereditary PAP, likely caused by the absence of receptor-mediated clearance. Although this serum GM-CSF test is not disease specific, the combination of serum GM-CSF autoantibody-negativity and elevated GM-CSF without underlying disease should prompt genetic evaluation for hereditary PAP (Figure 2)15. Whole blood GM-CSF signaling tests such as the GM-CSF induced STAT5 phosphorylation index test (STAT5-PI) may support the diagnosis of primary PAP15. Although elevated in a wide range of other lung diseases, serum levels of LDH, SP-A, SP-B, SP-D, KL-6, CEA, M-CSF and MCP-1 are elevated in PAP. Serum levels of LDH, KL-6 and CEA are reported to be correlated with some markers of lung function (PaO2, AaDO2, or DLCO%) 22,25. Cancer biomarkers other than CEA, such as NSE and Cyfra 21-1, are also elevated in PAP 23. The elevation of serum levels of SP-A, SP-D and KL-6 are also reported in secondary PAP 17. While most of these biomarkers are not very helpful in establishing a diagnosis, they may be useful in monitoring disease activity in patients with PAP.
Figure 5.
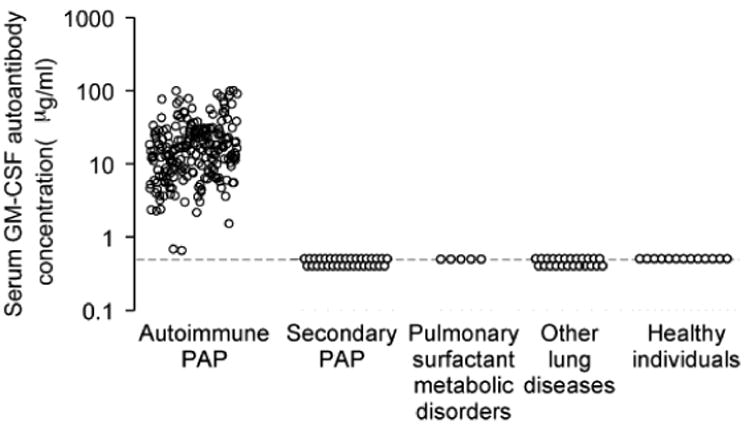
Serum GM-CSF autoantibody testing and diagnosis of autoimmune PAP. A serum level of GM-CSF autoantibody is specifically elevated in autoimmune PAP. The autoantibodies are not elevated in patients with other forms of PAP or other lung diseases or in normal persons. (From Trapnell BC, Luisetti M. Pulmonary alveolar proteinosis syndrome. In; Broaddus VC, Mason RJ, Ernst JD, et al, eds. Murray and Nadel's textbook of pulmonary medicine, 6th edition. Philadelphia: Elsevier Saunders; 2015; with permission.)
Natural History
In a Japanese cohort study, one-third of patients with autoimmune PAP were asymptomatic11. In contrast, 5% of patients were asymptomatic in an Italian study 23. The onset of disease is poorly studied because patients become symptomatic present for evaluation only after substantial alveolar filling with surfactant has occurred. The natural history of autoimmune PAP generally follows one of three patterns: spontaneous improvement (ranging from 5 to 7%), persistent, unremitting symptoms, or progressive deterioration with respiratory failure. Secondary infection occurs in primary and secondary PAP. The 5-year survival among 231 patients with PAP was 85% without therapy and 94% with whole lung lavage therapy 2. In this study, 72% of deaths were due to respiratory failure and 18% were due to uncontrolled infections. In the Japanese cohort study, the 5-year survival was 100% and significant infection was found in 4% of patients11. The clinical course of secondary PAP is strongly influenced by that of the underlying disease causing PAP and the prognosis is poorer than that of autoimmune PAP. Development of secondary PAP during the course of MDS is an important adverse prognostic risk factor in these patients 21.
Therapy
Current Therapy
Whole lung lavage (WLL) has been the standard first line therapy since the 1960s27. The WLL procedure is peformed under general anesthesia using a double lumen endotracheal tube to ventilate one lung while repeatedly filling and emptying the other with up to 50L of saline to physically remove surfactant from the lung. Although many PAP patients have been treated with WLL (28% in the Chinese cohort, 90% in the German cohort and 54% in the Italian cohort), the procedures are still not standardized and are highly operator-dependent2,22-24. Lobar and segmental lavage performed using a fiberoptic bronchoscope has also been reported. Although segmental BAL may be less effective compared to WLL because of the small lavage volume, one study reported that the two methods appear to be used at similar frequencies in the treatment of patients 24. For secondary PAP, approach to treatment is often dictated by the underlying disease. For instance, it has been reported that secondary PAP improves after hematopoietic stem cell transplantation in patients with hematologic diseases28. Therapy for the surfactant production disorders (congenital PAP) is supportive, although successful lung transplantation has been reported 29. There is no evidence that corticosteroids are effective for autoimmune PAP30.
Experimental Approaches
The first treatments with exogenous GM-CSF in PAP patients were administered subcutaneously 31. The use of subcutaneous GM-CSF in escalating doses for 6 to 12 months produced an overall response rate of 48% 32. In this study, 85% of patients receiving subcutaneous GM-CSF had local reactions at the site of injection and other minor adverse events. Aerosolized GM-CSF inhalation therapy has also been tested in PAP 33. In 35 patients with autoimmune PAP, 62% of patients receiving inhaled GM-CSF therapy had improvement in the A-aDO2 gradient, while serum GM-CSF autoantibody levels were unchanged34. Importantly, no serious adverse events occurred in this study34. Among these 35 patients, 66% had a durable response to the inhalation therapy and required no further treatments during the 30 month observation period. A low baseline vital capacity appeared to be a prognostic marker for disease recurrence 35. As another potential therapeutic strategy to reduce the level of GM-CSF autoantibodies, anti-B cell therapy with Rituximab for autoimmune PAP has been reported36-38. Plasmapheresis for autoimmune PAP is also a potential approach which can remove GM-CSF autoantibodies 39. Although some of these experimental approaches are promising, further efficacy and safety studies are necessary before conclusions can be drawn about their potential utility in the treatment of PAP patients.
Knowledge Gaps and Future Research
The critical role of GM-CSF signaling in primary PAP (autoimmune and hereditary PAP) pathogenesis has been revealed by effects of GM-CSF autoantibodies or GM-CSF receptor gene (CSF2RA or CSF2RB) mutations to disrupt the clearance of pulmonary surfactant by alveolar macrophages. However, the molecular mechanisms of PAP pathogenesis beyond the loss of GM-CSF signaling are largely unexplored. Further, the reasons that GM-CSF autoantibodies are produced in autoimmune PAP is not clear. Although a smoking history and dust or fume exposure are often observed in autoimmune PAP patients, the role of these inhalations in autoantibody production has not been elucidated. Although WLL is a standard therapy for PAP, no clinical practice guidelines have been established. Future studies are needed to define the optimal indications, timing and conduct of WLL procedure (e.g.-volume of saline infused, use of mechanical percussion, positioning of the patient etc.). In experimental approaches, a clinical study with inhaled GM-CSF in autoimmune PAP was shown to be safe and effective. However, the mechanisms underlying the therapeutic effect has not been determined. Further studies investigating the optimal dose, timing, duration of administration and long-term safety are necessary. In hereditary PAP, WLL is also a current standard therapy. Pulmonary macrophage transplantation and gene therapy have shown promising results in a mouse model of hereditary PAP 40 and represent promising future research directions.
Key Points.
Pulmonary alveolar proteinosis (PAP) is a syndrome characterized by the accumulation of surfactant in alveoli and terminal airways resulting in hypoxemic respiratory failure.
Surfactant is synthesized and secreted by alveolar type II epithelial cells, removed by uptake, recycling and catabolism in these epithelial cells and by uptake and catabolism in alveolar macrophages.
Autoimmune PAP is related to alveolar macrophage dysfunction due to the disruption of GM-CSF signaling caused by high level of anti GM-CSF autoantibody in the lungs.
Elevated serum GM-CSF autoantibody is diagnostic for autoimmune PAP in patients with typical history, radiologic findings, bronchoalveolar lavage cytology findings with a milky, turbid appearance and/or lung biopsy.
Whole lung lavage (WLL) is the standard first line therapy and GM-CSF inhalation for autoimmune PAP is promising therapy under clinical study.
Acknowledgments
The authors thank Paritha Arumugam and Elizabeth Kopras (Cincinnati Children's Hospital Medical Center) for reviewing this manuscript.
Footnotes
Disclosure statement: The Authors have nothing to disclose.
Contributor Information
Takuji Suzuki, Division of Pulmonary Biology, Cincinnati Children's Hospital Medical Center, Cincinnati, Ohio, USA.
Bruce C. Trapnell, Division of Pulmonary Biology, Cincinnati Children's Hospital Medical Center, Cincinnati, Ohio, USA.
References
- 1.Rosen SG, Castleman B, Liebow AA. Pulmonary alveolar proteinosis. The New England journal of medicine. 1958;258:1123–1142. doi: 10.1056/NEJM195806052582301. [DOI] [PubMed] [Google Scholar]
- 2.Seymour JF, Presneill JJ. Pulmonary alveolar proteinosis: progress in the first 44 years. American Joural of Respiratory and Critical Care Medicine. 2002;166:215–235. doi: 10.1164/rccm.2109105. [DOI] [PubMed] [Google Scholar]
- 3.Trapnell BC, Whitsett JA, Nakata K. Pulmonary alveolar proteinosis. The New England journal of medicine. 2003;349:2527–2539. doi: 10.1056/NEJMra023226. [DOI] [PubMed] [Google Scholar]
- 4.Whitsett JA, Wert SE, Weaver TE. Diseases of pulmonary surfactant homeostasis. Annual review of pathology. 2015;10:371–393. doi: 10.1146/annurev-pathol-012513-104644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dranoff G, et al. Involvement of granulocyte-macrophage colony-stimulating factor in pulmonary homeostasis. Science. 1994;264:713–716. doi: 10.1126/science.8171324. [DOI] [PubMed] [Google Scholar]
- 6.Stanley E, et al. Granulocyte/macrophage colony-stimulating factor-deficient mice show no major perturbation of hematopoiesis but develop a characteristic pulmonary pathology. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:5592–5596. doi: 10.1073/pnas.91.12.5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nishinakamura R, et al. Mice deficient for the IL-3/GM-CSF/IL-5 beta c receptor exhibit lung pathology and impaired immune response, while beta IL3 receptor- deficient mice are normal. Immunity. 1995;2:211–222. doi: 10.1016/1074-7613(95)90046-2. [DOI] [PubMed] [Google Scholar]
- 8.Robb L, et al. Hematopoietic and lung abnormalities in mice with a null mutation of the common beta subunit of the receptors for granulocyte-macrophage colony-stimulating factor and interleukins 3 and 5. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:9565–9569. doi: 10.1073/pnas.92.21.9565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kitamura T, et al. Idiopathic pulmonary alveolar proteinosis as an autoimmune disease with neutralizing antibody against granulocyte/macrophage colony-stimulating factor. The Journal of experimental medicine. 1999;190:875–880. doi: 10.1084/jem.190.6.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sakagami T, et al. Human GM-CSF autoantibodies and reproduction of pulmonary alveolar proteinosis. The New England journal of medicine. 2009;361:2679–2681. doi: 10.1056/NEJMc0904077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Inoue Y, et al. Characteristics of a large cohort of patients with autoimmune pulmonary alveolar proteinosis in Japan. American journal of respiratory and critical care medicine. 2008;177:752–762. doi: 10.1164/rccm.200708-1271OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martinez-Moczygemba M, et al. Pulmonary alveolar proteinosis caused by deletion of the GM-CSFRalpha gene in the X chromosome pseudoautosomal region 1. The Journal of experimental medicine. 2008;205:2711–2716. doi: 10.1084/jem.20080759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suzuki T, et al. Hereditary pulmonary alveolar proteinosis caused by recessive CSF2RB mutations. The European respiratory journal. 2011;37:201–204. doi: 10.1183/09031936.00090610. [DOI] [PubMed] [Google Scholar]
- 14.Suzuki T, et al. Familial pulmonary alveolar proteinosis caused by mutations in CSF2RA. The Journal of experimental medicine. 2008;205:2703–2710. doi: 10.1084/jem.20080990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suzuki T, et al. Hereditary pulmonary alveolar proteinosis: pathogenesis, presentation, diagnosis, and therapy. American journal of respiratory and critical care medicine. 2010;182:1292–1304. doi: 10.1164/rccm.201002-0271OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tanaka T, et al. Adult-onset hereditary pulmonary alveolar proteinosis caused by a single-base deletion in CSF2RB. J Med Genet. 2011;48:205–209. doi: 10.1136/jmg.2010.082586. [DOI] [PubMed] [Google Scholar]
- 17.Ishii H, et al. Clinical features of secondary pulmonary alveolar proteinosis: pre-mortem cases in Japan. The European respiratory journal. 2011;37:465–468. doi: 10.1183/09031936.00092910. [DOI] [PubMed] [Google Scholar]
- 18.Parenti G, et al. Lysinuric protein intolerance characterized by bone marrow abnormalities and severe clinical course. The Journal of pediatrics. 1995;126:246–251. doi: 10.1016/s0022-3476(95)70552-x. [DOI] [PubMed] [Google Scholar]
- 19.Hadchouel A, et al. Biallelic Mutations of Methionyl-tRNA Synthetase Cause a Specific Type of Pulmonary Alveolar Proteinosis Prevalent on Reunion Island. American journal of human genetics. 2015;96:826–831. doi: 10.1016/j.ajhg.2015.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hildebrandt J, et al. Characterization of CSF2RA mutation related juvenile pulmonary alveolar proteinosis. Orphanet journal of rare diseases. 2014;9:171. doi: 10.1186/s13023-014-0171-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ishii H, et al. Secondary pulmonary alveolar proteinosis complicating myelodysplastic syndrome results in worsening of prognosis: a retrospective cohort study in Japan. BMC pulmonary medicine. 2014;14:37. doi: 10.1186/1471-2466-14-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bonella F, et al. Pulmonary alveolar proteinosis: new insights from a single-center cohort of 70 patients. Respiratory medicine. 2011;105:1908–1916. doi: 10.1016/j.rmed.2011.08.018. [DOI] [PubMed] [Google Scholar]
- 23.Campo I, et al. Assessment and management of pulmonary alveolar proteinosis in a reference center. Orphanet journal of rare diseases. 2013;8:40. doi: 10.1186/1750-1172-8-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu Z, Jing J, Wang H, Xu F, Wang J. Pulmonary alveolar proteinosis in China: a systematic review of 241 cases. Respirology. 2009;14:761–766. doi: 10.1111/j.1440-1843.2009.01539.x. [DOI] [PubMed] [Google Scholar]
- 25.Presneill JJ, Nakata K, Inoue Y, Seymour JF. Pulmonary alveolar proteinosis. Clin Chest Med. 2004;25:593–613, viii. doi: 10.1016/j.ccm.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 26.Uchida K, et al. Standardized serum GM-CSF autoantibody testing for the routine clinical diagnosis of autoimmune pulmonary alveolar proteinosis. Journal of immunological methods. 2014;402:57–70. doi: 10.1016/j.jim.2013.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beccaria M, et al. Long-term durable benefit after whole lung lavage in pulmonary alveolar proteinosis. The European respiratory journal. 2004;23:526–531. doi: 10.1183/09031936.04.00102704. [DOI] [PubMed] [Google Scholar]
- 28.Cordonnier C, Fleury-Feith J, Escudier E, Atassi K, Bernaudin JF. Secondary alveolar proteinosis is a reversible cause of respiratory failure in leukemic patients. American journal of respiratory and critical care medicine. 1994;149:788–794. doi: 10.1164/ajrccm.149.3.8118651. [DOI] [PubMed] [Google Scholar]
- 29.Hamvas A, et al. Lung transplantation for treatment of infants with surfactant protein B deficiency. The Journal of pediatrics. 1997;130:231–239. doi: 10.1016/s0022-3476(97)70348-2. [DOI] [PubMed] [Google Scholar]
- 30.Akasaka K, et al. Outcome of corticosteroid administration in autoimmune pulmonary alveolar proteinosis: a retrospective cohort study. BMC pulmonary medicine. 2015;15:88. doi: 10.1186/s12890-015-0085-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seymour JF, Dunn AR, Vincent JM, Presneill JJ, Pain MC. Efficacy of granulocyte-macrophage colony-stimulating factor in acquired alveolar proteinosis. The New England journal of medicine. 1996;335:1924–1925. doi: 10.1056/NEJM199612193352513. [DOI] [PubMed] [Google Scholar]
- 32.Venkateshiah SB, et al. An open-label trial of granulocyte macrophage colony stimulating factor therapy for moderate symptomatic pulmonary alveolar proteinosis. Chest. 2006;130:227–237. doi: 10.1378/chest.130.1.227. [DOI] [PubMed] [Google Scholar]
- 33.Tazawa R, et al. Granulocyte-macrophage colony-stimulating factor and lung immunity in pulmonary alveolar proteinosis. American journal of respiratory and critical care medicine. 2005;171:1142–1149. doi: 10.1164/rccm.200406-716OC. [DOI] [PubMed] [Google Scholar]
- 34.Tazawa R, et al. Inhaled granulocyte/macrophage-colony stimulating factor as therapy for pulmonary alveolar proteinosis. American journal of respiratory and critical care medicine. 2010;181:1345–1354. doi: 10.1164/rccm.200906-0978OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tazawa R, et al. Duration of benefit in patients with autoimmune pulmonary alveolar proteinosis after inhaled granulocyte-macrophage colony-stimulating factor therapy. Chest. 2014;145:729–737. doi: 10.1378/chest.13-0603. [DOI] [PubMed] [Google Scholar]
- 36.Amital A, Dux S, Shitrit D, Shpilberg O, Kramer MR. Therapeutic effectiveness of rituximab in a patient with unresponsive autoimmune pulmonary alveolar proteinosis. Thorax. 2010;65:1025–1026. doi: 10.1136/thx.2010.140673. [DOI] [PubMed] [Google Scholar]
- 37.Borie R, Debray MP, Laine C, Aubier M, Crestani B. Rituximab therapy in autoimmune pulmonary alveolar proteinosis. The European respiratory journal. 2009;33:1503–1506. doi: 10.1183/09031936.00160908. [DOI] [PubMed] [Google Scholar]
- 38.Malur A, et al. Rituximab therapy in pulmonary alveolar proteinosis improves alveolar macrophage lipid homeostasis. Respiratory research. 2012;13:46. doi: 10.1186/1465-9921-13-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kavuru MS, Bonfield TL, Thomassen MJ. Plasmapheresis, GM-CSF, and alveolar proteinosis. American journal of respiratory and critical care medicine. 2003;167:1036–1037. doi: 10.1164/ajrccm.167.7.950. 1036; author reply. [DOI] [PubMed] [Google Scholar]
- 40.Suzuki T, et al. Pulmonary macrophage transplantation therapy. Nature. 2014;514:450–454. doi: 10.1038/nature13807. [DOI] [PMC free article] [PubMed] [Google Scholar]