

ABSTRACT
Tumor-associated inflammation is predictive of poor prognosis and drives a variety of tumorigenic phenotypes, including tumor proliferation and survival, angiogenesis, invasiveness, and metastasis. Here, we review mammalian data addressing the interaction of macroautophagy/autophagy with key signaling cascades associated with tumor inflammation. Although our understanding of this area remains incomplete, certain inflammatory pathways have emerged as important mediators of the crosstalk between autophagy and inflammation in tumors. Consistent with the multifaceted roles for autophagy in tumor cells, results to date support the hypothesis that inflammatory pathways can suppress or induce autophagy in a context-dependent manner; in turn, autophagy suppresses or promotes inflammation in cancers. Furthermore, emerging data suggest that autophagy may influence cytokine production and secretion via diverse mechanisms, which has implications for the immune and inflammatory microenvironment in tumors.
KEYWORDS: autophagy crosstalk, cytokines, HMGB-AGER/RAGE, inflammatory signaling, NFKB, ROS, toll-like receptor signaling, tumor microenvironment
Introduction
Inflammation is viewed as an important promoter of both tumor initiation and progression. Chronic inflammation due to infection is sufficient to drive formation of cancers, such as in gastric carcinoma and cervical carcinoma.1 Furthermore, solid tumors themselves have been described as “wounds that do not heal,” due to the activation of inflammatory related signaling, and the suppression of antitumor immunity.1,2 Inflammatory signaling can endorse multiple hallmarks of cancer,3 including evasion of apoptosis, introduction of DNA damage, crosstalk with oxidative stress pathways, tumor growth/proliferation, and metastasis. Interestingly, many of the signaling pathways that control inflammation during tumorigenesis are also known regulators of autophagy, a conserved lysosomal degradation process in which cells catabolize organelles and proteins in response to starvation or stress.
Although the cell autonomous functions of autophagy in tumor cells have been demonstrated to promote survival and metabolic adaptation by recycling essential metabolites and amino acids, it is now well recognized that autophagy can both impede and promote tumorigenesis.4 Notably, how this cellular self-eating pathway influences the inflammatory tumor microenvironment remains unclear; given the reciprocal interconnections between autophagy and inflammation, it is likely that autophagy influences the inflammatory response in cancer in diverse, multifaceted ways. Here, we focus on the regulation of autophagy via critical inflammatory signaling cascades, and autophagy during tumorigenesis.
Inflammatory cytokine signaling and autophagy in tumorigenesis
Cytokine signaling is involved in tumor-associated inflammation and has been implicated in promoting TIC (tumor initiating cell) self-renewal, tumor growth, angiogenesis, and metastasis.5 In tumors, some of the critical inflammatory cytokines include IL1 (interleukin 1), IL6 (interleukin 6), CXCL8/IL8 (C-X-C motif chemokine ligand 8), IL10 (interleukin 10), and INFG (interferon gamma), which function through conserved signaling cascades, including: 1) the activation of the JAK (Janus kinase), resulting in the phosphorylation-induced activation of STAT (signal transducer and activator of transcription)-dependent transcription; and 2) secondary effects, such as increased transcription of IRF1 (interferon regulatory factor 1), and increased IRF1-dependent transcription.6,7
Depending on the cancer model, cytokines have been found to either inhibit or enhance autophagy in tumors. For example, in lung carcinoma cell lines exposed to arsenic, oncogenic transformation correlates with sustained upregulation of IL6 and reduced autophagy.8 Furthermore, IL6-dependent transformation requires the inhibition of a BECN1/Beclin 1-BCL2 (B cell leukemia/lymphoma 2) complex, which is dependent on STAT3 signaling; accordingly, the enhancement of autophagy via BECN1 overexpression is sufficient to block transformation.8 In human melanoma cells, blocking IL1 via siRNA or a function-blocking antibody increases autophagic flux, while concomitantly decreasing melanoma cell growth, suggesting that IL1 may inhibit autophagy.9 In this model, IL1 inhibition also attenuates pro-inflammatory signaling including reactive oxygen species (ROS), reactive nitrogen species, phospho-NFKBIB/IκBβ (nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor, β; a marker of active NFKB [nuclear factor of kappa light polypeptide gene enhancer in B cells]), and MAPK8/c-Jun kinase activation; however, the precise role of autophagy in suppressing these pro-inflammatory signals remains an important question for future study.9
In contrast, another inflammatory cytokine, IFNG, may stimulate autophagy during tumorigenesis. Mice overexpressing Ifng in the stomach mucosa exhibit reduced gastric dysplasia and tumorigenesis driven by Helicobacter infection or overexpression of the cytokine IL1B.10 IFNG upregulates BECN1, which stimulates autophagy in the gastric epithelium as evidenced by increased autophagic flux and punctate GFP-LC3 (microtubule-associated protein 1 light chain 3/LC3).10 IFNG-induced autophagy in the gastric epithelium suppresses epithelial cell apoptosis, which is proposed to reduce the need for cell replacement; this leads to both reduced inflammation and decreased gastric progenitor cell proliferation and expansion.10 Similarly, IFNG treatment induces autophagy in both primary hepatocellular carcinoma (HCC) and HUH7 cells, which inhibits cell growth and promotes nonapoptotic cell death of HUH7 cells; accordingly, inhibiting autophagy by RNAi-mediated depletion of Becn1 or Atg5 (autophagy-related 5) reverses both of these phenotypes.11 Remarkably, IRF1 induced by cytokine signaling promotes autophagy, because shIRF-1 decreases autophagic flux.11
In contrast to the aforementioned studies in HCC, reciprocal connections between ATGs and IRF1 signaling have been implicated in the sensitivity to anti-estrogen therapies in hormone receptor-positive breast cancers. Nuclear IRF1 expression is associated with improved response to anti-estrogen therapies and prolonged survival, while ATG7 (autophagy-related 7), which inversely correlates with IRF1 expression in human tumor samples, is associated with anti-estrogen resistance.12 The inhibition of autophagy via siATG7 or siBECN1 induces the nuclear localization of IRF1 and promotes apoptosis in breast cancer cells.12 Conversely, silencing IRF1 is sufficient to stimulate autophagy.12
Autophagy may also modulate inflammatory cytokine release and secretion via diverse mechanisms (Fig. 1). For example, in murine peripheral blood monocytes, pharmacological inhibition of autophagy using 3-methyladenine (3-MA) increases IL1B (interleukin 1 β) release, while attenuating TNF/TNFα (tumor necrosis factor) release; here, the effects of autophagy on cytokines appear to be secondary to changes in cytokine gene transcription.13 A more direct role of autophagy in secretion has been observed in HRAS (HRas proto-oncogene, GTPase)-transformed mammary epithelial cells grown in 3-dimensional organotypic cultures. In these models, autophagy promotes invasive phenotypes, which requires the secretion of pro-migratory cytokines, including IL6.14 The inhibition of autophagy via ATG7 or ATG12 (autophagy related 12) shRNA reduces invasion in vitro and decreases lung metastasis in vivo in metastasis assays.14 Remarkably, autophagy inhibition in these models does not have an impact on either IL6 transcription or translation; rather, it leads to the diminished secretion of IL6 into the conditioned media.14 Finally, during RAS-mediated oncogene senescence, autophagy supports the protein translation of key inflammatory cytokines, such as IL6 and CXCL8, both of which are required for the senescence-associated secretory phenotype (SASP).15 Overall, these results suggest that autophagy can support the production and secretion of pro-inflammatory cytokines in tumor cells via diverse mechanisms.
Figure 1.
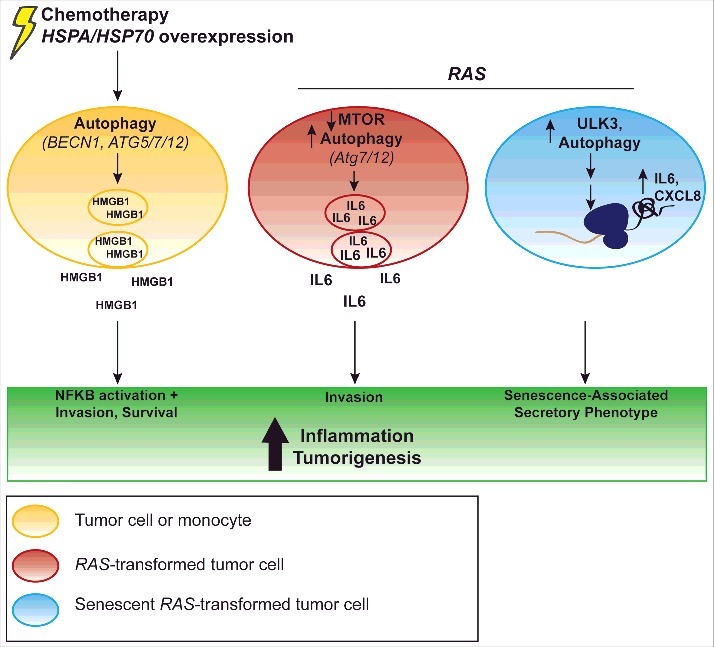
Autophagy can mediate pro-inflammatory cytokine production. HSPA/HSP70 overexpression (left) can induce BECN1 and MAPK8-dependent HMGB1 release, which promotes tumor cell invasiveness;43 this is present with NFKB activation.43 Similarly, targeted cell death of tumor cells induces ATG5-, ATG7-, and ATG12-dependent HMGB1 release, which is correlated with increased tumor cell survival.44 In monocytes, stimulation of the inflammasome elicits Atg5-dependent HMGB1 release as well.45 RAS transformation (center) can increase IL6 secretion dependent on ATG7 and ATG12.14 Activation of this signaling elicits increased invasion.14 Additionally, RAS oncogene-induced senescence (right) leads to inhibition of MTOR, increased ULK3 expression and increased LC3-II accumulation, which enhances IL6 and CXCL8 translation as part of the senescence-associated secretory phenotype.15
Autophagy and cytokine signaling in stromal constituents can also participate in feedback loops regulating tumorigenesis. Fibroblasts co-cultured with MCF-7 breast cancer cells produce increased levels of IL6, CXCL8, IL10, and IFNG, which are proposed to induce autophagic flux.16 In addition, murine mammary fat pads that display increased autophagy due the genetic loss of Cav1 (caveolin 1, caveolae protein) show an increase in CD3+ T cells, ADGRE1/F4/80+ macrophages, and PTPRC/CD45+ myeloid cells.16 Thus, autophagy may mediate a feed-forward inflammatory response between fibroblasts and tumor cells. However, autophagy may prevent inflammatory cytokine production in other cell types; for example, dendritic cells exposed to Kaposi sarcoma-associated herpes virus exhibit reduced autophagic flux along with a concomitant increase in the production of multiple inflammatory cytokines, including IL6, IL10, and IL23 (interleukin 23).17
The studies described above provide emerging insight into how cytokine signaling and autophagy pathways affect one another. Similar to its dual roles in cancer cell fate, autophagy appears to be a double-edged sword with respect to cytokine signaling, promoting cell death and suppressing tumor progression in certain instances, while enhancing pro-tumorigenic inflammatory-associated phenotypes in others.
Inflammatory ROS and autophagy in tumorigenesis
Reactive oxygen species include superoxide, hydroxyl, alkoxyl, and peroxyl free radicals, and oxides that are readily converted into radicals (i.e., hydrogen peroxide). ROS can produce DNA adducts and therefore mutations, damage proteins and mitochondria, recruit myeloid and lymphoid cells, act as second messengers to promote inflammation, and function as antimicrobial agents.18,19 In the tumor microenvironment or during chronic inflammation, ROS have been proposed to contribute to tumorigenesis by inducing mutations in tumor cells and promoting the recruitment of myeloid cells that contribute to an inflammatory tumor microenvironment.1 Notably, in solid tumors, ROS are frequently produced by infiltrating myeloid cells, which have been proposed to activate pro-inflammatory transcription via the NFKB pathway.1
Accordingly, ROS produced by both tumor cells and associated host inflammatory cells can interact with autophagy to promote cancer cell survival in response to chemotherapy or oncogenic and microenvironmental stresses. In non-small lung cancer cells, blocking autophagy increases ROS and mitochondrial oxidative stress in response to the cytotoxic chemotherapies cisplatin and etoposide, resulting in increased cell death and reduced proliferation.20 Furthermore, ROS induced by hypoxia can stimulate autophagy to promote the survival of breast, colon, melanoma, and ovarian cancer cells in vitro.21 Ras-driven tumors require autophagy to combat ROS and oxidative stress, maintain intact mitochondria and oxidative metabolism, and promote survival.22 It is unknown whether inflammation-associated ROS promote autophagy in these tumors. In HeLa cells, autophagy activation induces NOX (NADPH oxidase) and ROS production, resulting in increased pro-inflammatory STAT3 and IL6 transcription.23
Conversely, data link ROS to the induction of autophagic cell death, most notably in response to chemotherapy. Induction of NOX and ROS induces autophagy via MAPK8/JNK1 activation, which promotes necrosis in breast cancer cells.24 In this case, autophagy inhibits NFKB, part of a key inflammatory signaling pathway.24 The chemotherapeutic gemcitabine activates NFKB signaling and increases ROS levels, which synergize to induce autophagy-dependent cell death in pancreatic ductal adenocarcinoma (PDAC) cells.25 Currently, it remains unclear whether ROS associated with tumor inflammation in the absence of a chemotherapeutic stress can similarly stimulate autophagy-dependent cell death.
Autophagy-dependent regulation of ROS has also been implicated in the epithelial-mesenchymal transition (EMT), which is associated with invasive phenotypes in tumor cells. Autophagy-deficient ovarian cancer cells display increased ROS relative to ovarian cancer cells with higher basal autophagy.26 ATG7 depletion in ovarian cancer cells increases EMT marker expression, including VIM (vimentin) and ZEB1 (zinc finger E-box binding homeobox 1) proteins, as well as enhances motility and invasion in transwell assays; moreover, increased ROS correlates with EMT marker upregulation.26 Furthermore, reduced autophagy due to the knockdown of BECN1 increases ROS, NFKB activation, anchorage-independent growth, and EMT in gastric cancer cells; these phenotypes are blocked upon treatment with the anti-oxidant N-acetylcysteine, suggesting a critical functional role for ROS downstream of autophagy inhibition.27 Future studies are needed to evaluate how autophagy functions in relation to inflammation-associated ROS to promote EMT during carcinoma progression.
NFKB regulation of autophagy in cancer
NFKB is a master regulator of the inflammatory response. Many inflammatory cascades drive NFKB-dependent-transcription; NFKB-dependent transcription in turn promotes inflammatory programs and myeloid cell recruitment. Briefly, activation of this pathway elicits enhanced IKKs/inhibitory kappa kinases, i.e. CHUK and IKBKB activity, and phosphorylation, ubiquitination, and degradation of the inhibitory proteins NFKBIA/IκBα (nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor, α) and NFKBIB/IκBβ (nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor, β), which enables NFKB1 (nuclear factor kappa B subunit 1/p50-RELA/p65 (RELA proto-oncogene, NF-kB subunit) translocation to the nucleus and transcription (Fig. 2). With respect to tumorigenesis, NFKB signaling has context-dependent effects to inhibit antitumor immunity, increase pro-tumorigenic inflammation, and enhance tumor proliferation, TIC function, and angiogenesis.28 Additionally, NFKB interacts with autophagy to alter tumor cell survival and apoptosis.
Figure 2.

Interactions between autophagy and NFKB signaling in tumors. NFKB signaling is classically initiated by the phosphorylation and ubiquitination of NFKBIA/IκBα and NFKBIB/IκBβ via the IKKs (i.e., CHUK and IKBKB). The proteasomal degradation of NFKBIA and NFKBIB alleviates the inhibition of NFKB1/p50 and RELA/p65, leading to the transcription of pro-inflammatory genes as well as effectors of tumor cell survival. Autophagy and the cargo receptor SQSTM1 can stimulate IKK, and IKK can stimulate autophagy.31,32 Autophagy can also promote NFKB activation by contributing to NFKBIA degradation.22 In addition, SQSTM1 can inhibit or promote nuclear import of NFKB1/p50-RELA/p65 of NFKB.33,34 Autophagy may also directly contribute to NFKB1-RELA degradation.29
Autophagy can modulate NFKB signaling in both the stromal microenvironment constituents and in tumor cells. Conditioned media from mouse hepatoma cells induces TLR2 (toll-like receptor 2) signaling and NFKB transcriptional activity in macrophages.29 In this model, autophagy opposes NFKB activation by degrading RELA/p65 aggregates in tumor-associated macrophages in vivo, leading to reduced IL6 and IL12 (interleukin 12) secretion and promoting an M2 phenotype.29 In addition, HeLa cells undergoing ER stress display increased autophagy, which stimulates STAT3 phosphorylation and synergizes with NFKB to augment IL6 release.30 IL6 release promotes both cancer cell survival and endothelial cell migration.30 Genetic inhibition of autophagy via either shBECN1 or shATG5 attenuates both STAT3 and NFKB pathway activation, thereby abrogating IL6 expression.30 Taken together, these results indicate that autophagy can elicit both anti- and pro-inflammatory effects in the tumor microenvironment via its cell-specific effects on NFKB signaling.
IKKs, NFKBIA/IκBα, and NFKBIB/IκBβ, also interact with the autophagy pathway in cancer cells (Fig. 2). For example, in colorectal cancer cells, TNF treatment induces autophagic flux and the formation of autophagosomes that colocalize with NFKBIA.22 NFKBIA protein levels increase upon 3-MA or pepstatin A treatment to inhibit autophagy; additionally, 3-MA inhibits upregulation of NFKB-dependent transcripts such as Il8.22 Thus, autophagy may directly regulate NFKBIA degradation to sustain NFKB signaling. Conversely, activated IKK stimulates autophagic flux and LC3 puncta during nutrient starvation and matrix detachment in human cancer cell lines.31,32 Interestingly, autophagy induction in these models does not require RELA/p65 activity, suggesting that IKK complex activation promotes autophagy via an NFKB-independent mechanism; however, this does not exclude undefined feedback regulating the NFKB network.31 Exploring the significance of these pathways in vivo, and evaluating the impact on tumor-associated inflammation remain important issues for future study.
Importantly, several studies imply that the autophagy cargo receptor, SQSTM1/p62 (sequestosome 1), regulates NFKB signaling activation in cancer (Fig. 2). In a murine model of lung adenocarcinoma, genetic loss of Sqstm1 abrogates Ras-driven tumorigenesis.33 This is likely due to Sqstm1 loss inhibiting RELA/p65 nuclear localization and prosurvival NFKB activation, which inhibits pro-apoptotic ROS.33 Data suggest that RAS-dependent transcriptional upregulation of SQSTM1 is required for IKK phosphorylation and other ubiquitination events which promote NFKB activation.33 In addition, distinct interactions between SQSTM1 and NFKB have been delineated in cells following autophagy inhibition. The genetic loss of autophagy (Becn1 or Atg5) with SQSTM1 overexpression increases tumorigenesis in an immortalized baby mouse kidney cell model.34 The resulting increase in SQSTM1 protein levels is linked to the inhibition of NFKB signaling; indeed, Becn1+/− tumors with high SQSTM1 protein expression show reduced nuclear RELA/p65.34 Furthermore, SQSTM1-overexpressing tumors display downregulation of inflammatory gene signatures by microarray analysis, including altered toll-like receptor signaling, antigen presentation, and cytokine signaling.34 Similarly, IL6 luciferase reporter activity is reduced with SQSTM1 overexpression, and TNF/TNFα protein levels are upregulated as monitored by immunohistochemistry.34 In summary, it seems that SQSTM1 accumulation secondary to autophagy inhibition attenuates NFKB signaling.
Further defining the interactions between autophagy and NFKB signaling remains an important issue for further scrutiny, given the role of NFKB as a master regulator of inflammation. The data discussed here show that that NFKB signaling network members can modulate autophagy, while autophagy can alter NFKB signaling. To date, the interpretation of data with respect to the crosstalk of autophagy, NFKB, and tumor phenotypes has been challenging, since NFKB signaling can directly alter tumor proliferation and apoptosis. Future studies may benefit from more careful dissection of autophagy in genetically engineered tumor models.
TLRs and autophagy in tumorigenesis
TLRs act in the inflammatory response by binding microbe-associated particles (also referred to as pathogen-associated molecular patterns), and damage-associated molecular patterns (DAMPs), which are released in necrosis.35 Binding of TLRs activates pro-inflammatory transcriptional programs, such as NFKB- and IRF-dependent transcription, or signals through MAPK14/p38 (mitogen-activated protein kinase 14) and MAPK8/JNK1 to induce other transcription. In tumors, high TLR expression is correlated with poor patient prognosis, which could be due to TLR signaling blunting immune surveillance and increasing angiogenesis.35 Conversely, TLR activation can have antitumor properties in some contexts.35
Data suggest that activation of TLR-dependent signaling can stimulate autophagy. Pre-treatment of B16 melanoma cells with a TLR4 (toll-like receptor 4) and TLR9 (toll-like receptor 9) agonist prolongs rodent survival, and reduces lung metastasis which correlates with cleaved CASP3 (caspase 3) induction within the metastatic lesions.36 TLR4-TLR9 activation is present along with increased autophagic flux in tumors; furthermore, rapamycin treatment to induce autophagy elicits the same anti-metastasis and increased TUNEL phenotypes in this model, suggesting that autophagy promotes cell death.36 The stimulation of TLR4 and TLR9 also increases the M1:M2 ratio of the infiltrating macrophages, and reduces IFNG.36 Stimulation of TLR4-TLR9 also increase STAT1 phosphorylation, which inhibits pro-inflammatory STAT3.36 These data suggest that TLR-dependent autophagy synergizes with TLR stimulation of antitumor immunity to control tumorigenesis and metastasis in a time-dependent manner. It should also be considered that stimulation via lipid polysaccharide in these studies represents acute TLR activation, which may be different from prolonged TLR activation in a tumor. In contrast, human prostate cancer cell lines treated with TLR3 (toll-like receptor 3) agonists display increased prosurvival autophagy, together with reduced PtdIns3P (phosphatidylinositol 3-kinase) activation.37 SiTLR3, Baf-A, or chloroquine treatment reduce prostate cancer cell viability.37 However, given the pleiotropic effects of TLR signaling in tumorigenesis, it is unknown whether the TLR-induced autophagy-dependent cell death would represent the major phenotype in vivo. While TLR signaling can stimulate autophagy in a few different models via an undetermined mechanism, the interplay and relative significance of TLR-dependent autophagy and TLR-dependent changes to the tumor microenvironment remain open questions.
In addition to affecting tumor cell survival, TLR activation of autophagy in lung cancer has been linked to tumor cell invasiveness. In lung cancer cells, TLR3-TLR4 stimulation increases autophagic flux, which is present together with higher IL6 and VEGFA (vascular endothelial growth factor A) expression.38 Here, autophagy contributes to increased TLR-mediated migration in vitro in a wound scratch assay, which is blocked with siATG5 or siATG7.38 Whether TLR activation of autophagy promotes invasion and metastasis in vivo remains an interesting unanswered question.
Whereas data addressing interactions between autophagy and TLR signaling in cancer models are nascent, it is striking that these studies consistently report that stimulation of different TLR receptors induces autophagy. Data suggest that this signaling may regulate key aspects of tumor biology, including tumor cell survival, invasiveness, and metastasis. Future studies would benefit from elucidating mechanisms by which TLR signaling regulates autophagy, and evaluating whether inflammatory tumor microenvironments frequently drive TLR-dependent autophagy and determining the physiological consequences of this signaling.
HMGB1-AGER/RAGE and autophagy in tumors
HMGB1 (high motility group box 1) is a DAMP, released passively by necrotic cells, and actively during inflammation including by activated myeloid cells. It is a ligand for AGER/RAGE (advanced glycosylation end product specific receptor), TLR2, and TLR4. HMGB1-AGER can activate downstream targets including NFKB, to further promote inflammation. AGER is associated with increased tumorigenesis in cancers driven by chronic inflammation, such as gastric cancer. Additionally, HMGB1 can promote invasiveness by binding PLG (plasminogen) and promoting plasmin production and invasion.39
Several studies link HMGB1-AGER to the induction of autophagy. Targeted ablation of AGER in a Kras-driven murine PDAC model decreases autophagy, delays tumorigenesis, increases TUNEL, and decreases proliferation.40 AGER expression is required for induction of IL6 and STAT3 in pancreatic cancer.40 HMGB1 stimulates autophagic flux in leukemia cells; HMGB1-blocking antibodies increase chemotherapy sensitivity, similar to Baf-A or Becn1 shRNA treatment.41 Data suggest that dying leukemia cells produce HMGB1 to stimulate pro-survival autophagy.41 However, HMGB1 does not always have the same function with respect to tumors; indeed, one study found that HMGB1 with reduced cysteine residue 106 binds AGER but not TLR4, and stimulates autophagy and tumor cell survival, whereas HMGB1 harboring oxidized Cys106 stimulates CASP3 and CASP9 (caspase 9) in the presence of chemotherapy.42 The different functions of reduced versus oxidized HMGB1 may be due to differential regulation of HMGB1 nuclear import.
In addition to acting downstream of HMGB1-AGER, autophagy may regulate HMGB1 release (Fig. 1). HSPA/HSP70 (heat shock protein family A [Hsp70] member), another DAMP molecule, induces NFKB activation, HMGB1 secretion, and invasiveness of HCC cells.43 Autophagy may mediate HMGB1 release, as BECN1 shRNA blocks HMGB1 secretion; autophagy may coordinately induce HMGB1 release with MAPK8/JNK1.43 EGFR (epidermal growth factor receptor)-targeted cell death induces autophagy and HMGB1 release without necrosis in glioblastoma and other EGFR-positive cancer cells.44 ShRNA against ATG5, ATG7, or ATG12 blocks HMGB1 release and increases cell death.44 Autophagy can also modulate HMGB1 release in primary murine bone marrow monocytes: pharmacological stimulation of the inflammasome increases HMGB1 protein concentrations in the media, which is partially blocked by genetic ablation of Atg5.45 Although autophagy can promote the release of pro-inflammatory HMGB1 in multiple cell-based models, it is unknown if this occurs in the tumor microenvironment in vivo.
In summary, HMGB1-AGER signaling and autophagy interact during tumorigenesis, most notably, modulating the chemotherapeutic sensitivity of tumor cells, and other inflammatory cascades. Future studies are needed to corroborate whether and how autophagy directly regulates HMGB1 secretion, whether HMGB1-AGER signaling can engage in positive feedback, and whether HMGB1 release is consistently present with chemotherapy-induced autophagy in vivo.
Tumor cell autophagy and inflammatory cell recruitment
Tumor cell autophagy can have an impact on the recruitment of inflammatory cells to the tumor microenvironment, to modulate antitumor innate and adaptive immunity, and response to chemotherapy. For instance, in Kras-driven lung cancer, Ad-Cre;Atg5fl/fl tumors display upregulation of gene signatures associated with myeloid cell and lymphocyte activation, as well as increased regulatory T cell infiltration of tumors as observed by immunohistochemistry, compared with Ad-Cre;Atg5fl/+ tumors.46 Natural killer cell antitumor activity is also regulated by the autophagy status of tumor cells; in MCF7 breast cancer cells, hypoxia-induced autophagy reduces natural killer cell-dependent tumor cell death.47 Reduced natural killer cell-dependent apoptosis may be due to autophagy of GZMB (granzyme B; produced by natural killer cells to induce apoptosis), because GZMB colocalizes with GFP-LC3, and hypoxic MCF7 cells display reduced GZMB protein.47 Genetic depletion of BECN1 in hypoxic MCF7 cells restores tumor GZMB levels and reduces tumor volume, which is also observed with chloroquine treatment.47 It is unknown whether autophagy of GZMB occurs consistently with hypoxia, or whether nonhypoxic conditions can stimulate GZMB autophagy.
Autophagy may also affect antigen cross-presentation in tumors. FEMX melanoma cells with shBECN1 elicit reduced antigen uptake by T cells in vivo and in co-culture with dendritic and T cells.48 Pre-treatment of FEMX cells with rapamycin to induce autophagy stimulates antigen cross-presentation and slows tumorigenesis; while pre-treatment with 3-MA to inhibit autophagy increases tumorigenesis and decreases antigen uptake.48 These data fit with findings from a different study, where immunization of mice with autophagosome-enriched vesicles primes a broader set of T cells and increases rodent survival after inoculation with different sarcoma lines, relative to vaccination with irradiated whole cell lysates from chemically-derived sarcomas.49 Data suggest that Sqstm1 may be critical for autophagy-dependent antigen cross-presentation in this model.49
Tumor cell autophagy may also mediate immunogenic cell death in the context of chemotherapy. In a murine colon carcinoma model, Atg5 or Atg7 depletion reduces dendritic cell and T cell recruitment after chemotherapy treatment, leading to increased chemotherapy resistance.50 Additionally, Atg5 or Atg7 depletion reduces IFNG protein production by lymph nodes.50 In contrast, Atg7 or Atg12 depletion via shRNA in B16 melanoma or 4T1 breast cancer cells does not alter regulatory or helper T cell recruitment with or without doxorubicin treatment, in spite of reduced HMGB1 and CD274/PD-L1 secretion by autophagy-deficient tumor cells with chemotherapy treatment.51
Given the current intense interest in stimulating antitumor immunity using checkpoint inhibitors, these data with respect to autophagy and inflammation may be very clinically relevant. The current data again suggest complex roles for tumor cell autophagy and modulation of the inflammatory tumor microenvironment. On the one hand, tumor cell autophagy may increase antigen cross-presentation (which may extend survival),48,49 increase immunogenic cell death and chemotherapy efficacy,50 and decrease regulatory T cell presence to promote antitumor immunity.46 On the other hand, antitumor natural killer cell activity may be reduced by autophagy of tumor-killing proteins.47 Further studies in this area might elucidate more broadly how tumor cell autophagy alters innate immune cell recruitment and function, different aspects of immune surveillance and antitumor immunity, and how different contexts, for instance that may affect autophagy-mediated tumor cell death, affect the balance of inflammatory and anti-inflammatory cell recruitment and ultimately patient survival.
Conclusions
Here we have briefly overviewed how key inflammatory signals regulate autophagy, and how autophagy modulates inflammatory signaling in cancer. While autophagy is generally viewed as an anti-inflammatory mechanism, the findings discussed here highlight the divergent, context-specific functions of autophagy in inflammation-based signaling, which can influence tumorigenesis in a myriad of ways (summarized in Table 1). In addition to the results discussed here, autophagy regulates critical aspects of innate and adaptive immunity, including major histocompatibility class II presentation, that shape the tumor microenvironment; these functions of autophagy have been reviewed elsewhere.52,53 Going forward, it will be crucial to conduct studies in autochthonous cancer models in vivo to uncover whether and how autophagy mediates key inflammation-induced phenotypes during solid tumor progression, as well as in response to therapy. Furthermore, given the paradoxical roles of autophagy with respect to cell fate, additional work is needed to dissect how these inflammatory signaling cascades mediate autophagy-induced cell death versus survival in tumor cells and with chemotherapy treatment. Finally, elucidating the role of autophagy-dependent secretion in inflammatory cell recruitment and signaling and how such pathways affect cancer progression remains an important unanswered question.
Table 1.
Effects of autophagy on tumorigenesis and inflammation.
| Role of autophagy in tumorigenesis | Role of autophagy in inflammation | Model | Method for autophagy manipulation | Finding | Mechanistic role of autophagy | Ref. |
|---|---|---|---|---|---|---|
| Pro-tumorigenic | Pro-inflammatory | RAS-transformed mammary epithelial cells | shATG7, shATG12 | Autophagy induces tumor invasion via IL6 | Autophagy mediates IL6 secretion | 14 |
| Pro-tumorigenic | Autophagy correlated with inflammation; positive feedback loop proposed | MCF7 breast cancer cells co-cultured with fibroblasts | cav1−/− mouse mammary fat pad | Autophagy in CAFs is correlated with increased macrophage, myeloid, and T cell recruitment | Autophagy in fibroblasts stimulates IL6, CXCL8, and IFNG production | 16 |
| Pro-tumorigenic | Anti-inflammatory | Lung adenocarcinoma, other human cancer cell lines | Chloroquine, 3-MA, siATG7 | Autophagy promotes tumor cell survival | Autophagy eliminates ROS in situ and with chemo treatment | 20-21 |
| Pro-tumorigenic | Pro-inflammatory | HeLa carcinoma cells | 3-MA, rapamycin, shBECN1, shATG5 | Autophagy stimulates STAT3 and IL6 transcription | Autophagy induces NOX and ROS | 23 |
| Pro-tumorigenic | Pro-inflammatory | Lung cancer cell lines | Pep-A, 3-MA, siATG5, siATG7 | TLR3 agonists induce autophagy-dependent migration | TLR3-dependent autophagy promotes IL6 and VEGFA expression | 38 |
| Pro-tumorigenic | Pro-Inflammatory | H22 hepatocellular carcinoma overexpressing HSPA/HSP70, HeLa cells, glioblastoma cells, bone marrow macrophages | ShBECN1, siATG5, siATG12 | Increased tumor cell survival, decreased proliferation in chemo/stress contexts | Autophagy promotes HMGB1 release | 43-45 |
| Pro-tumorigenic | MCF7 breast cancer cells | Inducible BECN1−/−, hypoxia | Hypoxia-driven autophagy in tumor cells reduces natural killer cell-dependent tumor apoptosis | Autophagy may degrade GZMB produced by natural killer cells | 47 | |
| Anti-tumorigenic | Anti-inflammatory | Mice with Ifng-driven gastric cancer, gastric cancer cell lines; human hepatocellular carcinoma cells, cell lines | siBecn1, siAtg5 | IFNG reduces cell growth/increases apoptosis and/or reduces inflammation | IFNG stimulates autophagy via unknown mechanism to alter survival/proliferation | 10, 11 |
| Anti-tumorigenic | Anti-inflammatory | Ovarian cancer cell lines | Rapamycin, chloroquine, siAtg7 | Atg7 depletion stimulates ROS and EMT | Autophagy inhibits ROS accumulation to inhibit EMT/invasion | 26 |
| Anti-tumorigenic | Anti-inflammatory | Mice with Ras-driven lung adenocarcinoma | sqstm1−/− mouse | Autophagy inhibits ROS and apoptosis | SQSTM1 inhibits RELA nuclear localization | 33 |
| Anti-tumorigenic | Anti-inflammatory | Immortalized baby mouse kidney cells and tumors | atg5−/−; Becn1+/− | Autophagy promotes inflammatory gene signatures | BECN1 stimulates nuclear RELA localization and IL6- dependent transcription | 34 |
| Anti-tumorigenic | Anti-inflammatory | B16-F10 melanoma cells, i.v. injection/ metastasis to lung | Rapamycin | Increased survival, reduced metastasis, increased tumor apoptosis, reduced IFNG | TLR4-TLR9 activation synergizes with autophagy via an unknown mechanism to promote cell death | 36 |
| Anti-tumorigenic | Pro-inflammatory | FEMX melanoma cells in vivo | Rapamycin, 3-MA, wortmannin, shBECN1, siATG12 | Tumor cell autophagy depletion enhances antigen cross-presentation (dendritic cells/T cells) | SQSTM1/early steps of autophagy required optimal dendritic/T cell cross-presentation | 48,49 |
| n/a | Anti-inflammatory | Human peripheral blood mononuclear cells | 3-MA, serum starvation | 3-MA increases IL1B release, decreases TNF release | Autophagy alters cytokine gene transcription | 13 |
| No data; likely anti-tumorigenic | Anti-inflammatory | Murine hepatocellular carcinoma with bone marrow-derived macrophages | Baf-A, shAtg5 | Autophagy reduces IL6, IL12 secretion; promotes M2 phenotype | Autophagy degrades RELA | 29 |
Funding Statement
Grant support to J.D. includes the NIH (CA201849, CA126792, CA201849) the DOD BCRP (W81XWH-11-1-0130) and the Samuel Waxman Cancer Research Foundation.
Abbreviations
- 3-MA
3-methyladenine
- AGER
advanced glycation end product-specific receptor
- ATG
autophagy related
- Baf-A
bafilomycin A1
- BECN1
Beclin 1, autophagy related
- CAV1
caveolin 1, caveolae protein
- CASP3
caspase 3
- EMT
epithelial-mesenchymal transition
- HCC
hepatocellular carcinoma
- HMGB1
high mobility group box 1
- HSPA/HSP70
heat shock protein family A (Hsp70) member
- IRF
interferon regulatory factor
- IL
interleukin
- IFNG
interferon gamma
- JAK
Janus kinase
- MAP1LC3/LC3
microtubule-associated protein 1 light chain 3
- MAPK8
mitogen-activated protein kinase 8
- NFKB
nuclear factor of kappa light polypeptide gene enhancer in B cells
- NFKBIA
nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor, α
- NFKBIB
nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor, β
- NOX
NADPH oxidase
- PDAC
pancreatic ductal carcinoma
- ROS
reactive oxygen species
- RNS
reactive nitrogen species
- STAT
signal transducer and activator of transcription
- SQSTM1
sequestosome 1
- TIC
tumor initiating cell
- TNF
tumor necrosis factor
Disclosure of potential conflicts of interest
No potential conflicts of interest are disclosed.
References
- [1].Grivennikov SI, Greten FR, Karin M. Immunity, Inflammation, and Cancer. Cell. 2011;140:883-99. doi: 10.1016/j.cell.2010.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Tumors Dvorak HF.: Wounds that Do Not Heal. N Engl J Med. 1986;315:1650-9. doi: 10.1056/NEJM198612253152606. PMID:3537791 [DOI] [PubMed] [Google Scholar]
- [3].Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144:646-74. doi: 10.1016/j.cell.2011.02.013. PMID:21376230 [DOI] [PubMed] [Google Scholar]
- [4].Liu J, Debnath J. The Evolving, Multifaceted Roles of Autophagy in Cancer. Adv. Cancer Res; 2016; 130:1–53. [DOI] [PubMed] [Google Scholar]
- [5].Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436-44. doi: 10.1038/nature07205. PMID:18650914 [DOI] [PubMed] [Google Scholar]
- [6].Lin W, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Investig. 2007;117:1175-83. doi: 10.1172/JCI31537. PMID:17476347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kröger A, Köster M, Schroeder K, Hauser H, Mueller PP. Review: Activities of IRF-1. J Interf Cytokine Res. 2002;22:5-14. doi: 10.1089/107999002753452610 [DOI] [PubMed] [Google Scholar]
- [8].Qi Y, Zhang M, Li H, Frank JA, Dai L, Liu H, Zhang Z, Wang C, Chen G. Autophagy inhibition by sustained overproduction of IL6 contributes to arsenic carcinogenesis. Cancer Res. 2014;74:3740-52. doi: 10.1158/0008-5472.CAN-13-3182. PMID:24830721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Qin Y, Ekmekcioglu S, Liu P, Duncan LM, Lizée G, Poindexter N, Grimm E a. Constitutive aberrant endogenous interleukin-1 facilitates inflammation and growth in human melanoma. Mol Cancer Res. 2011;9:1537-50. doi: 10.1158/1541-7786.MCR-11-0279. PMID:21954434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tu SP, Quante M, Bhagat G, Takaishi S, Cui G, Yang XD, Muthuplani S, Shibata W, Fox JG, Pritchard DM, et al.. IFN-?? inhibits gastric carcinogenesis by inducing epithelial cell autophagy and T-cell apoptosis. Cancer Res. 2011;71:4247-59. doi: 10.1158/0008-5472.CAN-10-4009. PMID:21512143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Li P, Du Q, Cao Z, Guo Z, Evankovich J, Yan W, Chang Y, Shao L, Stolz DB, Tsung A, et al.. Interferon-γ induces autophagy with growth inhibition and cell death in human hepatocellular carcinoma (HCC) cells through interferon-regulatory factor-1 (IRF-1). Cancer Lett. 2012;314:213-22. doi: 10.1016/j.canlet.2011.09.031. PMID:22056812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Schwartz-Roberts JL, Cook KL, Chen C, Shajahan-Haq AN, Axelrod M, W??rri A, Riggins RB, Jin L, Haddad BR, Kallakury B V, et al.. Interferon regulatory factor-1 signaling regulates the switch between autophagy and apoptosis to determine breast cancer cell fate. Cancer Res. 2015;75:1046-55. doi: 10.1158/0008-5472.CAN-14-1851. PMID:25576084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Crişan TO, Plantinga TS, van de Veerdonk FL, Farcaş MF, Stoffels M, Kullberg BJ, van der Meer JWM, Joosten LAB, Netea MG. Inflammasome-independent modulation of cytokine response by autophagy in human cells. PLoS One. 2011;6(4):e18666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lock R, Kenific CM, Leidal AM, Salas E, Debnath J. Autophagy-dependent production of secreted factors facilitates oncogenic RAS-Driven invasion. Cancer Discov. 2014;4:466-79. doi: 10.1158/2159-8290.CD-13-0841. PMID:24513958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Young ARJ, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JFJ, Tavaré S, Arakawa S, Shimizu S, Watt FM, et al.. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23:798-803. doi: 10.1101/gad.519709. PMID:19279323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Martinez-Outschoorn UE, Whitaker-Menezes D, Lin Z, Flomenberg N, Howell A, Pestell RG, Lisanti MP, Sotgia F. Cytokine production and inflammation drive autophagy in the tumor microenvironment: Role of stromal caveolin-1 as a key regulator. Cell Cycle. 2011;10:1784-93. doi: 10.4161/cc.10.11.15674. PMID:21566463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Santarelli R, Gonnella R, Di Giovenale G, Cuomo L, Capobianchi A, Granato M, Gentile G, Faggioni A, Cirone M. STAT3 activation by KSHV correlates with IL-10, IL-6 and IL-23 release and an autophagic block in dendritic cells. Sci Rep. 2014;4:4241. doi: 10.1038/srep04241. PMID:24577500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ohshima H, Tatemichi M, Sawa T. Chemical basis of inflammation-induced carcinogenesis. Arch Biochem Biophys. 2003;417:3-11. doi: 10.1016/S0003-9861(03)00283-2. PMID:12921773 [DOI] [PubMed] [Google Scholar]
- [19].Wiseman H, Halliwell B. Damage to DNA by reactive oxygen and nitrogen species: role in inflammatory disease and progression to cancer. Biochem J. 1996;313 (Pt 1:17-29. doi: 10.1042/bj3130017. PMID:8546679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kaminskyy VO, Piskunova T, Zborovskaya IB, Tchevkina EM, Zhivotovsky B. Suppression of basal autophagy reduces lung cancer cell proliferation and enhances caspase-dependent and -independent apoptosis by stimulating ROS formation. Autophagy. 2012;8:1032-44. doi: 10.4161/auto.20123. PMID:22562073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Liu W, Glunde K, Bhujwalla ZM, Raman V, Sharma A, Phang JM. Proline oxidase promotes tumor cell survival in hypoxic tumor microenvironments. Cancer Res. 2012;72:3677-86. doi: 10.1158/0008-5472.CAN-12-0080. PMID:22609800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JMS, Karantza V, et al.. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25:460-70. doi: 10.1101/gad.2016311. PMID:21317241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Yoon S, Woo SU, Kang JH, Kim K, Kwon MH, Park S, Shin HJ, Gwak HS, Chwae YJ. STAT3 transcriptional factor activated by reactive oxygen species induces IL6 in starvation-induced autophagy of cancer cells. Autophagy. 2010;6:1125-38. doi: 10.4161/auto.6.8.13547. PMID:20930550 [DOI] [PubMed] [Google Scholar]
- [24].D'Anneo A, Carlisi D, Lauricella M, Puleio R, Martinez R, Di Bella S, Di Marco P, Emanuele S, Di Fiore R, Guercio A, et al.. Parthenolide generates reactive oxygen species and autophagy in MDA-MB231 cells. A soluble parthenolide analogue inhibits tumour growth and metastasis in a xenograft model of breast cancer. Cell Death Dis. 2013;4:e891. doi: 10.1038/cddis.2013.415. PMID:24176849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Donadelli M, Dando I, Zaniboni T, Costanzo C, Dalla Pozza E, Scupoli MT, Scarpa A, Zappavigna S, Marra M, Abbruzzese A, et al.. Gemcitabine/cannabinoid combination triggers autophagy in pancreatic cancer cells through a ROS-mediated mechanism. Cell Death Dis. 2011;2:e152. doi: 10.1038/cddis.2011.36. PMID:21525939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhao Z, Zhao J, Xue J, Zhao X, Liu P. Autophagy inhibition promotes epithelial-mesenchymal transition through ROS / HO-1 pathway in ovarian cancer cells. Am J Cancer Res. 2016;6:2162-77. [PMC free article] [PubMed] [Google Scholar]
- [27].Qin W, Li C, Zheng W, Guo Q, Zhang Y, Kang M, Zhang B, Yang B, Li B, Yang H, et al.. Inhibition of autophagy promotes metastasis and glycolysis by inducing ROS in gastric cancer cells. Oncotarget. 2015;6:39839-54. doi: 10.18632/oncotarget.5674. PMID:26497999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hoesel B, Schmid J a. The complexity of NF-κB signaling in inflammation and cancer. Mol Cancer [Internet]. 2013;12:86 Available from: Molecular Cancer. doi: 10.1186/1476-4598-12-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Chang C-P, Su Y-C, Hu C-W, Lei H-Y. TLR2-dependent selective autophagy regulates NF-κB lysosomal degradation in hepatoma-derived M2 macrophage differentiation. Cell Death Differ. 2012;20:515-23. doi: 10.1038/cdd.2012.146. PMID:23175187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yoon S, Woo SU, Kang JH, Kim K, Shin H-J, Gwak H-S, Park S, Chwae Y-J. NF-κB and STAT3 cooperatively induce IL6 in starved cancer cells. Oncogene. 2012;31:3467-81. doi: 10.1038/onc.2011.517. PMID:22105366 [DOI] [PubMed] [Google Scholar]
- [31].Criollo A, Senovilla L, Authier HELEN, Maiuri MC, Morselli E, Vitale I, Kepp O, Tasdemir E, Galluzzi L, Shen S, et al.. The IKK complex contributes to the induction of autophagy. EMBO J. 2009;29:619-31. doi: 10.1038/emboj.2009.364. PMID:19959994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chen N, Debnath J. IκB kinase complex (IKK) triggers detachment-induced autophagy in mammary epithelial cells independently of the PI3K-AKT-MTORC1 pathway. Autophagy. 2013;9:1214-27. doi: 10.4161/auto.24870. PMID:23778976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, Moscat J. The Signaling Adaptor p62 Is an Important NF-??B??Mediator in Tumorigenesis. Cancer Cell. 2008;13:343-54. doi: 10.1016/j.ccr.2008.02.001. PMID:18394557 [DOI] [PubMed] [Google Scholar]
- [34].Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, et al.. Autophagy Suppresses Tumorigenesis through Elimination of p62. Cell. 2009;137:1062-75. doi: 10.1016/j.cell.2009.03.048. PMID:19524509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ridnour LA, Cheng RYS, Switzer CH, Heinecke JL, Ambs S, Glynn S, Young HA, Trinchieri G, Wink DA. Molecular pathways: Toll-like receptors in the tumor microenvironment-poor prognosis or new therapeutic opportunity. Clin Cancer Res. 2013;19:1340-6. doi: 10.1158/1078-0432.CCR-12-0408. PMID:23271799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Yan J, Wang ZY, Yang HZ, Liu HZ, Mi S, Lv XX, Fu XM, Yan HM, Zhang XW, Zhan QM, et al.. Timing is critical for an effective Anti-Metastatic immunotherapy: The decisive role of IFN??/STAT1-Mediated activation of autophagy. PLoS One. 2011;6(9):e24705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Harashima N, Inao T, Imamura R, Okano S, Suda T, Harada M. Roles of the PI3K/Akt pathway and autophagy in TLR3 signaling-induced apoptosis and growth arrest of human prostate cancer cells. Cancer Immunol Immunother. 2012;61:667-76. doi: 10.1007/s00262-011-1132-1. PMID:22038398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zhan Z, Xie X, Cao H, Zhou X, Zhang XD, Fan H, Liu Z. Autophagy facilitates TLR4- and TLR3-triggered migration and invasion of lung cancer cells through the promotion of TRAF6 ubiquitination. Autophagy. 2014;10:257-68. doi: 10.4161/auto.27162. PMID:24321786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol. 2010;28:367-88. doi: 10.1146/annurev.immunol.021908.132603. PMID:20192808 [DOI] [PubMed] [Google Scholar]
- [40].Kang R, Loux T, Tang D, Schapiro NE, Vernon P, Livesey KM, Krasinskas A, Lotze MT, Zeh HJ. The expression of the receptor for advanced glycation endproducts (RAGE) is permissive for early pancreatic neoplasia. Proc Natl Acad Sci. 2012;109:7031-6. doi: 10.1073/pnas.1113865109. PMID:22509024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Liu L, Yang M, Kang R, Wang Z, Zhao Y, Yu Y, Xie M, Yin X, Livesey KM, Lotze MT, et al.. HMGB1-induced autophagy promotes chemotherapy resistance in leukemia cells. Leuk Off J Leuk Soc Am Leuk Res Fund, UK. 2011;25:23-31. doi: 10.1038/leu.2010.225 [DOI] [PubMed] [Google Scholar]
- [42].Tang D, Kang R, Cheh C-W, Livesey KM, Liang X, Schapiro NE, Benschop R, Sparvero LJ, Amoscato AA, Tracey KJ, et al.. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene. 2010;29:5299-310. doi: 10.1038/onc.2010.261. PMID:20622903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Gong W, Wang ZY, Chen GX, Liu YQ, Gu XY, Liu WW. Invasion potential of H22 hepatocarcinoma cells is increased by HMGB1-induced tumor NF-??B signaling via initiation of HSP70. Oncol Rep. 2013;30:1249-56. PMID:23836405 [DOI] [PubMed] [Google Scholar]
- [44].Thorburn J, Horita H, Redzic J, Hansen K, Frankel AE, Thorburn A. Autophagy regulates selective HMGB1 release in tumor cells that are destined to die. Cell Death Differ. 2009;16:175-83. doi: 10.1038/cdd.2008.143. PMID:18846108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. Embo J. 2011;30:4701-11. doi: 10.1038/emboj.2011.398. PMID:22068051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Rao S, Tortola L, Perlot T, Wirnsberger G, Novatchkova M, Nitsch R, Sykacek P, Frank L, Schramek D, Komnenovic V, et al.. A dual role for autophagy in a murine model of lung cancer. Nat Commun. 2014;5:3056. doi: 10.1038/ncomms4056. PMID:24445999 [DOI] [PubMed] [Google Scholar]
- [47].Baginska J, Viry E, Berchem G, Poli A, Noman MZ, van Moer K, Medves S, Zimmer J, Oudin A, Niclou SP, et al.. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc Natl Acad Sci U S A. 2013;110:17450-5. doi: 10.1073/pnas.1304790110. PMID:24101526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Li Y, Wang LX, Yang G, Hao F, Urba WJ, Hu HM. Efficient cross-presentation depends on autophagy in tumor cells. Cancer Res. 2008;68:6889-95. doi: 10.1158/0008-5472.CAN-08-0161. PMID:18757401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Twitty CG, Jensen SM, Hu HM, Fox BA. Tumor-derived autophagosome vaccine: Induction of cross-protective immune responses against short-lived proteins through a p62-dependent mechanism. Clin Cancer Res. 2011;17:6467-81. doi: 10.1158/1078-0432.CCR-11-0812. PMID:21810919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Michaud M, Martinis I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, Shen S, Kepp O, Scoazec M, Vacchelli E, et al.. Autophagy-Dependent Anticancer Immune Responses Induced by Chemotherapeutic Agents in Mice. Science (80-). 2011;334:1573-8. doi: 10.1126/science.1208347 [DOI] [PubMed] [Google Scholar]
- [51].Starobinets H, Ye J, Broz M, Barry K, Goldsmith J, Marsh T, Rostker F, Krummel M. Antitumor adaptive immunity remains intact following inhibition of autophagy and antimalarial treatment. J Clin Invest. 2016;126:1-13. doi: 10.1172/JCI85705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Crotzer VL, Blum JS. Autophagy and its role in MHC-mediated antigen presentation. J Immunol. 2009;182:3335-41. doi: 10.4049/jimmunol.0803458. PMID:19265109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol. 2007;7:767-77. doi: 10.1038/nri2161. PMID:17767194 [DOI] [PMC free article] [PubMed] [Google Scholar]