

ABSTRACT
Persistent inflammation within the respiratory tract underlies the pathogenesis of numerous chronic pulmonary diseases including chronic obstructive pulmonary disease, asthma and pulmonary fibrosis. Chronic inflammation in the lung may arise from a combination of genetic susceptibility and environmental influences, including exposure to microbes, particles from the atmosphere, irritants, pollutants, allergens, and toxic molecules. To this end, an immediate, strong, and highly regulated inflammatory defense mechanism is needed for the successful maintenance of homeostasis within the respiratory system. Macroautophagy/autophagy plays an essential role in the inflammatory response of the lung to infection and stress. At baseline, autophagy may be critical for inhibiting spontaneous pulmonary inflammation and fundamental for the response of pulmonary leukocytes to infection; however, when not regulated, persistent or inefficient autophagy may be detrimental to lung epithelial cells, promoting lung injury. This perspective will discuss the role of autophagy in driving and regulating inflammatory responses of the lung in chronic lung diseases with a focus on potential avenues for therapeutic targeting.
Abbreviations
- AR
allergic rhinitis
- AM
alveolar macrophage
- ATG
autophagy-related
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane conductance regulator
- COPD
chronic obstructive pulmonary disease
- CS
cigarette smoke
- CSE
cigarette smoke extract
- DC
dendritic cell
- IH
intermittent hypoxia
- IPF
idiopathic pulmonary fibrosis
- ILD
interstitial lung disease
- MAP1LC3B
microtubule associated protein 1 light chain 3 beta
- MTB
Mycobacterium tuberculosis
- MTOR
mechanistic target of rapamycin kinase
- NET
neutrophil extracellular traps
- OSA
obstructive sleep apnea
- PAH
pulmonary arterial hypertension
- PH
pulmonary hypertension
- ROS
reactive oxygen species
- TGFB1
transforming growth factor beta 1
- TNF
tumor necrosis factor
KEYWORDS: asthma, autophagy, chronic obstructive pulmonary disease (COPD), inflammation, pulmonary fibrosis, pulmonary hypertension, sleep apnea, tuberculosis
Autophagy
Autophagy is a normal physiological process in the body involved in cellular homeostasis and survival mechanisms in normal respiring cells. Autophagy is a fundamental intracellular process responsible for the lysosomal degradation of microorganisms (viruses, bacteria, fungi and protists/protozoa), damaged organelles and damaged proteins that cannot be degraded by the proteasome. There are 3 main types of autophagy: macroautophagy, microautophagy and chaperone-mediated autophagy, all of which differ in their delivery methods to the lysosome.1 Often referred to as ‘autophagy’, macroautophagy is the best-characterized form of autophagy and begins with the initiation phase or development of a phagophore. In a subsequent elongation phase, the phagophore membrane expands to surround, target and engulf cytoplasmic cargo to form an autophagic vacuole or autophagosome with a distinctive double-membraned structure. The formation of such double-membraned structures proceeds through the coordinated complex rearrangement of subcellular membranes around a specific target. Subsequently, the fusion of whole autophagosomes with lysosomes results in the formation of autolysosomes where an array of lysosomal degradative enzymes (e.g., cathepsins and other acid hydrolases) digest the encapsulated contents of autolysosomes. The digested contents are then released to the cytosol for reutilization in biosynthetic pathways.
Autophagosome formation is primarily executed by a series of autophagy-related (ATG) genes, regulated by the activation and assembly of signaling components in response to upstream environmental cues, including nutrient deprivation and energy loss or the accumulation of damaged substrates. Among these ATG proteins, BECN1/BECLIN 1 (the mammalian homolog of yeast Vps30/Atg6) and MAP1LC3B/LC3B (microtubule associated protein 1 light chain 3 beta) (MAP1LC3B/ATG8F), a homolog of yeast Atg8 represent the most well-known ATG proteins. BECN1 associates with a macromolecular complex that includes PIK3C3/VPS34 (phosphatidylinositol 3-kinase catalytic subunit type 3). The BECN1 complex produces phosphatidylinositol-3-phosphate (PtdIns3P), a second messenger that regulates phagophore nucleation. Upon the activation of macroautophagy, MAP1LC3B is conjugated to phosphatidylethanolamine and targeted to autophagic membranes.2 Discussing the regulation and execution of autophagy in a comprehensive manner goes beyond the scope of the current article and the reader is referred to other reviews focusing on the molecular mechanisms behind this homeostatic process.3,4
Although autophagy functions as a dynamic recycling system for the cell, it also has a broad range of other functions including: the maintenance of ATP and energy metabolism; cell survival in response to starvation, nutrient deprivation or growth factor depletion1 removal of dysfunctional and damaged organelles (e.g. mitochondria),5 which is important in protein turnover and organelle quality control; regulation of mitochondrial homeostasis and the execution of programmed cell death pathways (e.g. apoptosis).6 Finally and more importantly for this review, autophagy is critical in the regulation of innate and adaptive immune responses,7 in particular in the respiratory system. As described in the introduction to this thematic issue on autophagy and inflammation, autophagy plays a central role in host defense through several mechanisms including: control of adaptive immunity through regulation of antigen presentation and lymphocyte development, direct elimination of invading pathogens, modulation of cytokine signaling and induction of innate immunity.7
Autophagy and the inflammatory response of the lung to acute infection
Inflammation in the lung arises as a result of the persistent exposure of the respiratory tract to microbes, particles from the atmosphere, irritants, pollutants, allergens and pathogens. In response to such stimuli, the lung employs a number of defense mechanisms including: the epithelial barrier, of which airway epithelial cells lining the respiratory tract secrete numerous substances including mucins, lysozymes, defensins, siderophores and nitric oxide; the mucociliary escalator, a primary innate defense mechanism, in which motile-ciliated epithelial cells eliminate particles and pathogens trapped in mucus from the airways8 and the cells of the innate immune system, which are critical for the aggregation, trapping and killing of microbes and include macrophages, dendritic cells, monocytes, neutrophils, eosinophils, natural killer cells, and mast cells, all of which produce various inflammatory mediators such as reactive oxygen species (ROS) and cytokines (e.g. IL6 [interleukin 6], TNF [tumor necrosis factor], and IL1B).
Pathogen recognition receptors present on resident lung alveolar macrophages (AMs) and dendritic cells (DCs) recognize pathogen-associated molecular patterns and/or damage-associated molecular patterns and respond to infection or injury within the respiratory tract. The subsequent initiation of inflammatory cascades and the secretion of cytokines and other signaling molecules result in the recruitment of additional immune cells (i.e. neutrophils and monocytes). DCs also function as antigen-presenting cells that link the innate and adaptive immune responses, by phagocytosing microbes and migrating to regional lymph nodes to activate lymphocytes including T cells and B cells. AMs aid in microbial killing and the clearance of apoptotic cells during pulmonary infection and secrete pro- and anti-inflammatory cytokines such as IL6 and TNF to regulate inflammatory and repair responses.9 Neutrophils, which reside in pulmonary capillaries and interstitial space, are recruited into air spaces by chemokines during infection to kill ingested microbes with antimicrobial proteins and ROS, and they release chemotactic factors, which recruit more monocytes to the site of infection.
Lymphocytes are found throughout the airways and lung parenchyma and consist of thymus-dependent T cells (CD8+ killer cells and CD4+ helper cells [Th1 and Th2]) and bone marrow-dependent B cells. Activated Th1 cells produce pro-inflammatory cytokines (TNF and IFNG [interferon gamma]), whereas Th2 cells produce IL4, and IL13,9 which can stimulate B cells to produce IGES (immunoglobulin E concentration, serum) causing mast cell degranulation, and IL5, which stimulates eosinophils.10 Mast cells are also activated through their IGES receptor (FCER1) producing cytokines, leukotrienes and proteases.9,10
An immediate, strong, and highly regulated inflammatory defense mechanism is needed for the timely elimination of pathogens and noxious particles from the lung. In addition the successful maintenance of homeostasis within the respiratory system depends on proper crosstalk between the innate and adaptive immune systems. Autophagy plays an essential role in the inflammatory response of the lung to infection and stress. At baseline, autophagy in AMs is critical for inhibiting spontaneous pulmonary inflammation11 and is fundamental for airway mucus secretion by airway goblet cells.12 Autophagy-deficient mice (atg5−/− and atg7−/−) develop spontaneous sterile lung inflammation, characterized by marked recruitment of inflammatory cells submucosal thickening, goblet cell metaplasia, and increased collagen content.13 Similarly deletion of ATG5 in murine ITGAX/CD11c+ cells results in unprovoked spontaneous airway hyperactivity and severe neutrophilic lung inflammation.14
During acute infection of the lung, autophagy activation appears to be a protective mechanism involved in the host responses to both bacterial and viral infectious states. For example, in response to P. aeruginosa infection, autophagy deficiency (ER-Cre:Atg7fl/fl) in mice impairs pathogen clearance, increases neutrophilic inflammation, increases the production of IL1B resulting in severe lung injury and decreases survival.15 Similarly, respiratory syncytial virus-infected autophagy-deficient mice (map1l3b−/−) develop increased IL17A-dependent lung pathology upon infection16 and have increased Th2 cytokine production, mucus secretion, and lung infiltration of eosinophils and inflammatory DCs (Becn1+/−).17 Whereas autophagy in the infected lung appears to represent a protective response, autophagy, by virtue of excessive autophagosome accumulation, may play a maladaptive role in the late stage of sepsis, leading to acute lung injury.18 In addition, loss of autophagy (siRNA targeting PIK3C3) in macrophages during lipopolysaccharide-induced lung inflammation reduces lung and bronchoalveolar immune cell infiltration and air space cytokine concentration.19
In chronic lung diseases, where there is prolonged inflammation and stress, the role of autophagy is more complex. We will herein discuss the role of autophagy in driving and regulating inflammatory responses (Table 1) of the lung in chronic lung diseases (Fig. 1) with a focus on potential avenues for therapeutic targeting.
Table 1.
Autophagy and inflammation in chronic respiratory disease.
| Cell type | Disease | Autophagy protective |
Autophagy detrimental |
Therapy |
|---|---|---|---|---|
| Airway epithelial cell | COPD | Autophagy inhibition promotes secretory phenotype with IL8 secretion.42 Loss of autophagy enhances smoke-induced cell senescence and accumulation of ubiquitinated proteins and SQSTM1.35,42,43 |
Autophagy deficient cells protected from smoke-induced cilia shortening.8 Autophagy promotes cell death in vitro and in vivo.32–34,38,39 |
Agents that will enhance the clearance of aggregated/damaged proteins by autophagy and/or other proteostatic mechanisms; cysteamine51; 4-phenyl butyric acid (4-PBA)8; arachidonic acid-derived epoxyeicosatrienoic acids (EETs)52; lacCer-synthase inhibitors47; the HDAC6 inhibitor tubastatin 18; the mitophagy inhibitor Mdivi134; and the sodium channel inhibitor carbamazepine.36 |
| Asthma, allergic rhinitis and sinusitis | Genetic or pharmacological inhibition of autophagy prevents IL13-induced mucus12 and CCL26 secretion.67 Autophagy promotes TFGB1 pro-fibrotic responses.69 |
Therapies (e.g., monoclonal anti-IL5 antibody, anti-nerve growth antibody etc.) that have efficacious outcomes in murine asthma models appear to downregulate autophagy.62,65,70 | ||
| Interstitial lung disease | MTOR overactivation in type II alveolar epithelial cells have increased mortality and pulmonary fibrosis mice.102 | Rapamycin treatment has demonstrated antifibrotic abilities in murine bleomycin-mediated mortality and fibrosis95,102; however, IPF patients taking the rapamycin analog everolimus had progression of disease.110 | ||
| Cystic fibrosis | Cells from human CF patients display accumulated polyubiquitinated proteins, defective autophagy and the decreased clearance of aggresomes.114 | Inhibition of autophagosome-lysosome fusion (Baf-A1) reduced anti-inflammatory effects114 | Increasing autophagy prior to infection with B. cepacia inhibits B. cepacia replication.117 Restoration of autophagy may be a novel approach to the treatment of CF. |
|
| Neutrophil | COPD | Loss of autophagy associated with a compromised capability to ingest Staphylococcus aureus.50 | ||
| Asthma, allergic rhinitis and sinusitis | The activation of autophagy correlates with NET production in neutrophils.61 | Neutrophil autophagy and NETs enhance asthma severity by damaging airway epithelium and triggering inflammatory responses.61,71 | In general, therapies (e.g., monoclonal anti-IL5 antibody, anti-nerve growth antibody etc.) that have efficacious outcomes in murine asthma models appear to downregulate autophagy.62,65,70 However, the therapeutic targeting of autophagy in asthma must be approached with caution as autophagy may play a more protective role in airway inflammation and hyperreactivity in the context of innate immune cell signaling. | |
| Cystic fibrosis | Autophagy drives NET formation in neutrophils, which promotes lung fibrosis.97 | In vivo, the autophagy activator (MTOR inhibitor) rapamycin decreases bacterial burden in the lungs of CF mice and drastically reduces signs of lung inflammation.113,115 | ||
| Macrophage | COPD | Reduced autophagic flux, defective delivery of SQSTM1 to the lysosome, defective xenophagy.49 | ||
| Interstitial lung disease | Autophagy deficiency exacerbates inflammation and fibrosis following silica exposure104 and bleomycin challenge.13 | park2−/− AMs have increased macrophage apoptosis and are protected from pulmonary fibrosis105 | Targeting autophagy in IPF may offer therapeutic potential including the use of IL17A neutralizing antibodies,106MIR449A107 or PDGFR inhibitors.99 | |
| Cystic fibrosis | ΔF508 macrophages have fewer autophagosomes, less BECN1, accumulation of SQSTM1 and less clearance of Pseudomonas aeruginosa.115 | The IL1 receptor antagonist, which activates the autophagy-lysosomal degradation pathway, increases the microbicidal activity of macrophages.118 | ||
| Pulmonary tuberculosis | Induction of autophagy with starvation or treatment with rapamycin decreases the survival of intracellular MTB during in vitro studies.24 Atg5-myeloid lineage-deficient mice have an exaggerated inflammatory response to MTB and increased mortality when infected with highly virulent MTB strains.27 |
Depletion of BECN1 and ATG7 within infected macrophages enhances the inhibition of phagosomal-lysosomal fusion.25 HIV-infected macrophages that are co-infected with MTB develop increased MTB replication when autophagy is stimulated through inhibition of the MTORC1 pathway.28 |
||
| Fibroblast | Asthma, allergic rhinitis and sinusitis | Autophagosomes increase in fibroblasts in asthma patient64—autophagy may be a cellular mechanism that promotes TGFB1 airway remodeling. | ||
| Interstitial lung disease | Loss of autophagy renders fibroblasts apoptosis resistant.96 | Restoring autophagy may have beneficial effects in fibroblast responses. Rapamycin treatment demonstrates antifibrotic abilities in murine bleomycin-mediated mortality and fibrosis95,102; however, IPF patients taking the rapamycin analog everolimus have progression of disease.110 | ||
| Vascular endothelial cell | Pulmonary hypertension | Pulmonary artery endothelial cells have decreased proliferation.87 | Becn1+/− increased angiogenesis in pulmonary artery endothelial cells.89 | Rapamycin and/or rapalogs may have therapeutic effects in the development of PH in animal models.91 |
Figure 1.
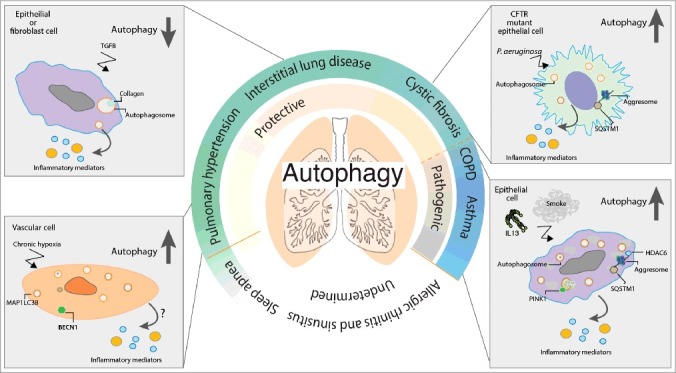
Autophagy and chronic lung disease. Autophagy plays an essential role in the inflammatory response of the lung to infection and stress. Autophagy is upregulated in response to a number of pathogenic stimuli including cigarette smoke, TGFB, IL13 and chronic hypoxia. The upregulation of autophagy may initially act as a prosurvival mechanism responsible for the clearance of damaged proteins or organelles. In the chronic lung diseases IPF, PH and CF, loss of autophagy drives lung inflammation and injury, suggesting autophagy plays a protective role. However, under certain circumstances, this naturally homeostatic cellular process may become overwhelmed or dysregulated and ultimately become unable to deal with the chronic burden and clearance of excessive autophagic targets. Such a burden may lead to the accumulation of aggresomes or damaged organelles with the concurrent appearance of increased autophagosomes, which may in turn be detrimental to the cell. Such cellular damage and stress caused by dysregulated autophagy drives lung inflammation and injury in a number of chronic lung diseases including COPD and asthma.
Pulmonary tuberculosis, autophagy and inflammation
Pulmonary tuberculosis, a disease caused by Mycobacterium tuberculosis (MTB), represents a chronic lung infection where long-term inflammation can lead to profound pathological consequences. The host immune response is critical in containing the MTB infection, as evidenced by the significant increase in incidence and mortality in immunocompromised hosts (i.e., HIV-infected patients).20 Retrospective analyses have described chronic obstructive ventilation defects, bronchiectasis, and emphysematous and fibrotic lung parenchymal alterations as the most common long-term sequelae of pulmonary tuberculosis following treatment.21,22 MTB is an obligate human pathogen that is transmitted through air droplets and is an intracellular bacterium that initially afflicts resident AMs and DCs localized to the distant alveoli. During the primary stage, the innate immune response is activated and inflammatory cells are recruited to the lungs. MTB that evades and destroys the AMs disseminates to the draining lymph nodes and triggers an antigen-specific Th1 T-cell response. Effector T-cells ultimately drive the formation of granulomas at the site of infection within the lung (latent MTB infection).23 It is this granulomatous inflammation that is thought to drive tissue injury, fibrosis, and, as such, the long-term clinical manifestations associated with pulmonary tuberculosis.22
It is well known that MTB replicates and persists within infected macrophages by preventing phagosome acidification and phagosome-lysosomal fusion. Autophagic processes have been linked to the clearance of MTB, whereby induction of autophagy with starvation, or treatment with rapamycin, decreases the survival of intracellular MTB during in vitro studies.24 In contrast, depletion of BECN1 and ATG7 within infected macrophages enhances the inhibition of phagosomal-lysosomal fusion.25 However, the role of autophagic proteins in the control of MTB revealed through in vivo studies is less clear.26 Atg5-myeloid lineage-deficient mice have an exaggerated inflammatory response to MTB and increased mortality when infected with highly virulent MTB strains.27 However, mice deficient in several other critical autophagy regulators do not reveal the same results.27 Additionally, recent in vitro studies revealed that HIV-infected macrophages that are co-infected with MTB develop increased MTB replication when autophagy is stimulated through inhibition of the MTOR (mechanistic target of rapamycin) kinase pathway.28 Whereas it was previously thought that the activation of autophagy represented a potential therapeutic strategy for MTB-infected patients, these recent data have called into question this possibility and limited further advancements.
Chronic obstructive pulmonary disease, autophagy and inflammation
Chronic obstructive pulmonary disease (COPD) is a debilitating irreversible inflammatory lung disease associated with cigarette smoking. COPD remains the third leading cause of death worldwide and is characterized by 3 major disease states: chronic bronchitis or excess mucus production in the proximal airways; emphysema or peripheral lung destruction and loss of alveolar attachments; and small airways disease characterized by inflammation and airway remodeling.29 Immune system disarray in response to the inhalation of cigarette smoke (CS) drives prolonged and exaggerated inflammation in the lung, which contributes to the development of COPD. Smokers have inflammation within their airways, which persists for many years even after smoking cessation and patients that develop COPD have prolonged exaggerated inflammatory responses that involve both the innate and adaptive immune systems.30 Upon smoke exposure AMs derived from circulating monocytes are thought to be the critical drivers of persistent inflammation in COPD and are thought to release chemokines that lead to the recruitment of CD8 lymphocytes, monocytes, and neutrophils into the bronchial epithelium.30 Repeated exposure to CS activates macrophages to release inflammatory mediators and the inflammatory cycle continues. Additionally, AMs secrete proteases and ELANE (elastase, neutrophil expressed) leading to degradation of ELN (elastin) and apoptosis of alveolar epithelial cells.
Epithelial cells lining the airways and alveoli represent a primary target of inhaled CS. Numerous reports document aberrant activation of autophagy,31–34 and selective autophagy including mitophagy,34,35 ciliophagy (selective removal of cilia components)8 and aggrephagy36 in lung epithelial cells of human COPD patients, in murine models and in cell culture model systems. The presence of polymorphisms in the autophagy gene ATG16L1 is a significant risk factor for susceptibility to COPD37 and autophagy is also increased in lung epithelium from patients with a genetic variant of emphysema, namely α1-antitrypsin deficiency (a1-AT), whose etiology is independent of smoke or particle inhalation.32
In general, autophagy is activated in the lung epithelium in response to CS. However, in the COPD community, the interpretation and consequent downstream interpretations of increased autophagy and its role in the pathogenesis of the disease is debated. Specifically, a number of studies demonstrate the activation of autophagy and selective autophagy as detrimental mechanisms in an epithelial cell in response to smoke.8,31–34 Specifically, these studies show that CS increases autophagosomal turnover (flux) and promotes epithelial cell death in vitro (5–20% cigarette smoke extract or CSE) and in vivo,32–34,38,39 which may in turn initiate and exaggerate airway inflammation40 and mucus hyperproduction.41 Consistently, loss of the autophagy genes Map1lc3b, Pink1 and Hdac6 protect mice from smoke-induced emphysema and impairment of mucociliary clearance.8,34 Conversely, in vitro studies using lower doses of smoke extract (0.5–1% CSE) have shown that loss of autophagy (PINK1 siRNA and chemical inhibition) enhances smoke-induced epithelial cell senescence, mitochondrial ROS production and the accumulation of ubiquitinated proteins along with accumulation of SQSTM1 (sequestosome 1),35,42,43 suggesting a more protective role for autophagy in epithelial cells in the pathogenesis of COPD. In these studies chemical activation of autophagy (determined by an accumulation of MAP1LC3B and SQSTM1) also protects cells in vitro42 and in vivo from smoke exposure.36 However, in the above studies caution must be exerted when interpreting autophagy-based activity on SQSTM1 expression. Smoke alone transcriptionally activates SQSTM144 and SQSTM1 can form insoluble aggregates itself.4 Assessment of flux independently of SQSTM1 expression, through lysosomal blockade or through the use of genetic approaches such as knock down of ATG7 or ATG5 is imperative to understanding the role of autophagy in the epithelial cell response to smoke.
While the above studies, in spite of obvious differences in the concentrations of smoke extract used, may appear to have led to opposing views in the interpretation of increasing levels of autophagy, a unifying hypothesis may involve a combination of both sides. Specifically, autophagy may be activated in an epithelial cell in response to increased accumulation of ubiquitinated proteins and aggresome bodies. Consistently, lung tissues from COPD patients demonstrate increased aggresome formation and SQSTM1 accumulation.8,36 In situations of low smoke concentrations or low stress burden, enhancing autophagy by promoting the clearance of these aggresome bodies may be beneficial.8,36 However, upon saturation of normal autophagic clearance mechanisms (such as with a high dose of smoke in vitro), in addition to the loss of proteasomal activity45,46 exposure to cigarette smoke may overwhelm the cells’ natural proteostatic mechanisms. Such an impairment of proteostasis with excessive activation of autophagy may lead to the accumulation of aggregated proteins and SQSTM1, leading to inefficient autophagy, epithelial cell dysfunction and death.8,32,33
While smoke-induced autophagy in epithelial cells appears to promote airway inflammation (IL6 production and lymphocyte recruitment into the lung),40,47,48 and impairment of mucociliary clearance function,8,34 little is known about the role of autophagy in the inflammatory responses of infiltrating leukocytes into the lung in response to smoke. Autophagy is increased in AMs and neutrophils in response to smoke. However, AMs isolated from the lungs of active smokers have a reduced ability to deliver bacteria to the lysosome (defective xenophagy) when compared to nonsmoker controls.49 Bacterial xenophagy is the macroautophagic removal of cytoplasmic bacteria, that is, bacteria that have escaped the phagosomal compartment upon phagocytosis.4 In addition, smoke-exposed neutrophils that have increased autophagy have compromised capability to ingest the respiratory pathogen, Staphylococcus aureus.50 Further extensive studies are required to determine if the smoke-induced autophagy observed in lung leukocytes is a protective or detrimental mechanism for the lung.
It appears that the most therapeutically viable mechanism to target autophagy in COPD involves agents that will enhance the clearance or the recognition of aggregated damaged proteins by autophagy and/or other proteostatic mechanisms. In murine models, a number of studies have attempted to therapeutically alleviate the aberrant autophagy observed during smoke exposure: these include the antioxidant drug, cysteamine51; the chemical chaperone 4-phenyl butyric acid8; arachidonic acid-derived epoxyeicosatrienoic acids (anti-inflammatory as well as regulating autophagy)52; lacCer-synthase inhibitors47; the HDAC6 (histone deacetylase 6) inhibitor tubastatin;18 the mitophagy inhibitor Mdivi134; and the sodium channel inhibitor carbamazepine.36 However, all of these studies involved a prophylactic treatment approach for the duration of smoke exposure. In addition, use of the MTOR inhibitor rapamycin demonstrates that while enhancing autophagy may be beneficial during CS exposure (reducing alveolar inflammation), at baseline, autophagy enhancement by rapamycin heightens the number of apoptotic and inflammatory cells in control mice, highlighting that the timing and lung cell targets of autophagy activation are crucial to define its beneficial and pathological roles in disease.53 Further studies are required to more extensively evaluate the role of these agents in treating the aberrant autophagy observed in COPD.
Asthma, allergic rhinitis, and sinusitis, autophagy and inflammation
Asthma is a complex disorder of the airways involving chronic airway inflammation, bronchial hyper-reactivity, mucus overproduction, airway wall remodeling and airway narrowing that affects 300 million people worldwide. Genetic susceptibility and environmental influences are important risk factors in the development of asthma,54 which ultimately presents as a dysregulated immune response to airborne allergens leading to a chronic inflammatory response. The most common entity is allergic-type asthma that is often associated with its upper airway counterpart allergic rhinitis (AR). AR symptoms include sneezing, rhinorrhea and pruritus as well as nasal congestion, lower respiratory tract infections and sinusitis.55 Both asthma and AR are disease states where environmental stimuli (i.e., house dust mite, animal dander, tree pollen, or fungal spores) activate DC and airway epithelial cells to initiate a Th2 immune response.56 This leads to the recruitment of secondary effector cells such as mast cells, basophils and neutrophils9 to the airways or nasal mucosa. Mediators and cytokines released from these recruited immune cells result in the increased production of IGES promoting eosinophil adhesion,10 increased mucus production and fibrosis,57 goblet cell hyperplasia, vasodilation with increased airway hyperresponsiveness and tissue damage.58
While there is little known about the role of autophagy in AR or sinusitis, autophagy seems to play a role in asthma pathogenesis; autophagy markers are increased in sputum granulocytes, fibroblasts, neutrophils, peripheral blood eosinophils and peripheral blood monocytes of patients with asthma.59–62 In addition, genetic variants in the autophagy gene ATG5 have been associated with the promotion of airway remodeling and loss of lung function in asthma.63,64 In a similar manner, inhalation of OVA (ovalbumin, murine asthma model) in mice increases autophagy in airway tissues.65,66 Little evidence exists on the regulation of autophagy in other murine asthma models including house dust mite antigen or other antigens. However, cell culture model systems suggest that in the lung epithelium, such an activation of autophagy may be detrimental and promote disease progression. Genetic (ATG5 and ATG14) or pharmacological (3-methyladenine or bafilomycin A1 (Baf-A1) inhibition of autophagy in cultured epithelial cells treated with IL13 results in less mucus secretion12 and less CCL26/EOTAXIN-3 (C-C motif chemokine ligand 26; a potent eosinophil chemokine) secretion,67 respectively, suggesting that autophagy may promote the epithelial Th2 response in asthma. Autophagy may also promote airway epithelial cell IL18 secretion in response to Alternaria alternata, an outdoor allergen that causes allergic airway diseases,68 as well as well as promoting a profibrotic response of primary human airway smooth muscle cells to TGFB1 (transforming growth factor beta 1).69 Consistently, autophagy inhibition (3-methyladenine and ATG5 shRNA treatment) in a murine OVA model of asthma reduces airway responsiveness, eosinophilia, and inflammation.62
In general, therapies (e.g., monoclonal anti-IL5 antibody, anti-nerve growth antibody, astragalin, etc.) that have efficacious outcomes in murine asthma models appear to downregulate autophagy.62,65,70 However, the therapeutic targeting of autophagy in asthma must be approached with caution as autophagy may play a more protective role in airway inflammation and hyperreactivity in the context of innate immune cell signaling. Whereas the activation of autophagy correlates with neutrophil extracellular traps (NETs) production in neutrophils61 and genetic polymorphisms of ATG5 and ATG7 contribute to neutrophilic airway inflammation in the pathogenesis of adult asthma,71 severe lung inflammation impairs autophagy in lung ITGAX/CD11c+ cells, and loss of autophagy in these cells induces unprovoked spontaneous airway hyperactivity and severe IL17A-mediated neutrophilic lung inflammation.14
Obstructive sleep apnea syndrome, autophagy and inflammation
Obstructive sleep apnea (OSA) is a breathing disorder that occurs when recurrent upper airway obstruction during sleep leads to episodes of apnea, hypopnea, and/or respiratory effort awakenings. These respiratory disturbances often lead to sleep fragmentation, hypoxemia, hypercapnia, and increased sympathetic activity along with an increased risk of hypertension, congestive heart failure, type II diabetes, stroke, and premature death.72 This is an area of growing public health concern as the most current data show that the prevalence of moderate to severe OSA ranges from 10–17% in men and 3–9% in women, a notable increase of more than 25% over the past 2 decades.73
The underlying pathological mechanisms driving the systemic dysfunction seen in patients with OSA are complex and not fully understood, but the general consensus is that the effects are linked to the regular events of intermittent hypoxia (IH) experienced by patients. In vivo experiments link IH in abnormal glucose metabolism and obesity. However, there is accumulating evidence that highlights the role of IH in driving local and systemic inflammation that causes the OSA-related cardio-metabolic disease.74 These studies report increases in leukocyte-endothelial cellular interactions and T cell activation, which are postulated to drive vascular inflammation and remodeling. IH is a known activator of the NFKB1/2 (nuclear factor kappa B subunit 1/2) pathways, which ultimately serves as a potent inflammatory activator resulting in the release of TNF, IL6, IL8, and CCL2/MCP-1 (C-C motif chemokine ligand 2). There is human data that suggest increased circulating ROS in patients with OSA, but the studies are limited.75 While more studies are needed, it is understood that systemic inflammation plays a role in the pathogenesis of the disease processes associated with OSA.
Recently, the role of autophagy in the pathogenesis of OSA has been queried. Animal studies have linked the regulation of autophagy and chronic intermittent hypoxia.76 Using a chronic IH rat model, investigators demonstrated that increases in autophagy, via administration of melatonin, protect against cardiac changes commonly observed in OSA.77 Interestingly, an additional study tested the role of autophagy in driving insulin resistance in response to chronic intermittent hypoxia and found that these processes were not causatively linked.78 These data suggest that autophagy may play a role in the systemic processes driven by chronic intermittent hypoxia experienced by OSA patients, but more studies are required to define which processes rely on autophagic control (i.e., cardiac health vs. insulin resistance). This is an important area of ongoing research, as supplementing our current management strategies with autophagy activators/inhibitors would provide advanced methods of managing the systemic complications of this disease.
Pulmonary hypertension, autophagy and inflammation
Pulmonary arterial hypertension (PAH) is a severely debilitating and fatal disease characterized by pulmonary vascular remodeling, leading to a sustained rise in pulmonary artery pressure, which causes right ventricular failure and death. Variants of pulmonary hypertension (PH) affect more than 100 million people worldwide and the current treatments focus on encouraging vasodilation, but fail to halt or reverse ongoing vascular remodeling.79 The small distal arterioles are the targets of the disease and the vascular wall represents a pro-proliferative/anti-apoptotic and hypermetabolic state, akin to a malignant tumor microenvironment, leading to occlusion of the blood vessels over time. The role of inflammation in the development of PAH is supported by the observation that perivascular inflammatory infiltrates, consisting of dysregulated lymphocytes, AMs, DCs, and mast cells, frequently oppose the pulmonary arterial vascular lesions found in human PAH tissue samples.80 The extent of perivascular inflammation is linked with enhanced intima and media remodeling, reflective of a more severe disease state.81 The extracellular matrix also perpetuates the chronic inflammatory state and vascular remodeling found in PH.82
In addition to the local inflammatory process, a systemic inflammatory profile of elevated circulating levels of pro-inflammatory cytokines and chemokines including IL6, TNF, IL1B, IL18, and CCL2 in PAH patients are described.83 This pro-inflammatory state is correlated with poorer outcomes, a concept that is mimicked in in vivo studies.84,85 Further investigations revealed a potential pathogenic role for inflammasome activation in the development of PH when mice lacking the inflammasome adaptor protein PYCARD/ASC (PYD and CARD domain containing) were found to have attenuated hypoxia-induced remodeling.86
Several studies have probed the role of autophagy in driving the pathogenesis of PH and the conclusions remain controversial. It is well documented that there is an elevated incidence of autophagy detected in human PAH lung tissue samples.87 In the chronic hypoxia mouse model, protein markers of autophagy (MAP1LC3B) and autophagosomes in total lung tissue are increased. map1lc3b−/− mice have more severe observable PH after exposure to chronic hypoxia compared to wild-type mice, and loss of BECN1, results in increased angiogenesis in pulmonary artery endothelial cells from fetal lambs with persistent pulmonary hypertension.88,89 In contrast, inhibition of autophagy by treating rats with chloroquine blocks the development of PAH in the monocrotaline rat model.90 Further studies are needed to clarify the conditions where autophagy protects against or augments the pathogenesis of PH.
Interestingly, there have been several studies examining the role of rapamycin and/or rapalogs, MTOR inhibitors and activators of autophagy, in the prevention of the development of PH in animal models.91 Furthermore, following the use of one of the rapalogs, everolimus, in the treatment of patients with severe PH secondary to chronic thromboembolic disease there was improvement of exercise tolerance and a decrease in pulmonary vascular resistance.92 Whether these observed improvements are a consequence of autophagy-mediated inhibition of inflammasome activity remains to be clarified.
Interstitial lung disease, autophagy and inflammation
Interstitial lung diseases (ILDs) include a wide spectrum of disorders whose pathogenesis still is not fully understood. Suggested risk factors for the initiation and progression of ILDs include genetic and epigenetic abnormalities as well as infection or environmental exposures (such as cigarette smoking or silica exposure). Idiopathic pulmonary fibrosis (IPF) is the most frequent, severe and most studied form of ILDs. IPF is characterized by the aberrant deposition of extracellular matrix, as a result of repetitive injury to the alveolar epithelium. Epithelial cells release mediators that activate fibroblast proliferation leading to the presence of abundant foci of highly activated fibroblasts and myofibroblasts that are resistant to apoptosis and lead to extensive lung remodeling. While IPF is an epithelial-driven disease it also displays elements of a pathological adaptive immune response, including dysregulated T- and B-cell responses. Inflammation-driven pathways are also thought to be significant pathogenic drivers in ILD, and current treatment approaches predominantly consist of immunomodulatory agents.93 The inflammatory pathways involved in the pathogenesis of the multiple forms of ILD are not entirely understood, but the notion that common pathways are triggered with injury/disease, which lead to destruction of the alveolar-capillary basement membrane and the development of fibrosis is appreciated. In particular, it is thought that the resolution of ongoing inflammatory processes through phagocytosis and apoptosis is inappropriately halted. As a result, activation of cytokines, chemokines, and growth factors persist and remain unchecked.94
Autophagic pathways (including macroautophagy and mitophagy) are decreased in lung epithelial cells and lung fibroblasts from IPF patients,95–99 in murine models of IPF (bleomycin, TGFB1-adenoviral and silicosis models)98 and in fibroblasts activated with TGFB1.95 In general, autophagy appears to be a protective mechanism controlling the development of fibrosis in the lung by regulating collagen degradation and autophagy-associated cell death. Consistently, autophagy (atg4b−/−) and mitophagy deficient (pink1−/−, park2−/−) mice display significantly higher inflammatory responses (increased neutrophilic infiltration and significant alterations in proinflammatory cytokines) to bleomycin-induced lung fibrosis.98–101 Similarly mice with MTOR overactivation in type II alveolar epithelial cells (conditional and inducible TSC1 [tuberous sclerosis 1]) knockdown mice SPCrtTA/TetO-Cre/Tsc1fx/+) have increased mortality and pulmonary fibrosis compared with control mice.102
Fibrosis may represent the final step induced by a chronic inflammatory state, where fibrosis is triggered by cytokines, chemokines and growth factors released by lymphocytes or macrophages. In general, autophagy appears to be a protective mechanism in infiltrating immune cells in the lung in IPF. Neutrophil activation by fibrosis-related agents and the release of NETs plays an important role in the inflammation observed in IPF. Autophagy regulates NET formation by neutrophils in response to pro-fibrotic stimuli, and both autophagy and NETs are involved not only in inflammation but also in the ensuing fibrosis.103 Autophagy deficiency in macrophages (Atg5fl/fl/Lyz2Cre mice and Atg7fl/fl/Lyz2cre) exacerbates inflammation and fibrosis following silica exposure104 and bleomycin challenge.13 However, the activation of mitophagy in AMs may promote apoptosis resistance during the fibrotic process. Specifically, mitophagy is activated in AMs from IPF patients and from bleomycin-treated mice, and mitophagy deficient AMs (park2−/−) mice have increased macrophage apoptosis and are protected from pulmonary fibrosis.105 Taken together the above studies indicate that pro-fibrotic AMs may require autophagy for polarization and apoptosis resistance, whereas anti-fibrotic macrophages generating pro-inflammatory cytokines require autophagy for protection. More extensive studies are required to explore the role of autophagy in macrophage polarization in IPF.
Targeting autophagy in IPF may offer therapeutic potential including the use of IL17A neutralizing antibodies,106 MIR449A (microRNA 449a),107 or PDGFRB (platelet derived growth factor receptor beta) inhibitors.99 In addition, MAP1LC3B overxpression in vitro protects against the development of Hermansky-Pudlak syndrome, a rare autosomal recessive disorder associated with the development of pulmonary fibrosis.108 Rapamycin treatment has demonstrated antifibrotic abilities in murine bleomycin-mediated mortality and fibrosis95,102. However, rapamycin appears to enhance the effects induced by silicon dioxide109 and IPF patients taking the rapamycin analog everolimus have progression of disease.110 Again, further studies are required to evaluate the benefits of targeting autophagy in IPF and whether or not these targeting strategies should be cell type-specific.
Cystic fibrosis and noncystic fibrosis bronchiectasis, autophagy and inflammation
Cystic fibrosis (CF) is a fatal autosomal recessive disease, which is caused by mutation in the CFTR (cystic fibrosis transmembrane conductance regulator) gene. CF is characterized by abnormally viscous mucus, which obstructs the airways, resulting in recurrent pulmonary infections. The progressive obliteration of the airways, leading to the bronchiectasis found in CF, is a consequence of chronic neutrophilic inflammation as a result of persistent infection. Noncystic fibrosis bronchiectasis is a distinct clinical entity, but patients’ respiratory systems are also complicated by cough, sputum, and recurrent chest infections. Unlike CF, the causes of non-CF bronchiectasis are many, and include chronic or post-infection states, chronic immunodeficiency, foreign body or aspiration, rheumatological disorders, inflammatory bowel disease, and disorders of ciliary dysfunction.111 The pathogenesis of both disease processes depends on persistent colonization of the respiratory tract with pathogenic bacteria, which triggers a profound immune response coordinated by airway epithelial cells through the release of cytokines and chemokines (IL8, and TNF). As a result, immune cells, namely neutrophils, macrophages, and lymphocytes, migrate to the airways and release ELANE and metalloproteinases. These proteases break down mucosal barriers, increase mucus secretion, and further impair clearance.112
Pseudomonas aeruginosa (P. aeruginosa) is a bacterial pathogen, which is the leading cause of morbidity and mortality among CF patients. Autophagy is required for an effective immune response against P. aeruginosa infection in vivo. Whereas P. aeruginosa induces autophagy in mast cells in culture, inhibition of autophagy (with chloroquine) in murine models inhibits the clearance of intracellular P. aeruginosa from the lung.113 Lung epithelial cells from human CF patients displaying the CFTR mutation display accumulated polyubiquitinated proteins, defective autophagy and the decreased clearance of aggresomes. Such dysfunctional autophagosome clearance contributes to the heightened inflammatory responses from CFTR mutant cells.114 Loss of BECN1 has been associated with such defects in autophagosomal clearance. Defective autophagy due to decreased levels of BECN1 renders murine lungs more susceptible to infection by P. aeruginosa and by Burkholderia cenocepacia (B. cepacia).113,115,116 Infection by B. cepacia is a particularly lethal threat to CF patients because it causes severe and persistent lung inflammation and is resistant to nearly all available antibiotics. Increasing autophagy prior to infection with B. cepacia inhibits B. cepacia replication.117
In vivo, rapamycin decreases bacterial burden in the lungs of CF mice and drastically reduces signs of lung inflammation.113,115 Similarly the IL1RN (interleukin 1 receptor antagonist), which activates the autophagy-lysosomal degradation pathway, increases the microbicidal activity of macrophages.118 Restoration of autophagy may be a novel approach to the treatment of CF and may pave the way for the development of a new class of drugs that, by enhancing autophagy or by enhancing BECN1 levels, could be effective treatments for CF.116
Conclusion
It is clear that autophagy has a critical role in both the normal functioning of the inflammatory response system of the lung, as well as in the development and pathogenesis of numerous chronic lung diseases. However, what is also clear is that autophagy, while protective in pro-inflammatory immune cells and in fibroblasts, may promote or exacerbate pathogenic mechanisms when activated in cells such as epithelial cells or in polarized pro-fibrotic macrophages. Targeting autophagy for therapeutic exploitation in chronic lung disease will therefore depend on the exact nature of the autophagic defect present in each cell type. In studies discussed in this review, the exact autophagic defects contributing to chronic lung disease pathogenesis have yet to be fully elucidated. This can be attributed in part to the lack of extensive autophagic flux assays and to the limited specificity of current autophagy modulators in targeting one cell type. In addition, when interpreting the genetic experiments carried out in these studies it is important to consider the fact that several components of the autophagic machinery operate at the interface of multiple cellular processes mediating autophagy-independent functions.119 Similarly, interpreting the chemical-autophagy modulator experiments carried out in these studies requires consideration of the fact that these chemicals may work through off-target autophagy-independent mechanisms, with many FDA-approved drugs being able to activate or inhibit autophagy to some extent. In spite of great potential, no interventions that are specifically aimed at modulating autophagy are currently available for use in humans.119
Evaluating the real impact of autophagy activators or inhibitors in chronic lung diseases where the autophagic response in different compartments of the lung may have opposite effects is challenging. Addressing this complexity by studying the effects of highly targeted autophagy modulators in disease models with cell-specific autophagic defects is key to the development of clinically viable strategies to activate or inhibit autophagy.119
Funding Statement
This work was supported by the American Lung Association of the Northeast [grant number RG-348928]; HHS | NIH | National Heart, Lung, and Blood Institute (NHBLI) [grant number P01-HL114501], [grant number R01-HL079904], [grant number P01-HL105339], [grant number K99-HL125899], [grant number R01-HL055330].
Acknowledgments
This work was supported by the US National Institute of Health–National Heart, Lung and Blood Institute; K99-HL125899 (S.M.C.), P01-HL114501 (A.M.K.C.), R01-HL055330 (A.M.K.C.), R01-HL079904 (A.M.K.C.) and P01-HL105339 (to Edwin K. Silverman) and by an American Lung Association Biomedical Research grant RG-348928 (S.M.C.).
References
- 1.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–741. doi: 10.1016/j.cell.2011.10.026. PMID:22078875 [DOI] [PubMed] [Google Scholar]
- 2.Mizumura K, Cloonan SM, Haspel JA, Choi AMK. The emerging importance of autophagy in pulmonary diseases. Chest. 2012;142(5):1289–1299. doi: 10.1378/chest.12-0809. PMID:23131937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Choi AMK, Ryter SW, Levine B. Autophagy in human health and disease. New England Journal of Medicine. 2013;368(7):651–662. doi: 10.1056/NEJMra1205406. PMID:23406030 [DOI] [PubMed] [Google Scholar]
- 4.Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, Choi AM, Chu CT, Codogno P, Colombo MI, et al. Molecular definitions of autophagy and related processes. The EMBO Journal. 2017;36(13):1811–1836. doi: 10.15252/embj.201796697. PMID:28596378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nature Reviews. Molecular Cell Biology. 2011;12(1):9–14. doi: 10.1038/nrm3028. PMID:21179058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galluzzi L, Vicencio JM, Kepp O, Tasdemir E, Maiuri MC, Kroemer G. To die or not to die: That is the autophagic question. Current Molecular Medicine. 2008;8(2):78–91. doi: 10.2174/156652408783769616. PMID:18336289 [DOI] [PubMed] [Google Scholar]
- 7.Deretic V, Levine B. Autophagy, immunity, and microbial adaptations. Cell Host & Microbe. 2009;5(6):527–549. doi: 10.1016/j.chom.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lam HC, Cloonan SM, Bhashyam AR, Haspel JA, Singh A, Sathirapongsasuti JF, Cervo M, Yao H, Chung AL, Mizumura K, et al. Histone deacetylase 6-mediated selective autophagy regulates COPD-associated cilia dysfunction. J Clin Invest. 2013;123(12):5212–5230. doi: 10.1172/JCI69636. PMID:24200693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moldoveanu B, Otmishi P, Jani P, Walker J, Sarmiento X, Guardiola J, Saad M, Yu J. Inflammatory mechanisms in the lung. Journal of Inflammation Research. 2009;2:1–11. PMID:22096348 [PMC free article] [PubMed] [Google Scholar]
- 10.Holgate ST. Pathogenesis of asthma. Clinical and Experimental Allergy: Journal of the British Society for Allergy and Clinical Immunology. 2008;38(6):872–897. doi: 10.1111/j.1365-2222.2008.02971.x. PMID:18498538 [DOI] [PubMed] [Google Scholar]
- 11.Kanayama M, He YW, Shinohara ML. The lung is protected from spontaneous inflammation by autophagy in myeloid cells. Journal of Immunology (Baltimore, Md.: 1950). 2015;194(11):5465–5471. doi: 10.4049/jimmunol.1403249. PMID:25911758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dickinson JD, Alevy Y, Malvin NP, Patel KK, Gunsten SP, Holtzman MJ, Stappenbeck TS, Brody SL, et al. IL13 activates autophagy to regulate secretion in airway epithelial cells. Autophagy. 2016;12(2):397–409. doi: 10.1080/15548627.2015.1056967. PMID:26062017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abdel Fattah E, Bhattacharya A, Herron A, Safdar Z, Eissa NT. Critical role for IL-18 in spontaneous lung inflammation caused by autophagy deficiency. Journal of Immunology (Baltimore, Md.: 1950). 2015;194(11):5407–5416. doi: 10.4049/jimmunol.1402277. PMID:25888640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suzuki Y, Maazi H, Sankaranarayanan I, Lam J, Khoo B, Soroosh P, Barbers RG, James Ou JH, Jung JU, Akbari O, et al. Lack of autophagy induces steroid-resistant airway inflammation. Journal of Allergy and Clinical Immunology. 2016;137(5):1382–1389.e1389. doi: 10.1016/j.jaci.2015.09.033. PMID:26589586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pu Q, Gan C, Li R, Li Y, Tan S, Li X, Wei Y, Lan L, Deng X, Liang H, et al. Atg7 deficiency intensifies inflammasome activation and pyroptosis in pseudomonas sepsis. J Immunol. 2017;198(8):3205–3213. doi: 10.4049/jimmunol.1601196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reed M, Morris SH, Owczarczyk AB, Lukacs NW. Deficiency of autophagy protein Map1-LC3b mediates IL-17-dependent lung pathology during respiratory viral infection via ER stress-associated IL-1. Mucosal Immunology. 2015;8(5):1118–1130. doi: 10.1038/mi.2015.3. PMID:25669150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reed M, Morris SH, Jang S, Mukherjee S, Yue Z, Lukacs NW. Autophagy-inducing protein beclin-1 in dendritic cells regulates CD4 T cell responses and disease severity during respiratory syncytial virus infection. Journal of Immunology (Baltimore, Md.: 1950). 2013;191(5):2526–2537. doi: 10.4049/jimmunol.1300477. PMID:23894198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lo S, Yuan SS, Hsu C, Cheng YJ, Chang YF, Hsueh HW, Lee PH, Hsieh YC, et al. Lc3 over-expression improves survival and attenuates lung injury through increasing autophagosomal clearance in septic mice. Annals of Surgery. 2013;257(2):352–363. doi: 10.1097/SLA.0b013e318269d0e2. PMID:22968077 [DOI] [PubMed] [Google Scholar]
- 19.Guo L, Stripay JL, Zhang X, Collage RD, Hulver M, Carchman EH, Howell GM, Zuckerbraun BS, Lee JS, Rosengart MR, et al. CaMKIalpha regulates AMP kinase-dependent, TORC-1-independent autophagy during lipopolysaccharide-induced acute lung neutrophilic inflammation. Journal of Immunology (Baltimore, Md.: 1950). 2013;190(7):3620–3628. doi: 10.4049/jimmunol.1102975. PMID:23447692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kwan CK, Ernst JD. HIV and tuberculosis: A deadly human syndemic. Clin Microbiol Rev. 2011;24(2):351–376. doi: 10.1128/CMR.00042-10. PMID:21482729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Byrne AL, Marais BJ, Mitnick CD, Lecca L, Marks GB. Tuberculosis and chronic respiratory disease: A systematic review. Int J Infect Dis. 2015;32:138–146. doi: 10.1016/j.ijid.2014.12.016. PMID:25809770 [DOI] [PubMed] [Google Scholar]
- 22.Hunter RL. Pathology of post primary tuberculosis of the lung: an illustrated critical review. Tuberculosis (Edinb). 2011;91(6):497–509. doi: 10.1016/j.tube.2011.03.007. PMID:21733755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sasindran SJ, Torrelles JB. Mycobacterium tuberculosis infection and inflammation: What is beneficial for the host and for the bacterium? Front Microbiol. 2011;2:2. doi: 10.3389/fmicb.2011.00002. PMID:21687401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119(6):753–766. doi: 10.1016/j.cell.2004.11.038. PMID:15607973 [DOI] [PubMed] [Google Scholar]
- 25.Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science (New York, N.Y.). 2006;313(5792):1438–1441. doi: 10.1126/science.1129577. PMID:16888103 [DOI] [PubMed] [Google Scholar]
- 26.Behar SM, Baehrecke EH. Tuberculosis: Autophagy is not the answer. Nature. 2015;528(7583):482–483. doi: 10.1038/nature16324. PMID:26649822 [DOI] [PubMed] [Google Scholar]
- 27.Kimmey JM, Huynh JP, Weiss LA, Park S, Kambal A, Debnath J, Virgin HW, Stallings CL. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature. 2015;528(7583):565–569. doi: 10.1038/nature16451. PMID:26649827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andersson AM, Andersson B, Lorell C, Raffetseder J, Larsson M, Blomgran R. Autophagy induction targeting mTORC1 enhances Mycobacterium tuberculosis replication in HIV co-infected human macrophages. Scientific Reports. 2016;6:28171. doi: 10.1038/srep28171. PMID:27302320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burney PG, Patel J, Newson R, Minelli C, Naghavi M. Global and regional trends in COPD mortality, 1990–2010. Eur Respir J. 2015;45(5):1239–1247. doi: 10.1183/09031936.00142414. PMID:25837037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brusselle GG, Joos GF, Bracke KR. New insights into the immunology of chronic obstructive pulmonary disease. Lancet (London, England). 2011;378(9795):1015–1026. doi: 10.1016/S0140-6736(11)60988-4. PMID:21907865 [DOI] [PubMed] [Google Scholar]
- 31.An CH, Wang XM, Lam HC, Ifedigbo E, Washko GR, Ryter SW, Choi AM. TLR4 deficiency promotes autophagy during cigarette smoke-induced pulmonary emphysema. American Journal of Physiology. Lung Cellular and Molecular Physiology. 2012;303(9):L748–757. doi: 10.1152/ajplung.00102.2012. PMID:22983353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen ZH, Kim HP, Sciurba FC, Lee SJ, Feghali-Bostwick C, Stolz DB, Dhir R, Landreneau RJ, Schuchert MJ, Yousem SA, Nakahira K, et al. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PloS One. 2008;3(10):e3316. doi: 10.1371/journal.pone.0003316. PMID:18830406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen ZH, Lam HC, Jin Y, Kim HP, Cao J, Lee SJ, Ifedigbo E, Parameswaran H, Ryter SW, Choi AM. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc Natl Acad Sci USA. 2010;107(44):18880–18885. doi: 10.1073/pnas.1005574107. PMID:20956295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T, Glass K, Owen CA, Mahmood A, Washko GR, et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest. 2014;124(9):3987–4003. doi: 10.1172/JCI74985. PMID:25083992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ito S, Araya J, Kurita Y, Kobayashi K, Takasaka N, Yoshida M, Hara H, Minagawa S, Wakui H, Fujii S, et al. PARK2-mediated mitophagy is involved in regulation of HBEC senescence in COPD pathogenesis. Autophagy. 2015;11(3):547–559. doi: 10.1080/15548627.2015.1017190. PMID:25714760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tran I, Ji C, Ni I, Min T, Tang D, Vij N. Role of cigarette smoke-induced aggresome formation in chronic obstructive pulmonary disease-emphysema pathogenesis. American Journal of Respiratory Cell and Molecular Biology. 2015;53(2):159–173. doi: 10.1165/rcmb.2014-0107OC. PMID:25490051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen CZ, Ou CY, Wang RH, Lee CH, Lin CC, Chang HY, Hsiue TR. Association of Egr-1 and autophagy-related gene polymorphism in men with chronic obstructive pulmonary disease. Journal of the Formosan Medical Association = Taiwan yi zhi. 2015;114(8):750–755. doi: 10.1016/j.jfma.2013.07.015. PMID:24012056 [DOI] [PubMed] [Google Scholar]
- 38.Wang G, Zhou H, Strulovici-Barel Y, Al-Hijji M, Ou X, Salit J, Walters MS, Staudt MR, Kaner RJ, Crystal RG. Role of OSGIN1 in mediating smoking-induced autophagy in the human airway epithelium. Autophagy. 2017;13(7):1205–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hou HH, Cheng SL, Chung KP, Kuo MY, Yeh CC, Chang BE, Lu HH, Wang HC, Yu CJ, et al. Elastase induces lung epithelial cell autophagy through placental growth factor: A new insight of emphysema pathogenesis. Autophagy. 2014;10(9):1509–1521. doi: 10.4161/auto.29190. PMID:24988221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li D, Hu J, Wang T, Zhang X, Liu L, Wang H, Wu Y, Xu D, Wen F, et al. Silymarin attenuates cigarette smoke extract-induced inflammation via simultaneous inhibition of autophagy and ERK/p38 MAPK pathway in human bronchial epithelial cells. Scientific Reports. 2016;6:37751. doi: 10.1038/srep37751. PMID:27874084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou JS, Zhao Y, Zhou HB, Wang Y, Wu YF, Li ZY, Xuan NX, Zhang C, Hua W, Ying SM, et al. Autophagy plays an essential role in cigarette smoke-induced expression of MUC5AC in airway epithelium. American Journal of Physiology. Lung Cellular and Molecular Physiology. 2016;310(11):L1042–1052. doi: 10.1152/ajplung.00418.2015. PMID:27036871 [DOI] [PubMed] [Google Scholar]
- 42.Fujii S, Hara H, Araya J, Takasaka N, Kojima J, Ito S, Minagawa S, Yumino Y, Ishikawa T, Numata T, et al. Insufficient autophagy promotes bronchial epithelial cell senescence in chronic obstructive pulmonary disease. Oncoimmunology. 2012;1(5):630–641. doi: 10.4161/onci.20297. PMID:22934255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roscioli E, Tran HB, Jersmann H, Nguyen PT, Hopkins E, Lester S, Farrow N, Zalewski P, Reynolds PN, Hodge S. The Uncoupling of Autophagy and Zinc Homeostasis in Airway Epithelial Cells as a Fundamental Contributor to COPD. Am J Physiol Lung Cell Mol Physiol. 2017;313(3):L453–L465. doi: 10.1152/ajplung.00083.2017. [DOI] [PubMed] [Google Scholar]
- 44.Zhu L, Barrett EC, Xu Y, Liu Z, Manoharan A, Chen Y. Regulation of Cigarette Smoke (CS)-Induced Autophagy by Nrf2. PloS One. 2013;8(4):e55695. doi: 10.1371/journal.pone.0055695. PMID:23585825 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45.Yamada Y, Tomaru U, Ishizu A, et al. Decreased proteasomal function accelerates cigarette smoke-induced pulmonary emphysema in mice. Laboratory Investigation; A Journal of Technical Methods and Pathology. 2015;95(6):625–634. doi: 10.1038/labinvest.2015.43. PMID:25915723 [DOI] [PubMed] [Google Scholar]
- 46.van Rijt SH, Keller IE, John G, Ito T, Kiuchi T, Ono A, Miyajima S, Nagai K, Higashi T, Matsuno Y, et al. Acute cigarette smoke exposure impairs proteasome function in the lung. American Journal of Physiology. Lung Cellular and Molecular Physiology. 2012;303(9):L814–823. doi: 10.1152/ajplung.00128.2012. PMID:22962013 [DOI] [PubMed] [Google Scholar]
- 47.Bodas M, Min T, Vij N. Lactosylceramide-accumulation in lipid-rafts mediate aberrant-autophagy, inflammation and apoptosis in cigarette smoke induced emphysema. Apoptosis: An International Journal on Programmed Cell Death. 2015;20(5):725–739. doi: 10.1007/s10495-015-1098-0. PMID:25638276 [DOI] [PubMed] [Google Scholar]
- 48.Bodas M, Patel N, Silverberg D, Walworth K, Vij N. Master autophagy regulator transcription factor EB regulates cigarette smoke-induced autophagy impairment and chronic obstructive pulmonary disease-emphysema pathogenesis. Antioxid Redox Signal. 2017;27(3):150–167. doi: 10.1089/ars.2016.6842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Monick MM, Powers LS, Walters K, Lovan N, Zhang M, Gerke A, Hansdottir S, Hunninghake GW, et al. Identification of an autophagy defect in smokers' alveolar macrophages. Journal of Immunology (Baltimore, Md.: 1950). 2010;185(9):5425–5435. doi: 10.4049/jimmunol.1001603. PMID:20921532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guzik K, Skret J, Smagur J, Bzowska M, Gajkowska B, Scott DA, Potempa JS, et al. Cigarette smoke-exposed neutrophils die unconventionally but are rapidly phagocytosed by macrophages. Cell Death & Disease. 2011;2:e131. doi: 10.1038/cddis.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vij N, Chandramani P, Westphal CV, Hole R, Bodas M. Cigarette smoke induced autophagy-impairment accelerates lung aging, COPD-emphysema exacerbations and pathogenesis. Am J Physiol Cell Physiol. 2016:ajpcell 00110 02016. doi: 10.1152/ajpcell.00110.2016. PMID:27413169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li Y, Yu G, Yuan S, Tan C, Lian P, Fu L, Hou Q, Xu B, Wang H. Cigarette smoke-induced pulmonary inflammation and autophagy are attenuated in ephx2-deficient mice. Inflammation. 2017;40(2):497–510. doi: 10.1007/s10753-016-0495-z. PMID:28028752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yoshida T, Mett I, Bhunia AK, Bowman J, Perez M, Zhang L, Gandjeva A, Zhen L, Chukwueke U, Mao T, et al. Rtp801, a suppressor of mTOR signaling, is an essential mediator of cigarette smoke-induced pulmonary injury and emphysema. Nature Medicine. 2010;16(7):767–773. doi: 10.1038/nm.2157. PMID:20473305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nelson HS. The importance of allergens in the development of asthma and the persistence of symptoms. Disease-a-Month: DM. 2001;47(1):5–15. doi: 10.1067/mda.2000.da0470005. PMID:11182682 [DOI] [PubMed] [Google Scholar]
- 55.Bousquet J, van Cauwenberge P, Khaltaev N. Allergic rhinitis and its impact on asthma. J Allergy Clin Immunol. 2017;140(4):950–958. doi: 10.1016/j.jaci.2017.03.050. PMID:11707753 [DOI] [PubMed] [Google Scholar]
- 56.Hammad H, Lambrecht BN. Dendritic cells and epithelial cells: Linking innate and adaptive immunity in asthma. Nature Reviews. Immunology. 2008;8(3):193–204. doi: 10.1038/nri2275. PMID:18301423 [DOI] [PubMed] [Google Scholar]
- 57.Lambrecht BN, Hammad H. The immunology of asthma. Nature Immunology. 2015;16(1):45–56. doi: 10.1038/ni.3049. PMID:25521684 [DOI] [PubMed] [Google Scholar]
- 58.Bradding P, Walls AF, Holgate ST. The role of the mast cell in the pathophysiology of asthma. Journal of Allergy and Clinical Immunology. 117(6):1277–1284. doi: 10.1016/j.jaci.2006.02.039. PMID:16750987 [DOI] [PubMed] [Google Scholar]
- 59.Poon AH, Chouiali F, Tse SM, Litonjua AA, Hussain SN, Baglole CJ, Eidelman DH, Olivenstein R, Martin JG, Weiss ST, et al. Genetic and histologic evidence for autophagy in asthma pathogenesis. The Journal of Allergy and Clinical Immunology. 2012;129(2):569–571. doi: 10.1016/j.jaci.2011.09.035. PMID:22040902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ban GY, Pham DL, Trinh TH, Lee SI, Suh DH, Yang EM, Ye YM, Shin YS, Chwae YJ, Park HS. Autophagy mechanisms in sputum and peripheral blood cells of patients with severe asthma: A new therapeutic target. Clinical and Experimental Allergy: Journal of the British Society for Allergy and Clinical Immunology. 2016;46(1):48–59. doi: 10.1111/cea.12585. PMID:26112695 [DOI] [PubMed] [Google Scholar]
- 61.Pham DL, Ban GY, Kim SH, Shin YS, Ye YM, Chwae YJ, Park HS. Neutrophil autophagy and extracellular DNA traps contribute to airway inflammation in severe asthma. Clinical and Experimental Allergy: Journal of the British Society for Allergy and Clinical Immunology. 2017;47(1):57–70. doi: 10.1111/cea.12859. PMID:27883241 [DOI] [PubMed] [Google Scholar]
- 62.Liu JN, Suh DH, Trinh HK, Chwae YJ, Park HS, Shin YS. The role of autophagy in allergic inflammation: A new target for severe asthma. Experimental & Molecular Medicine. 2016;48(7):e243. doi: 10.1038/emm.2016.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Martin LJ, Gupta J, Jyothula SS, Butsch Kovacic M, Biagini Myers JM, Patterson TL, Ericksen MB, He H, Gibson AM, Baye TM, et al. Functional variant in the autophagy-related 5 gene promotor is associated with childhood asthma. PloS One. 2012;7(4):e33454. doi: 10.1371/journal.pone.0033454. PMID:22536318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Poon A, Eidelman D, Laprise C, Hamid Q. ATG5, autophagy and lung function in asthma. Autophagy. 2012;8(4):694–695. doi: 10.4161/auto.19315. PMID:22498476 [DOI] [PubMed] [Google Scholar]
- 65.Cao Z, Pan P, Tan H, Tan Q, Wang Z, Su X, Hu C. [Anti-nerve growth factor antibody reduces airway hyperresponsiveness in a mouse model of asthma by down-regulating the level of autophagy in lungs]. Zhonghua Jie he he hu xi za zhi = Zhonghua jiehe he huxi zazhi = Chinese Journal of Tuberculosis and Respiratory Diseases. 2014;37(7):507–511. PMID:25262691 [PubMed] [Google Scholar]
- 66.Cho I-H, Choi Y-J, Gong J-H, Shin D, Kang M-K, Kang Y-H. Astragalin inhibits autophagy-associated airway epithelial fibrosis. Respiratory Research. 2015;16(1):51. doi: 10.1186/s12931-015-0211-9. PMID:25895672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dickinson JD, Alevy Y, Malvin NP, Patel KK, Gunsten SP, Holtzman MJ, Stappenbeck TS, Brody SL. Autophagy. 2016;12(2):397–409. doi: 10.1080/15548627.2015.1056967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Murai H, Okazaki S, Hayashi H, Kawakita A, Hosoki K, Yasutomi M, Sur S, Ohshima Y. Alternaria extract activates autophagy that induces IL-18 release from airway epithelial cells. Biochemical and Biophysical Research Communications. 2015;464(4):969–974. doi: 10.1016/j.bbrc.2015.05.076. PMID:26032499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ghavami S, Yeganeh B, Serebrin A, Mutawe MM, Sharma P, McNeill KD, et al. Autophagy regulates TGF-beta1 induced fibrosis in human airway smooth muscle cells. Am J Respir Crit Care Med. 2011;183:A2110. [Google Scholar]
- 70.Cho IH, Choi YJ, Gong JH, Shin D, Kang MK, Kang YH. Astragalin inhibits autophagy-associated airway epithelial fibrosis. Respir Res. 2015;16:51. doi: 10.1186/s12931-015-0211-9. PMID:25895672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pham DL, Kim SH, Losol P, Yang EM, Shin YS, Ye YM, Park HS. Association of autophagy related gene polymorphisms with neutrophilic airway inflammation in adult asthma. The Korean Journal of Internal Medicine. 2016;31(2):375–385. doi: 10.3904/kjim.2014.390. PMID:26701229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stansbury RC, Strollo PJ. Clinical manifestations of sleep apnea. J Thorac Dis. 2015;7(9):E298–310. PMID:26543619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Franklin KA, Lindberg E. Obstructive sleep apnea is a common disorder in the population-a review on the epidemiology of sleep apnea. J Thorac Dis. 2015;7(8):1311–1322. PMID:26380759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ryan S. Adipose tissue inflammation by intermittent hypoxia: mechanistic link between obstructive sleep apnoea and metabolic dysfunction. J Physiol. 2017;595(8):2423–2430. doi: 10.1113/JP273312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kent BD, Ryan S, McNicholas WT. Obstructive sleep apnea and inflammation: relationship to cardiovascular co-morbidity. Respir Physiol Neurobiol. 2011;178(3):475–481. doi: 10.1016/j.resp.2011.03.015. PMID:21439407 [DOI] [PubMed] [Google Scholar]
- 76.Xie S, Deng Y, Pan YY, Ren J, Jin M, Wang Y, Wang ZH, Zhu D, Guo XL, Yuan X, et al. Chronic intermittent hypoxia induces cardiac hypertrophy by impairing autophagy through the adenosine 5'-monophosphate-activated protein kinase pathway. Archives of Biochemistry and Biophysics. 2016;606:41–52. doi: 10.1016/j.abb.2016.07.006. PMID:27412517 [DOI] [PubMed] [Google Scholar]
- 77.Xie S, Deng Y, Pan YY, Wang ZH, Ren J, Guo XL, Yuan X, Shang J, Liu HG. Melatonin protects against chronic intermittent hypoxia-induced cardiac hypertrophy by modulating autophagy through the 5' adenosine monophosphate-activated protein kinase pathway. Biochemical and Biophysical Research Communications. 2015;464(4):975–981. doi: 10.1016/j.bbrc.2015.06.149. PMID:26188509 [DOI] [PubMed] [Google Scholar]
- 78.Pauly M, Assense A, Rondon A, Thomas A, Dubouchaud H, Freyssenet D, Benoit H, Castells J, Flore P. High intensity aerobic exercise training improves chronic intermittent hypoxia-induced insulin resistance without basal autophagy modulation. Scientific Reports. 2017;7:43663. doi: 10.1038/srep43663. PMID:28255159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Farber HW, Miller DP, Poms AD, Badesch DB, Frost AE, Muros-Le Rouzic E, Romero AJ, Benton WW, Elliott CG, McGoon MD, et al. Five-Year outcomes of patients enrolled in the REVEAL Registry. Chest. 2015;148(4):1043–1054. doi: 10.1378/chest.15-0300. PMID:26066077 [DOI] [PubMed] [Google Scholar]
- 80.Tamosiuniene R, Nicolls MR. Regulatory T cells and pulmonary hypertension. Trends Cardiovasc Med. 2011;21(6):166–171. doi: 10.1016/j.tcm.2012.05.004. PMID:22814424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stacher E, Graham BB, Hunt JM, Gandjeva A, Groshong SD, McLaughlin VV, Jessup M, Grizzle WE, Aldred MA, Cool CD, et al. Modern age pathology of pulmonary arterial hypertension. American Journal of Respiratory and Critical Care Medicine. 2012;186(3):261–272. doi: 10.1164/rccm.201201-0164OC. PMID:22679007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.El Kasmi KC, Stenmark KR. Contribution of metabolic reprogramming to macrophage plasticity and function. Semin Immunol. 2015;27(4):267–275. doi: 10.1016/j.smim.2015.09.001. PMID:26454572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rabinovitch M, Guignabert C, Humbert M, Nicolls MR. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ Res. 2014;115(1):165–175. doi: 10.1161/CIRCRESAHA.113.301141. PMID:24951765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, Waxman AB. Interleukin-6 overexpression induces pulmonary hypertension. Circ Res. 2009;104(2):236–244, 228p following 244. doi: 10.1161/CIRCRESAHA.108.182014. PMID:19074475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Savale L, Tu L, Rideau D, Izziki M, Maitre B, Adnot S, Eddahibi S. Impact of interleukin-6 on hypoxia-induced pulmonary hypertension and lung inflammation in mice. Respir Res. 2009;10:6. doi: 10.1186/1465-9921-10-6. PMID:19173740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cero FT, Hillestad V, Sjaastad I, Yndestad A, Aukrust P, Ranheim T, Lunde IG, Olsen MB, Lien E, Zhang L, et al. Absence of the inflammasome adaptor ASC reduces hypoxia-induced pulmonary hypertension in mice. American Journal of Physiology. Lung Cellular and Molecular Physiology. 2015;309(4):L378–387. doi: 10.1152/ajplung.00342.2014. PMID:26071556 [DOI] [PubMed] [Google Scholar]
- 87.Lee SJ, Smith A, Guo L, Alastalo TP, Li M, Sawada H, Liu X, Chen ZH, Ifedigbo E, Jin Y, et al. Autophagic protein LC3B confers resistance against hypoxia-induced pulmonary hypertension. American Journal of Respiratory and Critical Care Medicine. 2011;183(5):649–658. doi: 10.1164/rccm.201005-0746OC. PMID:20889906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Teng RJ, Du J, Welak S, Guan T, Eis A, Shi Y, Konduri GG. Cross talk between NADPH oxidase and autophagy in pulmonary artery endothelial cells with intrauterine persistent pulmonary hypertension. American Journal of Physiology. Lung Cellular and Molecular Physiology. 2012;302(7):L651–663. doi: 10.1152/ajplung.00177.2011. PMID:22245997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lee SJ, Kim HP, Jin Y, Choi AM, Ryter SW. BECN1 deficiency is associated with increased hypoxia-induced angiogenesis. Autophagy. 2011;7(8):829–839. doi: 10.4161/auto.7.8.15598. PMID:21685724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Long L, Yang X, Southwood M, Lu J, Marciniak SJ, Dunmore BJ, Morrell NW. Chloroquine prevents progression of experimental pulmonary hypertension via inhibition of autophagy and lysosomal bone morphogenetic protein type II receptor degradation. Circ Res. 2013;112(8):1159–1170. doi: 10.1161/CIRCRESAHA.111.300483. PMID:23446737 [DOI] [PubMed] [Google Scholar]
- 91.Spiekerkoetter E, Tian X, Cai J, Sudheendra D, Li CG, El-Bizri N, Sawada H, Haghighat R, Chan R, Haghighat L, et al. FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J Clin Invest. 2013;123(8):3600–3613. doi: 10.1172/JCI65592. PMID:23867624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Seyfarth HJ, Hammerschmidt S, Halank M, Neuhaus P, Wirtz HR. Everolimus in patients with severe pulmonary hypertension: A safety and efficacy pilot trial. Pulm Circ. 2013;3(3):632–638. doi: 10.1086/674311. PMID:24618547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.O'Dwyer DN, Ashley SL, Moore BB. Influences of innate immunity, autophagy, and fibroblast activation in the pathogenesis of lung fibrosis. American Journal of Physiology. Lung Cellular and Molecular Physiology. 2016;311(3):L590–601. doi: 10.1152/ajplung.00221.2016. PMID:27474089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wuyts WA, Agostini C, Antoniou KM, Bouros D, Chambers RC, Cottin V, Egan JJ, Lambrecht BN, Lories R, Parfrey H.et al. The pathogenesis of pulmonary fibrosis: A moving target. Eur Respir J. 2013;41(5):1207–1218. doi: 10.1183/09031936.00073012. PMID:23100500 [DOI] [PubMed] [Google Scholar]
- 95.Patel AS, Lin L, Geyer A, Haspel JA, An CH, Cao J, Rosas IO, Morse D. Autophagy in idiopathic pulmonary fibrosis. PloS One. 2012;7(7):e41394. doi: 10.1371/journal.pone.0041394. PMID:22815997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nho RS, Hergert P. IPF fibroblasts are desensitized to type I collagen matrix-induced cell death by suppressing low autophagy via aberrant Akt/mTOR kinases. PloS One. 2014;9(4):e94616. doi: 10.1371/journal.pone.0094616. PMID:24728102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Araya J, Kojima J, Takasaka N, Ito S, Fujii S, Hara H, Yanagisawa H, Kobayashi K, Tsurushige C, Kawaishi M, et al. Insufficient autophagy in idiopathic pulmonary fibrosis. American Journal of Physiology. Lung Cellular and Molecular Physiology. 2013;304(1):L56–69. doi: 10.1152/ajplung.00213.2012. PMID:23087019 [DOI] [PubMed] [Google Scholar]
- 98.Sosulski ML, Gongora R, Danchuk S, Dong C, Luo F, Sanchez CG. Deregulation of selective autophagy during aging and pulmonary fibrosis: the role of TGFbeta1. Aging Cell. 2015;14(5):774–783. doi: 10.1111/acel.12357. PMID:26059457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kobayashi K, Araya J, Minagawa S, Hara H, Saito N, Kadota T, Sato N, Yoshida M, Tsubouchi K, Kurita Y, et al. Involvement of PARK2-Mediated mitophagy in idiopathic pulmonary fibrosis pathogenesis. Journal of Immunology (Baltimore, Md.: 1950). 2016;197(2):504–516. doi: 10.4049/jimmunol.1600265. PMID:27279371 [DOI] [PubMed] [Google Scholar]
- 100.Patel AS, Song JW, Chu SG, Mizumura K, Osorio JC, Shi Y, El-Chemaly S, Lee CG, Rosas IO, Elias JA, et al. Epithelial cell mitochondrial dysfunction and PINK1 are induced by transforming growth factor-beta1 in pulmonary fibrosis. PloS One. 2015;10(3):e0121246. doi: 10.1371/journal.pone.0121246. PMID:25785991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cabrera S, Maciel M, Herrera I, Nava T, Vergara F, Gaxiola M, López-Otín C, Selman M, Pardo A. Essential role for the ATG4B protease and autophagy in bleomycin-induced pulmonary fibrosis. Autophagy. 2015;11(4):670–684. doi: 10.1080/15548627.2015.1034409. PMID:25906080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gui YS, Wang L, Tian X, Li X, Ma A, Zhou W, Zeng N, Zhang J, Cai B, Zhang H, et al. mTOR overactivation and compromised autophagy in the pathogenesis of pulmonary fibrosis. PloS One. 2015;10(9):e0138625. doi: 10.1371/journal.pone.0138625. PMID:26382847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chrysanthopoulou A, Mitroulis I, Apostolidou E, Arelaki S, Mikroulis D, Konstantinidis T, Sivridis E, Koffa M, Giatromanolaki A, Boumpas DT, et al. Neutrophil extracellular traps promote differentiation and function of fibroblasts. The Journal of Pathology. 2014;233(3):294–307. doi: 10.1002/path.4359. PMID:24740698 [DOI] [PubMed] [Google Scholar]
- 104.Jessop F, Hamilton RF, Rhoderick JF, Shaw PK, Holian A. Autophagy deficiency in macrophages enhances NLRP3 inflammasome activity and chronic lung disease following silica exposure. Toxicology and Applied Pharmacology. 2016;309:101–110. doi: 10.1016/j.taap.2016.08.029. PMID:27594529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Larson-Casey JL, Deshane JS, Ryan AJ, Thannickal VJ, Carter AB. Macrophage akt1 kinase-mediated mitophagy modulates apoptosis resistance and pulmonary fibrosis. Immunity. 2016;44(3):582–596. doi: 10.1016/j.immuni.2016.01.001. PMID:26921108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mi S, Li Z, Yang HZ, Liu H, Wang JP, Ma YG, Wang XX, Liu HZ, Sun W, Hu ZW. Blocking IL-17A promotes the resolution of pulmonary inflammation and fibrosis via TGF-beta1-dependent and -independent mechanisms. Journal of Immunology (Baltimore, Md.: 1950). 2011;187(6):3003–3014. doi: 10.4049/jimmunol.1004081. PMID:21841134 [DOI] [PubMed] [Google Scholar]
- 107.Han R, Ji X, Rong R, Li Y, Yao W, Yuan J, Wu Q, Yang J, Yan W, Han L, et al. MiR-449a regulates autophagy to inhibit silica-induced pulmonary fibrosis through targeting Bcl2. Journal of Molecular Medicine (Berlin, Germany). 2016;94(11):1267–1279. doi: 10.1007/s00109-016-1441-0. PMID:27351886 [DOI] [PubMed] [Google Scholar]
- 108.Ahuja S, Knudsen L, Chillappagari S, Henneke I, Ruppert C, Korfei M, Gochuico BR, Bellusci S, Seeger W, Ochs M, et al. MAP1LC3B overexpression protects against hermansky-pudlak syndrome type-1-induced defective autophagy in vitro. American Journal of Physiology. Lung Cellular and Molecular Physiology. 2016;310(6):L519–531. doi: 10.1152/ajplung.00213.2015. PMID:26719147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liu H, Cheng Y, Yang J, Wang W, Fang S, Zhang W, Han B, Zhou Z, Yao H, Chao J, et al. BBC3 in macrophages promoted pulmonary fibrosis development through inducing autophagy during silicosis. Cell Death & Disease. 2017;8(3):e2657. doi: 10.1038/cddis.2017.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Malouf MA, Hopkins P, Snell G, Glanville AR. An investigator-driven study of everolimus in surgical lung biopsy confirmed idiopathic pulmonary fibrosis. Respirology (Carlton, Vic.). 2011;16(5):776–783. doi: 10.1111/j.1440-1843.2011.01955.x. PMID:21362103 [DOI] [PubMed] [Google Scholar]
- 111.Pasteur MC, Bilton D, Hill AT, British Thoracic Society Bronchiectasis non CFGG British thoracic society guideline for non-CF bronchiectasis. Thorax. 2010;65 Suppl 1:i1–58. doi: 10.1136/thx.2010.136119. PMID:20627931 [DOI] [PubMed] [Google Scholar]
- 112.King PT. The pathophysiology of bronchiectasis. Int J Chron Obstruct Pulmon Dis. 2009;4:411–419. doi: 10.2147/COPD.S6133. PMID:20037680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Junkins RD, Shen A, Rosen K, McCormick C, Lin TJ. Autophagy enhances bacterial clearance during P. aeruginosa lung infection. PloS One. 2013;8(8):e72263. doi: 10.1371/journal.pone.0072263. PMID:24015228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mayer ML, Blohmke CJ, Falsafi R, Fjell CD, Madera L, Turvey SE, Hancock RE. Rescue of dysfunctional autophagy attenuates hyperinflammatory responses from cystic fibrosis cells. Journal of Immunology (Baltimore, Md.: 1950). 2013;190(3):1227–1238. doi: 10.4049/jimmunol.1201404. PMID:23264659 [DOI] [PubMed] [Google Scholar]
- 115.Abdulrahman BA, Khweek AA, Akhter A, Caution K, Kotrange S, Abdelaziz DH, Newland C, Rosales-Reyes R, Kopp B, McCoy K, et al. Autophagy stimulation by rapamycin suppresses lung inflammation and infection by Burkholderia cenocepacia in a model of cystic fibrosis. Autophagy. 2011;7(11):1359–1370. doi: 10.4161/auto.7.11.17660. PMID:21997369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Luciani A, Villella VR, Esposito S, Brunetti-Pierri N, Medina D, Settembre C, Gavina M, Pulze L, Giardino I, Pettoello-Mantovani M, et al. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nature Cell Biology. 2010;12(9):863–875. doi: 10.1038/ncb2090. PMID:20711182 [DOI] [PubMed] [Google Scholar]
- 117.Al-Khodor S, Marshall-Batty K, Nair V, Ding L, Greenberg DE, Fraser ID. Burkholderia cenocepacia J2315 escapes to the cytosol and actively subverts autophagy in human macrophages. Cellular Microbiology. 2014;16(3):378–395. doi: 10.1111/cmi.12223. PMID:24119232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Iannitti RG, Napolioni V, Oikonomou V, De Luca A, Galosi C, Pariano M, Massi-Benedetti C, Borghi M, Puccetti M, Lucidi V, et al. IL-1 receptor antagonist ameliorates inflammasome-dependent inflammation in murine and human cystic fibrosis. Nature Communications. 2016;7:10791. doi: 10.1038/ncomms10791. PMID:26972847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Galluzzi L, Bravo-San Pedro JM, Levine B, Green DR, Kroemer G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nature Reviews. Drug Discovery. 2017;16(7):487–511. doi: 10.1038/nrd.2017.22. PMID:28529316 [DOI] [PMC free article] [PubMed] [Google Scholar]