

ABSTRACT
Macroautophagy/autophagy is a fundamental intracellular degradation process with multiple roles in immunity, including direct elimination of intracellular microorganisms via ‘xenophagy.’ In this review, we summarize studies from the fruit fly Drosophila melanogaster and the nematode Caenorhabditis elegans that highlight the roles of autophagy in innate immune responses to viral, bacterial, and fungal pathogens. Research from these genetically tractable invertebrates has uncovered several conserved immunological paradigms, such as direct targeting of intracellular pathogens by xenophagy and regulation of autophagy by pattern recognition receptors in D. melanogaster. Although C. elegans has no known pattern recognition receptors, this organism has been particularly useful in understanding many aspects of innate immunity. Indeed, work in C. elegans was the first to show xenophagic targeting of microsporidia, a fungal pathogen that infects all animals, and to identify TFEB/HLH-30, a helix-loop-helix transcription factor, as an evolutionarily conserved regulator of autophagy gene expression and host tolerance. Studies in C. elegans have also highlighted the more recently appreciated relationship between autophagy and tolerance to extracellular pathogens. Studies of simple, short-lived invertebrates such as flies and worms will continue to provide valuable insights into the molecular mechanisms by which autophagy and immunity pathways intersect and their contribution to organismal survival.
Abbreviations
- Atg
autophagy related
- BECN1
Beclin 1
- CALCOCO2
calcium binding and coiled-coil domain 2
- Cry5B
crystal toxin 5B
- Daf
abnormal dauer formation
- DKF-1
D kinase family-1
- EPG-7
Ectopic P Granules-7
- FuDR
fluorodeoxyuridine
- GFP
green fluorescent protein
- HLH-30
Helix Loop Helix-30
- Imd
immune deficiency
- ins-18
INSulin related-18; LET-363, LEThal-363
- lgg-1
LC3, GABARAP and GATE-16 family-1
- MAPK
mitogen-activated protein kinase
- MATH
the meprin and TRAF homology
- MTOR
mechanistic target of rapamycin
- NBR1
neighbor of BRCA1 gene 1
- NFKB
nuclear factor of kappa light polypeptide gene enhancer in B cells
- NOD
nucleotide-binding oligomerization domain containing
- OPTN
optineurin
- PAMPs
pathogen-associated molecular patterns
- Park2
Parkinson disease (autosomal recessive, juvenile) 2, parkin
- pdr-1
Parkinson disease related
- PFTs
pore-forming toxins
- PGRP
peptidoglycan-recognition proteins
- PIK3C3
phosphatidylinositol 3- kinase catalytic subunit type 3
- pink-1
PINK (PTEN-I induced kinase) homolog
- PRKD
protein kinase D; PLC, phospholipase C
- PRKN
parkin RBR E3 ubiquitin protein ligase
- PRRs
pattern-recognition receptors
- PtdIns3P
phosphatidylinositol-3-phosphate
- rab-5
RAB family-5
- RB1CC1
RB1-inducible coiled-coil 1
- RNAi
RNA interference
- sqst
SeQueSTosome related
- SQSTM1
sequestosome 1
- TBK1
TANK-binding kinase 1
- TFEB
transcription factor EB
- TGFB/TGF-β
transforming growth factor beta
- TLRs
toll-like receptors
- unc-51
UNCoordinated-51
- VPS
vacuolar protein sorting; VSV, vesicular stomatitis virus
- VSV-G
VSV surface glycoprotein G
- Wipi2
WD repeat domain, phosphoinositide interacting 2
KEYWORDS: autophagy, C. elegans, D. melanogaster, epithelial immunity, infection, extracellular pathogens, innate immunity, pattern recognition receptors, xenophagy
Introduction to autophagy
Early studies of autophagy were mainly restricted to its role in nonselective recycling of intracellular material to the lysosome in yeast responding to starvation conditions.1,2 However, it is now clear that autophagy has a range of specialized functions, including selective elimination of large endogenous material, such as damaged organelles (e.g., mitophagy), as well as exogenous material, such as invading pathogens (xenophagy). During autophagy, these ‘cargo’ are selectively recognized and sequestered within double-membrane vesicles called autophagosomes, which subsequently fuse with acidic lysosomes containing hydrolases used for degradation of cargo. This review will focus on macroautophagy (hereafter referred to as autophagy) in 2 invertebrate model organisms: the nematode Caenorhabditis elegans and the fruit fly Drosophila melanogaster.
Autophagy proceeds through at least 5 sequential steps: (1) initiation, (2) double-membrane nucleation and formation of a pre-autophagosome or phagophore, (3) phagophore elongation and sequestration of cytoplasmic cargo, (4) autophagosome fusion with a lysosome to form an autolysosome, and (5) cargo degradation in the autolysosome (Fig. 1). Several conserved autophagy-related (Atg) proteins, many of which have clear homologs in C. elegans and D. melanogaster (Table 1), function as macromolecular complexes at the different steps of the autophagy process (Fig. 1). Specifically, activation of the Atg1/ULK1 initiation complex, which contains Atg101 and Atg13 (and RB1CC1 in mammals, Atg17 in flies, and EPG-7 in worms), allows creation of a phagophore by the Vps34/PIK3C3-Vps30/BECN1 and phosphatidylinositol 3-phosphate (PtdIns3P)-binding complexes. Phagophore elongation is mediated by 2 ubiquitin-like conjugation systems. The first involves covalent conjugation of the ubiquitin-like protein Atg12 to Atg5 by the E1- and E2-like enzymes Atg7 and Atg10, respectively. The Atg12–Atg5 conjugate then promotes conjugation (possibly via its E3-like ligase activity) of phosphatidylethanolamine (PE) to cytosolic Atg8/LC3 (referred to as Atg8-I), which is formed by cleavage of the ubiquitin-like protein Atg8/LC3 by the protease Atg4. Processed and PE-conjugated Atg8/LC3 (referred to as Atg8-II/LC3-II) associates with the phagophore membrane, where it facilitates elongation and cargo recognition, and may regulate fusion with the lysosome (Fig. 1). Of note, some of the autophagy proteins mentioned above, including Atg8/LC3, also have nonautophagy roles.3 A more detailed discussion of the different steps of autophagy can be found in Klionsky et al.4
Figure 1.
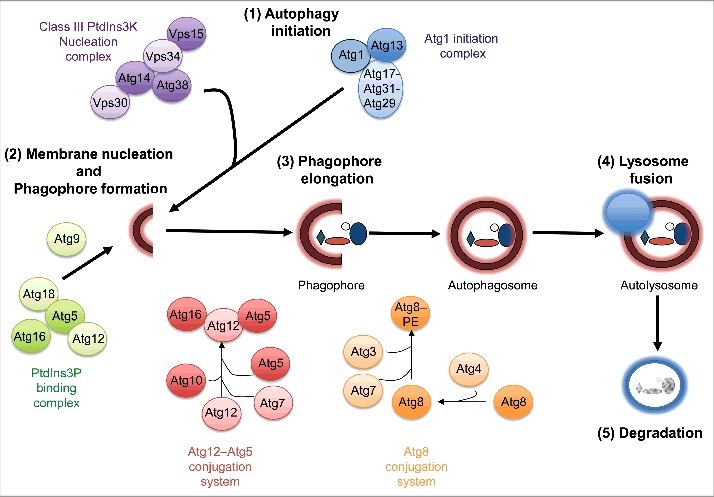
Overview of the macroautophagy process. Macroautophagy (referred to as autophagy) proceeds through at least 5 discrete steps: initiation, membrane nucleation and phagophore formation, phagophore elongation, lysosome fusion, and degradation. These steps are executed by at least 5 protein complexes: Atg1/ULK1 initiation complex, class III PtdIns 3-kinase nucleation complex, PtdIns3P-binding complex, Atg12 conjugation system, and Atg8/LC3 conjugation system. See text for details. Figure is modified from Gelino et al.53
Table 1.
Autophagy-related genes linked to immunity in D. melanogaster and C. elegans.
|
D. melanogaster |
S. cerevisiae/H. sapiens |
Pathogen |
Reference |
| Atg1 | Atg1/ULK1 | Wolbachia, VSV | 19,21 |
| Atg6 | VPS30/BECN1 | VSV | 21 |
| Atg2 | ATG2 | VSV | 21 |
| Atg18 | WIPI2 | VSV | 21 |
| Atg9 | ATG9 | VSV | 21 |
| Atg12 | ATG12 | E. coli, VSV | 16,21 |
| Atg7 | ATG7 | E. coli, VSV | 16,21 |
| Atg4 | ATG4 | VSV | 21 |
| Atg5 | ATG5 | L. monocytogenes, E.coli | 14,16 |
| Atg8 | MAP1LC3 | VSV | 21 |
| Park | PRKN | M.marinum, | 18 |
| |
|
S. enterica |
|
|
C. elegans |
S. cerevisiae/H. sapiens |
Pathogen |
Reference |
| unc-51 | Atg1/ULK1 | S. aureus | 41 |
| atg-13 | ATG13 | S. aureus | 19,21 |
| bec-1 | Vps30/BECN1 | S. enterica, | 34,44,45 |
| P. aeruginosa, | |||
| Cry5B | |||
| vps-34 | Vps34/PIK3C3 | S. aureus | 41 |
| atg-2 | ATG2 | S. aureus | 19,21 |
| atg-18 | WIPI2 | N. parisii, | 40,44 |
| Cry5B | |||
| lgg-3 | ATG12 | Cry5B | 44 |
| atg-7 | ATG7 | S. enterica | 34 |
| atg-16.2 | ATG16L1 | S. aureus | 19,21 |
| atg-4.1/2 | ATG4 | Cry5B | 44 |
| lgg-1 | MAP1LC3 | S. aureus, | 34,40,41,44,45 |
| S. enterica, | |||
| P. aeruginosa, | |||
| N. parisii, | |||
| Cry5B | |||
| lgg-2 | MAP1LC3 | S. aureus | 41 |
| sqst-1 | SQSTM1 | N. parisii | 40 |
Autophagy-related genes with reported roles in anti-viral, -bacterial, and -fungal immunity in D. melanogaster and C. elegans are shown, together with the yeast/human homologs. VSV, vesicular stomatitis virus; Cry5B, pore-forming toxin from B. thuringiensis. See Fig. 1 for a functional overview of autophagy genes, and the text for details.
The selectivity of autophagy is achieved by receptors that specifically recognize certain types of substrates (cargo), such as aggregated proteins or damaged organelles, and concomitantly interact with lipidated Atg8/LC3, thus bringing the cargo to the phagophore. Several cargo receptors recognize polyubiquitinated substrates. For example, SQSTM1/p62, CALCOCO2/NDP52 and OPTN/Optineurin recognize ubiquitinated mitochondria and facilitate their degradation by mitophagy.5 Clearance of damaged mitochondria protects the cells from potentially toxic components, such as reactive oxygen species released by breakdown of mitochondrial membrane integrity. The ability of autophagic vesicles to engulf such bulky cargo several microns in diameter is also exploited by the cell to clear intracellular microbes, a process termed xenophagy. The involvement of autophagy in host defense is an exciting area of research that is addressing a number of key questions. For example, how are microbes selected for autophagic clearance? Does autophagy play roles in host immunity beyond xenophagic degradation of the pathogen? How does the host use this intracellular degradation pathway to fight infection by extracellular pathogens?
Some of these questions are addressed in several excellent reviews on autophagy and infection.6-8 Here, we summarize some of the insights gained from studies of D. melanogaster and C. elegans, 2 powerful model systems that have enabled important discoveries about how metazoans use autophagy to protect against microbial infections. We define ‘microbe’ as an entity that causes infection in D. melanogaster or C. elegans; specifically, viruses, bacteria, and eukaryotic single-celled organisms. Notably, D. melanogaster and C. elegans both lack an adaptive immune system, relying instead on innate immune responses mediated by dedicated ‘professional’ (D. melanogaster) or ‘nonprofessional’ (C. elegans) immune cells. In D. melanogaster, hemocytes are one type of professional immune cell, which are analogous to macrophages and can eliminate microbial pathogens by phagocytosis. In contrast, C. elegans appears to lack professional immune cells and instead relies on epithelial cells for immune defense. Interestingly, autophagy is also important in mammalian epithelial cells, although its role has been studied more extensively in professional immune cells.9-11 Thus, D. melanogaster and C. elegans provide the opportunity to explore the contribution of autophagy to innate immunity, particularly epithelial defense, in the absence of confounding effects of adaptive immunity. Indeed, studies in D. melanogaster provided some of the earliest examples of how host pattern-recognition receptors (PRRs) promote autophagy, while studies in C. elegans provided the first example of xenophagy targeting microsporidia, and the first description of the conserved role of the transcription factor TFEB/HLH-30 in immunity. Several recent studies in C. elegans have begun to elucidate the involvement of autophagy in promoting host tolerance (i.e., the ability to limit detrimental impact on the host) rather than resistance (i.e., the ability to limit pathogen burden) to infection by extracellular pathogens. Both model organisms are genetically tractable at the whole organism and cell- and tissue-specific levels and additionally have short life cycles, which facilitate analysis of the role of autophagy on organismal survival. Within this context, we discuss how key components of innate immune signaling pathways in flies and worms interface with autophagy to regulate host defense against various types of microbial infection.
Autophagy and immunity in Drosophila melanogaster
Studies in flies and mice in the 1990s uncovered the central paradigm that animals use PRRs to detect pathogen-associated molecular patterns (PAMPs), which are molecules produced specifically by microbes.12 Most PAMPs may be more aptly named microbe-associated molecular patterns in that they are common to both pathogenic and non-pathogenic microbes. The 2 major immune detection systems in the fly, Toll and Imd pathways, recognize PAMPs derived from Gram-positive bacteria and fungi and from Gram-negative bacteria, respectively. Both the Toll and Imd pathways activate transcription factors orthologous to mammalian NFKB/NF-κB, arguably the central transcription factor in mammalian immunity.12 The Toll-NFKB pathway was first defined for its role in D. melanogaster embryonic development, and the subsequent discovery of its involvement in fly immunity occurred concurrent with the finding that a PAMP-toll-like receptor (TLR)-NFKB pathway is also involved in microbial defense in the mouse. PAMP-PRR signaling is not the only aspect of microbial immunity that is conserved in D. melanogaster and mice, as both species also use xenophagic turnover of pathogens to control intracellular infection. This topic was previously reviewed,13 and, here, we summarize the most recent reports of how autophagy components and PRRs interface to detect and control intracellular pathogen infections in D. melanogaster (Fig. 2).
Figure 2.
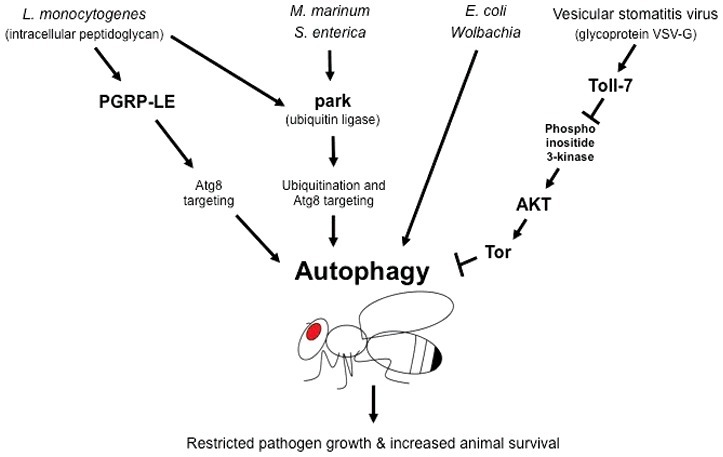
Pathogen responses linked to autophagy in Drosophila melanogaster. Autophagy is linked to defense against several intracellular pathogens in the fruit fly D. melanogaster. Intracellular peptidoglycan of the bacterium Listeria monocytogenes binds to the peptidoglycan recognition receptor PGRP-LE in hemocytes, which induces autophagy and clears the pathogen via Atg8 targeting. Infection by Mycobacterium marinum, Salmonella enterica, Escherichia coli and Wolbachia, a common insect pathogen, is also cleared by autophagy. In particular, clearance of M. marinum and S. enterica involves the ubiquitin ligase PARK2/park and Atg8 targeting. Clearance of viral infections has also been linked to autophagy. Specifically, binding of the vesicular stomatitis virus (VSV)-G glycoprotein to the pattern recognition receptor Toll-7 inhibits activation of Tor through the phosphoinositide 3-kinase-Akt pathway. Because Tor negatively regulates autophagy, its inhibition by VSV-G signaling results in induction of autophagy, which restricts viral replication and promotes organismal survival. See text for details and Table 1 for autophagy genes linked to immunity in D. melanogaster.
Xenophagy in D. melanogaster
One of the first reports that xenophagy can control infection in D. melanogaster came from studies of the bacterium Listeria monocytogenes.14 This pathogen causes food-borne illness in humans and is a facultative intracellular pathogen, meaning that it can replicate extracellularly or intracellularly. PRRs such as Toll receptors and TLRs are expressed both on the cell surface as well as intracellularly and are thus poised to detect both types of pathogens. Studies from Yano et al. demonstrated that PGRP-LE, an intracellular PRR in D. melanogaster, detects a peptidoglycan PAMP produced by L. monocytogenes and triggers LC3/Atg8 targeting to the bacteria followed by xenophagic engulfment.14,15 Concurrently, a separate study indicated that autophagy components are important for D. melanogaster defense against infection with Escherichia coli. RNAi knock-down of autophagy components caused an increased pathogen load and decreased survival upon E. coli infection, although not a decreased lifespan overall.16 Notably, studies of xenophagy against L. monocytogenes reported that xenophagy is induced independently of the Toll and Imd pathways, and the noncanonical pathway triggered by PGRP-LE has yet to be defined.14 Specifically, these studies showed that flies defective in the autophagy component Atg5 or in PGRP-LE carry a higher L. monocytogenes load and survive for shorter times after infection as compared to wild-type flies. Interestingly, a pathway with parallels to the D. melanogaster PRGP-LE–xenophagy pathway was subsequently found in mammals. Mammalian NOD1 and NOD2, which are intracellular receptors distinct from TLRs, also recognize peptidoglycans and trigger xenophagy in defense against intracellular pathogens.17 Although NOD2 directly recruits ATG16L1 to bacteria at the site of membrane entry, it is less clear how bacteria are captured for xenophagy after escape into the cytosol.7,8 Of note, variants of the NOD2 and ATG16L1 genes are associated with Crohn disease, a serious inflammatory bowel disease in humans.11
Several studies have suggested that some of the same autophagy machinery involved in mitophagy may also be important for xenophagy. PRKN/PARK2/parkin is an E3 ubiquitin ligase with a well-characterized role in conjugating ubiquitin to the surface of damaged mitochondria, thereby recruiting the autophagy machinery via ubiquitin-binding receptor proteins such as BNIP3. The finding that park/PARK2-deficient flies and mice have defects in pathogen clearance was therefore particularly exciting.18 These animals carry higher pathogen loads than wild-type flies or mice upon infection with several facultative intracellular pathogens, including L. monocytogenes, Mycobacterium spp. (M. tuberculosis in mice and M. marinum in flies), and Salmonella enterica. Moreover, park-mutant flies succumb earlier to infection than wild-type flies, highlighting the importance of park for long-term health. In mice, PARK2 is required for ubiquitination of M. tuberculosis and recruitment of ubiquitin-recognition receptors, including SQSTM1, NBR1, CALCOCO2, and phospho-TBK1, as well as the autophagy proteins Atg8/LC3 and Atg12. The park protein is also required for ultimate microbial targeting to lysosomes and subsequent degradation.18 However, the mechanisms by which park and other ubiquitin ligases recognize pathogen substrates to be targeted for ubiquitination and subsequent autophagic degradation remain poorly understood.8
Autophagy plays a similarly protective role in flies infected with the obligate intracellular Gram-negative bacterium Wolbachia,19 which is commonly harbored by insects and nematodes. Because Wolbachia is vertically transmitted (i.e., it is transmitted through the mother to progeny through the eggs, and undergoes its entire life cycle inside the fly), it has a very close association with its host. Given that mitochondria were thought to be derived from an internalized bacterium that became an obligate intracellular microbe and eventually an organelle,20 it is interesting to note that an obligate, intracellular microbe like Wolbachia would be targeted for xenophagic clearance as well.
Autophagy is also required for D. melanogaster defense against viral infection. Shelly et al. found that the mammalian viral pathogen vesicular stomatitis virus (VSV), can also infect D. melanogaster and trigger an anti-viral response.21 In this study, inhibition of the autophagy-related genes Atg1/Ulk1, Atg5, Atg8a/Lc3, and Atg18/Wipi2 in D. melanogaster S2 cells was found to increase the VSV infection rate. Similarly, RNAi knockdown of Atg18/Wipi2 in adult flies increases viral replication and decreases the survival of infected flies. Whereas earlier studies in other systems had been able to detect viruses within autophagic vesicles, this study in D. melanogaster was the first to demonstrate that autophagy plays an active role in antiviral immunity. Interestingly, Shelly et al. found that autophagy induction is independent of VSV replication and can be triggered solely by the VSV surface glycoprotein G (VSV-G), which activates autophagy via the nutrient-sensing phosphoinositide 3-kinase-Akt signaling pathway.21 Subsequent studies by Nakamoto et al. clarified how infected flies recognize VSV-G. Surprisingly, they found that VSV-G is detected by a cell-surface Toll receptor, indicating that the virus is sensed extracellularly. This study was pivotal in shedding light on the function of another one of the 9 Toll receptors expressed in D. melanogaster. After the initial discovery that a Toll receptor senses Gram-positive bacterial and fungal infections, the role of the other 8 Toll receptors had remained unclear. Nakamoto et al. showed that VSV replication and mortality are higher in Toll-7 mutants compared with wild-type flies, thus identifying Toll-7 as a VSV-G receptor capable of triggering antiviral autophagy.22 Interestingly, Moy et al. showed that D. melanogaster Toll-7 also directs antiviral immunity against arthropod-borne viruses that are more pathogenic to humans and those regulate autophagy defense against these viruses via toll-like receptor signaling in mammalian cells as well.23
As summarized here, studies in D. melanogaster have been instrumental in establishing PGRP-LE and Toll-7 as PRRs important for autophagy induction in response to intracellular bacterial and viral infections and in discovering the conserved function of the ubiquitin ligase park in targeting intracellular bacterial pathogens.
Autophagy and immunity in Caenorhabditis elegans
The microscopic nematode C. elegans is another common invertebrate model organism for studies of innate immunity, in part because of the extensive genetic toolbox available for this animal.24,25 In contrast to work in D. melanogaster, which has concentrated on systemic immunity and signaling by professional immune cells, most studies in C. elegans have focused on intestinal epithelial immunity to oral infections. C. elegans is notable among models for studying immunity in that it is devoid of any specialized immune cells and instead relies on nonprofessional cells, such as epithelial cells, for defense. Fortunately, the transparent body plan of C. elegans facilitates direct observation of microbes and their interactions with host epithelial cells during infection, which has been useful for the study of both extracellular and intracellular pathogens. C. elegans contains 20 nonrenewing intestinal epithelial cells similar in structure and function to their mammalian counterparts.26 In addition, C. elegans intestinal epithelial cells, like their mammalian counterparts, are nonphagocytic indicating that pathogen uptake occurs through a mechanism distinct from that of D. melanogaster hemocytes or mouse macrophages. Another intriguing mechanistic difference is that NFKB appears to have been lost from the evolutionary lineage that gave rise to C. elegans.26 This feature provides the advantage that it permits dissection of the roles of transcription factors other than NFKB, which has been extensively studied in many species. Indeed, studies in C. elegans have identified several novel transcription factors with roles in immune defense, and it appears unlikely that a single factor plays the dominant role, as is the case for NFKB in flies and mammals. Instead, studies in C. elegans suggest that distinct transcription factors may be important for defense against different pathogens,27,28 or even against different virulence factors from the same pathogen. For example, C. elegans displays several context-dependent modes of defense against the bacterial pathogen Pseudomonas aeruginosa, each involving distinct pathogen virulence factors and different host pathways.29,30
How does C. elegans detect pathogens? Surprisingly, no PRRs have yet been identified in C. elegans. Moreover, the single C. elegans Toll-like receptor, TOL-1, which was identified by sequence homology, does not appear to play a canonical role in pathogen defense. However, like its fly ortholog, tol-1 does have a role in early development.25 Intriguingly, tol-1 also plays a role in the development of sensory neurons important for behavioral avoidance of pathogens.25 Several reports suggest that C. elegans does not detect the pathogen or its products per se, but rather senses the physiological consequences of pathogenic attack, or ‘patterns of pathogenesis’.24,31 For example, C. elegans intestinal cells sense P. aeruginosa indirectly through the effects of the bacterial product exotoxin A, which is endocytosed by host cells and causes a block in mRNA translation.24 Interestingly, it is the translational inhibition, not the toxin itself, that is sensed and leads to increased protein levels of the bZIP transcription factor ZIP-2 through an unknown mechanism. In turn, ZIP-2-mediated gene expression reduces pathogen load and increases host survival. Several C. elegans transcription factors have been shown to mediate defense by upregulating expression of large classes of predicted antimicrobial genes, including secreted C-type lectins, lysozymes, and lipases.24,25 The molecular connections between the pathogenic triggers and host signaling pathways are still being defined, but the major pathway controlling expression of antimicrobial genes appears to be the conserved MAPK (mitogen-activated protein kinase)/p38 pathway. This pathway controls C. elegans resistance to most pathogens tested, although it appears to activate different transcription factors in response to different pathogens.27,28,32 Another pathway that is involved in C. elegans immunity is the DAF-2/DAF-16 insulin-like signaling pathway, which also has roles in development, metabolism, and longevity.33 How autophagy is involved in C. elegans defense via these signaling pathways is only beginning to be investigated. Below, we summarize the findings of several reports that not only suggest a key role for autophagy in C. elegans antiviral, -bacterial, and -fungal responses, but also highlight several novel autophagy regulators and immunological concepts, especially with respect to defense against extracellular pathogens (Fig. 3).
Figure 3.
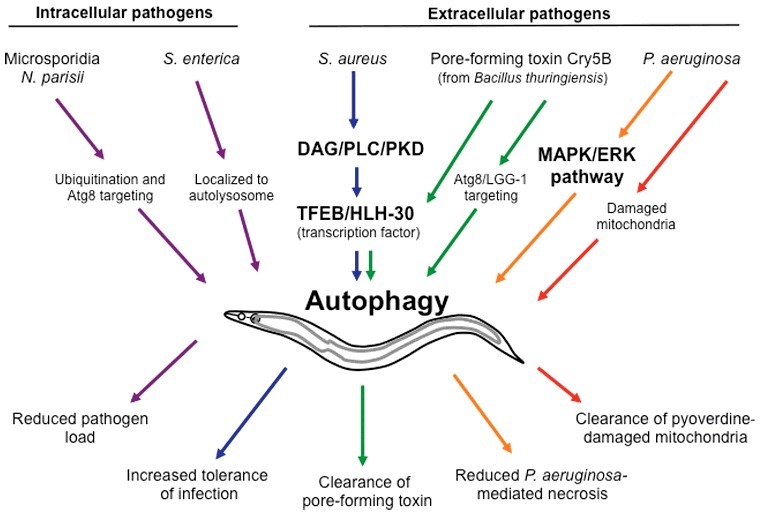
Pathogen responses linked to autophagy in Caenorhabditis elegans. Autophagy plays crucial roles in the defense against both intracellular and extracellular pathogens in the nematode C. elegans. The microsporidian pathogen Nematocida parisii is targeted for ubiquitination and recruitment of Atg8/LGG-1, which likely results in xenophagy. Replication of Salmonella enterica is restricted by localization to lysosomes. Staphylococcus aureus and the Bacillus thuringiensis pore-forming toxin Cry5B both induce transcription of autophagy-related genes via TFEB/HLH-30. Pseudomonas aeruginosa-induced necrosis of C. elegans is reduced by activation of autophagy through the MAPK/ERK signaling pathway. Autophagy (mitophagy) also clears mitochondria damaged by the P. aeruginosa virulence factor pyoverdine, thereby reducing mortality. See text for details and Table 1 for autophagy genes linked to immunity in C. elegans.
Xenophagy in C. elegans
Most studies of bacterial infection in C. elegans have been performed by feeding worms with clinically relevant bacteria that accumulate in the intestinal lumen, inflict damage, and ultimately kill the host. All C. elegans bacterial pathogens characterized to date (including facultative intracellular bacteria such as S. enterica and L. monocytogenes) appear not to enter cells but remain in the intestinal lumen during the active stage of the infection (although for some bacterial pathogens, such as P. aeruginosa, invasion of intestinal cells in wild-type animals has been seen later in infection, when there is extensive intestinal tissue damage). The reason for this restriction to the lumen early during infection was proposed by Jia et al.34 whose work suggested that autophagy may prevent S. enterica intracellular persistence and replication. Specifically, intact bacteria were not observed with electron microscopy within intestinal cells in control animals, whereas RNAi of the autophagy components VPS30/Becn1/bec-1 and ATG8/lgg-1 resulted in what appeared to be intracellular bacteria and an expanded bacterial population in the intestinal lumen. Notably, however, the electron microscopy lacked any specific markers to definititvely identify the bacteria, and the authors have been unable to observe fluorescently tagged S. enterica intracellularly in later work.35 Irrespective of bacterial localization following infection, these studies suggest that RNAi-mediated inhibition of autophagy genes reduces the survival of infected C. elegans, indicating a role for this pathway in defense against S. enterica infection. The authors also examined S. enterica infection in C. elegans daf-2/IGF-1 receptor mutants, and showed that these mutants require autophagy genes for increased survival upon infection.34 Taken together, these studies support a role for autophagy in limiting bacterial proliferation and reducing lumenal bacterial load. Although further details remain to be investigated, it is possible that autophagy exerts these effects via xenophagy, possibly in combination with regulating secretion of antimicrobial molecules by intestinal cells, similar to the behavior of Paneth cells in the mouse intestine.36,37
The first pathogen shown to normally reside and replicate in the intestinal cells of wild-type C. elegans is Nematocida parisii. N. parisii defines a new genus and species in the Microsporidia phylum, which contains diverse obligate intracellular pathogens that can infect many animals, including humans. Microsporidia have been isolated from wild-caught nematodes around the world,38,39 suggesting that they exert evolutionary pressure on their hosts. Microsporidia deploy a unique invasion strategy involving an infection apparatus called a polar tube, which delivers a parasite cell called the sporoplasm directly into the host cell, where it replicates in the host cytoplasm. Not surprisingly, the cytoplasmic location makes the sporoplasms vulnerable to targeting by the autophagy machinery, as shown by Bakowski et al.40 These authors performed transcriptional profiling studies of infected C. elegans and found that N. parisii induced a gene set distinct from that induced by extracellular pathogens such as P. aeruginosa and Staphylococcus aureus but nearly identical to that induced by Orsay virus, another natural obligate intracellular pathogen of C. elegans that also replicates in the intestinal cells.
The gene set commonly upregulated by Orsay virus and N. parisii infection is enriched for functional domains associated with ubiquitin-mediated proteolysis, including the ubiquitin ligase adapter F-box and MATH domain genes. Following on this lead, the authors showed that ubiquitin and Atg8/LGG-1 are colocalized to N. parisii early during the parasite replication phase within the intestinal cells. Furthermore, knockdown of key autophagy components, such as ATG8/lgg-1 and Sqstm1/sqst-1, increases the pathogen load; conversely, activation of autophagy by blocking the nutrient-sensor and negative autophagy regulator MTOR/LET-363 increases Atg8/LGG-1 targeting and reduces the pathogen load. Notably, the increase in pathogen load by knockdown of autophagy is relatively small, raising the possibility that N. parisii might actively suppress autophagy. Indeed, the authors showed that treatment of C. elegans with either the DNA synthesis inhibitor fluorodeoxyuridine (FuDR) or the antimicrosporidia drug fumagillin, both of which slow pathogen growth, increases the efficiency of ubiquitin targeting to parasite cells. Collectively, these results suggest that ubiquitination and autophagy play a role in controlling intestinal infection with N. parisii and provide the first demonstration of direct localization of autophagy machinery to intracellular pathogens in C. elegans.40 These studies are also the first to demonstrate xenophagy of a microsporidia species, and this remains one of the few innate immune strategies known to defend against microsporidia infection.
HLH-30/TFEB and tolerance of C. elegans to infection
Studies in C. elegans have been instrumental in identifying a new transcription factor, the helix-loop-helix transcription factor HLH-30 (ortholog of TFEB) as a key regulator of the innate immune response to the extracellular bacterium S. aureus.41 TFEB had previously been shown to control expression of certain autophagy and lysosomal genes in response to nutritional stress in mammalian cells.42 A search for transcription factors regulating expression of genes induced by infection of C. elegans with S. aureus identified HLH-30 as a candidate. Notably, S. aureus infection induces translocation of TFEB/HLH-30 to the nucleus, where it regulates ∼80% of the transcriptional immune response to infection.41 The affected genes included components of conserved signaling pathways (e.g., MAPK/JNK, MAPK/p38, TGFB/TGF-β, INS/INS-18), antimicrobial genes (e.g., lysozymes, C-type lectins), and autophagy and lysosomal genes. Consistent with these observations, Visvikis et al. demonstrated that TFEB/HLH-30 plays an important role in the immune response to S. aureus via upregulation of autophagy genes. They showed that expression of Atg8/LGG-1 is significantly induced in C. elegans intestinal cells by S. aureus infection, although—intriguingly—the bacteria remain extracellular. RNAi of ATG8/lgg-1, ATG1/Ulk1/unc-51, or VPS34/pik3c3/vps-34 autophagy genes decrease the survival rate of infected wild-type animals, but not of Tfeb/hlh-30 mutants, indicating a requirement for TFEB/HLH-30-regulated autophagy in the antibacterial defense. Similar observations have been made in murine macrophages infected with S. aureus, suggesting that TFEB is a conserved transcriptional regulator of immune responses.41
In mammals, phosphorylation of TFEB by MTOR and MAPK1/ERK2 inhibits TFEB nuclear translocation under basal conditions, but depletion of intracellular amino acids induces TFEB dephosphorylation and transport into the nucleus. To identify the relevant regulatory enzymes, Najibi et al. screened an RNAi library targeting most C. elegans protein kinases and phosphatases for genes capable of regulating nuclear localization of the reporter protein HLH-30::GFP. They found that a novel PRKD (protein kinase D), called DKF-1, is required for TFEB/HLH-30 nuclear localization in response to S. aureus infection.43 Furthermore, knockdown of dkf-1 increases the susceptibility to infection of wild-type animals but not of Tfeb/hlh-30 mutants, suggesting that dkf-1 and Tfeb/hlh-30 act in the same pathway. Additional experiments placed phospholipase C (PLC) upstream of PRKD in the C. elegans pathway and also showed that the PLC-PRKD-TFEB pathway is conserved in murine macrophages.
Bacterial pathogens generate virulence factors known as pore-forming toxins (PFTs) that damage host cellular membranes. A recent report showed that the PFTs Cry5B and Cry21A, produced by the extracellular Gram-positive bacterium Bacillus thuringiensis, induce autophagy in C. elegans via TFEB/HLH-30.44 Specifically, C. elegans fed with E. coli expressing Cry5B were examined by electron microscopy and by fluorescence microscopy to detect autophagy marker proteins. Cry5B feeding not only increased the abundance of autophagic vesicles but also induced the nuclear translocation of HLH-30::GFP in intestinal cells. Moreover, GFP-tagged Atg8/LGG-1 colocalizes with rhodamine-labeled Cry5B proteins within intestinal cells, suggesting that Cry5B is degraded by autophagy. Consistent with this observation, inhibition of VPS30/Becn1/bec-1, ATG4/Atg4/atg-4.1/2, Atg8/lgg-1/2/3, Wipi2/atg-18, and Tfeb/hlh-30 decreases the survival of animals fed E. coli expressing Cry5B, but not animals fed E. coli expressing vector control. Thus, in contrast to the classical definition of xenophagy as an intracellular microbe-targeting pathway, targeting of Cry5B appears to be an example of xenophagy of intracellular toxins delivered by extracellular microbes. Interestingly, transcriptomic analysis of the Cry5B-fed C. elegans revealed that HLH-30/TFEB also regulates the transcription of membrane-repair genes (e.g., the small GTPase rab-5). Autophagy genes are required for the repair of membrane pores induced by Cry5B,44 suggesting an additional function for autophagy in host defense to PFT-producing bacteria. Among the genes induced by Cry5B are a number related to C-type lectins, lysosomes, peroxisomes, and heat shock proteins, which is in agreement with the overall function of TFEB/HLH-30 in regulating expression of cytoprotective and antimicrobial genes in defense against S. aureus infection.41 Taken together, these data demonstrate the essential role of transcriptional regulation of autophagy in the multi-step defense of C. elegans against Cry5B and S. aureus. Additionally, it appears that defense against extracellular bacteria such as S. aureus also involves xenophagic clearance of toxins delivered into the host cells. Further work will shed light on this important defense mechanism.
Nonxenophagic roles for autophagy-mediated defense in C. elegans
As noted above, autophagy is required for the repair of membrane pores caused by Cry5B, highlighting the role of this pathway not only in clearing the pathogen/toxin but also in minimizing their deleterious effects and maintaining host fitness during infection. In this regard, Zou et al.45 showed that autophagy controlled C. elegans infection with the extracellular bacterium P. aeruginosa not by blocking intestinal accumulation but rather by inhibiting pathogen-induced necrosis of intestinal cells. Although autophagy interfaces with apoptosis,2 relatively little is known about its connection to necrosis. Zou et al. found that autophagy is regulated via the MAPK/ERK pathway. Although bec-1 RNAi reduces the survival of infected animals, concomitant knockdown of bec-1 and the necrosis-related genes asp-3 and asp-4 rescue survival.45 Thus, autophagy increases the host tolerance not by clearing the extracellular pathogen but by dampening its deleterious effects on the host.
Another nonxenophagic mechanism by which autophagy contributes to C. elegans immune defense is by clearing organelles damaged by pathogen products, as recently reported by Kirienko et al. In this study, the authors used a model of P. aeruginosa infection of C. elegans that involves different host and pathogen factors than the P. aeruginosa model described above. The P. aeruginosa virulence factor pyoverdine is an iron-chelating siderophore that disrupts iron metabolism and mitochondrial homeostasis in the host,46 possibly by inducing iron scavenging resulting in a ‘hypoxic crisis.’ Exposure of C. elegans to P. aeruginosa or to partially purified pyoverdine causes the appearance of fragmented mitochondria with large, punctate bodies, in contrast to the long-branched tubular appearance of healthy mitochondria. Genetic mutation and RNAi experiments showed that knockdown of the autophagy-related genes VPS30/Becn1/bec-1 and lgg-1/ATG8 or of the mitophagy regulators pink-1 and Park2/pdr-1 reduces the survival of P. aeruginosa-infected C. elegans. Intriguingly, the pyoverdine-induced mitophagy response also promotes C. elegans resistance to P. aeruginosa. Taken together, these studies illustrate the essential nonxenophagic contributions of autophagy to the antimicrobial response of C. elegans.
Conclusions and future directions
As summarized above, studies in the metazoans D. melanogaster and C. elegans have identified multiple links between the cellular recycling process of autophagy and the innate immune responses to diverse microbes. Seminal discoveries on PRR-mediated activation of autophagy were made in D. melanogaster, including the function of PGRP-LE in triggering xenophagy in response to intracellular bacteria. Somewhat surprisingly, viral glycoproteins appear to be sensed by the cell-surface receptor Toll-7 in flies, indicating that autophagy is triggered before viral invasion and replication. Ultimately, however, viral burden is likely lowered intracellularly through xenophagy. This strategy may be a form of ‘priming,’ allowing the host to upregulate defense mechanisms before extensive damage has been inflicted. Given recent findings about the complement system in D. melanogaster regulating autophagy in neighboring cells, perhaps antimicrobial autophagy responses might even be established in a cell nonautonomous fashion.47
C. elegans also uses xenophagic elimination of intracellular microbes, and autophagy machinery can be targeted to virulence factors secreted by extracellular microbes invading this organism. The B. thuringiensis Cry5B PFT disrupts host cell membrane integrity and is then endocytosed as part of the host membrane-repair process. In the cytoplasm, Cry5B colocalizes with the autophagy machinery and is subsequently cleared from the cell. These observations suggest that the definition of xenophagy could be expanded to include the targeting and degradation of intracellular microbial factors as well as the intact microbes themselves. In this case, the distinction between extracellular and intracellular pathogens is blurred, because the virulence factor is delivered into the host cell and targets core intracellular processes but the pathogen itself is extracellular. Given that hundreds of such virulence factors are deployed by pathogens to attack host cells,48 it seems likely that autophagy may target a vast array of molecules in a similar manner. For example, P. aeruginosa appears to secrete many virulence factors in addition to pyoverdine and exotoxin A during C. elegans infection. It will be interesting to determine whether exotoxin A induces autophagy, as pyoverdine does.
The autophagy regulator TFEB/HLH-30 was first shown to be important for pathogen responses in C. elegans and then subsequently confirmed in mammals. While it is clear that TFEB/HLH-30 controls clearance of the B. thuringiensis Cry5B virulence factor via autophagy, it is not clear how it orchestrates C. elegans defense against the extracellular pathogen S. aureus. Loss of hlh-30/Tfeb does not appear to affect S. aureus pathogen load but does reduce host survival, suggesting that TFEB/HLH-30 affects host tolerance to infection. This model is supported by the observation that the survival advantage conferred by overexpression of Tfeb/hlh-30 requires the conserved autophagy machinery. Perhaps autophagy clears an unidentified virulence factor produced by S. aureus, as with B. thuringiensis and Cry5B. Alternatively, or in addition, autophagy may degrade a damaged host factor or organelle to improve cellular health. This novel function for autophagy in controlling host tolerance has been demonstrated for several infectious agents in C. elegans; for example, pyoverdine-induced mitophagy during P. aeruginosa infection, as described above. Autophagy also appears to block necrosis induced by P. aeruginosa. Although the studies of P. aeruginosa mitophagy and necrosis used different models, it is tempting to speculate that the pathways could be linked, perhaps through triggering of necrosis by reactive oxygen species released from damaged mitochondria. Further work will be necessary to determine whether and how the various autophagic responses are linked, and whether they promote tolerance to infection by targeting host or pathogen factors.
C. elegans also provided the first example of xenophagic targeting of microsporidia, a phylum of fungal-like pathogens that infect virtually all animals. Unlike most intracellular bacteria, which replicate inside a specialized membrane-enclosed compartment inside the cell, microsporidia resemble viruses in that they replicate in the cytosol, where they can be directly targeted by the host xenophagic response. Studies in C. elegans have identified a novel role for xenophagy in resistance to N. parisii infection, although host defense is ultimately unsuccessful in controlling infection in the laboratory strain of C. elegans.
In closing, we note that evidence is accumulating for the involvement of different subsets of autophagy components in processes other than canonical autophagy. For example, Atg8/LC3-associated phagocytosis, first observed in murine macrophages, involves some, but not all, of the components involved in canonical autophagy,49 although this process has not been investigated extensively in D. melanogaster or C. elegans.50 Likewise, a novel role for autophagy in unconventional secretion has recently been described in yeast and in mice, and it will be interesting to determine whether this also occurs in the model organisms discussed here.51 Furthermore, a recent study in mammalian cells found that Atg8/LC3 can recruit innate immune proteins for a type of antiviral defense that appears to not involve fusion with the lysosome.52 These new insights will likely stimulate further analysis of autophagy proteins also in D. melanogaster and C. elegans, to determine whether they act in canonical autophagy, noncanonical autophagy, or some other process to provide immune defense. Overall, it seems likely that different versions of autophagy may have evolved for different purposes. We speculate that this versatility may be especially important for defense against pathogens, which must evolve mechanisms to evade or suppress autophagy for their survival. Therefore, if one component of the host autophagy pathway is inhibited by a pathogen, alternative mechanisms may evolve to restore functional autophagy. In this way, new autophagy pathways could be built. Further work in model organisms such as D. melanogaster and C. elegans will help investigate this interesting idea.
Funding Statement
CJK was funded by the Taiwanese Ministry of Science and Technology grant (105-2917-I-006-012). MH was funded by NIH/NIA grant AG038664 and by a Julie Martin Mid-Career Award in Aging Research from The Ellison Medical Foundation/AFAR. ERT was funded by NIH grants GM114139 and AG052622 and by a Burroughs Wellcome Fund fellowship.
Note
Nomenclature: In the Introduction, yeast genes/proteins are stated first, followed by the mammalian name, if different. The nomenclature for other model organisms is used subsequently, where applicable.
Disclosure of potential conflicts of interests
No potential conflicts of interest were disclosed.
Acknowledgements
We thank Dr. Anne O'Rourke for comments on the manuscript.
References
- 1.Zimmermann A, Kainz K, Andryushkova A, Hofer S, Madeo F, Carmona-Gutierrez D. Autophagy: one more Nobel Prize for yeast. Microb Cell. 2016;3:579–581. doi: 10.15698/mic2016.12.544. PMID:28357329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yin Z, Pascual C, Klionsky DJ. Autophagy: machinery and regulation. Microb Cell. 2016;3:588–596. doi: 10.15698/mic2016.12.546. PMID:28357331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ktistakis NT, Tooze SA. Digesting the Expanding Mechanisms of Autophagy. Trends Cell Biol. 2016;26:624–635. doi: 10.1016/j.tcb.2016.03.006. PMID:27050762 [DOI] [PubMed] [Google Scholar]
- 4.Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1–222. doi: 10.1080/15548627.2015.1100356. PMID:26799652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2014;7:279–296. doi: 10.4161/auto.7.3.14487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller C, Celli J. Avoidance and subversion of eukaryotic homeostatic autophagy mechanisms by bacterial pathogens. J Mol Biol. 2016;428:3387–3398. doi: 10.1016/j.jmb.2016.07.007. PMID:27456933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sorbara MT, Girardin SE. Emerging themes in bacterial autophagy. Curr Opin Microbiol. 2015;23:163–170. doi: 10.1016/j.mib.2014.11.020. PMID:25497773 [DOI] [PubMed] [Google Scholar]
- 8.Randow F, Youle R. Self and nonself: how autophagy targets mitochondria and bacteria. Cell Host Microbe. 2014;15:403–411. doi: 10.1016/j.chom.2014.03.012. PMID:24721569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Benjamin JL, Sumpter R, Jr., Levine B, Hooper LV. Intestinal epithelial autophagy is essential for host defense against invasive bacteria. Cell Host Microbe. 2013;13:723–734. doi: 10.1016/j.chom.2013.05.004. PMID:23768496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gallo RL, Hooper LV. Epithelial antimicrobial defence of the skin and intestine. Nat Rev Immunol. 2012;12:503–516. doi: 10.1038/nri3228. PMID:22728527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kmiec Z, Cyman M, Slebioda TJ. Cells of the innate and adaptive immunity and their interactions in inflammatory bowel disease. Adv Med Sci. 2017;62:1–16. doi: 10.1016/j.advms.2016.09.001. PMID:28126697 [DOI] [PubMed] [Google Scholar]
- 12.Ishii KJ, Koyama S, Nakagawa A, Coban C, Akira S. Host innate immune receptors and beyond: making sense of microbial infections. Cell Host Microbe. 2008;3:352–363. doi: 10.1016/j.chom.2008.05.003. PMID:18541212 [DOI] [PubMed] [Google Scholar]
- 13.Moy RH, Cherry S. Antimicrobial autophagy: a conserved innate immune response in Drosophila. J Innate Immun. 2013;5:444–455. doi: 10.1159/000350326. PMID:23689401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yano T, Mita S, Ohmori H, Oshima Y, Fujimoto Y, Ueda R, Takada H, Goldman WE, Fukase K, Silverman N, et al. Autophagic control of listeria through intracellular innate immune recognition in drosophila. Nat Immunol. 2008;9:908–916. doi: 10.1038/ni.1634. PMID:18604211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaneko T, Yano T, Aggarwal K, Lim JH, Ueda K, Oshima Y, Peach C, Erturk-Hasdemir D, Goldman WE, Oh BH, et al. PGRP-LC and PGRP-LE have essential yet distinct functions in the drosophila immune response to monomeric DAP-type peptidoglycan. Nat Immunol. 2006;7:715–723. doi: 10.1038/ni1356. PMID:16767093 [DOI] [PubMed] [Google Scholar]
- 16.Ren C, Finkel SE, Tower J. Conditional inhibition of autophagy genes in adult Drosophila impairs immunity without compromising longevity. Exp Gerontol. 2009;44:228–235. doi: 10.1016/j.exger.2008.10.002. PMID:18955126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, Yuan L, Soares F, Chea E, Le Bourhis L, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11:55–62. doi: 10.1038/ni.1823. PMID:19898471 [DOI] [PubMed] [Google Scholar]
- 18.Manzanillo PS, Ayres JS, Watson RO, Collins AC, Souza G, Rae CS, Schneider DS, Nakamura K, Shiloh MU, Cox JS. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature. 2013;501:512–516. doi: 10.1038/nature12566. PMID:24005326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Voronin D, Cook D, Steven A, Taylor M. Autophagy regulates Wolbachia populations across diverse symbiotic associations. Proc Natl Acad Sci U S A. 2012;109:E1638–46. PMID:22645363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Archibald JM. Endosymbiosis and Eukaryotic Cell Evolution. Curr Biol. 2015;25:R911–21. doi: 10.1016/j.cub.2015.07.055. PMID:26439354 [DOI] [PubMed] [Google Scholar]
- 21.Shelly S, Lukinova N, Bambina S, Berman A, Cherry S. Autophagy is an essential component of Drosophila immunity against vesicular stomatitis virus. Immunity. 2009;30:588–598. doi: 10.1016/j.immuni.2009.02.009. PMID:19362021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakamoto M, Moy Ryan H, Xu J, Bambina S, Yasunaga A, Shelly Spencer S, Gold B, Cherry S. Virus Recognition by Toll-7 Activates Antiviral Autophagy in Drosophila. Immunity. 2012;36:658–667. doi: 10.1016/j.immuni.2012.03.003. PMID:22464169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moy RH, Gold B, Molleston JM, Schad V, Yanger K, Salzano MV, Yagi Y, Fitzgerald KA, Stanger BZ, Soldan SS, et al. Antiviral autophagy restrictsRift Valley fever virus infection and is conserved from flies to mammals. Immunity. 2014;40:51–65. doi: 10.1016/j.immuni.2013.10.020. PMID:24374193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cohen LB, Troemel ER. Microbial pathogenesis and host defense in the nematode C. elegans. Curr Opin Microbiol. 2015;23:94–101. doi: 10.1016/j.mib.2014.11.009. PMID:25461579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ewbank JJ, Pujol N. Local and long-range activation of innate immunity by infection and damage in C. elegans. Curr Opin Immunol. 2016;38:1–7. doi: 10.1016/j.coi.2015.09.005. PMID:26517153 [DOI] [PubMed] [Google Scholar]
- 26.Irazoqui JE, Urbach JM, Ausubel FM. Evolution of host innate defence: insights from Caenorhabditis elegans and primitive invertebrates. Nat Rev Immunol. 2010;10:47–58. doi: 10.1038/nri2689. PMID:20029447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shivers RP, Pagano DJ, Kooistra T, Richardson CE, Reddy KC, Whitney JK, Kamanzi O, Matsumoto K, Hisamoto N, Kim DH. Phosphorylation of the conserved transcription factor ATF-7 by PMK-1 p38 MAPK regulates innate immunity in Caenorhabditis elegans. PLoS Genet. 2010;6:e1000892. doi: 10.1371/journal.pgen.1000892. PMID:20369020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoeven R, McCallum KC, Cruz MR, Garsin DA. Ce-Duox1/BLI-3 generated reactive oxygen species trigger protective SKN-1 activity via p38 MAPK signaling during infection in C. elegans. PLoS Pathog. 2011;7:e1002453. doi: 10.1371/journal.ppat.1002453. PMID:22216003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tan M, Mahajan-Miklos S, Ausubel F. Killing of Caenorhabditis elegans by Pseudomonas aeruginosa used to model mammalian bacterial pathogenesis. Proc Natl Acad Sci U S A. 1999;96:715–720. doi: 10.1073/pnas.96.2.715. PMID:9892699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kirienko NV, Kirienko DR, Larkins-Ford J, Wahlby C, Ruvkun G, Ausubel FM. Pseudomonas aeruginosa disrupts Caenorhabditis elegans iron homeostasis, causing a hypoxic response and death. Cell Host Microbe. 2013;13:406–416. doi: 10.1016/j.chom.2013.03.003. PMID:23601103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vance RE, Isberg RR, Portnoy DA. Patterns of pathogenesis: discrimination of pathogenic and nonpathogenic microbes by the innate immune system. Cell Host Microbe. 2009;6:10–21. doi: 10.1016/j.chom.2009.06.007. PMID:19616762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim DH. Bacteria and the aging and longevity of Caenorhabditis elegans. Annu Rev Genet. 2013;47:233–246. doi: 10.1146/annurev-genet-111212-133352. PMID:24274752 [DOI] [PubMed] [Google Scholar]
- 33.Murphy CT, Hu PJ. Insulin/insulin-like growth factor signaling in C. elegans. WormBook. 2013;26:1–43. doi: 10.1895/wormbook.1.164.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jia K, Thomas C, Akbar M, Sun Q, Adams-Huet B, Gilpin C, Levine B. Autophagy genes protect against Salmonella typhimurium infection and mediate insulin signaling-regulated pathogen resistance. Proc Natl Acad Sci U S A. 2009;106:14564–14569. doi: 10.1073/pnas.0813319106. PMID:19667176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Curt A, Zhang J, Minnerly J, Jia K. Intestinal autophagy activity is essential for host defense against Salmonella typhimurium infection in Caenorhabditis elegans. Dev Comp Immunol. 2014;45:214–218. doi: 10.1016/j.dci.2014.03.009. PMID:24674884 [DOI] [PubMed] [Google Scholar]
- 36.Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, Kishi C, Kc W, Carrero JA, Hunt S, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259–263. doi: 10.1038/nature07416. PMID:18849966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bel S, Pendse M, Wang Y, Li Y, Ruhn KA, Hassell B, Leal T, Winter SE, Xavier RJ, Hooper LV. Paneth cells secrete lysozyme via secretory autophagy during bacterial infection of the intestine. Science. 2017;357:1047–1052. doi: 10.1126/science.aal4677. PMID:28751470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Troemel ER, Félix MA, Whiteman NK, Barrière A, Ausubel FM. Microsporidia are natural intracellular parasites of the nematode Caenorhabditis elegans. PLoS Biol. 2008;6:2736–2752. doi: 10.1371/journal.pbio.0060309. PMID:19071962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang G, Sachse M, Prevost MC, Luallen RJ, Troemel ER, Felix MA. A large collection of novel nematode-infecting microsporidia and their diverse interactions with caenorhabditis elegans and other related nematodes. PLoS Pathog. 2016;12:e1006093. doi: 10.1371/journal.ppat.1006093. PMID:27942022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bakowski MA, Desjardins CA, Smelkinson MG, Dunbar TL, Lopez-Moyado IF, Rifkin SA, Cuomo CA, Troemel ER. Ubiquitin-mediated response to microsporidia and virus infection in C. elegans. PLoS Pathog. 2014;10:e1004200. doi: 10.1371/journal.ppat.1004200. PMID:24945527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Visvikis O, Ihuegbu N, Labed SA, Luhachack LG, Alves AM, Wollenberg AC, Stuart LM, Stormo GD, Irazoqui JE. Innate host defense requires TFEB-mediated transcription of cytoprotective and antimicrobial genes. Immunity. 2014;40:896–909. doi: 10.1016/j.immuni.2014.05.002. PMID:24882217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Settembre C, Di Malta C, Polito V, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433. doi: 10.1126/science.1204592. PMID:21617040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Najibi M, Labed SA, Visvikis O, Irazoqui JE. An Evolutionarily Conserved PLC-PKD-TFEB Pathway for Host Defense. Cell Rep. 2016;15:1728–1742. doi: 10.1016/j.celrep.2016.04.052. PMID:27184844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen HD, Kao CY, Liu BY, Huang SW, Kuo CJ, Ruan JW, Lin YH, Huang CR, Chen YH, Wang HD, et al. HLH-30/TFEB-mediated autophagy functions in a cell-autonomous manner for epithelium intrinsic cellular defense against bacterial pore-forming toxin in C. elegans. Autophagy. 2017;13:371–385. doi: 10.1080/15548627.2016.1256933. PMID:27875098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zou CG, Ma YC, Dai LL, Zhang KQ. Autophagy protects C. elegans against necrosis during Pseudomonas aeruginosa infection. Proc Natl Acad Sci U S A. 2014;111:12480–12485. doi: 10.1073/pnas.1405032111. PMID:25114220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kirienko NV, Ausubel FM, Ruvkun G. Mitophagy confers resistance to siderophore-mediated killing by Pseudomonas aeruginosa. Proc Natl Acad Sci U S A. 2015;112:1821–1826. doi: 10.1073/pnas.1424954112. PMID:25624506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin L, Rodrigues F, Kary C, Contet A, Logan M, Baxter RHG, Wood W, Baehrecke EH. Complement-related regulates autophagy in neighboring cells. Cell. 2017;170:158-171 e8. doi: 10.1016/j.cell.2017.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Isaac DT, Isberg R. Master manipulators: an update on Legionella pneumophila Icm/Dot translocated substrates and their host targets. Future Microbiol. 2014;9:343–359. doi: 10.2217/fmb.13.162. PMID:24762308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, Komatsu M, Tanaka K, Cleveland JL, Withoff S, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450:1253–1257. doi: 10.1038/nature06421. PMID:18097414 [DOI] [PubMed] [Google Scholar]
- 50.Fazeli G, Trinkwalder M, Irmisch L, Wehman AM. C. elegans midbodies are released, phagocytosed and undergo LC3-dependent degradation independent of macroautophagy. J Cell Sci. 2016;129:3721–3731. doi: 10.1242/jcs.190223. PMID:27562069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu J, Debnath J. The evolving, multifaceted roles of autophagy in cancer. Adv Cancer Res. 2016;130.1–53. doi: 10.1016/bs.acr.2016.01.005. PMID:27037750 [DOI] [PubMed] [Google Scholar]
- 52.Biering SB, Choi J, Halstrom RA, Brown HM, Beatty WL, Lee S, McCune BT, Dominici E, Williams LE, Orchard RC, et al. Viral replication complexes are targeted by LC3-Guided interferon-inducible GTPases. Cell Host Microbe. 2017;22:74–85 e7. doi: 10.1016/j.chom.2017.06.005. PMID:28669671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gelino S, Hansen M. Autophagy – An emerging anti-aging mechanism. J Clin Exp Pathol. 2012;s4.1–12. PMID:23505614 [DOI] [PMC free article] [PubMed] [Google Scholar]