

ABSTRACT
Macroautophagy/autophagy is a homeostatic process with multiple effects on immunity. One of the pivotal contributions of autophagy in immunity is the cell autonomous control of inflammation. This property leads to systemic consequences and thereby influences the development of innate and adaptive immunity, which promotes or suppresses pathology in various disease contexts. In this review we focus on the intersections between autophagy and inflammasome activation, autophagy and interferons, and autophagy and inflammation in association with infection.
KEYWORDS: Autophagy, microbes mitochondria, inflamamsome, interferon, lysosome, phagosome
Abbreviations
- CD
Crohn disease
- FA
Fanconi anemia
- GWAS
genome-wide association studies
- Mtb
Mycobacterium tuberculosis
- PAMPs
pathogen-associated molecular patterns
- ROS
reactive oxygen species
- SLR
SQSTM1-like receptor
Introduction
As a metabolic, cytoplasmic quality control and general homeostatic process, autophagy is primarily cytoprotective, tissue protective and anti-inflammatory. Its manifestations in adaptive immunity and in sterile and infection-associated inflammation, cytokine- and innate immune cell-processes intended to help clear cell-damaging sterile irritants or invading pathogens, are numerous.1,2 The general theme of the present review is how autophagy balances and modulates immune activation to avoid excessive inflammation, with a focus on single-cell level intracellular processes that may serve as initial stimuli to trigger or skew broader tissue responses. An emerging concept underlying this theme is the important intricate relationship between endomembrane and organelle homeostasis and inflammatory outputs (Fig. 1) as a key determinant of innate immunity.
Figure 1.
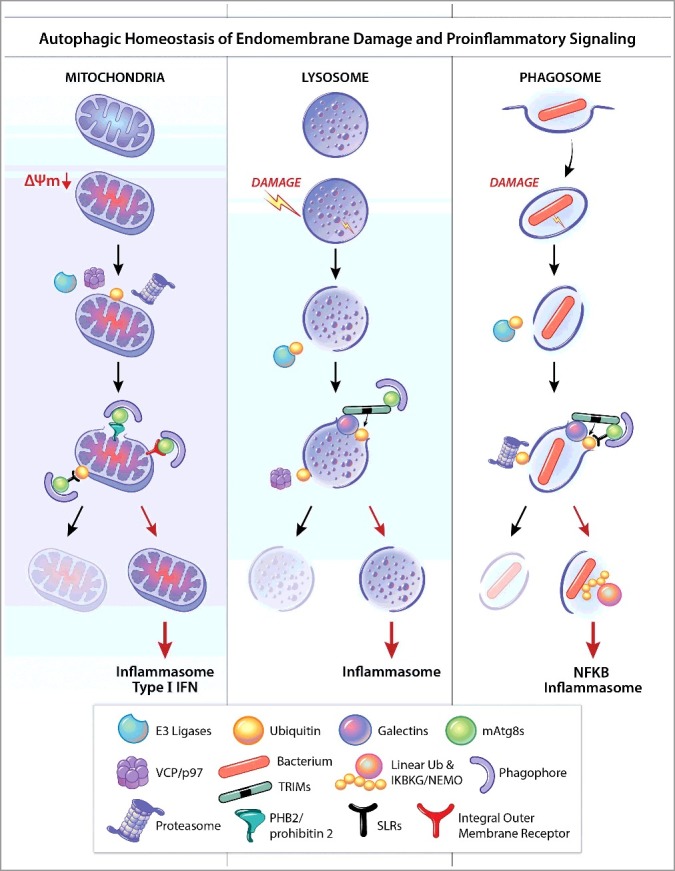
Common themes in autophagic homeostasis of endomembrane damage and proinflammatory signaling. Organelles such as mitochondria (A), lysosomes (B), and phagosomes (C), can be depolarized (ΔΨm), perforated, or otherwise damaged. E3 ligases, such as PRKN/PARK2/parkin, SMURF1, etc., can ubiquitinate targets (K48, K63) on damaged organelles. The AAA-ATPase VCP/p97 can either turn over ubiquitin (K48) by recruiting deubiquitinases to permit further steps in lysosomal homeostasis, or unfold targets such as MFNs (mitofusins) on mitochondria to present them to the proteasome for degradation. K48 ubiquitin also presents phagosomal membrane to proteasomes thus contributing to membrane processing or further damage. Galectins can recognize membrane tears and bind to exposed lumenal glycans while in turn binding to receptors such as SLRs (e.g., LGALS8-CALCOCO2/NDP52), which deliver organelles to phagophores or receptor-regulators (e.g., LGALS3-TRIM16 or LGALS8-TRIM16) that function as receptors and in addition assemble and promote ubiqitination and activation of regulatory ATG factors. SLRs, or other types of receptor such as PHB2 (exposed on the inner membrane of ruptured mitochondria) or integral outer membrane receptors on mitochondria bind Atg8-family proteins (e.g., LC3) via their LC3-interacting region motifs. Damaged organelles are either removed (note crescents representing phagophores, i.e. autophagosome precursors) or otherwise repaired, and if not, they activate inflammasomes (and potentially type I IFNs) as described in the text. If bacteria are not removed along with the remnants of their vacuoles/phagosomes (e.g., via LGALS8), linear ubiquitin chains are formed to activate IKBKG/NEMO and NFKB, leading to inflammation. These processes share common principles and contribute to either suppression of pro-inflammatory responses (by repair or removal of offending organelles and bacteria) or, when they are overwhelmed or otherwise fail, elicit inflammatory cytokines as a second line of defense but at a cost due to associated tissue damage. mATG8s, mammalian Atg8-family proteins.
Genetic links between autophagy and inflammatory diseases
The strong connections between inflammatory diseases—which encompass a vast variety of diverse disorders with dysregulated inflammatory responses causing tissue pathology in a wide spectrum of human organs—and alterations in autophagy were initially gleaned from genome-wide association studies (GWAS) examining associations between genetic polymorphisms and predisposition for a range of human diseases.3 Most notably, early studies connected Crohn disease (CD) susceptibility with polymorphisms in genes encoding ATG16L1 and IRGM, whose products interact to control autophagy in coordination with NOD2, a familial risk factor in CD.4 Links between CD (or another form of inflammatory bowel disease, ulcerative colitis) and other autophagy-related genetic polymorphisms span a full range of genes that function in different steps of autophagy and which may, in some cases, also have additional functions.1 These include, for example, ULK1, which acts at an early step in the autophagy regulatory cascade5 and selective autophagy factors, such as SMURF1,6–9 and CALCOCO2/NDP52.10
Other connections have been reported between several autophagy loci and genetic predispositions for chronic inflammatory disorders and autoimmune diseases. These include systemic lupus erythematosus (IRGM,11 ATG5,12 PRDM1-ATG513; and DRAM114), asthma (ATG515), rheumatoid arthritis (albeit somewhat marginally significant ATG5 rs6568431 association16), Vici syndrome (EPG517), celiac disease18 and other conditions (CLEC16A, ULK3, and MIR's that target autophagy genes),18–21 multiple sclerosis (CLEC16A21), and other neurological disorders, including amyotrophic lateral sclerosis and frontotemporal dementia, especially in patients with stronger neuroinflammatory components (where connections to autophagy have been made through mutations in TBK122). CLEC16A is also genetically linked to type 1 diabetes mellitus, a condition associated with immune infiltration, and plays a role in mitophagy and autolysosome function.23 The latter function of CLEC16A is interesting as it points to stages beyond autophagosome-lysosome fusion. Finally, a recent study has connected the autosomal dominant mutations in the gene (MEFV/PYRIN/TRIM20) responsible for familial Mediterranean fever to the control of selective autophagy.24 Thus, autophagy and inflammation show genetic links in a broad spectrum of human disorders that have inflammatory and/or autoimmune components.
Autophagy and the inflammasome
Inflammasomes, which come in several variants, are cytosolic responders to microbial products and sterile endogenous agonists. Once activated, inflammasomes proteolytically process pivotal pro-inflammatory cytokines including IL1B/IL-1β.25 A canonical inflammasome consists of pro-CASP1 (caspase 1), PYCARD/ASC adaptor, and one of the proteins sensing diverse endogenous agonists (often associated with mitochondrial reactive oxygen species [ROS] or lysosomal damage) or bacterial products such as NLRP1, NLRP3, NLRC4, or cytosolic DNA sensors such as AIM2 and IFI16.25 Once inflammasome components assemble, activated CASP1 processes cytosolic pro-IL1B into mature IL1B ready to be secreted from the cells.25 Noncanonical inflammasomes do not depend on ASC and NLRPs, and instead the agonists, such as cytosolic LPS, directly activate CASP4/CASP11 (mouse) or CASP4 and CASP5 (human) resulting in proteolytic processing of GSDMD (gasdermin D) causing a type of cell death termed pyroptosis. There are crossovers between canonical and noncanonical inflammasomes as CASP1 can also proteolytically activate GSDMD.26
The intersections between autophagy and the inflammasome are numerous and represent some of the earliest and best examples of the anti-inflammatory role of autophagy. The initial proof-of-principle for a genetic linkage between autophagy gene mutation and inflammation, including the role of IL1B, processed by activated inflamamsomes as described above, came from studies modeling Crohn disease in mice lacking functional ATG16L1 in hematopoietic cells,27 and in mice expressing a whole body hypomorphic allele of Atg16l1.28 Mice lacking ATG16L1 in hematopoietic cells show elevated levels of IL1B and IL18, the key pro-inflammatory cytokines triggered by inflammasome activation, and pathology in these mice can be countered by blocking antibodies against IL1B and IL18. Thus, this study,27 provided the first in vivo evidence that an autophagy gene can function to control inflammasome activation. More recently, macrophage-specific deletion of autophagy genes in mice has been shown to lead to inflammasome-mediated IL1B release and uveitis, an inflammation-mediated eye disease often observed in patients with CD.29
Several early reports30,31 describe modes of indirect suppression of inflammasome activation by autophagy. Because autophagy removes damaged or irreversibly depolarized mitochondria, intact autophagy is necessary to prevent the accumulation of depolarized mitochondria (a process called mitophagy) that release endogenous inflammasome agonists such as ROS and oxidized mitochondrial DNA.30,31 These earlier findings have been strengthened by recent new lines of evidence linking intact mitophagy to prevention of inflammasome activation. For example, macrophage-specific deletion of the gene encoding the autophagy adaptor SQSTM1/p62 results in accumulation of damaged mitochondria, excessive inflammasome activation-IL1B-dependent inflammation, and macrophage death.32 Similarly, in response to treatment with inflammasome activators, bone marrow-derived macrophages from mice lacking the Fanconi anemia (FA) gene, Fancc (a newly identified mediator of selective autophagy) accumulate damaged mitochondria and have increased mitochondrial ROS-dependent inflammasome activation.33 Intriguingly, a common naturally-occurring mutation in FANCC associated with a milder clinical phenotype preserves the mitophagy function but not the DNA damage repair function of FANCC. This observation suggests that defects in mitophagy—and consequent aberrant inflammasome activation—may underlie some of the pathology in patients with FA (a congenital disease) or oncogenesis in patients with tumors due to mutations in the FA gene family.
In addition to preventing inflammasome activation by removing damaged mitochondria, autophagy has also recently been shown to play a more direct role in regulation of the inflammasome. Notably, individual inflammasome components are substrates for autophagic degradation, thus representing another modality by which autophagy prevents excessive inflammasome activation. AIM2 is removed by autophagy in a process involving SQSTM1 recruitment to K63-ubiquitinated PYCARD/ASC.34
Several newly described autophagy receptors35 from the family of TRIM proteins play a role in autophagic degradation of a number of individual inflammasome components. For example, MEFV/TRIM20, targets pro-CASP1, NLRP1, and NLRP3 for autophagic degradation.24 MEFV/TRIM20 recognizes these targets via its SPRY domain, has 3 LC3-interacting region motifs allowing it to interact with various members of the mammalian homologs of yeast Atg8 (LC3s and GABARAPs), and assembles locally the core autophagy regulators ULK1, BECN1/Beclin 1 and ATG16L1.24 Moreover, mutations in MEFV/TRIM20 associated with familial Mediterranean fever, diminish interactions between MEFV/TRIM20 and the core regulators of autophagy.24 In addition, TRIM11 acts as a receptor for AIM2; AIM2 binds to the PRY-SPRY domain of TRIM11; and TRIM11 auto-polyubiquitinates and recruits SQSTM1 to eliminate AIM2 via SQSTM1-dependent selective autophagy.36
However, autophagy does not exclusively play a negative role in the regulation of inflammasomes. Despite its general suppressive effects on the inflammasome itself, autophagy components play a positive role in the unconventional secretion of IL1B37-39 from the cytosol into the extracellular milieu where this cytokine exerts its biological activity. Thus, autophagy, in its engagement with inflammasomes and their substrates, appears to play a balancing act in supporting productive inflammatory responses while simultaneously preventing excessive inflammatory responses and tissue damage.
Autophagy intersects with type I and type II IFN
IFNs (interferons) are immunomodulators secreted by immune and other cells in response to pathogens or tumors. Type I IFN is often associated with induction of protective antiviral states but may be counterproductive in certain bacterial infections at least in part due to inhibition of IL1 production.40 Autophagy factors can directly suppress activation of protein complexes that stimulate type I IFN production. Type I IFN activation can occur through several signaling pathways. These come from toll-like receptor (TLR) and other signaling molecules that activate NFKB/NF-κB, including intracellular nucleic acid sensors such as DDX58/RIG-I that interact with mitochondrial-localized MAVS and activate TBK1 kinase, and a second messenger cGAMP (generated by CGAS/cGAMP synthase in response to cytosolic DNA) which binds to the ER adaptor protein TMEM173/STING, which in turn promotes dimerization of IRF3 and its phosphorylation by TBK1. IRF3 (and IRF7) then activates transcription of type I IFN genes.
Early studies41 indicated that the ATG12–ATG5 complex inhibits DDX58/RIG-I signaling responsible for type I IFN induction. This was due to direct binding of ATG12–ATG5 to the CARD domains of DDX58/RIG-I and MAVS. Additionally, ATG9A negatively controls trafficking of the ER-associated TMEM173/STING and inhibits activation of TBK1. The absence of autophagy may also amplify DDX58/RIG-I like receptor signaling through increased MAVS levels on accumulating mitochondria, and, as is the case with the inflammasome, increases in depolarized mitochondria pools in the absence of mitophagy lead to increased ROS and enhanced DDX58/RIG-I like receptor outputs.42 A very recent study indicates that MAVS itself is a direct target for autophagic removal coordinated by the BST2/tetherin-recruited E3 ligase MARCH8 and CALCOCO2/NDP52.43
Other recent studies have shown that individual components of type I IFN activation pathways are targets for degradation by selective autophagy. TRIM21 targets both IKBKB/IKKβ (of the NFKB activation pathway)44 and IRF3 dimers (but not its inactive monomers)24 for selective degradation through autophagy. TRIM21, similarly to MEFV/TRIM20, binds the core autophagy machinery components including mammalian Atg8-family members, ULK1, BECN1, ATG16L1, and SQSTM1.24 Finally, CGAS, a cytosolic DNA sensor45 that activates type I IFN via the production of cGAMP, which activates TMEM173 and TBK1, itself is a substrate for selective SQSTM1-dependent autophagy.46 Interestingly, a TRIM (TRIM14) negatively controls this process. Unlike TRIM11, MEFV/TRIM20 and TRIM21, which promote degradation of the components of the inflammasome or type I IFN activation pathways, TRIM14 recruits a deubiquitinase to CGAS thus protecting it from autophagic degradation. TRIM14 itself is a member of type I interferon-stimulated genes, and thus its increased expression in response to viral infections amplifies the cellular capacity to produce type I IFN when needed.46
Type II interferon, IFNG/IFN-γ, a key cytokine associated with protective Th1 responses against intracellular bacteria, also intersects with autophagy. IFNG acts upstream of autophagy by activating systems that modulate autophagy. This regulation can occur through an IFNG stimulated kinase, DAPK1, which in turn phosphorylates and activates BECN1.47 However, this can also proceed through immunity-related GTPases. For example, IRGM1, a mouse paralog of human IRGM, is IFNG inducible.48 IRGM organizes core autophagy factors including ULK1, BECN1 and ATG16L1, and links them with receptors of innate immunity.49 IFNG may also act through increased expression of TRIM subsets. Among the TRIMs potently activated by IFNG is MEFV/TRIM20, which targets inflammasome components for selective autophagy.24 The subsequent degradation can contribute at least in part to the tapering/balancing effects of IFNG on inflammation.50 UBQLN (ubiquilin) also seems to work in selective autophagy upon IFNG activation by promoting ubiquitination of targets.51
Autophagy may reduce triggers of inflammation during bacterial infection
Several organelles in eukaryotic cells, such as mitochondria, may originate from bacterial endosymbionts. Thus, it is not surprising to observe parallels between autophagic control of bacteria and autophagic removal of a range of endomembranous organelles during sterile homeostasis.9,52–61 These phenomena include cooperation between several quality control systems and autophagy, and the examples thus far include mitochondria, lysosomes and phagosomes. These organelles, when damaged by crystals such as silica or monosodium uric acid,62 cholesterol crystals,63 and possibly protein fibrils/amyloid,64–66 and bacteria or viruses, become dysfunctional and all are capable of triggering inflammasome activation.62 This property is not limited to mitochondria alone, because many classical agonists of the inflammasome such as silica, alum, and monosodium urate crystals, are primarily lysosome damaging agents.62 Thus, we speculate that by limiting consequences of damage to lysosomes, endosomes, and phagosomes58,59,61,67 (potentially including pathogen-containing phagosomes) as well as mitochondria, autophagy may reduce triggers of cell-autonomous inflammation.
A parallel between mitophagy and autophagic elimination of bacteria68 includes participation of the same ubiquitin E3 ligase PRKN/PARK2/parkin and SQSTM1-like receptors (SLRs) in autophagic defense against Mycobacterium tuberculosis (Mtb)57 and in mitophagy.55 Recent studies continue to expand these parallels and have revealed a set of multilayered similarities between autophagic elimination of pathogens and removal of damaged organelles, including participation of E3 ligases (which mediate K48 ubiquitination, in addition to K63 ubiqutination, which is more classically associated with autophagy), ubiquitination-deubiquitination cycles possibly contributing to quality control, ubiquitin-directed VCP/p97 unfoldase/segregase action on damaged proteins, and proteasomal degradation of such substrates, all functioning in concert with or in advance of mitophagy52–57 and autophagic elimination of damaged lysosomes or phagosomes.9, 58–61
K48 ubiquitination can precede or act in parallel during elimination of damaged lysosomes60 or mitochondria.52–54 Both damaged lysosomes60 and mitochondria52 engage VCP/p97, and, mitochondria (but not lysosomes, where deubiquitinases carry out deubiquitination of K48 linkages60) engage the proteasome contributing to progression toward mitophagy.52–54,56 In mitophagy, these activities expose the inner core of the organelle allowing the inner mitochondrial mitophagy receptor, PHB2 (prohibitin 2), to bind LC3-II and target mitochondria for autophagic degradation.56 Similar K48 and proteasome requirements (with the SMURF1 E3 ligase generating K48 ubiquitin linkages) have been recently reported9 to act in combination with PARK2-K63-SLR to mediate autophagic targeting of Mtb phagosomes to the lysosome. These relationships are illustrated in Fig. 1.
Autophagy, galectins, and inflammation
In addition to the above autophagy-related processes that control inflammation, galectins (a group of cytosolic lectins) play a role in the detection of endomembrane injury and subsequent triggering of autophagy aimed at protection against sterile or infection-associated phagosomal61,69–72 and lysosomal58,59,61,67 membrane damage. Galectins can react to membrane damage and form intracellular puncta in response to lysosomal damaging agents such as polymers of Leu-Leu-OMe/LLOMe that poke membrane holes67,73 and when cells take up inanimate objects such as latex beads69 coated with transfection reagents.59 Galectins recognize membrane damage mediated by bacterial secretory system effector proteins in phagosomes/vacuoles in cells infected with Shigella,71 Listeria,70 Legionella,74,75 Yersinia,75 and Salmonella..59,70,72 An in vitro role for LGALS8 (galectin 8) has been reported in autophagic control of Salmonella-containing vacuoles72 and viruses.76, 77
Recent studies have placed LGALS8 at the crossroads of autophagy and inflammation.39, 78 First, LGALS8-marked membranes shield escaping bacteria from recruiting linear ubiquitin chain assembly complex/LUBAC, an E3 ligase that generates linear polyubiquitin chains, which, in addition to contributing to autophagic processes, activate IKBKG/NEMO and NFKB that in turn trigger inflammation.78 Second, LGALS8 participates in secretion of IL1B in response to lysosomal damage.39 These studies expand the effects of intracellular galectins to triggers and mechanisms of extracellular proinflammatory cytokine activation and delivery.
Another lectin, LGALS3 (galectin 3), plays a role in autophagic control of M. tuberculosis both in infected macrophages and in a mouse model of tuberculosis.61 LGALS3 controls autophagic responses to endomembrane (e.g. lysosomal and phagosomal) damage in cooperation with TRIM16 leading to the assembly of core ATG factors ATG16L1, ULK1 and BECN1.61 LGALS3 has been implicated in recognition of Legionella74, 75 and Yersinia75 vacuolar damage, but connections with autophagy have not been investigated For the majority of galectins (i.e. with the exception of LGALS361), their in vivo role remains to be established with respect to their control of autophagic functions in relationship to innate immunity and inflammation.
The complex interplay of autophagy and infection-associated inflammation in animals
All of the above processes are eventually linked to potential activation of proinflammatory cytokines. Thus, the selective autophagic removal of intracellular bacteria and endomembranous organelles is likely important not only for the direct benefits of removing unwanted cargo, but also for decreasing inflammasome and NFKB activation and reducing inflammation in tissues.
The links between defects in autophagy or autophagy genes (either core components of the machinery or targeting factors) and excessive inflammation have become evident from in vivo studies using murine models of Mtb infection. Increased parameters of lung inflammation9,79,80 have been detected in Mtb-infected mice with defective Atg5 in the myeloid lineage (Atg5fl/fl Lys2/LysM-Cre mice)79,80 and whole-body smurf1 knockout mice.9 The types of cytokines detected in each case have some similarities, as IL17 is elevated in both Atg5fl/fl Lys2-Cre79 and whole-body smurf1 knockout mice.9 Neutrophilic infiltration and elevated IL1A/IL-1α or IL1B are detected in conditional knockout mice with the ATG5 defect in the myeloid lineage (Atg5fl/fl Lys2-Cre mice)79, 80 but not in the study with smurf1 knockout mice, where mononuclear inflammatory cells dominate instead of neutrophils9 or in studies with myeloid-specific knockout of other core autophagy machinery components.81 Neutrophilic infiltration in Mtb-infected Atg5fl/fl Lys2-Cre mice has been shown81 to be responsible for the early lethality of such mice,79,80 and a neutrophil-driven transcriptional signature82 is considered to be a hallmark of active tuberculosis in humans. However, not all effects may be attributable to macroautophagy in the studies reported.81 An in vivo role for LGALS3 in protection against Mtb in a mouse model of tuberculosis has been reported, in keeping with its in vitro role in autophagic killing of Mtb,61 and this may also include regulation of inflammatory components secondary to phagosomal damage.
Type I interferon is another signature of active disease in Mtb-infected patients82 and is considered as a potentially counterproductive host response in bacterial infections (in contrast to its usual protective role in viral infections). However, it may also act as a balancer by suppressing excessive IL1B responses.83 Bacterial DNA released from phagosomes harboring Mtb and Legionella pneumophila can be recognized by CGAS to induce type I interferon, proposed to be damaging or at least counterproductive for the host during bacterial infection.84 However, mb21d1/CGAS knockout mice do not survive better as one might expect, but rather succumb to disease during the chronic phase of tuberculosis infection.85 Perhaps the duality of the roles for MB21D1/CGAS in inducing type I IFN responses and connecting to the core autophagy apparatus86 while sensing bacterial presence and targeting the autophagic response to control intracellular bacteria84,85 may result in a net protective effect of MB21D1/CGAS against Mtb infection in mice.
While the widely held view is that defective autophagy results in excessive inflammation that contributes to disease pathology, there are some recent reports—where in the context of certain animal models of viral infection—that the “hyperinflammatory state” observed in the mice with myeloid-specific deletion of autophagy genes can be protective. For example, myeloid-specific deletion of several autophagy genes (e.g., Rb1cc1/Fip200, Atg5, Atg7, and Atg14) results in enhanced basal lung inflammation and resistance to influenza infection.87 Similarly, myeloid-specific deletion of Rb1cc1/Fip200, Becn1, Atg14, Atg3, Atg5, Atg7, or Atg16l1 results in increased systemic inflammation (IFNG-dependent) in chronic herpesvirus infection, which prevents viral reactivation from latency.88 These studies lead to the intriguing hypothesis that the normal homeostatic mechanisms that limit basal inflammation may actually promote certain viral infections. However, it should be cautioned that one cannot extrapolate from the effects of experimental deletion of autophagy genes prior to infection to effects of autophagy pathway inhibition on established infection.
Autophagy may reduce triggers of inflammation during viral infections
During viral infections, autophagy may reduce inflammation through mitophagy and consequent effects on inflammasome activation. For example, in influenza A infection, a NOD2-RIPK2-ULK1 pathway is important for mitophagy to prevent excessive inflammasome activation.89 In addition, we speculate that the clearance of pathogen-associated molecular patterns (PAMPs) by selective autophagy of viruses (virophagy) may, in concert with damage-associated molecular patterns generated during infection, prevent inflammasome activation.
One potentially clinically relevant observation is that bone marrow failure (a condition triggered largely by excessive inflammasome activation) in children with FA syndrome is commonly triggered by viral infections; experimentally, in Fancc-deficient mice which are defective in both mitophagy and virophagy, bone marrow failure triggered by inflammasome activators can be prevented by anti-oxidants. Thus, the “perfect storm” for inflammatory diseases may be concurrent defects in mitophagy and pathogen autophagy, leading to cellular accumulation of PAMPs and damage-associated molecular patterns (including mitochondrial ROS), and the resulting inflammasome activation.
Conclusions
There is a growing list of host factors that function dually in autophagic and other responses to endomembrane damage and intracellular pathogens. This encompasses mitophagy and pathogen-induced autophagy (either bacterial- and/or viral-induced autophagy) including: PRKN/PARK2/parkin,55,57 CALCOCO2/NDP52,55,90,91 OPTN (optineurin),55,92,93 TBK1,91,92,94–98 and SQSTM1,57,80,95,99,100 several of which have been previously reviewed by Randow and Youle101 and are updated here; and SMURF1,6,9 FANCC,33 and PEX13.6,102 Similarly, autophagic and other homeostatic responses to lysosomal damage and damage to phagosomal/vacuolar membranes harboring bacteria or viruses, recognized and initiated by galectins, is an emerging example of organelle damage as a signal, parallel to that of mitochondrial damage. Specific galectins (LGALS3 and LGALS8) and other parts of the autophagic machinery respond to phagosomal,61,69–72 endosomal, and lysosomal58,59,61,67 membrane damage, including sterile damage58,59,61,67,69 or rupture imposed by bacteria59,61,70–72 ,74,78 or viruses.76,77
A further important cellular benefit in all of the above processes may be to coordinate the control of not only different damaged host membranes but also microbial PAMPs to prevent excessive inflammation. The roles of autophagy in immune regulation in inflammation independent of the processes discussed above, extend to its intrinsic effects on differentiation of immune cells, their polarization, and the function of immune networks, and include inflammatory M1 versus M2 macrophages, and Th1 versus Treg cells (not covered here).
In summary, autophagy represents an anti-inflammatory mechanism; it protects against endomembrane damage triggered by various agents of endogenous or infectious origin and prevents unnecessary or excessive inflammation. We propose that autophagy supports productive, and prevents unnecessarily over-exuberant, inflammatory responses, thus playing a balancing act intended to avoid excessive tissue damage and ensure a measured response. Defining the molecular and cellular aspects of this concept may allow us to harness this aspect of autophagy for clinical purposes. A better understanding of the connections between autophagy and the immune response may have broad applications as the pathology associated with numerous diseases involves some form of inflammation.
Funding Statement
HHS | NIH | National Institute of Allergy and Infectious Diseases (NIAID) [grant number AI42999], HHS | NIH | National Institute of General Medical Sciences (NIGMS) [grant number GM121176], HHS | NIH | National Institute of Allergy and Infectious Diseases (NIAID) [grant number AI109725], HHS | NIH | National Institute of Allergy and Infectious Diseases (NIAID) [grant number AI111935]
Acknowledgments
We thank numerous colleagues for their contributions to the field of immune and inflammatory manifestations of autophagy and apologize for the limited referencing defined by the scope of the review. Funding sources: NIH R01 AI042999, R01 AI111935, and 1P20GM121176-01 Center of Biomedical Research Excellence for Autophagy, Inflammation and Metabolism (AIM) in Disease to V.D. and NIH U19-AI109725 to B.L.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Virgin HW, Levine B. Autophagy genes in immunity. Nat Immunol 2009;10:461–470. doi: 10.1038/ni.1726. PMID:19381141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol 2013;13:722–737. doi: 10.1038/nri3532. PMID:24064518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 2007;447:661–678. doi: 10.1038/nature05911. PMID:17554300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chauhan S, Mandell MA, Deretic V. Mechanism of action of the tuberculosis and Crohn disease risk factor IRGM in autophagy. Autophagy 2015:0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Henckaerts L, Cleynen I, Brinar M, John JM, Van Steen K, Rutgeerts P, Vermiere S.. Genetic variation in the autophagy gene ULK1 and risk of Crohn's disease. Inflammatory Bowel Diseases 2011;17:1392–1397. doi: 10.1002/ibd.21486. PMID:21560199 [DOI] [PubMed] [Google Scholar]
- 6.Orvedahl A, Sumpter R, Jr, Xiao G, Ng A, Zou Z, Tang Y, et al. . Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 2011;480:113–117. doi: 10.1038/nature10546. PMID:22020285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. . Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012;491:119–124. doi: 10.1038/nature11582. PMID:23128233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Franke A, Balschun T, Sina C, Ellinghaus D, Hasler R, Mayr G, et al. . Genome-wide association study for ulcerative colitis identifies risk loci at 7q22 and 22q13 (IL17REL). Nat Genet 2010;42:292–294. doi: 10.1038/ng.553. PMID:20228798 [DOI] [PubMed] [Google Scholar]
- 9.Franco LH, Nair VR, Scharn CR, Xavier RJ, Torrealba JR, Shiloh MU, et al. . The ubiquitin ligase Smurf1 functions in selective autophagy of Mycobacterium tuberculosis and anti-tuberculous host defense. Cell Host Microbe 2017;21:59–72. doi: 10.1016/j.chom.2016.11.002. PMID:28017659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ellinghaus D, Zhang H, Zeissig S, Lipinski S, Till A, Jiang T, et al. . Association between variants of PRDM1 and NDP52 and Crohn's disease, based on exome sequencing and functional studies. Gastroenterology 2013;145:339–347. doi: 10.1053/j.gastro.2013.04.040. PMID:23624108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramos PS, Criswell LA, Moser KL, Comeau ME, Williams AH, Pajewski NM, et al. . A comprehensive analysis of shared loci between systemic lupus erythematosus (SLE) and sixteen autoimmune diseases reveals limited genetic overlap. PLoS Genetics 2011;7:e1002406. doi: 10.1371/journal.pgen.1002406. PMID:22174698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harley JB, Alarcon-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, Moser KL, et al. . Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nature Genetics 2008;40:204–210. doi: 10.1038/ng.81. PMID:18204446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Han JW, Zheng HF, Cui Y, Sun LD, Ye DQ, Hu Z, et al. . Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nature Genetics 2009;41:1234–1237. doi: 10.1038/ng.472. PMID:19838193 [DOI] [PubMed] [Google Scholar]
- 14.Yang W, Tang H, Zhang Y, Tang X, Zhang J, Sun L, et al. . Meta-analysis followed by replication identifies loci in or near CDKN1B, TET3, CD80, DRAM1, and ARID5B as associated with systemic lupus erythematosus in Asians. American Journal of Human Genetics 2013;92:41–51. doi: 10.1016/j.ajhg.2012.11.018. PMID:23273568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin LJ, Gupta J, Jyothula SS, Butsch Kovacic M, Biagini Myers JM, Patterson TL, et al. . Functional variant in the autophagy-related 5 gene promotor is associated with childhood asthma. PloS one 2012;7:e33454. doi: 10.1371/journal.pone.0033454. PMID:22536318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Orozco G, Eyre S, Hinks A, Bowes J, Morgan AW, Wilson AG, et al. . Study of the common genetic background for rheumatoid arthritis and systemic lupus erythematosus. Ann Rheum Dis 2011;70:463–468. doi: 10.1136/ard.2010.137174. PMID:21068098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cullup T, Kho AL, Dionisi-Vici C, Brandmeier B, Smith F, Urry Z, et al. . Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nat Genet 2013;45:83–87. doi: 10.1038/ng.2497. PMID:23222957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Comincini S, Manai F, Meazza C, Pagani S, Martinelli C, Pasqua N, et al. . Identification of autophagy-related genes and their regulatory miRNAs associated with celiac disease in children. Int J Mol Sci 2017;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ricano-Ponce I, Zhernakova DV, Deelen P, Luo O, Li X, Isaacs A, et al. . Refined mapping of autoimmune disease associated genetic variants with gene expression suggests an important role for non-coding RNAs. J Autoimmun 2016;68:62–74. doi: 10.1016/j.jaut.2016.01.002. PMID:26898941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Redmann V, Lamb CA, Hwang S, Orchard RC, Kim S, Razi M, et al. . Clec16a is critical for autolysosome function and Purkinje cell survival. Sci Rep 2016;6:23326. doi: 10.1038/srep23326. PMID:26987296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schuster C, Gerold KD, Schober K, Probst L, Boerner K, Kim MJ, et al. . The Autoimmunity-associated gene CLEC16A modulates thymic epithelial cell autophagy and alters T cell selection. Immunity 2015;42:942–952. doi: 10.1016/j.immuni.2015.04.011. PMID:25979422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahmad L, Zhang SY, Casanova JL, Sancho-Shimizu V. Human TBK1: A gatekeeper of neuroinflammation. Trends Mol Med 2016;22:511–527. doi: 10.1016/j.molmed.2016.04.006. PMID:27211305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soleimanpour SA, Gupta A, Bakay M, Ferrari AM, Groff DN, Fadista J, et al. . The diabetes susceptibility gene Clec16a regulates mitophagy. Cell 2014;157:1577–1590. doi: 10.1016/j.cell.2014.05.016. PMID:24949970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kimura T, Jain A, Choi SW, Mandell MA, Schroder K, Johansen T, et al. . TRIM-mediated precision autophagy targets cytoplasmic regulators of innate immunity. J Cell Biol 2015;210:973–989. doi: 10.1083/jcb.201503023. PMID:26347139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell 2014;157:1013–1022. doi: 10.1016/j.cell.2014.04.007. PMID:24855941 [DOI] [PubMed] [Google Scholar]
- 26.Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, et al. . Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015;526:666–671. doi: 10.1038/nature15541. PMID:26375259 [DOI] [PubMed] [Google Scholar]
- 27.Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, et al. . Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008;456:264–268. doi: 10.1038/nature07383. PMID:18849965 [DOI] [PubMed] [Google Scholar]
- 28.Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, et al. . A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008;456:259–263. doi: 10.1038/nature07416. PMID:18849966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Santeford A, Wiley LA, Park S, Bamba S, Nakamura R, Gdoura A, et al. . Impaired autophagy in macrophages promotes inflammatory eye disease. Autophagy 2016;12:1876–1885. doi: 10.1080/15548627.2016.1207857. PMID:27463423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011;469:221–225. doi: 10.1038/nature09663. PMID:21124315 [DOI] [PubMed] [Google Scholar]
- 31.Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. . Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 2011;12:222–230. doi: 10.1038/ni.1980. PMID:21151103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, et al. . NF-kappaB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 2016;164:896–910. doi: 10.1016/j.cell.2015.12.057. PMID:26919428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sumpter R, Jr., Sirasanagandla S, Fernandez AF, Wei Y, Dong X, Franco L, et al. . Fanconi anemia proteins function in mitophagy and immunity. Cell 2016;165:867–881. doi: 10.1016/j.cell.2016.04.006. PMID:27133164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, et al. . Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol 2012;13:255–263. doi: 10.1038/ni.2215. PMID:22286270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kimura T, Mandell M, Deretic V. Precision autophagy directed by receptor-regulators. Journal of Cell Science 2016;In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu T, Tang Q, Liu K, Xie W, Liu X, Wang H, et al. . TRIM11 suppresses AIM2 inflammasome by degrading AIM2 via p62-dependent selective autophagy. Cell Rep 2016;16:1988–2002. doi: 10.1016/j.celrep.2016.07.019. PMID:27498865 [DOI] [PubMed] [Google Scholar]
- 37.Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J 2011;30:4701–4711. doi: 10.1038/emboj.2011.398. PMID:22068051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang M, Kenny S, Ge L, Xu K, Schekman R. Translocation of interleukin-1beta into a vesicle intermediate in autophagy-mediated secretion. eLife 2015;4. doi: 10.7554/eLife.11205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kimura T, Jia J, Kumar S, Choi SW, Gu Y, Mudd M, et al. . Dedicated SNAREs and specialized TRIM cargo receptors mediate secretory autophagy. EMBO J 2017;36:42–60. doi: 10.15252/embj.201695081. PMID:27932448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Forster I, et al. . Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 2011;34:213–223. doi: 10.1016/j.immuni.2011.02.006. PMID:21349431 [DOI] [PubMed] [Google Scholar]
- 41.Jounai N, Takeshita F, Kobiyama K, Sawano A, Miyawaki A, Xin KQ, et al. . The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc Natl Acad Sci U S A 2007;104:14050–14055. doi: 10.1073/pnas.0704014104. PMID:17709747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tal MC, Sasai M, Lee HK, Yordy B, Shadel GS, Iwasaki A. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc Natl Acad Sci U S A 2009;106:2770–2775. doi: 10.1073/pnas.0807694106. PMID:19196953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jin S, Tian S, Luo M, Xie W, Liu T, Duan T, et al. . Tetherin suppresses type I interferon signaling by targeting MAVS for NDP52-mediated selective autophagic degradation in human cells. Mol Cell 2017. doi: 10.1016/j.molcel.2017.09.005. [DOI] [PubMed] [Google Scholar]
- 44.Niida M, Tanaka M, Kamitani T. Downregulation of active IKK beta by Ro52-mediated autophagy. Molecular immunology 2010;47:2378–2387. doi: 10.1016/j.molimm.2010.05.004. PMID:20627395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013;339:786–791. doi: 10.1126/science.1232458. PMID:23258413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen M, Meng Q, Qin Y, Liang P, Tan P, He L, et al. . TRIM14 inhibits cGAS degradation mediated by selective autophagy receptor p62 to promote innate immune responses. Mol Cell 2016. doi: 10.1016/j.molcel.2016.08.025. [DOI] [PubMed] [Google Scholar]
- 47.Zalckvar E, Berissi H, Mizrachy L, Idelchuk Y, Koren I, Eisenstein M, et al. . DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-X(L) and induction of autophagy. EMBO Rep. 2009. doi: 10.1038/embor.2008.246. PMID:19180116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. PMID:15607973 [DOI] [PubMed] [Google Scholar]
- 49.Chauhan S, Mandell MA, Deretic V. IRGM governs the core autophagy machinery to conduct antimicrobial defense. Mol Cell 2015;58:507–521. doi: 10.1016/j.molcel.2015.03.020. PMID:25891078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nandi B, Behar SM. Regulation of neutrophils by interferon-gamma limits lung inflammation during tuberculosis infection. J Exp Med. 2011;208:2251–2262. doi: 10.1084/jem.20110919. PMID:21967766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sakowski ET, Koster S, Portal Celhay C, Park HS, Shrestha E, Hetzenecker SE, et al. . Ubiquilin 1 promotes IFN-gamma-induced xenophagy of Mycobacterium tuberculosis. PLoS Pathog 2015;11:e1005076. doi: 10.1371/journal.ppat.1005076. PMID:26225865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, et al. . Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010;191:1367–1380. doi: 10.1083/jcb.201007013. PMID:21173115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, et al. . Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet 2011;20:1726–1737. doi: 10.1093/hmg/ddr048. PMID:21296869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yoshii SR, Kishi C, Ishihara N, Mizushima N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem. 2011;286:19630–19640. doi: 10.1074/jbc.M110.209338. PMID:21454557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, et al. . The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015;524:309–314. doi: 10.1038/nature14893. PMID:26266977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wei Y, Chiang WC, Sumpter R, Jr, Mishra P, Levine B. Prohibitin 2 Is an inner mitochondrial membrane mitophagy receptor. Cell 2017;168:224–238 e10. doi: 10.1016/j.cell.2016.11.042. PMID:28017329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Manzanillo PS, Ayres JS, Watson RO, Collins AC, Souza G, Rae CS, et al. . The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature 2013;501:512–516. doi: 10.1038/nature12566. PMID:24005326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maejima I, Takahashi A, Omori H, Kimura T, Takabatake Y, Saitoh T, et al. . Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J 2013;32:2336–2347. doi: 10.1038/emboj.2013.171. PMID:23921551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fujita N, Morita E, Itoh T, Tanaka A, Nakaoka M, Osada Y, et al. . Recruitment of the autophagic machinery to endosomes during infection is mediated by ubiquitin. J Cell Biol 2013;203:115–128. doi: 10.1083/jcb.201304188. PMID:24100292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Papadopoulos C, Kirchner P, Bug M, Grum D, Koerver L, Schulze N, et al. . VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy. EMBO J 2017;36:135–150. doi: 10.15252/embj.201695148. PMID:27753622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chauhan S, Kumar S, Jain A, Ponpuak M, Mudd MH, Kimura T, et al. . TRIMs and galectins globally cooperate and TRIM16 and galectin-3 co-direct autophagy in endomembrane damage homeostasis. Dev Cell 2016;39:13–27. doi: 10.1016/j.devcel.2016.08.003. PMID:27693506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schroder K, Tschopp J. The inflammasomes. Cell 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. PMID:20303873 [DOI] [PubMed] [Google Scholar]
- 63.Razani B, Feng C, Coleman T, Emanuel R, Wen H, Hwang S, et al. . Autophagy links inflammasomes to atherosclerotic progression. Cell Metab 2012;15:534–544. doi: 10.1016/j.cmet.2012.02.011. PMID:22440612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, et al. . Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat Immunol 2010;11:897–904. doi: 10.1038/ni.1935. PMID:20835230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. . NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature 2013;493:674–678. doi: 10.1038/nature11729. PMID:23254930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Parry TL, Melehani JH, Ranek MJ, Willis MS. Functional amyloid signaling via the inflammasome, necrosome, and signalosome: New therapeutic targets in heart failure. Front Cardiovasc Med 2015;2:25. doi: 10.3389/fcvm.2015.00025. PMID:26664897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Aits S, Kricker J, Liu B, Ellegaard AM, Hamalisto S, Tvingsholm S, et al. . Sensitive detection of lysosomal membrane permeabilization by lysosomal galectin puncta assay. Autophagy 2015;11:1408–1424. doi: 10.1080/15548627.2015.1063871. PMID:26114578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Deretic V. Autophagy of intracellular microbes and mitochondria: two sides of the same coin? F1000 Biol Rep 2010;2. doi: 10.3410/B2-45. PMID:20948788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Garin J, Diez R, Kieffer S, Dermine JF, Duclos S, Gagnon E, et al. . The phagosome proteome: insight into phagosome functions. J Cell Biol 2001;152:165–180. doi: 10.1083/jcb.152.1.165. PMID:11149929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Paz I, Sachse M, Dupont N, Mounier J, Cederfur C, Enninga J, et al. . Galectin-3, a marker for vacuole lysis by invasive pathogens. Cell Microbiol 2010;12:530–544. doi: 10.1111/j.1462-5822.2009.01415.x. PMID:19951367 [DOI] [PubMed] [Google Scholar]
- 71.Dupont N, Lacas-Gervais S, Bertout J, Paz I, Freche B, Van Nhieu GT, et al. . Shigella phagocytic vacuolar membrane remnants participate in the cellular response to pathogen invasion and are regulated by autophagy. Cell Host Microbe 2009;6:137–149. doi: 10.1016/j.chom.2009.07.005. PMID:19683680 [DOI] [PubMed] [Google Scholar]
- 72.Thurston TL, Wandel MP, von Muhlinen N, Foeglein A, Randow F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 2012;482:414–418. doi: 10.1038/nature10744. PMID:22246324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Thiele DL, Lipsky PE. Mechanism of L-leucyl-L-leucine methyl ester-mediated killing of cytotoxic lymphocytes: dependence on a lysosomal thiol protease, dipeptidyl peptidase I, that is enriched in these cells. Proc Natl Acad Sci U S A 1990;87:83–87. doi: 10.1073/pnas.87.1.83. PMID:2296607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Creasey EA, Isberg RR. The protein SdhA maintains the integrity of the Legionella-containing vacuole. Proc Natl Acad Sci U S A 2012;109:3481–3486. doi: 10.1073/pnas.1121286109. PMID:22308473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Feeley EM, Pilla-Moffett DM, Zwack EE, Piro AS, Finethy R, Kolb JP, et al. . Galectin-3 directs antimicrobial guanylate binding proteins to vacuoles furnished with bacterial secretion systems. Proc Natl Acad Sci U S A 2017;114:E1698–E1706. doi: 10.1073/pnas.1615771114. PMID:28193861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Montespan C, Marvin SA, Austin S, Burrage AM, Roger B, Rayne F, et al. . Multi-layered control of Galectin-8 mediated autophagy during adenovirus cell entry through a conserved PPxY motif in the viral capsid. PLoS Pathog 2017;13:e1006217. doi: 10.1371/journal.ppat.1006217. PMID:28192531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Staring J, von Castelmur E, Blomen VA, van den Hengel LG, Brockmann M, Baggen J, et al. . PLA2G16 represents a switch between entry and clearance of Picornaviridae. Nature 2017;541:412–416. doi: 10.1038/nature21032. PMID:28077878 [DOI] [PubMed] [Google Scholar]
- 78.Noad J, von der Malsburg A, Pathe C, Michel MA, Komander D, Randow F. LUBAC-synthesized linear ubiquitin chains restrict cytosol-invading bacteria by activating autophagy and NF-kappaB. Nat Microbiol 2017;2:17063. doi: 10.1038/nmicrobiol.2017.63. PMID:28481331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Castillo EF, Dekonenko A, Arko-Mensah J, Mandell MA, Dupont N, Jiang S, et al. . Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc Natl Acad Sci USA 2012;109:E3168–E3176. doi: 10.1073/pnas.1210500109. PMID:23093667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Watson RO, Manzanillo PS, Cox JS. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell 2012;150:803–815. doi: 10.1016/j.cell.2012.06.040. PMID:22901810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kimmey JM, Huynh JP, Weiss LA, Park S, Kambal A, Debnath J, et al. . Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature 2015;528:565–569. doi: 10.1038/nature16451. PMID:26649827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T, et al. . An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 2010;466:973–977. doi: 10.1038/nature09247. PMID:20725040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ludigs K, Parfenov V, Du Pasquier RA, Guarda G. Type I IFN-mediated regulation of IL-1 production in inflammatory disorders. Cell Mol Life Sci.: CMLS 2012;69:3395–3418. doi: 10.1007/s00018-012-0989-2. PMID:22527721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Watson RO, Bell SL, MacDuff DA, Kimmey JM, Diner EJ, Olivas J, et al. . The cytosolic sensor cGAS detects Mycobacterium tuberculosis DNA to induce type I interferons and activate autophagy. Cell Host Microbe 2015;17:811–819. doi: 10.1016/j.chom.2015.05.004. PMID:26048136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Collins AC, Cai H, Li T, Franco LH, Li XD, Nair VR, et al. . Cyclic GMP-AMP synthase is an innate immune DNA sensor for Mycobacterium tuberculosis. Cell Host Microbe 2015;17:820–828. doi: 10.1016/j.chom.2015.05.005. PMID:26048137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liang Q, Seo GJ, Choi YJ, Kwak MJ, Ge J, Rodgers MA, et al. . Crosstalk between the cGAS DNA sensor and Beclin-1 autophagy protein shapes innate antimicrobial immune responses. Cell Host Microbe 2014;15:228–238. doi: 10.1016/j.chom.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lu Q, Yokoyama CC, Williams JW, Baldridge MT, Jin X, DesRochers B, et al. . Homeostatic control of innate lung inflammation by Vici syndrome gene Epg5 and additional autophagy genes promotes influenza pathogenesis. Cell Host Microbe 2016;19:102–113. doi: 10.1016/j.chom.2015.12.011. PMID:26764600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Park S, Buck MD, Desai C, Zhang X, Loginicheva E, Martinez J, et al. . Autophagy genes enhance murine gammaherpesvirus 68 reactivation from latency by preventing virus-induced systemic inflammation. Cell Host Microbe 2016;19:91–101. doi: 10.1016/j.chom.2015.12.010. PMID:26764599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lupfer C, Thomas PG, Anand PK, Vogel P, Milasta S, Martinez J, et al. . Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nat Immunol 2013. doi: 10.1038/ni.2563. PMID:23525089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.von Muhlinen N, Thurston T, Ryzhakov G, Bloor S, Randow F. NDP52, a novel autophagy receptor for ubiquitin-decorated cytosolic bacteria. Autophagy 2010;6:288–289. doi: 10.4161/auto.6.2.11118. PMID:20104023 [DOI] [PubMed] [Google Scholar]
- 91.Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW. The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell 2015;60:7–20. doi: 10.1016/j.molcel.2015.08.016. PMID:26365381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, et al. . Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 2011;333:228–233. doi: 10.1126/science.1205405. PMID:21617041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wong YC, Holzbaur EL. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci U S A 2014;111:E4439–E4448. doi: 10.1073/pnas.1405752111. PMID:25294927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pilli M, Arko-Mensah J, Ponpuak M, Roberts E, Master S, Mandell MA, et al. . TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 2012;37:223–234. doi: 10.1016/j.immuni.2012.04.015. PMID:22921120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Matsumoto G, Shimogori T, Hattori N, Nukina N. TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum Mol Genet 2015;24:4429–4442. doi: 10.1093/hmg/ddv179. PMID:25972374 [DOI] [PubMed] [Google Scholar]
- 96.Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, et al. . Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A 2016;113:4039–4044. doi: 10.1073/pnas.1523926113. PMID:27035970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Thurston TL, Boyle KB, Allen M, Ravenhill BJ, Karpiyevich M, Bloor S, et al. . Recruitment of TBK1 to cytosol-invading Salmonella induces WIPI2-dependent antibacterial autophagy. EMBO J 2016;35:1779–1792. doi: 10.15252/embj.201694491. PMID:27370208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Moore AS, Holzbaur EL. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc Natl Acad Sci U S A 2016;113:E3349–E3358. doi: 10.1073/pnas.1523810113. PMID:27247382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ponpuak M, Davis AS, Roberts EA, Delgado MA, Dinkins C, Zhao Z, et al. . Delivery of cytosolic components by autophagic adaptor protein p62 endows autophagosomes with unique antimicrobial properties. Immunity 2010;32:329–341. doi: 10.1016/j.immuni.2010.02.009. PMID:20206555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, et al. . PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 2010;12:119–131. doi: 10.1038/ncb2012. PMID:20098416 [DOI] [PubMed] [Google Scholar]
- 101.Randow F, Youle RJ. Self and nonself: how autophagy targets mitochondria and bacteria. Cell Host Microbe 2014;15:403–411. doi: 10.1016/j.chom.2014.03.012. PMID:24721569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lee MY, Sumpter R, Jr, Zou Z, Sirasanagandla S, Wei Y, Mishra P, et al. . Peroxisomal protein PEX13 functions in selective autophagy. EMBO Rep 2017;18:48–60. doi: 10.15252/embr.201642443. PMID:27827795 [DOI] [PMC free article] [PubMed] [Google Scholar]