

Abstract
Hyperimmunoglobulin E syndromes (HIES) are rare primary immunodeficiency diseases characterized by markedly elevated serum immunoglobulin (Ig) E, recurrent pneumonia, and chronic eczema. To date, information about pediatric HIES is limited. We aimed to evaluate the spectrum of clinical and immunological features in pediatric patients with HIES in China.
We retrospectively reviewed the cases of 4 pediatric patients with HIES followed at the Guangzhou Women and Children's Medical Center from May 2013 to September 2017. We analyzed clinical presentation, laboratory data, immunological evaluations, imagenological characteristics, treatment, response to therapy, genetic and bronchoalveolar lavage fluid (BALF) findings, and prognosis.
The common clinical features of the patients were recurrent respiratory and mucocutaneous infections and eczematoid skin lesions. In 3 of 4 patients, BALF and transbronchial lung biopsy (TBLB) demonstrated fungal pneumonia with organisms including invasive Aspergillus and Penicillium marneffei. Elevated serum IgG and IgM were detected in 3 and 2 cases, respectively, while CD4+ T and CD19+ B cells were slightly reduced in only 1 patient. Nitroblue tetrazolium tests (NBTs) were normal in all patients, and reduced natural killer cell counts were identified in 3 patients. A novel missense mutation in exon 17 (c.1593A>T, p.K531N) was identified in the signal transducer and activator of transcription 3 (STAT3) gene that has not been reported previously. One patient had 3 homozygous nonsynonymous variations of the complement receptor 2 (CR2) gene distributed in exons 10 (c.1916G>A, p.S639N) and 11 (c.1987T>C, p.S663P and c.2012G>A, p.R671H) with high frequency.
This case series suggests that fungi are important respiratory pathogens in children with HIES and should be considered in cases of pneumonia in this population. The NIH scoring system does not allow diagnostic certainty, particularly in infants, because some of the common manifestations of HIES may not develop until the patient matures. Pulmonary complications must be identified in the early stage of the disease to treat them effectively. In addition, we report a mutation in STAT3 that has not been identified previously.
Keywords: children, fungi, hyper-IgE syndromes, immunodeficiency, mutation
1. Introduction
Hyperimmunoglobulin E syndromes (HIES) are rare primary immunodeficiencies characterized by markedly elevated immunoglobulin (Ig) E levels, recurrent bacterial infections (especially staphylococcal skin abscesses), chronic eczema, and recurrent pulmonary infections.[1–3] In addition to these immunologic features, a characteristic facial appearance, scoliosis, retained primary teeth, joint hyperextensibility, bone fractures following minimal trauma, and craniosynostosis are the main nonimmunologic manifestations.[2,3] There are 2 forms characterized by clinical features and genetics, autosomal dominant (AD)-HIES and autosomal recessive (AR)-HIES.[2,4] Although the National Institutes of Health (NIH) developed a clinical HIES scoring system in 1999,[5] obtaining this diagnosis in young children can be challenging as symptoms accumulate over time along with confounding clinical signs. HIES occurs in individuals from diverse ethnic backgrounds and does not seem to be more common in specific populations.[3,6] In this study, we analyzed 4 patients with HIES confirmed by NIH Score, genetic analysis, or both to determine the clinical and immunological features of pediatric HIES in China.
2. Case reports
We present a case series of 4 pediatric patients (P1-P4) with HIES who were followed at the Guangzhou Women and Children's Medical Center from May 2013 to September 2017. For each patient, clinical presentation, laboratory data, immunological evaluations, imagenological characteristics, treatment, response to therapy, genetic data, bronchoalveolar lavage fluid (BALF) findings, and outcome were retrospectively recorded. Serum IgE was detected with rate nephelometry using an Immage 800 (Beckman Coulter, Inc., Brea, CA), the cut-off value for an elevated IgE level was set with ages (age <1 year: 15 IU/mL, age 1–5 years: 60 IU/mL, age 5–9 years: 100 IU/mL, age 10–15 years: 200 IU/mL).[7,8] No patient had human immunodeficiency virus infection on serological tests (ribonucleic acid polymerase chain reaction testing for this virus was not performed at our hospital at the time of patient admission). No patient had a family history of immunodeficiency or consanguineous marriage. All patients were serum-specific antibody-negative for some common viruses, including cytomegalovirus (CMV), Epstein–Barr virus (EBV), and other herpes viruses. In addition, tuberculosis and mycoplasma smears and cultures were negative in all patients. We assessed all patients at their latest visit per the NIH HIES scoring system. Using a candidate gene approach, we performed targeted region sequencing of the signal transducer and activator of transcription 3 (STAT3), dedicator of cytokinesis 8 (DOCK8), and tyrosine kinase 2 (TYK2) genes in 2 patients and whole exome sequencing in another. Consent for genetic diagnosis was obtained from the patients’ parents, and the Ethics Committee of Guangzhou Women and Children's Medical Center, Guangzhou Medical University approved the study.
2.1. Patient 1
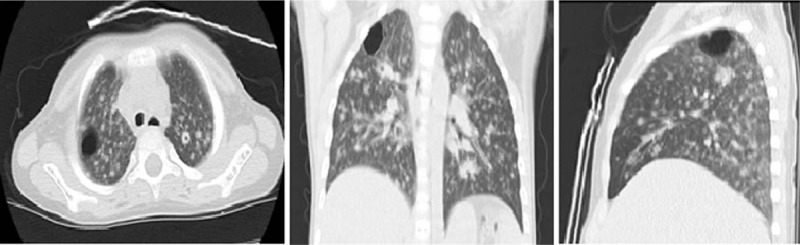
A 13-year-old boy (P1) with an asymmetrical face, high palate, and thoracolumbar scoliosis presented with a 20-day history of trachyphonia and dyspnea, but no fever. His medical history was significant for recurrent eczema and thrush since infancy, 2 lower airway infections, and several upper respiratory tract infections. He had no food allergies. His serum IgE was markedly elevated (5310–7660 IU/mL). High-resolution computed tomography (HRCT) examination revealed diffuse incipient lesions and a thin-walled cystic lesion in the left upper lobe of the lung (Fig. 1). Electronic bronchoscopy showed granulation tissue in his throat (Fig. 2). BALF cultures yielded Penicillium marneffei (P. marneffei) and Acinetobacter baumannii (A. baumannii). Further, he had papular lesions with central umbilication on his lips as typical of P. marneffei infection (Fig. 3). The patient required mechanical ventilation on 2 occasions due to complications of acute respiratory distress syndrome and upper airway obstruction by granulation tissue. We administered amphotericin B, voriconazole, and imipenem-cilastatin sodium intravenously for 2 weeks and itraconazole orally for 2 months as antifungal agents with good clinical and image response (Fig. 4). At the same time, trimethoprim-sulfamethoxazole was given as prophylaxis. HIES was diagnosed on the basis of his NIH Score and STAT3 mutation, but a similar mutation was not detected in his parents.
Figure 1.
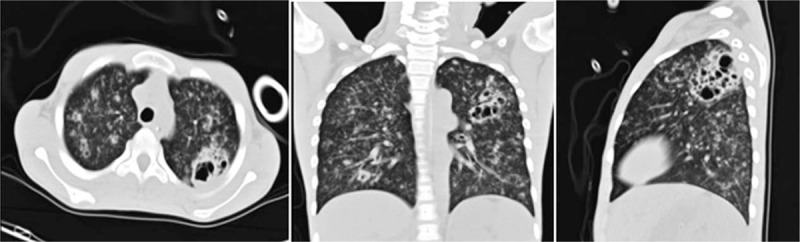
High-resolution chest computed tomography lung windows on the day of admission reveal diffuse incipient lesions and a cystic lesion in the left upper lobe of Patient 1.
Figure 2.
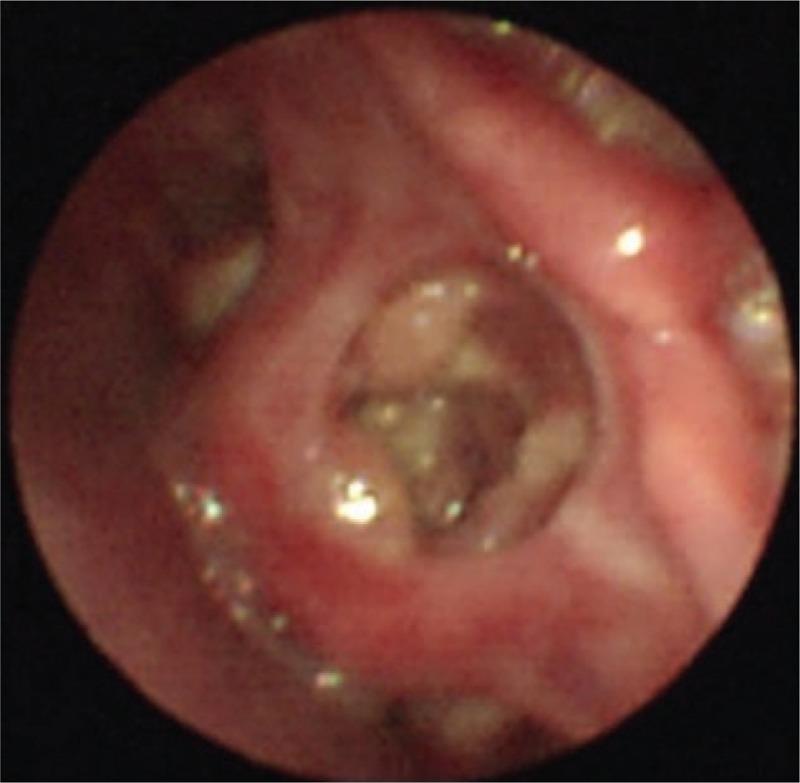
Electronic bronchoscopy shows granulation tissue in the throat in Patient 1.
Figure 3.
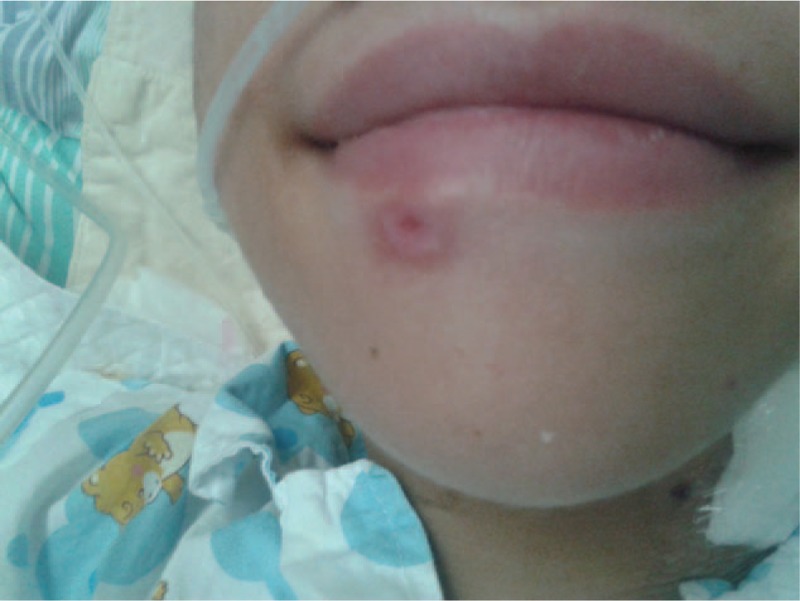
Penicillium marneffei positive facial skin lesions with central umbilication in Patient 1.
Figure 4.
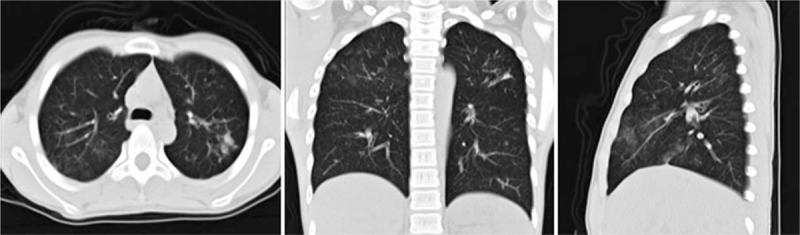
High-resolution chest computed tomography lung windows reveal a resolving pulmonary inflammatory infiltration and no cystic lesions after antifungal treatment in Patient 1.
2.2. Patient 2
In January 2014, a 3-year-old boy (P2) was hospitalized due to fever and cough for 20 days. The patient had a dermatologist-documented history of recurrent eczema and cold abscesses since infancy and recurrent lower respiratory tract infections since the age of 8 months. As an infant, he was diagnosed with newborn rash and thrush. The patient had allergies to many foods and mites documented by blood serum allergen tests. Serum IgE was repeatedly measured at over 4000 IU/mL (4840–5130 IU/mL). We found cold abscesses in the skin of his left knee joint medially and left instep (Fig. 5). His admission HRCT examination revealed an upper right lung lobe tissue shadow. BALF and cold abscess cultures all yielded Staphylococcus aureus (S. aureus), and his respiratory symptoms and pulmonary lesions resolved after treatment with cefoperazone (Sulperazon; Pfizer Inc., New York City, NY). However, the cold abscesses showed no improvement. A genetic evaluation revealed 3 homozygous nonsynonymous variations in his complement receptor 2 (CR2) gene. The nonimmunological features of HIES had not yet appeared given the patient's age at presentation.
Figure 5.
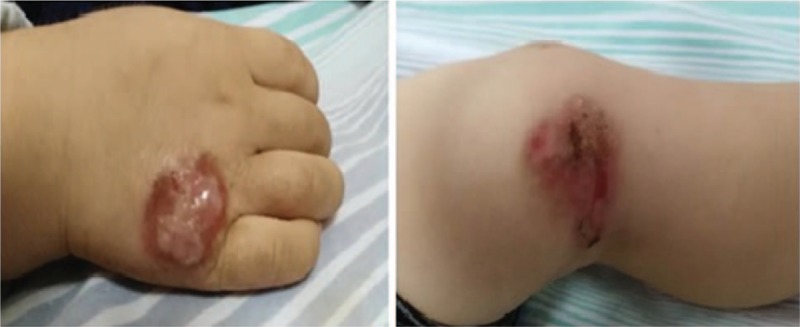
Medial left knee joint and left instep skin cold abscesses in Patient 2.
2.3. Patient 3
A 3-year-old girl (P3) presented with a 2-month history of coughing, wheezing, and intermittent fevers. The physical examination revealed miliaria pustulosa on her face and head (Fig. 6). In addition, she had facial asymmetry with a prominent forehead and chin, coarse skin, and a high arched palate. Nonimmunological features of HIES, such as scoliosis, skeletal fractures, and vascular abnormalities were not identified. The patient's medical history was significant for recurrent eczema and thrush from infancy, S. aureus pneumonia and tympanitis, and upper respiratory tract infections at least 4 times yearly. She had been diagnosed with pneumonia twice. Her serum IgE was significantly elevated (4090–10,200 IU/mL). The bronchoscopy showed granulomatous hyperplasia of all principal bronchi (Fig. 7), while cystic structures were identified in the right upper lung lobe on HRCT (Fig. 8). BALF cultures yielded Haemophilus influenzae (H. influenzae) and microscopic examination of her transbronchial lung biopsy (TBLB) showed Aspergillus hyphae (Fig. 9). She was diagnosed with acute respiratory distress syndrome and required mechanical ventilation for 15 days. Closed thoracic drainage was performed to treat a pneumothorax. While in the pediatric intensive care unit, S. aureus was detected in her BALF and skin. She recovered after 3 weeks of vancomycin and Sulperazon and 3 months of voriconazole and itraconazole as therapeutic and prophylactic treatments, respectively. Unfortunately, genetic studies were not performed in this case. The patient was diagnosed with HIES based on her clinical features and the NIH scoring system.
Figure 6.
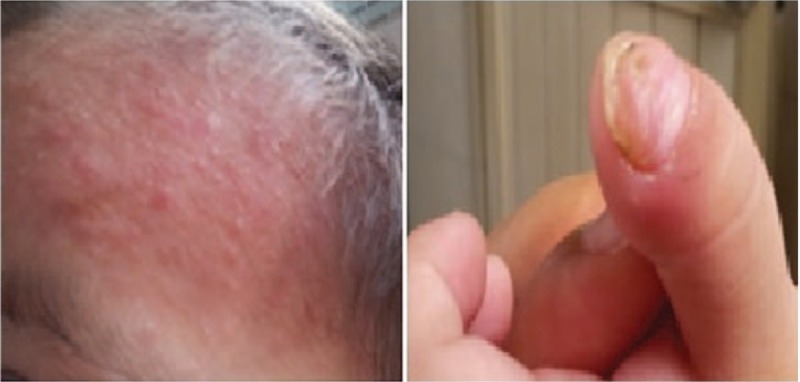
Head and facial miliaria pustulosa in Patient 3.
Figure 7.
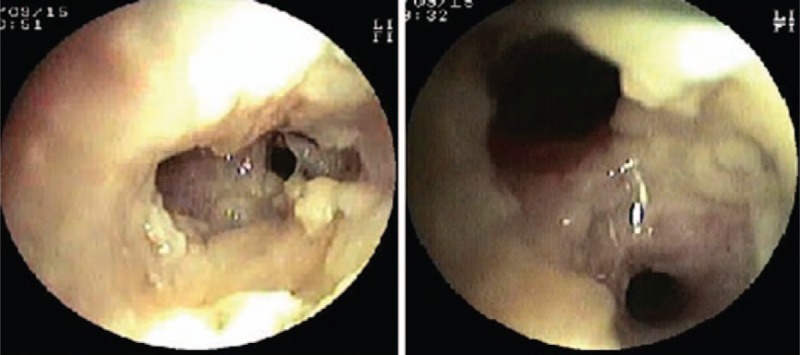
Bronchoscopy shows granulomatous hyperplasia in all principal bronchi in Patient 3.
Figure 8.
High-resolution chest computed tomography lung windows on the day of admission reveal a thin-walled cystic lesion in the right upper lobe in Patient 3.
Figure 9.
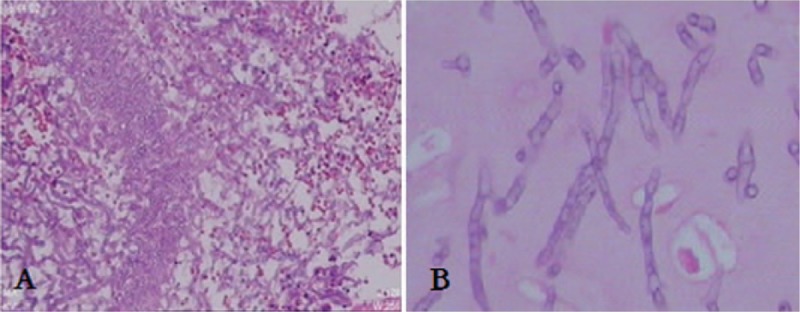
(A) A transbronchial lung biopsy (TBLB) in Patient 3 shows granulation tissue (hematoxylin and eosin (HE), ×100), (B) TBLB reveals Aspergillus hyphae (HE, ×400).
2.4. Patient 4
A 7-year-old girl (P4) presented with a pustular or eczematoid eruption on the scalp and face. She also had a low-grade fever. Her medical history was significant for recurrent eczema, oral candidiasis, sinopulmonary infections since infancy, newborn rash, and otitis media. Her surgical history included pulmonary lobectomies for treatment of lung abscesses. The first resection involved the right lobe and was performed at another hospital less than 3 years before the current admission. She required a second operation to remove a large, thick-walled cavity in the left lower lobe (Fig. 10). Cultures of her BALF yielded H. influenzae, and microscopic examination of her TBLB revealed A. hyphae. A diagnosis of invasive Aspergillus was confirmed by surgery. The patient had nonimmunological features of HIES, including retained primary teeth, scoliosis, and skeletal fractures (Fig. 11). Vascular anomalies and tumors were absent. The patient's serum IgE was notably elevated (17,400–23,600 IU/mL). The diagnosis of HIES was confirmed by genetic studies showing a STAT3 mutation. During the follow-up period, the patient developed a cervical lymphatic abscess followed by a liver abscess. Both were positive for S. aureus and required surgical intervention.
Figure 10.
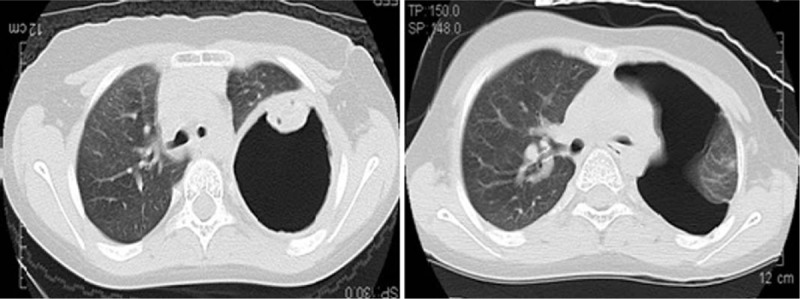
High-resolution chest computed tomography lung windows reveal a huge thick-walled cavity in the left lower lobe in Patient 4.
Figure 11.
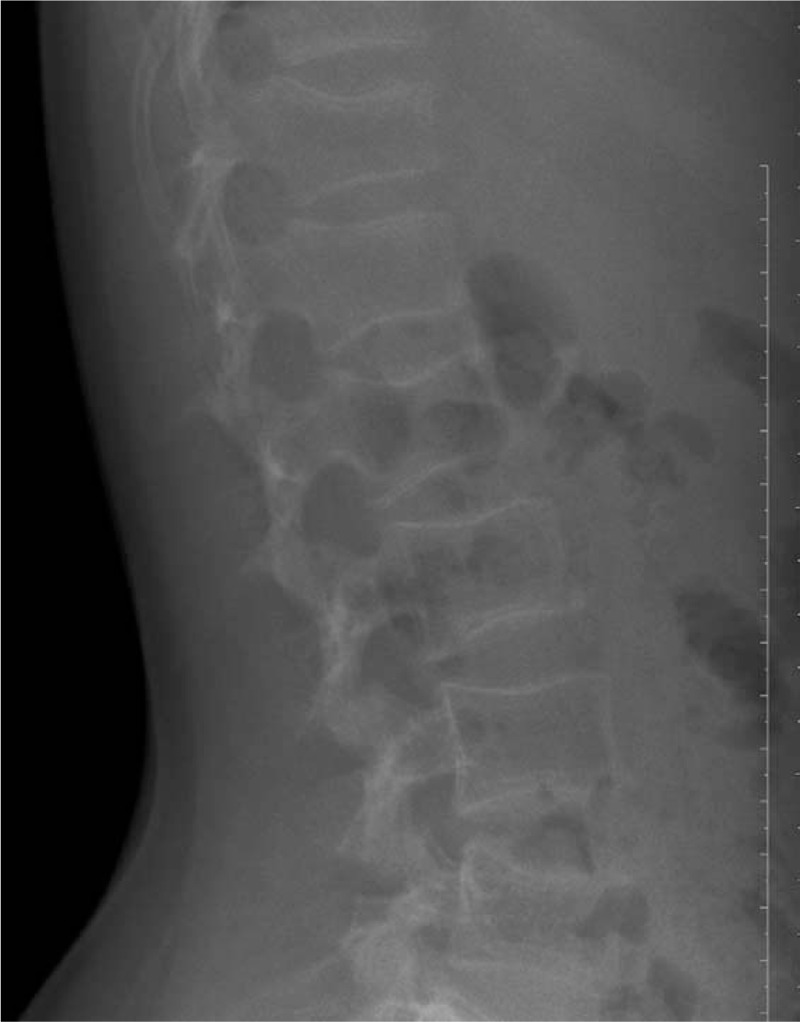
Lateral views of the lumbar spine show lumbar vertebrae osteoporosis in Patient 4.
3. Results
3.1. Clinical features and NIH score
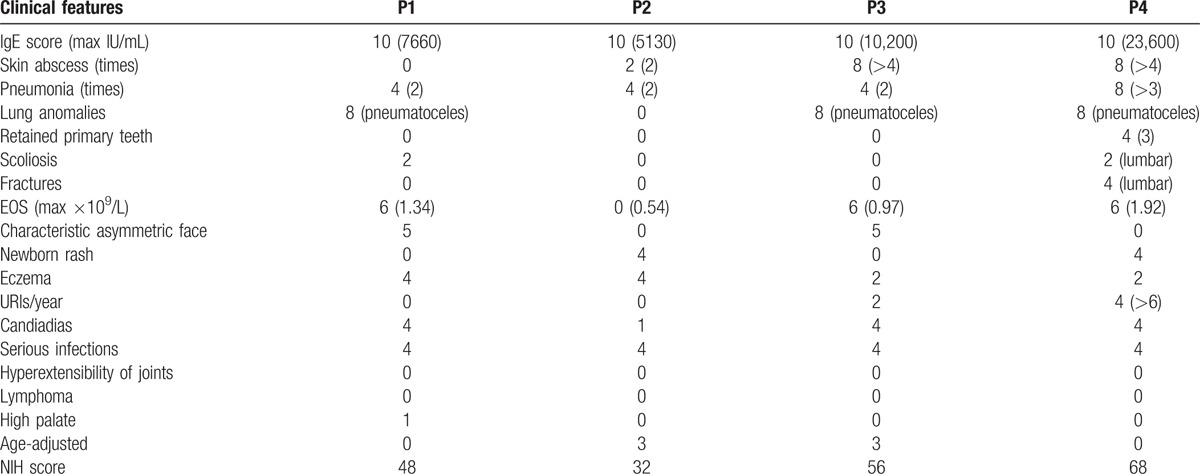
The immunologic and nonimmunologic features of the patients are summarized in Tables 1 and 2 (NIH SCORE). The most common features of HIES were recurrent respiratory infections, eczematoid skin lesions of varying degree, mucocutaneous infections, otitis media (P3 and P4), and food allergies (P2). The most common presenting features, displayed by all patients, were immunologic manifestations such as elevated serum IgE, pneumonia, cutaneous manifestations, and eosinophilia. At presentation, P3 and P4 had facial and scalp miliaria pustulosa, while P2 and P4 had cutaneous abscesses. P4 had lung and liver abscesses, while cyst-forming pneumonia was identified in 3 cases (P1, P3, and P4). Our patients did not demonstrate many of the nonimmunological manifestations typical of HIES, although P1 and P3 displayed the characteristic asymmetric facial features. Lumbar scoliosis was found in P1 and P4, while retained primary teeth and skeletal fractures were seen only in P4. There were no deaths, vascular complications, severe neurological deficits, or malignancy in the 4 patients during the follow-up period.
Table 1.
Clinical features and laboratory findings of patients diagnosed HIES.
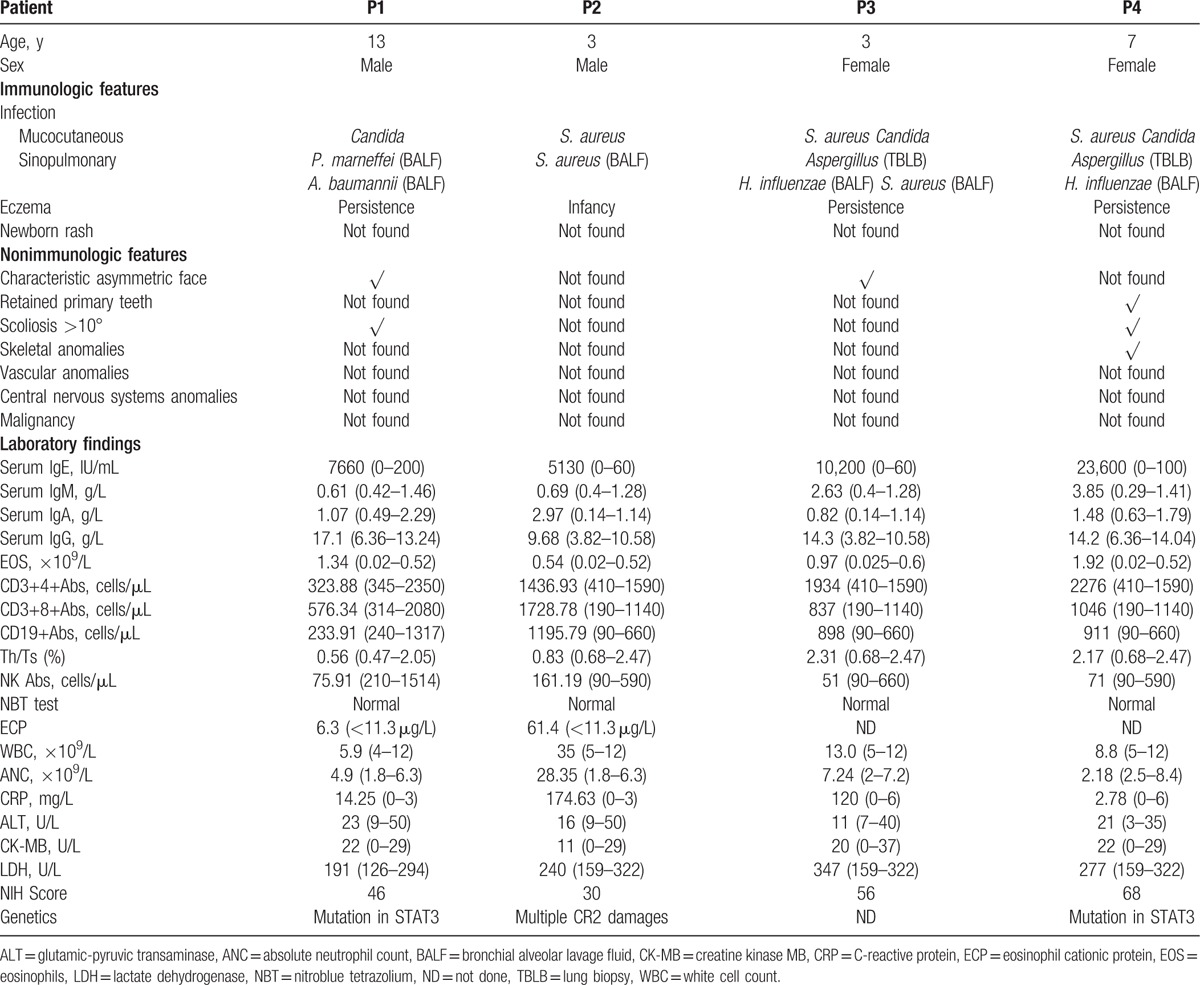
Table 2.
NIH SCORE of patients.
3.2. Infection and pathogens
All 4 patients had at least 1 episode of pneumonia, and 3 experienced fungal infection with cavitation on HRCT (P1, P3, and P4). However, fever was not a common symptom. Most pathogens that caused pneumonia were found in BALF and TBLB specimens. Organisms included fungi such as invasive Aspergillus (P3, P4) and P. marneffei (P1) as well as S. aureus (P2, P3), H. influenzae (P3, P4), and A. baumannii (P1). S. aureus was the most common bacteria recovered from the skin and deep-seated abscesses. Mucocutaneous pathogens were typically S. aureus (P2, P3, and P4) and Candida yeasts (P1, P3, and P4), while 1 patient (P4) had a liver abscess due to S. aureus. Clinical symptoms and the appearance of the lung on imaging studies were improved after antimicrobial and antifungal treatment in P1, P2, and P3. Unfortunately, the skin cold abscess showed no improvement in P2. P4 underwent lobectomy twice because of severe pulmonary infections.
3.3. Hematological and immunological parameters
The hematological and immunological profiles of the 4 patients at the time of diagnosis are summarized in Table 1. Leucocytes and neutrophils ranged from normal to markedly elevated in patients with acute infections. The level of serum C-reactive protein increased by varying degrees, especially in P2 and P3. Alanine aminotransferase and creatine kinase MB were normal in all patients, and lactate dehydrogenase was slightly increased in P3. One of the clearest manifestations of HIES in laboratory tests of the patients was markedly elevated serum IgE in serial measurements (4090–23,600 IU/mL). P3 and P4 showed a slight elevation in serum IgM. P1, P3, and P4 had an increased IgG level, although all had a normal IgA. P2 alone had a normal IgG and elevated IgA. Eosinophilia (>0.52 × 109/L) was detected in all patients, and the count of eosinophils varied from 0.54 × 109/L to 1.92 × 109/L. High CD3+4+ and CD3+8+ T cell counts were identified in P4 and P2, respectively. CD19+ B cell counts were elevated in P2, P3, and P4. Only P1 showed slightly decreased CD4+ T and CD19+ B cell counts. Nitroblue tetrazolium tests were normal in all patients. All patients had a reversed CD4/CD8 T lymphocyte ratio. Reduced natural killer cell counts were identified in 3 of 4 patients (P1, P3, and P4).
3.4. Mutation studies
Targeted region sequencing of candidate genes demonstrated that 2 patients (P1and P4) had AD-HIES, as heterozygous STAT3 mutations were identified in both. A novel missense mutation of STAT3 in exon 17 (c.1593A>T, p.K531N) was detected in P1, that, to our knowledge, has not yet been reported. This defect was predicted as a disease-causing mutation by multiple software applications, including PolyPhen2, Mutation Taster, Protein Variation Effect Analyzer, and SIFT. We also identified a known pathogenic mutation (c.1144C>T, p.R382W)7 in exon 13 in P4. As both P1 and P4 had HIES and a STAT3 defect with no family history of the disease, the parents of both patients were recruited for mutation analysis to evaluate the inheritance. Expectedly, the parents of P1 and P4 did not carry the same mutation found in their children, indicating that both c.1593A>T and c.1144C>T mutations were de novo mutations. Moreover, P2 had 3 homozygous nonsynonymous variations of the CR2 gene distributed in exons 10 (c.1916G>A, p.S639N) and 11 (c.1987T>C, p.S663P and c.2012G>A, p.R671H) that appeared with high frequency in the 1000 Genome Project.
4. Discussion
Here, we present our analysis of four Chinese patients with confirmed HIES. HIES is a complex primary immunodeficiency syndrome characterized by both immunologic and nonimmunologic manifestations.[1–3] Clinically, in patients with HIES, the most frequently found immunological abnormalities are eczematoid rashes, skin abscesses, recurrent respiratory infections, markedly elevated serum IgE, mucocutaneous candidiasis, and eosinophilia.[2] The nonimmunologic manifestations include craniofacial, musculoskeletal, dental, and vascular abnormalities.[2] There is no specific unique immunological or molecular marker of HIES.[9] In our patients, the suspicion of immunodeficiency was raised primarily because of elevated serum IgE and severe and recurrent respiratory tract infections or skin abscesses. The NIH scoring system and genetic evaluation facilitated the diagnosis in these cases. The family history was noncontributory in all of our patients, and each presented with at least 1 clinical finding (recurrent eczema, pneumonia, or cold abscess) that had been present since infancy, although no patient was diagnosed before the end of the first year of life similar to that of previous report.[1–3,10] Thus, our cases illustrate that young children with HIES are likely to experience a delay in diagnosis even after the disease becomes symptomatic. Further, our findings demonstrate the possibility that the serum IgE may not reach the diagnostic threshold (>2000 IU/mL) and recurrent respiratory infections may not be present in very young children.
HIES is distinguished from many other primary immunodeficiencies by its many nonimmunologic features.[11] However, except in P3 and P4, these signs were largely absent in our cases. P3 only had the characteristic facial features, and P4 had scoliosis, retained primary teeth, and skeletal fractures. Whereas, hypertension, vascular system abnormalities, severe food allergies, and central nervous system (CNS) findings were absent in all patients. Thus, in pediatric HIES, nonimmunological manifestations, including craniofacial and vascular system abnormalities and malignant tumors, are rare and may appear gradually over time. By the age of 13 years, P1 had developed some, but not all of these features. P2 had classic signs of HIES and an atypical genetic defect. However, his NIH score was low, possibly because he was too young to manifest the nonimmunologic features of HIES fully. Therefore, the NIH scoring system does not allow diagnostic certainty in infants and small children because some disease manifestations may not be present in young patients.
Acute and recurrent infections, often pulmonary or mucocutaneous, were diagnosed in all of our cases. P1, P3, and P4 had fungal lung infections accompanied by cavitary lesions, presumably due to dysregulated mediators of inflammation with resultant local tissue destruction. P3 and P4 had confirmed invasive aspergillosis. Furthermore, P4 underwent pulmonary lobectomy to treat extensive lesions in the lung. While HIES-related P. marneffei infections are very rare, P1 had both P. marneffei and A. baumannii infections in the lungs. Therefore, pulmonary fungal infections may be a significant cause of morbidity in our pediatric patients with HIES, which unlike previous reports, the lung pathogens are mainly bacterial.[10,12] Different regions and climates may lead to those dissimilarities. As in previously reported cases,[2,4,13] in our patients, mucocutaneous infections were typically caused by S. aureus (P2, P3, P4). On the contrary, all of our patients had eczema, and P3 and P4 also had S. aureus-related pustular or eczematoid eruptions on the scalp and face, which is a finding that has not previously been documented in HIES cases. Both P2 and P4 developed cold abscesses. Therefore, this cutaneous manifestation could be an early diagnostic feature of HIES, especially in a patient with elevated serum IgE and recurrent respiratory infections.
Patients with HIES usually have a markedly elevated serum IgE, and a level >2000 IU/mL has been arbitrarily selected as diagnostic of HIES.[1,2,14] Serum IgE levels were very high in all of our patients. This elevation in serum IgE tended to cause serious infection in our cases, a finding that contrasts with previously published observations that serum IgE level is not related to disease severity.[2,15,16] Eosinophilia was also detected in all patients but did not correlate with the elevation in IgE. Further, eosinophil cationic protein did not consistently correlate with eosinophil count or serum IgE in our patients. The specific roles of serum IgE, eosinophils, and eosinophil cationic protein in HIES remain uncertain. We observed no consistent abnormality in the number or distribution of lymphocyte subpopulations or serum Ig in our case series. Serum IgG, IgA, and IgM are typically normal in HIES patients according to previous reports.[17,18] We found a slight increase in serum IgG and IgM in some of our patients, while CD4+ T and CD19+ B cells were slightly reduced only in P1, a finding consistent with those of previous reports.[19] Other patients had increased CD19+ B and increased or normal CD4+ and CD8+ lymphocyte counts. At the same time, most patients had a reduced natural killer cell count. Hence, cellular immunity dysfunction could also be an important factor that predisposes patients to HIES-related infections. It is regrettable that flow cytometric analysis of Th17 cells was not detected in our patients. Further research is necessary to evaluate immunologic parameters in HIES and their effects.
Most cases of AD-HIES are caused by heterozygous mutations in STAT3, whereas homozygous mutations in DOCK8 are responsible for many cases of AR-HIES. In addition to these mutations, recently, homozygous mutations in phosphoglucomutase 3 (PGM3) have been described in some cases.[20,21] In our series, all cases were from nonconsanguineous families. Thus, an AD mode of inheritance can be assumed. Targeted region sequencing confirmed a diagnosis of AD-HIES in P1 and P4 with heterozygous missense mutations in STAT3 exons 17 (c.1593A>T, p.K531N) and 13 (c.1144C>T, p.R382W), respectively. In both patients, the defect was demonstrated to be a de novo mutation on further pedigree analysis. The c.1144C>T mutation in STAT3 is a known pathogenic mutation,[22] whereas the c.1593A>T mutation has not been previously reported. Moreover, P2 had 3 homozygous nonsynonymous variations of CR2 in exon 10 (c.1916G>A, p.S639N) and 11 (c.1987T>C, p.S663P and c.2012G>A, p.R671H). As these appear in the 1000 Genome Project with a high frequency, the 3 variations are probably disease susceptibility loci rather than pathogenic mutations. The relationship between HIES clinical phenotypes and the CR2 gene requires further evaluation.
The mainstay of HIES therapy centers on proper skin care to prevent infections and aggressive treatment of those that do occur.[13] Most of our patients had been diagnosed with severe infections, especially in the lung, but all of them survived after timely and effective treatment. As a congenital immunodeficiency, HIES frequently leads to severe opportunistic infections without typical inflammatory features. A detailed history, thorough physical examination, and pertinent diagnostic studies facilitate the identification of the causative pathogen. Antimicrobial medications should be given early, provide broad-spectrum coverage, and be administered for a prolonged period to control infection in patients with HIES. In cases of severe or lethal infection, it is vital to perform the appropriate surgery in a timely fashion. Prophylactic antibiotics are used to decrease the frequency of pneumonia with the goal of preventing parenchymal lung damage and include those targeted against S. aureus and fungi.[2,4,15,18] In our patients with fungal lung infections, anti-Aspergillus therapy (itraconazole or voriconazole) was used, and none had developed a recurrent fungal infection at the time of their last follow-up examination.
5. Conclusion
These 4 cases demonstrate that fungi are important pathogens in children with HIES and can cause significant morbidity in this population. The question of when HIES should be suspected and further work-up initiated is not fully answered, particularly in infants. The NIH scoring system does not allow diagnostic certainty in infants because some disease manifestations may not be present in very young patients. Cutaneous manifestations could be an early indicator of HIES, especially in patients with high serum IgE and recurrent respiratory infections. Pulmonary complications must be identified early to treat them effectively, and molecular and genetic diagnostic tests are important for all cases of suspected HIES. Further, we report a novel HIES-associated STAT3 mutation. This finding will facilitate the correlation of genotype with phenotype in Chinese children with HIES and indicates the importance of evaluating the prevalence of STAT3 mutations in cases of pediatric HIES. We also used targeted region sequencing studies to identify the correlation between multiple CR2 gene defects and HIES.
Author contributions
Data curation: D. Zhang.
Formal analysis: L. Huang.
Investigation: G. Lu, D. Yang, Y. Xie.
Methodology: J. Yu.
Resources: L. Huang.
Writing – original draft: H. Fan, L. Huang.
Writing – review & editing: G. Lu, Y. Lin.
Footnotes
Abbreviations: A. baumannii = Acinetobacter baumannii, AD-HIES = autosomal dominant hyper-IgE syndrome, AR–HIES = autosomal recessive hyper-IgE syndrome, BALF = bronchoalveolar lavage fluid, CR2 = complement receptor 2, DOCK8 = dedicator of cytokinesis 8, H. influenzae = Haemophilus influenzae, HIES = hyper-IgE syndromes, HRCT = high-resolution computed tomography, IgE = immunoglobulin E, NIH = National Institutes of Health, P. marneffei = Penicillium marneffei, S. aureus = Staphylococcus aureus, STAT3 = signal transducer and activator of transcription, TBLB = transbronchial lung biopsy, TYK2 = tyrosine kinase 2.
HF and LH contributed equally to this article.
The authors report no conflicts of interest.
References
- [1].Freeman AF, Holland SM. Clinical manifestations of hyper IgE syndromes. Dis Markers 2010;29:123–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Yong PF, Freeman AF, Engelhardt KR, et al. An update on the hyper-IgE syndromes. Arthritis Res Ther 2012;14:228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Szczawinska-Poplonyk A, Kycler Z, Pietrucha B, et al. The hyperimmunoglobulin E syndrome: clinical manifestation diversity in primary immune deficiency. Orphanet J Rare Dis 2011;6:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Farmand S, Sundin M. Hyper-IgE syndromes: recent advances in pathogenesis, diagnostics and clinical care. Curr Opin Hematol 2015;22:12–22. [DOI] [PubMed] [Google Scholar]
- [5].Grimbacher B, Holland SM, Gallin JI, et al. Hyper-IgE syndrome with recurrent infections: an autosomal dominant multisystem disorder. N Engl J Med 1999;340:692–702. [DOI] [PubMed] [Google Scholar]
- [6].Moin M, Farhoudi A, Movahedi M, et al. The clinical and laboratory survey of Iranian patients with hyper-IgE syndrome. Scand J Infect Dis 2006;38:898–903. [DOI] [PubMed] [Google Scholar]
- [7].Dati F, Ringel KP. Reference values for serum IgE in healthy non-atopic children and adults. Clin Chem 1982;28:1556. [Google Scholar]
- [8].Wittig HJ, Belloit J, De Fillippi I, et al. Age-related serum immunoglobulin E levels in healthy subjects and in patients with allergic disease. J Allergy Clin Immunol 1980;66:305–13. [DOI] [PubMed] [Google Scholar]
- [9].Saghafi S, Pourpak Z, Glocker C, et al. The diagnosis of hyper immunoglobulin e syndrome based on project management. Iran J Allergy Asthma Immunol 2015;14:126–32. [PubMed] [Google Scholar]
- [10].Zhang LY, Tian W, Shu L, et al. Clinical features, STAT3 gene mutations and Th17 cell analysis in nine children with hyper-IgE syndrome in mainland China. Scand J Immunol 2013;78:258–65. [DOI] [PubMed] [Google Scholar]
- [11].Freeman AF, Holland SM. Clinical manifestations, etiology, and pathogenesis of the hyper-IgE syndromes. Pediatr Res 2009;65:R32–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Xie L, Hu X, Yang L, et al. Hyper-IgE syndrome with STAT3 mutation: a case report in mainland China. Clin Dev Immunol 2010;2010:204–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Minegishi Y, Saito M. Cutaneous manifestations of hyper IgE syndrome. Allergol Int 2012;61:191–6. [DOI] [PubMed] [Google Scholar]
- [14].Hagl B, Heinz V, Schlesinger A, et al. Key findings to expedite the diagnosis of hyper-IgE syndromes in infants and young children. Pediatr Allergy Immunol 2016;27:177–84. [DOI] [PubMed] [Google Scholar]
- [15].Sowerwine KJ, Holland SM, Freeman AF. Hyper-IgE syndrome update. Ann N Y Acad Sci 2012;1250:25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Berger M, Kirkpatrick CH, Goldsmith PK, et al. IgE antibodies to Staphylococcus aureus and Candida albicans in patients with the syndrome of hyperimmunoglobulin E and recurrent infections. J Immunol 1980;125:2437–43. [PubMed] [Google Scholar]
- [17].Lee WI, Huang JL, Lin SJ, et al. Clinical, immunological and genetic features in Taiwanese patients with the phenotype of hyper-immunoglobulin E recurrent infection syndromes (HIES). Immunobiology 2011;216:909–17. [DOI] [PubMed] [Google Scholar]
- [18].Chandesris MO, Melki I, Natividad A, et al. Autosomal dominant STAT3 deficiency and hyper-IgE syndrome: molecular, cellular, and clinical features from a French national survey. Medicine (Baltimore) 2012;91:e1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Siegel AM, Heimall J, Freeman AF, et al. A critical role for STAT3 transcription factor signaling in the development and maintenance of human T cell memory. Immunity 2011;35:806–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zhang Y, Yu X, Ichikawa M, et al. Autosomal recessive phosphoglucomutase 3 (PGM3) mutations link glycosylation defects to atopy, immune deficiency, autoimmunity, and neurocognitive impairment. J Allergy Clin Immunol 2014;133:1400–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yang L, Fliegauf M, Grimbacher B. Hyper-IgE syndromes: reviewing PGM3 deficiency. Curr Opin Pediatr 2014;26:697–703. [DOI] [PubMed] [Google Scholar]
- [22].Minegishi Y, Saito M, Tsuchiya S, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature 2007;448:1058–62. [DOI] [PubMed] [Google Scholar]