

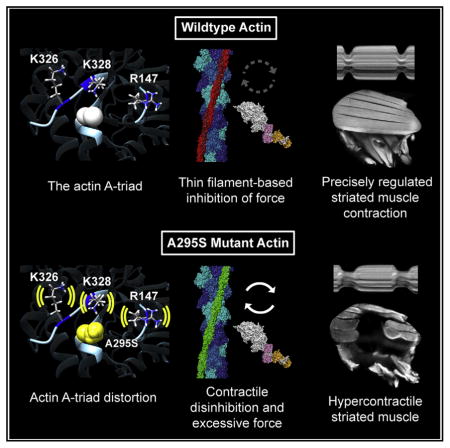
SUMMARY
Striated muscle contraction is regulated by the movement of tropomyosin over the thin filament surface, which blocks or exposes myosin binding sites on actin. Findings suggest that electrostatic contacts, particularly those between K326, K328, and R147 on actin and tropomyosin, establish an energetically favorable F-actin-tropomyosin configuration, with tropomyosin positioned in a location that impedes actomyosin associations and promotes relaxation. Here, we provide data that directly support a vital role for these actin residues, termed the A-triad, in tropomyosin positioning in intact functioning muscle. By examining the effects of an A295S α-cardiac actin hypertrophic cardiomyopathy-causing mutation, over a range of increasingly complex in silico, in vitro, and in vivo Drosophila muscle models, we propose that subtle A-triad-tropomyosin perturbation can destabilize thin filament regulation, which leads to hypercontractility and triggers disease. Our efforts increase understanding of basic thin filament biology and help unravel the mechanistic basis of a complex cardiac disorder.
In Brief
Viswanathan et al. demonstrate that the conserved actin A-triad, composed of K326, K328, and R147, normally biases tropomyosin to a position that impedes actomyosin associations along resting striated muscle thin filaments. The proximally located actin A295S hypertrophic cardiomyopathy mutation distorts A-triad-tropomyosin associations, which promotes contractile disinhibition, hypercontraction, and disease pathogenesis.
INTRODUCTION
Striated muscle contraction results from transient interactions between myosin-containing thick and actin-containing thin filaments. Contractile regulation, throughout the animal kingdom, is achieved by Ca2+-dependent modulation of myosin cross-bridge cycling on actin by the thin filament troponin-tropomyosin complex (Lehman et al., 1994; Tobacman, 1996; Cammarato et al., 2004; Lehman, 2016). The complex consists of an elongated tropomyosin (Tm) dimer and the troponin C (TnC, calcium binding), troponin I (TnI, inhibitory), and troponin T (TnT, Tm binding) subunits of troponin (Tn). Tm is a modular protein consisting of seven tandem pseudo-repeating motifs designed to bind seven successive actin monomers along the thin filament (Brown et al., 2005; Hitchcock-DeGregori, 2008; Li et al., 2011). The continuous Tn-Tm complexes adopt various states characterized by different average Tm positions that govern the access of myosin binding sites and hence force production (Tobacman, 1996; Lehman, 2016). Under low Ca2+ conditions, TnI binds to actin and constrains Tn-Tm to the B state, in which Tm sterically blocks and limits myosin binding. Upon activation, Ca2+ binds to TnC, which triggers TnI release from actin and Tm movement away from myosin binding sites, resulting in the C state. Initial myosin binding further displaces Tm, which increases myosin accessibility along actin to establish the open (M) state and promotes cooperative activation of contraction. Tm can oscillate dynamically between the states at all Ca2+ levels, and it is the average azimuthal location of this equilibrium that is normally determined by Tn, Ca2+, and myosin (McKillop and Geeves, 1993; Maytum et al., 2003; Pirani et al., 2005).
The association of Tm with actin is largely electrostatic (Brown et al., 2005; Li et al., 2011; Barua et al., 2013). Models of filamentous actin and Tm (F-actin-Tm), in the absence of Tn (i.e., the A state), have been proposed based on molecular evolutionary and mutational analysis, computational chemistry, and electron microscopy (EM) reconstructions (Barua et al., 2011, 2013; Li et al., 2011; von der Ecken et al., 2015). These models define a conserved F-actin-Tm binding interface that is characterized by clusters of charged residues on F-actin, which make favorable contacts with each consecutive Tm pseudo-repeat. The electrostatic interactions place the dimer in a location that is close to its B-state position on regulated F-actin-Tn-Tm filaments when pinned down by TnI at low Ca2+ and establish an energetically stable conformation (Li et al., 2011; Barua et al., 2013; Lehman et al., 2013). In particular, K326, K328, and R147 on actin appear to interact with acidic residues of each repetitive motif along Tm’s entire length, associations that in muscle are likely essential for helping establish an energy basin to bias Tm to an inhibitory position (Orzechowski et al., 2014).
Because precisely coordinated communication among individual components is compulsory for proper contractile regulation, it follows that mutations at or near thin filament subunit interfaces can disrupt its function and initiate myopathy. For example, the loci of many of the ~100 thin filament mutations that cause hypertrophic cardiomyopathy (HCM) are found at intermolecular surfaces (Tardiff, 2011). HCM is a clinically diverse, autosomal dominant disease of cardiac muscle that afflicts 1:500 of the general population (Maron, 2002). It is characterized by abnormal thickening of the heart, myocellular disarray, arrhythmias, and altered Ca2+ homeostasis. However, the earliest signs of disease are hyperdynamic contractile properties and diastolic dysfunction, which precede ventricular hypertrophy and have been shown to be essential for HCM pathobiology (Green et al., 2016). Diastolic dysfunction is associated with elevated filling pressures due to impaired relaxation and increased myocardial stiffness (Kass et al., 2004; Campbell and Sorrell, 2015).
In 1999, the first α-cardiac actin (ACTC1) mutation linked to HCM was reported (Mogensen et al., 1999). Expression of the ACTC1 A295S variant engendered highly penetrant yet heterogeneous disease phenotypes among family members. Only one of 13 individuals was asymptomatic, whereas nine were phenotypically assigned with major HCM and three were phenotypically assigned with minor HCM. Despite its identification nearly two decades ago, efforts to characterize A295S actin have been limited and failed to resolve any disruptions in in vitro transcription and/or translation, folding efficiency, or actomyosin ATPase activity of mutant proteins (Vang et al., 2005; Dahari and Dawson, 2015). Overall, such clinical heterogeneity, the absence of mutation-induced in vitro effects, and a lack of animal models limits meaningful genotype-phenotype correlations for predicting patient outcomes and our understanding of the molecular basis of disease (Tardiff, 2011). However, model organisms that minimize confounding genetic and environmental factors to curtail phenotypic variability, and permit analysis from the molecular through the organ level, can help resolve the intricate origins of myopathies.
Here, we used Drosophila melanogaster as our primary experimental model for comprehensive in vivo, in situ, in vitro, and in silico analysis of the ACTC1 A295S HCM-causing mutation. We tested the hypothesis that the mutation perturbs critical associations between nearby actin residues K326, K328, and R147, termed the A-triad, and Tm, which disrupts contractile inhibition, stimulates hyperdynamic and excessive tension generation, and consequently initiates disease. The variant would therefore be expected to yield a molecular phenotype characterized by inadequate Tm-based blocking of myosin binding on thin filaments. When expressed in the fly heart, A295S mutant actin significantly decreased cardiac output, prolonged systolic intervals, and elicited diastolic dysfunction by increasing actively cycling cross-bridges without affecting the resting cellular Ca2+ milieu. A295S actin expression in the indirect flight muscle (IFM) also induced hypercontraction and enhanced Ca2+ sensitivity of reconstituted thin filament sliding. Computational modeling indicated that the ACTC1 A295S HCM mutation changed the orientation of A-triad residues and their interactions with Tm, which would destabilize the energetically favored inhibitory A/B state and may bias Tm toward the C-state configuration. Demonstration that possible subtle changes in F-actin-Tm interactions promote discernable contractile disinhibition emphasizes the value of Drosophila models for providing a mechanistic basis for proximal causes of myocardial disease. Thus, our efforts increase understanding of basic thin filament biology and help elucidate the pathogenesis of a complex cardiac disorder.
RESULTS
The A295S Mutation Resides Proximal to the Highly Conserved Actin A-Triad
Human and Drosophila actins are encoded by six highly homologous genes. As found in vertebrates, flies express cardiac and skeletal muscle-specific isoforms. Act57B is one of two sarcomeric actin genes expressed in the adult fly heart, while Act88F encodes all sarcomeric actin of the IFM (Cammarato et al., 2011a; Shah et al., 2011; Hiromi and Hotta, 1985). The cardiac and skeletal isoforms differ in only a few amino acids within and between species (Figure 1A). K326, K328, and R147, which based on F-actin-Tm models, make multiple favorable electrostatic contacts with Tm, are exceptionally well conserved. Analysis of more than 800 actin sequences, from vertebrates and invertebrates, revealed 100% fidelity of these basic residues (Figure S1). This supports a vital role for these amino acids in actin function and potentially, in striated muscle thin filaments, in Tn-Tm-mediated regulation of contraction. To emphasize the importance of these residues in establishing the A state of F-actin-Tm and, in principle, in contractile regulation of striated muscle, we have designated these amino acids as the A-triad. A295 is centered just behind the A-triad in the actin subunit structure (Figure 1B). Therefore, amino acid substitutions at this locus may induce propagated effects that influence the association of the A-triad with corresponding residues along Tm and influence its capacity to impede myosin binding.
Figure 1. The Actin A-Triad Is Highly Conserved and Forms Critical Electrostatic Interactions with Tropomyosin.

(A) Multiple sequence alignment of muscle actins from Homo sapiens and Drosophila melanogaster. ACTC, human α-cardiac muscle actin; ACTA, human a[α]-skeletal muscle actin; Act57B, Drosophila cardiac actin; Act88F, Drosophila IFM actin. Residues are shaded based on degree of conservation. An asterisk indicates positions that have identical residues, a colon indicates substitution with high structural similarity, and a period indicates substitution of low structural similarity. R147, K326, and K328, which form the A-triad, are shaded blue, and A295 is shaded yellow.
(B) Molecular model of Tm (brown) on actin (blue) (Li et al., 2011), illustrating electrostatic interactions between the A-triad and E139 and E142 of Tm. Inset: A295 is centered at the base of, and just behind, the actin A-triad.
See also Figure S1.
A295S Actin Incorporates Uniformly along Drosophila Cardiac Thin Filaments
Drosophila has a linear cardiac tube that consists of a single layer of cardiomyocytes and is highly reminiscent of the primitive vertebrate embryonic heart (Figure 2A) (Miller, 1950). The relative simplicity of the fly heart, combined with an array of tools to manipulate the fly genome, provide unique opportunities to examine how thin filament mutations affect cardiac muscle and drive disease. Using the PhiC31 integrase system (Groth et al., 2004), we generated several transgenic lines that permit examination of the muscle-specific effects of wild-type versus A295S mutant actin expression at multiple levels. PhiC31-mediated transgenesis ensured each gene integrated at an identical, predetermined genomic location. Thus, our results are directly comparable and phenotypic differences in control versus mutant flies were immediately attributed to the single amino acid substitution in actin. Heart-specific expression of transgenes was achieved via the Gal4-upstream activating sequence (UAS) expression system (Brand and Perrimon, 1993). Flies harboring the Hand4.2-Gal4 driver were crossed with flies containing constructs consisting of an UAS followed by an Act57B transgene. The progenies inherit both genes and express the UAS-Act57B transgene exclusively in the cardiac tube, which was confirmed by direct visualization of fluorescent signals from Hand4.2-Gal4 > UAS-Act57BGFP.WT (abbr.Hand > Act57BGFP.WT) flies (Figure 2A). Closer examination revealed clear, striated GFP signals from the hearts of Hand > Act57BGFP.WT and Hand > Act57BGFP.A295S Drosophila (Figures 2B and 2C). At 100× magnification, GFP signals from both lines were seen overlapping congruently with tetramethylrhodamine (TRITC)-phalloidin actin signals (Figures 2C and 2D). Act57BGFP.A295S actin was not excluded from sarcomeres and did not accumulate in localized sub-sarcomeric regions. These data illustrate homogeneous co-polymerization of Act57BGF-P.A295S mutant actin with endogenous cardiac actin evenly, along the entire length of the thin filaments, in a manner indistinguishable from that of Act57BGFP.WT actin.
Figure 2. Transgenic Actin Integrates Uniformly along Drosophila Cardiac Thin Filaments.

(A) Cardiac-restricted expression of UAS-Act57BGFP.WT actin was achieved via the Hand4.2-Gal4 driver. GFP fluorescence emanated from cardiomyocytes of the heart tube (10× magnification). Scale bar, 100 μm.
(B) Confocal micrographs taken at 40×magnification reveal Act57BGFP.WT and Act57BGFP.A295S transgenic actin incorporated discretely along cardiac myofibrils, as evident by striated GFP signals. Scale bar, 25 μm.
(C) At 100× magnification, GFP-labeled actin appeared to overlap completely with TRITC-phalloidin (T-Ph) signals. Scale bar, 5 μm.
(D) Fluorescent intensity line scans of cardiac myofibrils (imaged at 100×) illustrate well-aligned TRITC-phalloidin (red) and GFP (green) signals, indicating that the transgenic actin co-polymerized with endogenously expressed actin evenly along the length of cardiac thin filaments. The index of correlation, calculated as the fraction of positively correlated TRITC-phalloidin and GFP pixels from multiple Hand > Act57BGFP.WT and Hand > Act57BGFP.A295S sarcomeres (n = 85–97), was 0.82 and 0.78, respectively, reflecting a high degree of overlapping signals.
The ACTC1 A295S HCM Mutation Induces Cardiomyopathy in Drosophila
High-speed video microscopy and semi-automated motion analysis were used to investigate the consequences of the HCM actin mutation on Drosophila cardiac tubes. We analyzed beating hearts of females expressing UAS-Act57BWT versus UAS-Act57BA295S transgenes that lacked GFP to eliminate confounding effects caused by the N-terminally located moiety (Figure S2A). M-mode kymograms, which illustrate heart wall movement over time, suggested mutation-induced alterations in cardiac dimensions and contractile dynamics in three-week-old flies (Figures 3A and 3B). Hand > Act57BA295S hearts were characterized by restricted diameters and prolonged systolic periods relative to controls.
Figure 3. Expression of A295S Actin Reduces Cardiac Output and Engenders Restrictive Physiology with Prolonged Periods of Systolic Tension Development.

(A) M-mode kymograms generated from high-speed videos of beating three-week-old Hand > Act57BWT and Hand > Act57BA295S hearts. Vertical red lines delineate diastolic diameters and terminate at opposing edges of the heart wall.
(B) Horizontal lines demarcate SIs (top) and complete HPs (bottom) for each genotype. Relative to transgenic Act57BWT-, Act57BA295S-expressing cardiac tubes displayed restricted diastolic diameters and prolonged systolic periods.
(C) Hand > Act57BA295S Drosophila exhibited highly significant alterations in several cardiac functional parameters relative to Hand > Act57BWT and Hand × yw control flies. Decreased output, cardiac dimensions, fractional shortening, and extended periods of systole were observed in the mutants at all ages studied.
Data are presented as mean ± SEM (n = 35–46 for each genotype and age group). See also Figure S2 and Tables S1 and S2.
We quantified the effects of Act57BA295S mutant actin expression on several cardiac parameters relative to wild-type actin expression and to endogenous actin expression in non-transgenic controls (the progeny of Hand × yw), from one through seven weeks of age (Figure 3C; Table S1). As shown previously, cardiac output, diastolic and systolic diameters, and fractional shortening progressively decreased among all genotypes over time (Blice-Baum et al., 2017). However, Hand > Act57BA295S Drosophila exhibited significantly diminished output relative to controls. This reduction was due to restricted diastolic and systolic diameters. Moreover, the effect of mutant actin expression on diastolic diameters was greater than that on systolic diameters, which resulted in a significant decrease in fractional shortening versus controls. Overall, as observed in a cohort of HCM patients with myosin or TnI mutations (Kubo et al., 2007), Hand > Act57BA295S hearts demonstrated a restrictive phenotype and were unable to adequately relax during diastole.
We also evaluated the myogenic cardiac contractile dynamics for Hand > Act57BA295S relative to Hand > Act57BWT and non-transgenic actin controls. The heart period, which is the combined length of time required for a single diastolic and ensuing systolic event of the cardiac cycle, significantly increased with age for all lines but did not differ among genotypes (Figure 3C). However, normalization of the systolic interval to the heart period (SI/HP) for each line, which illustrates the proportion of time spent generating active tension during the cardiac cycle, revealed the A295S actin mutation induced an increase in the SI/HP ratio relative to controls at all ages.
Thus, analogous to what is seen during early stages of HCM, which are characterized by diastolic dysfunction and hyper-dynamic contractile properties (Green et al., 2016), the A295S actin mutation perturbs the heart’s ability to reestablish resting diastolic volumes and it prolongs tension-generating periods. All preceding findings were verified using a second, independent pair of transgenic Drosophila lines (Figures S2B and S2C; Table S2).
A295S Actin Reduces Heart Diameters during Diastole by Increasing Ca2+-Independent Actomyosin Associations
Altered Ca2+ homeostasis is frequently observed in HCM, and diastolic dysfunction, a hallmark of HCM, can be partly attributed to elevated diastolic Ca2+ levels that promote actomyosin associations and impair relaxation. The inherent nature of the Drosophila heart tube, which is formed by opposing rows of cardiomyocytes joined by intercellular junctions (Miller, 1950), provides unique opportunities to assess diastolic properties with single-cell resolution, in situ, in the native context of the functioning organ (Viswanathan et al., 2014; Kaushik et al., 2015). To determine a contribution of potentially elevated diastolic Ca2+ to shortened Hand > Act57BA295S myocytes and thus to reduced cardiac diameters, we incubated live hearts of all genotypes in a solution containing 10 mM EGTA and 100 μM EGTA,AM. EGTA,AM is a cell-permeant acetoxymethyl ester of the Ca2+ chelator EGTA, which passively enters cells, is hydrolyzed by intracellular esterases, and produces EGTA. Upon EGTA/EGTA,AM incubation, rhythmic contractions ceased (Figure 4A). Relative to those established during diastole, all lines experienced a slight yet significant ~2.5% increase in diameters upon chelation of extra- and intracellular Ca2+ (Figures 4B and 4C). Therefore, minor amounts of intracellular diastolic Ca2+ likely trigger actomyosin interactions and produce slightly contracted cardiomyocytes. Comparison of the responses (Figure 4C), however, revealed no significant difference in the average change in cardiac diameters among Hand × yw (Δ = 1.70 ± 0.08 μm), Hand > Act57BWT (Δ = 1.88 ± 0.09 μm), and Hand > Act57BA295S (Δ = 1.85 ± 0.06 μm). This implies that a similar diastolic Ca2+ level is shared among the genotypes, which contributes equally to resting tension, and that the significantly restricted Hand > Act57BA295S heart diameters do not result from elevated resting Ca2+ causing excessive myocyte shortening. Unperturbed Ca2+ handling was also supported by qPCR analysis (Figure S3), which revealed no differences in transcript levels of L-type Ca2+ channels, ryanodine receptors, sarco/endoplasmic reticulum calcium ATPase (SERCA), Na+/Ca2+ exchangers, or inositol triphosphate (IP3) receptors between the lines. Thus, the cardiomyocyte Ca2+-handling biosignature appeared normal following Act57BA295S actin expression.
Figure 4. Excessive Ca2+-Independent Actomyosin Associations during Diastole Promote Enhanced Myocyte Shortening and Incomplete Relaxation of Hand > Act57BA295S Cardiomyocytes.

(A) M modes generated from the same region of a three-week-old Hand > Act57BWT heart reveal graded responses of cardiac diameters following exposure to distinct small-molecule compounds. Red arrowheads indicate the position of the heart wall edges during diastole, upon incubation with EGTA/EGTA,AM, and finally, following the addition of blebbistatin. Incubation with EGTA/EGTA,AM resulted in complete cessation of wall motion. Relative to the diameter during diastole, each treatment induced a slight increase in diameter across the heart tube.
(B) Significant, incremental increases in cardiac diameters were verified for all genotypes following extra- and intracellular Ca2+ chelation and upon blebbistatin incubation.
(C) The average change in diameter across the heart wall in response to EGTA/EGTA,AM was similar among all lines.
(D) Blebbistatin treatment prompted a significantly greater response across the wall of Hand > Act57BA295S hearts relative to that observed for Hand × yw and Hand > Act57BWT hearts.
Data are presented as mean ± SEM (n = 21). ***p ≤ 0.001. See also Figure S3.
Blebbistatin, a small-molecule inhibitor of several striated muscle myosins, impedes actomyosin interaction in cardiac preparations from multiple species, including flies (Fedorov et al., 2007; Viswanathan et al., 2014). Subsequent analysis verified that the addition of blebbistatin to EGTA/EGTA,AM-treated hearts evoked a second significant increase in diameters across the wall of all genotypes (Figure 4B). This suggests that the initial chelation of intracellular Ca2+ does not induce complete relaxation of Drosophila cardiomyocytes and that under such Ca2+-free conditions, Tm-based steric prevention of actomyosin interactions is incomplete. Hence, in addition to those stimulated by trace amounts of free Ca2+, a small population of residual cross-bridges is actively cycling, generating force, reducing cardiomyocyte length, and establishing basal myocardial tone during diastole. To evaluate the relative proportion of these Ca2+-independent, unimpeded, force-generating, diastolic cross-bridges, we compared the blebbistatin-induced diameter changes among all lines. The average cardiac diameters for Hand × yw and Hand > Act57BWT following blebbistatin treatment were ~2% greater (Δ = 1.61 ± 0.05 and 1.72 ± 0.10 μm, respectively) than those determined during EGTA/EGTA,AM incubation (Figures 4B–4D). However, the average cardiac diameter for Hand > Act57BA295S following blebbistatin treatment was ~8% greater (Δ = 5.47 ± 0.26 μm) than that determined during EGTA/EGTA,AM incubation (Figures 4B–4D). Comparing the diameter changes among the genotypes (Figure 4D) revealed the response to blebbistatin was significantly greater for Hand > Act57BA295S hearts relative to that for controls. These observations are consistent with diastolic dysfunction and restrictive physiology that are unrelated to resting Ca2+ levels but rather due to excessively disinhibited Ca2+-independent cross-bridge cycling, enhanced basal tension, and incomplete relaxation for Hand > Act57BA295S mutant hearts. Thus, the A295S actin mutation could destabilize Tm positioning such that it inadequately blocks myosin binding at rest, causing excessive tension and myocyte shortening that restricts proper filling, even in the absence of disrupted Ca2+ handling.
A295S Actin Impairs Drosophila Flight Ability and Promotes Skeletal Muscle Hypercontraction
Drosophila IFMs are well suited for mechanical and structural analyses, are exquisitely sensitive to sarcomeric mutations, and provide sufficient material for biochemical and biophysical investigation (Beall and Fyrberg, 1991; Bing et al., 1998; Razzaq et al., 1999; Cammarato et al., 2004, 2008). To study the effects of the HCM mutation on the IFM, we created Act88FWT and Act88FA295S transgenic flies. Different doses of transgenic versus endogenous actin were achieved by back-crossing the transgene into a flightless, Act88F null strain. Compared to w1118 non-transgenic and Act88FWT/+ controls, Act88FA295S/+ heterozygous flies showed a slight but significant decrease in flight ability, as determined by standard metrics (Figure S4A). The capacity for typical upward flight appeared blunted in the mutants. Act88FWT/+ Drosophila also displayed modest flight impairment relative to w1118.
To enhance our ability to characterize organismal behavior, we employed digital inline holography to comprehensively phenotype flight kinematics (Figure S5). We computed three-dimensional trajectories for ascending w1118, Act88FWT/+, and Act88FA295S/+ Drosophila and compared free-flying velocity and acceleration parameters (Figure 5A). Both Act88FWT/+ (0.42 ± 0.01 m/s, 27.02 ± 1.15 rad/s) and Act88FA295S/+ (0.36 ± 0.02 m/s, 22.90 ± 1.40 rad/s) transgenic lines displayed significantly lower overall and angular velocities relative to w1118 (0.45 ± 0.01 m/s, 31.51 ± 1.22 rad/s) (Figure 5B). However, Act88FA295S/+ (3.38 ± 0.32 m/s2) exhibited significantly reduced acceleration relative to both Act88FWT/+ (4.60 ± 0.26 m/s2) and w1118 (5.00 ± 0.27 m/s2). Likewise, centripetal acceleration for Act88FA295S/+ (8.43 ± 0.74 m/s2) was significantly lower than that for Act88FWT/+ (11.38 ± 0.57 m/s2) and w1118 (14.48 ± 0.68 m/s2). Moreover, because the body weights of all genotypes were identical (Figure S4B), and given that the mutants exhibited reduced acceleration, A295S actin expression appears to compromise the amount of IFM force development during upward flight. Despite clear flight impairment for Act88FA295S/+ heterozygotes, no discernable gross morphological disruptions were observed in their IFMs, relative to w1118 and Act88FWT/+, that could account for the altered behavior (Figure 6A).
Figure 5. A295S Actin Impairs Drosophila Flight Kinematics.

(A) Representative flight trajectories, and instantaneous acceleration values at every sampled point, for w1118, heterozygous Act88FWT/+, and heterozygous Act88FA295/+. Act88FA295S/+ mutants are characterized by low acceleration values over the length of their flight paths.
(B) Probability distribution plots of velocity, angular velocity, acceleration, and centripetal acceleration for w1118 (gray, n = 80), Act88FWT/+ (black, n = 90), and Act88FA295S/+ (red, n = 30) flies. While no difference in mean velocity or angular velocity was detected between Act88FWT/+ and Act88FA295S/+, both were significantly lower for each line relative to w1118. Moreover, no difference in acceleration during ascending flight was observed for w1118 versus Act88FWT/+, while acceleration for the mutant line was significantly lower than that for both. Similarly, centripetal acceleration for Act88FA295S/+ was significantly lower than that for w1118 and Act88FWT/+.
See also Figures S4 and S5.
Figure 6. A295S Actin Promotes Indirect Flight Muscle Hyper-contraction without Altering Thin Filament Length.

(A) The dorsal longitudinal IFMs (DLMs) of two-day-old Act88FWT/+ and Act88FA295S/+ heterozygotes and of Act88FWT/Act88FWT homozygotes were indistinguishable from those of w1118 controls. Act88FA295S/Act88FA295S homozygotes, however, displayed hypercontracted and torn DLMs.
(B) The IFM thin filament lengths did not significantly differ among the lines. Data are presented as scatterplots that display the mean ± SEM for each genotype (n = 222–254).
(C) Averaged composite images of sarcomeres (n = 47–89) from randomly selected myofibrils substantiated the similarities in thin filament lengths among the genotypes. Z and M denote Z- and M-line locations, respectively.
See also Figure S4.
Homozygous expression of wild-type transgenic actin in Act88FWT/Act88FWT Drosophila did not rescue the flightless phenotype associated with the Act88F null strain (Figure S4A), potentially due to an insufficient degree of transgene expression (Beall et al., 1989). IFM development and morphology, however, were indistinguishable between Act88FWT/Act88FWT and w1118 non-transgenic controls (Figure 6A). Act88FA295S/Act88FA295S Drosophila were similarly flightless (Figure S4A), yet their IFMs exhibited destructive hypercontraction (Figure 6A), a well-characterized phenotype that results from excessive and/or unregulated force generation (Beall and Fyrberg, 1991; Cammarato et al., 2004; Viswanathan et al., 2014). This degenerative disorder was observed with 100% penetrance in Act88FA295S/Act88FA295S homozygotes, where all endogenous wild-type IFM actin was replaced by the mutant variant, a finding consistent with impaired Tm-based inhibition of actomyosin interactions.
In an attempt to discern the mechanistic root of ACTC1-based cardiomyopathies, differences in the intrinsic biophysical properties of mutant actins, including folding and polymerization characteristics, have been investigated (Vang et al., 2005; Mundia et al., 2012; Muller et al., 2012). Despite a lack of supportive in vitro evidence, the A295S substitution was predicted to alter actin stability and/or polymerization properties (Muller et al., 2012; Mundia et al., 2012), which in vivo, could result in aberrant thin filament lengths. Thin filament length is an important determinant of force production (de Winter et al., 2016). Mutations in skeletal muscle α-actin ACTA1 that cause nemaline myopathy have been shown to alter thin filament length and force generation. Altered filament lengths may therefore contribute to the pathogenesis of A295S-mediated HCM. We measured thin filament lengths from confocal micrographs of TRITC-phalloidin-labeled IFM myofibrils (Figure S4C). Average thin filament length from two-day-old w1118 (1.38 ± 0.01 μm), Act88FWT/+ (1.39 ± 0.01 μm), Act88FA295S/+ (1.39 ± 0.01 μm), Act88FWT/Act88FWT (1.39 ± 0.01 μm), and Act88FA295S/Act88FA295S (1.41 ± 0.01 μm) Drosophila did not significantly differ (Figure 6B). These results were corroborated visually by image averaging of the TRITC-phalloidin actin signals from randomly selected IFM sarcomeres of each genotype (Figure 6C), and they are consistent with unimpaired thin filament formation and stability, in vivo, irrespective of actin variant dose. Thus, disrupted thin filament length is likely not a contributing factor to ACTC1 A295S-mediated pathology.
The A295S Actin Mutation Enhances Thin Filament Ca2+ Sensitivity
To investigate the impact of the HCM mutation on molecular performance, homogeneous populations of transgenic actin were purified from the IFMs of Act88FWT/Act88FWT and Act88FA295S/Act88FA295S Drosophila (Razzaq et al., 1999). Fluorescently labeled F-actin movement over immobilized rabbit skeletal myosin was studied in an in vitro motility assay (Kron and Spudich, 1986). Unregulated velocities for Act88FWT (2.77 ± 0.08 μm/s) and Act88FA295S (2.80 ± 0.04 μm/s) F-actin were identical (Figure 7A), which suggests the A295S lesion does not overtly influence actomyosin associations. We next reconstituted thin filaments using bovine cardiac Tn-Tm, in conjunction with rabbit skeletal, Act88FWT, or Act88FA295S actin, to resolve potential differences in regulated motility (Liang et al., 2003). As previously reported, motility parameters of rabbit and Act88FWT F-actin and reconstituted thin filaments were indistinguishable (Figure S6) (Bing et al., 1998), which justifies the use of IFM actin for investigating the molecular effects of human disease-causing mutations. Reconstituted Act88FA295S thin filaments showed a leftward shift in the velocity:pCa (−log10of the calcium concentration) relationship relative to Act88FWT thin filaments (Figures 7B and 7C). The half-maximal pCa50 activation value was significantly higher for Act88FA295S (6.23 ± 0.03) versus Act88FWT (6.09 ± 0.05) regulated thin filaments, consistent with increased Ca2+ sensitivity. No significant differences, however, were observed in maximal sliding speed (3.26 ± 0.08 versus 3.23 ± 0.11 μm/s) or in the cooperativity of Ca2+ activation (n, Hill coefficient 1.77 ± 0.24 versus 1.26 ± 0.16) between mutant and control filaments. These data support an A295S actin-mediated gain in molecular thin filament function due to disrupted Tn-Tm-mediated regulation.
Figure 7. The A295S Actin Mutation Augments Thin Filament Ca2+ Sensitivity and Distorts the F-Actin-Tropomyosin Energy Landscape.

(A) Act88FWT and Act88FA295S F-actin, propelled by rabbit psoas muscle myosin, moved at identical velocities, which suggests the A295S mutation does not markedly affect actomyosin interactions. Average velocities for each of three biological replicate experiments (n = 3) were determined from 20 to 25 filaments per experiment. Bar graphs depict weighted mean velocity ± SEM.
(B) Act88FWT and Act88FA295S-containing thin filaments were reconstituted using bovine cardiac Tn-Tm. Three individual actin preparations were assayed twice (n = 6), and average velocities at each Ca2+ concentration, derived from 20 to 25 filaments per experiment, were pooled, plotted as a function of [Ca2+], and fit to the Hill equation. Act88FWT and Act88FA295S thin filament maximum velocities (Vmax), derived from the fits, were indistinguishable and used to normalize each pooled filament velocity. Normalized velocities ± SEM versus corresponding pCa values are presented.
(C) The pCa50 of reconstituted thin filaments was significantly higher for Act88FA295S relative to Act88FWT filaments, indicative of augmented Ca2+sensitivity. Bar graphs depict pCa50 values ± SEM calculated from the fits. *p = 0.015.
(D) Electrostatic interaction energy landscapes of Tm rotated and translated over the surface of wild-type F-actin (left) and A295S mutant F-actin (right). The color is scaled as indicated on the right axis. Energy values in each graph are relative to the minimum point in the landscape, which was set to zero. The total minimum energy for the wild-type is higher than that for the mutant (−2,165 versus −2,221 kcal/mol, respectively). The minima of the landscapes are denoted with a white X. The position of Tm in the Li et al. (2011) A/B state is indicated by a white circle for reference (Orzechowski et al., 2014). Contours on the plots start at 100 kcal/mol and increase by 200 kcal/mol.
See also Figures S6 and S7 and Tables S3 and S4.
The A295S Actin Mutation Shifts the Electrostatic Equilibrium Position of Tropomyosin toward the Closed State
An in silico approach was undertaken to identify molecular changes that may account for the hypercontractility of muscles and elevated Ca2+ sensitivity of thin filaments containing A295S actin. Landscapes of the electrostatic interaction energy between Tm and F-actin, which sample Tm energies over a range of azimuthal rotations and longitudinal translations around the actin filament axis, were calculated (Figure 7D). The electrostatic interaction energy landscape for wild-type F-actin-Tm was characterized by a broad basin ~7 Å high and 8° wide. The basin exhibited a predominantly uniform and flat energy distribution. For example, the energy difference between the minimum configuration (X in Figure 7D) and that of a previously assigned A/B state (circle in Figure 7D) (Li et al., 2011; Orzechowski et al., 2014), both of which reside within the broad energy well, is miniscule. This implies that in the absence of Tn, Tm could access numerous locations within the basin with little energetic impediment. The A295S actin mutation, however, substantially changed the breadth of the F-actin-Tm energetic landscape. The energy basin of the mutant was narrower (~5 Å high and 4° wide) and slightly deeper than that of wild-type, which resulted in a basin approximately 60% smaller in area. Therefore, Tm likely has fewer locations over the surface of A295S F-actin that are accessible with small energetic impediments. The mutant landscape also predicts that, on average, equilibrium positions of Tm are shifted upward toward the filament pointed end and azimuthally over the F-actin surface toward the actin inner domain, closer to the C state.
The minimized structures show that the total electrostatic energy difference observed between wild-type and mutant F-actin-Tm results from the cumulative effects of several small changes in the interactions of the A-triad with pseudo-repeats along Tm’s entire length (Figure S7; Table S3). For instance, on average, the salt bridges formed between Tm and K328 and K326 on actin are 0.58 and 0.17 Å shorter in the mutant compared to wild-type, respectively (Table S3). Summation of the individual interaction values among R147, K326, and K328 of actin and acidic residues of Tm (Table S4) highlights that these associations, and in particular that of K328, account for nearly the entire difference in total electrostatic energy between wild-type and A295S F-actin-Tm. This suggests that the A-triad has a significant influence on, and is a major determinant of, Tm positioning. The A295S mutation appears to change A-triad-Tm interactions and thus the F-actin-Tm energetic landscape. The characteristic broad and flat energy basin, which helps properly position Tm for effective myosin blocking, is perturbed. Such mutation-induced effects in muscle could promote contractile disinhibition.
DISCUSSION
The A-triad is an ancient surface feature of actin. It is composed of K326, K328, and R147, which are among a large number of amino acids displaying 100% sequence identity (Figure S1). The extreme degree of conservation is believed to result from evolutionary constraints imposed by actin’s interactions with a multitude of binding partners, which specify its function over a range of physiological processes (Gunning et al., 2015; Pollard, 2016). Here we provide in vivo evidence for a vital role of the A-triad residues in the fundamental process of Tn-Tm-mediated regulation of striated muscle contraction.
Previously, energy landscape determinations for Tm positioned over the surface of F-actin revealed the A-triad helps establish a broad energy basin associated with Tm favorably situated to block myosin binding (Orzechowski et al., 2014). This configuration is close to that of the B state of Tn-Tm-regulated thin filaments but on average has greater positional variance (Lehman et al., 2009, 2013; Orzechowski et al., 2014). We have now extended the in silico and in vitro findings and present data that support a direct role for these ground-state A-triad-Tm associations in steric hindrance of actomyosin cross-bridge cycling in the physiological context of muscle. We propose that in addition to TnI-actin binding, A-triad-Tm contacts are essential for force inhibition and that posttranslational modifications (Viswanathan et al., 2015) or mutations to or around the A-triad can modulate contraction and can initiate disease.
The a[α]-cardiac actin A295S substitution, the first reported ACTC1 mutation linked to HCM, occurs proximal to the A-triad (Mogensen et al., 1999). To unravel its molecular origin, we generated several transgenic Drosophila models of this highly penetrant disease. Our studies rely on technologies that provide distinct advantages over classical methodologies for investigating myopathies across a host of scales. While mouse models have been employed for such purposes, confounding genetic modifiers and environmental factors can influence and conceal pathology. For example, the clinical profile associated with the ACTC1 A331P HCM mutation includes ventricular fibrillation, cardiac arrest, repolarization abnormalities, and hypertrophied walls, yet mice expressing the variant showed no phenotypic disruptions or signs of disease (Olson et al., 2000; Toko et al., 2010). Although the fly heart is less complex in gross structure relative to that of mammals, the ultrastructural and proteomic makeup are highly conserved (Cammarato et al., 2011a). Defects in myofilament proteins, αB-crystallin, and the receptor tyrosine kinase pathway result in cardiac phenotypes remarkably similar to those characterized in human cardiomyopathy, suggesting conserved pathological responses also exist in Drosophila and that, accordingly, the fly can serve as an efficient translational disease model (Viswanathan et al., 2014; Cammarato et al., 2008; Xie et al., 2013; Yu et al., 2013).
Our study employed the powerful Drosophila PhiC31 integrase-mediated transgenesis system to create HCM thin filament mutants (Groth et al., 2004). Integrase technology eliminates genetic variability and controls for expression inconsistency and modifying factors to help elucidate critical genotype-phenotype information that often eludes other models. In addition, flies permit evaluation of muscle performance from hundreds of animals, with tissue-specific expression, for exceptionally well-powered and highly controlled analyses. When expressed in the Drosophila heart, A295S actin incorporated uniformly along the length of thin filaments, reduced cardiac volumes especially at rest, and prolonged the duration of systolic tension development (Figures 2 and 3). Restricted volumes and extended tension periods are consistent with impaired relaxation and the hyperdynamic contractile properties that precede myocardial hypertrophy (Green et al., 2016) and that stimulate specific remodeling cascades (Davis et al., 2016). Diastolic dysfunction was further investigated by comparing relaxation responses in cardiac diameters to EGTA/EGTA,AM and subsequently to blebbistatin for control versus mutant hearts (Figure 4). Changes in diameter are directly proportional to changes in opposing cell lengths. Diastole is normally characterized by slightly shortened myocytes with elevated basal stiffness (Viswanathan et al., 2014) due to minor contributions from Ca2+-induced and, apparently, Ca2+-independent cross-bridge cycling (Figure 4). However, our data suggest that the excessively shortened cardiomyocytes in quiescent Hand > Act57BA295S hearts were not the result of elevated diastolic Ca2+ but rather were due to poor thin filament blocking of, and a disproportionately heightened number of disinhibited, actively cycling cross-bridges.
Heterozygous A295S actin expression in the IFM led to flight impairment, characterized by reduced acceleration (Figure 5), which was likely secondary to force-induced microscopic damage. Such small hypercontractures can compromise performance at the level of the whole muscle and result in lower total IFM force generation during flight. Homozygous expression caused extensive macroscopic IFM damage and supported a variant-driven “gain of sarcomeric function,” which resulted in a total lack of force production at the organismal level. The fibers were torn apart and displayed the typical hypercontraction phenotype (Figure 6) associated with thin filament mutations that weaken Tm’s ability to effectively block myosin cross-bridge cycling (Beall and Fyrberg, 1991; Cammarato et al., 2004; Viswanathan et al., 2014). The A295S actin mutation did not influence in vivo thin filament lengths or maximum F-actin sliding velocity (Figures 6 and 7). These findings suggest that changes in force due to aberrant filament lengths or, as originally proposed, directly due to molecular deficiencies in actomyosin interactions (Mogensen et al., 1999) are not primary determinants of pathology. However, A295S-containing thin filaments, reconstituted from Drosophila actin and vertebrate cardiac Tn-Tm, displayed a significant increase in Ca2+ sensitivity, which is consistent with other HCM-inducing thin filament lesions (Tardiff, 2011) and with pathogenesis linked to impaired contractile regulation.
Disease-causing mutations frequently occur in residues at the F-actin-Tm interface. Such mutations likely interfere with electrostatic interactions between the proteins and distort the normal thin filament energy landscape. Because Tm location on F-actin is not fixed and can oscillate among the A/B, C, and M positions in either the absence or the presence of Ca2+, low-energy barriers are believed to separate distinct energy minima associated with each thin filament regulatory state (McKillop and Geeves, 1993; Maytum et al., 2003; Pirani et al., 2005). Therefore, relatively minor changes in surface interactions between F-actin and Tm could effectively alter these barriers to dramatically tip the balance between regulatory states (Lehman et al., 2000; Pirani et al., 2005; Lehman, 2016). Energy landscape determinations predict that the ACTC1 A295S HCM mutation induces numerous minor changes in the side-chain orientations of neighboring A-triad residues, which perturb its interaction with Tm and the aggregate F-actin-Tm energetic topology. Relative to the broad and flat energy basin found with wild-type F-actin-Tm, which helps establish the normal A/B state, the electrostatic energy landscape of A295S F-actin-Tm has a narrow and slightly deeper basin with a discrete minimum centered away from the A/B position and closer to the C position. Consequently, this may bias Tm away from locations impeding cross-bridge-actin interactions, thus promoting C-state occupancy for ACTC1 A295S-containing thin filaments.
Our data, from the molecular through the tissue level, support a single unifying mechanism that explains the most proximal effects of the ACTC1 A295S substitution and the development of HCM. In addition, they highlight a ubiquitous role for the neighboring A-triad in thin filament regulation and contractile relaxation of striated muscle. A295S-mediated pathology appears to result from propagated effects that structurally rearrange the A-triad and thus deform the energy basin that helps pin Tm in an inhibitory position. In our disease model, lower B-state occupancy of mutant thin filaments resulted in additional, unimpeded cross-bridge cycling, even in the absence of Ca2+, accounting for the restricted diastolic diameters of fly hearts. Furthermore, such inordinate myosin binding enhances Tn’s affinity for Ca2+ (Tobacman, 1996; Moss et al., 2004; Hinken and Solaro, 2007). The thin filament would accordingly be primed and require less Ca2+-dependent movement of Tm and TnC-Ca2+ binding for activation. This is consistent with our findings of increased A295S thin filament Ca2+ sensitivity, which even in the absence of changes in Ca2+ handling, could prompt early onset and delay the completion of systole. In accordance with our results, these changes in vivo would manifest as prolonged SIs. Finally, altering intermolecular electrostatic A-triad-Tm interactions may influence deactivation. The A-triad likely facilitates the return of Tm to the energy basin that helps constrain it to an inhibitory position (Orzechowski et al., 2014), which may also assist with TnI-actin docking and thus expedite the termination of force production. Disrupted A-triad-Tm interactions could thereby account for the inability of IFM thin filaments, in our mutant model, to properly turn off and inhibit tension, leading to destructive myosin-based force generation (Beall and Fyrberg, 1991). Therefore, our findings increase comprehension of basic thin filament biology, demonstrate how the ACTC1 A295S mutation triggers the early signs of HCM, and support the mechanistic model in which mutations that initiate hyperdynamic contraction and impaired relaxation result in pathological remodeling of the human heart (Green et al., 2016; Davis et al., 2016).
EXPERIMENTAL PROCEDURES
Multiple Sequence Alignment of Actins
Amino acid sequence comparison of human and Drosophila cardiac and skeletal actins was performed using the Clustal Omega multiple sequence alignment program.
Structural Modeling
F-actin-Tm models (Li et al., 2011) were generated using Chimera v.1.9 (Pettersen et al., 2004).
Construction of UAS-Act57B and Act88F Transgenes and Transgenic Drosophila
The pUASp.Act57BGFP.WT actin construct was provided by Dr. Katja Röper (Medical Research Council, Cambridge, UK). From this, the Act57BGFP.WT and Act57BWT cDNA sequences were spliced into the pUASTattB vector, behind 5 tandem UAS repeats (5×UAS), using the KpnI and XbaI and the NotI and XbaI restriction sites, respectively. UAS-Act57BGFP.A295S and UAS-Act57BA295S transgenes were generated by site-directed mutagenesis using the QuikChange Site-directed mutagenesis kit (Agilent Technologies) and custom synthesized primers:
Forward primer: 5′-AAGGACCTGTACTCCAACATCGTCATG-3′
Reverse primer: 5′-CATGACGATGTTGGAGTACAGGTCCTT-3′
The pattB Act88FWT genomic construct was provided by Dr. József Mihály (Hungarian Academy of Sciences, Budapest, Hungary). The pattB Act88FA295S mutant construct was created by Genetic Services (Sudbury, MA).
Transgenic Drosophila were generated using the PhiC31 integrase system as previously delineated (Viswanathan et al., 2015).
Drosophila Stocks and Husbandry
Flies were raised at 25°C on a standard cornmeal-yeast-sucrose-agar medium. The Hand4.2-Gal4 and the Act88F null (ry506 KM88 es) lines were obtained from Drs. Rolf Bodmer (Sanford Burnham Prebys Medical Discovery Institute, La Jolla, CA) and John Sparrow (University of York, York, UK), respectively. For all Gal4-UAS crosses, Hand4.2-Gal4 virgin females were mated with UAS-Actin males.
Act88F transgenic Drosophila had an endogenous actin gene (+) on each third chromosome, plus a transgene (Act88FWT or Act88FA295S) inserted into each second chromosome (i.e., Act88FWT/Act88FWT;+/+ or Act88FA295S/Act88FA295S;+/+). The transgenes were crossed into the KM88 background by standard mating schemes using balancer chromosomes to generate Act88FWT/Act88FWT;KM88/KM88 and Act88FA295S/Act88FA295S;KM88/KM88 homozygous flies (designated as Act88FWT/Act88FWT and Act88FA295S/Act88FA295S, respectively). To generate heterozygotes, with one transgenic and one endogenous Act88F gene, homozygotes were crossed with w1118 to create Act88FWT;KM88/+ and Act88FA295S;KM88/+ (designated as Act88FWT/+ and Act88FA295S/+).
Confocal Microscopy of the Fly Heart
Hand > Act57BGFP.WT and Hand > Act57BGFP.A295S cardiac tubes were prepared as detailed by Alayari et al. (2009) and imaged with a Leica TCS SPE RGBV confocal microscope.
To characterize the degree of overlap of GFP-labeled and TRITC-phalloidin-stained actin, images were analyzed using the ImageJ co-localization plugin. The average index of correlation (Jaskolski et al., 2005) was calculated for cardiac sarcomeres of each genotype.
Cardiac Physiological Analysis
One-, three-, five-, and seven-week-old, female Hand ×yw, Hand > Act57BWT, and Hand > Act57BA295S semi-intact Drosophila hearts were prepared under artificial hemolymph (AH) as described by Vogler and Ocorr (2009). Physiological parameters were assessed as formerly outlined (Cammarato et al., 2015; Blice-Baum et al., 2017), and significant differences between genotype and age, and interaction effects, were determined using two-way ANOVAs with Bonferroni post hoc tests.
Measurement of EGTA/EGTA,AM- and Blebbistatin-Induced Changes in Cardiac Dimensions
Semi-intact hearts of three-week-old Hand × yw, Hand > Act57BWT, and Hand > Act57BA295S females were imaged and filmed as described by Viswanathan et al. (2014). Beating hearts were recorded for 10 s in AH at various focal depths to resolve clear edges along the length of each tube. After initial filming, the AH was replaced with AH containing 10 mM EGTA and 100 μM EGTA,AM (AAT Bioquest). Following 30 min (25°C) to chelate extra- and intra-cellular Ca2+, the quiescent hearts were filmed as before and then incubated for 30 min in the same EGTA/EGTA,AM solution supplemented with 100 μM blebbistatin (Cayman Chemical). The hearts were re-imaged and filmed for a final time. Individual frames that clearly resolved the same region of both heart wall edges from videos taken under each condition were analyzed using HCImage Live software. Three distinct diameter measurements were made at identical locations along each tube from images obtained during diastole and under each treatment condition. These values were averaged for each animal and then for each genotype. The stepwise effects of EGTA/EGTA,AM and blebbistatin treatment on cardiac dimensions were evaluated using repeated-measures ANOVAs with Bonferroni multiple comparison tests of the matched groups. One-way ANOVAs followed by Bonferroni multiple comparison tests were used to distinguish significant differences in the cardiac responses (i.e., diameter changes) to EGTA/EGTA,AM and then to blebbistatin among genotypes.
Holography
Cinematic digital inline holography (Katz and Sheng, 2010) was implemented to track flight trajectories of two-day-old w1118 non-transgenic actin control, Act88FWT/+, and Act88FA295S/+ male and female Drosophila. 30 flies of a given genotype were released from the bottom of a 44.5 × 44.5 × 44.5 cm3 transparent acrylic enclosure maintained at 24°C. A halogen lamp was anchored above the chamber to entice upward flight. Data acquisition and analysis methods are described in Supplemental Experimental Procedures, including sketches of the optical setup in Figure S5A and of the analysis in Figure S5B. Briefly, a sample volume of 11.6 × 11.6 × 44.5 cm3 was illuminated by a collimated pulsed neodymium-doped yttrium aluminum garnet (Nd:YAG) laser beam. The interference of light scattered from the flies with the undisturbed portions of the beam, the hologram, was recorded by a 2,016 × 2,016 pixels complementary metal-oxide semiconductor (CMOS) camera operating at 400 frames/s. Each run lasted 7.82 s, providing 3,128 holograms. The holograms were reconstructed numerically to obtain the intensity fields in a series of parallel planes. Multi-step detection procedures subsequently identified the in-focus location of each fly and tracked its displacement in time. The trajectories were used to calculate the kinematic properties of flight, including the velocity and acceleration vectors, as well as the angular velocity and the centripetal acceleration component, the latter signifying the fly’s maneuverability. Significance was assessed by non-parametric Kruskal-Wallis one-way ANOVAs followed by Dunn’s multiple comparison tests.
Indirect Flight Muscle and Sarcomere Imaging
Fluorescent microscopy was performed as in Viswanathan et al. (2015) to examine gross IFM morphology.
Myofibril imaging was carried out as previously described (Haigh et al., 2010; Cammarato et al., 2011b). Thin filament lengths were measured using ImageJ. Significance was assessed via the Kruskal-Wallis one-way ANOVA with Dunn’s post hoc test.
For image averaging of sarcomeres, confocal micrographs of IFM myofibrils were imported into ImageJ. Individual myofibrils were straightened to fill a rectangular box by drawing a segmented line, of a width equal to the myofibril width, through its center and executing the Straighten command. Single sarcomeres were cropped and combined into an image stack. Images were aligned in x and y coordinate space using the plugin Template Matching → Align Slices in Stack (https://sites.google.com/site/qingzongtseng/template-matching-ij-plugin). A reference image was selected, and all images in the stack were translated to align in approximately the same position as the reference. Following automated image alignment, average sarcomere structures were generated by executing the Z Project command with the average intensity projection type.
Actin Purification from Drosophila Indirect Flight Muscle
Act88FWT and Act88FA295S transgenic actin from ~40 IFMs was purified according to Razzaq et al. (1999).
In Vitro Motility
Sliding of Act88FWT and Act88FA295S F-actin (Kron and Spudich, 1986) and of Act88FWT and Act88FA295S thin filaments (Liang et al., 2003), over full-length rabbit skeletal myosin, was measured via standard modification of the in vitro motility assay. Thin filaments were reconstituted using bovine cardiac Tm and Tn. F-actin velocities measured in each of three replicate experiments were averaged, and the average velocities were pooled and weighted based on the number of filaments analyzed in each experiment. Significance was assessed by an unpaired t test. For Ca2+-regulated motility experiments, average filament velocities at each distinct [Ca2+] were pooled and weighted based on the number of filaments analyzed for each of six replicate experiments. Pooled velocity:Ca2+ data were re-plotted and fit to the Hill equation, and significant differences in fit parameters (Vmax, pCa50, and cooperativity) were determined via extra sum of squares F tests. See Supplemental Experimental Procedures for further details.
Determination of F-Actin-Tropomyosin Energy Landscapes
Energy landscapes for Tm and wild-type or A295S mutant F-actin were calculated as previously published (Rynkiewicz et al., 2015) with minor modifications. Briefly, a model of Tm and F-actin was constructed. Tm was then azimuthally rotated and longitudinally translated to the grid points to be tested. This step can result in poor contacts that are unresolvable through minimization. Therefore, before minimization, Tm and actin side chains in proximity in the model were shrunk, and they were allowed to relax back during minimization. Constraints were applied in the minimization to keep the distance to actin and the overall shape of Tm consistent with known values for a Tm cable. All calculations were performed with CHARMM v.35b (Brooks et al., 2009).
Statistical Methods
Statistical analysis was performed using GraphPad Prism 7.
Supplementary Material
Highlights.
The highly conserved actin A-triad consists of residues K326, K328, and R147
A-triad-tropomyosin interactions along thin filaments promote muscle relaxation
Drosophila cardiomyopathy models illustrate the A-triad’s significance in vivo
Distortion of the A-triad can trigger hypercontraction and disease
Acknowledgments
The authors thank Drs. Marek Orzechowski (Boston University School of Medicine) for preliminary molecular dynamics simulations and Aditi Madan (Johns Hopkins University) for technical assistance. This work was supported by NSF CBET-1438203 (to J.K.), ONR N000141512404 (to J.K.), and NIH grants T32HL007227-38 (to W.S.), R37HL036153 (to W.L.), R56HL124091 (to A.C.), and R01HL124091 (to A.C.).
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and four tables and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2017.08.070.
AUTHOR CONTRIBUTIONS
M.C.V., W.S., M.J.R., J.K., W.L., and A.C. conceived and designed the experiments. M.C.V., W.S., M.J.R., K.A., and J.G. acquired the data. M.C.V., W.S., M.J.R., K.A., J.G., J.K., W.L., and A.C. analyzed and interpreted the data. M.C.V., W.S., M.J.R., K.A., J.G., J.K., W.L., and A.C. drafted and revised the manuscript.
References
- Alayari NN, Vogler G, Taghli-Lamallem O, Ocorr K, Bodmer R, Cammarato A. Fluorescent labeling of Drosophila heart structures. J Vis Exp. 2009;32:1423. doi: 10.3791/1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barua B, Pamula MC, Hitchcock-DeGregori SE. Evolutionarily conserved surface residues constitute actin binding sites of tropomyosin. Proc Natl Acad Sci USA. 2011;108:10150–10155. doi: 10.1073/pnas.1101221108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barua B, Fagnant PM, Winkelmann DA, Trybus KM, Hitchcock-De-Gregori SE. A periodic pattern of evolutionarily conserved basic and acidic residues constitutes the binding interface of actin-tropomyosin. J Biol Chem. 2013;288:9602–9609. doi: 10.1074/jbc.M113.451161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beall CJ, Fyrberg E. Muscle abnormalities in Drosophila melanogaster heldup mutants are caused by missing or aberrant troponin-I isoforms. J Cell Biol. 1991;114:941–951. doi: 10.1083/jcb.114.5.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beall CJ, Sepanski MA, Fyrberg EA. Genetic dissection of Drosophila myofibril formation: effects of actin and myosin heavy chain null alleles. Genes Dev. 1989;3:131–140. doi: 10.1101/gad.3.2.131. [DOI] [PubMed] [Google Scholar]
- Bing W, Razzaq A, Sparrow J, Marston S. Tropomyosin and troponin regulation of wild type and E93K mutant actin filaments from Drosophila flight muscle. Charge reversal on actin changes actin-tropomyosin from on to off state. J Biol Chem. 1998;273:15016–15021. doi: 10.1074/jbc.273.24.15016. [DOI] [PubMed] [Google Scholar]
- Blice-Baum AC, Zambon AC, Kaushik G, Viswanathan MC, Engler AJ, Bodmer R, Cammarato A. Modest overexpression of FOXO maintains cardiac proteostasis and ameliorates age-associated functional decline. Aging Cell. 2017;16:93–103. doi: 10.1111/acel.12543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Brooks BR, Brooks CL, Mackerell AD, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, et al. CHARMM: the biomolecular simulation program. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JH, Zhou Z, Reshetnikova L, Robinson H, Yammani RD, Tobacman LS, Cohen C. Structure of the mid-region of tropomyosin: bending and binding sites for actin. Proc Natl Acad Sci USA. 2005;102:18878–18883. doi: 10.1073/pnas.0509269102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cammarato A, Hatch V, Saide J, Craig R, Sparrow JC, Tobacman LS, Lehman W. Drosophila muscle regulation characterized by electron microscopy and three-dimensional reconstruction of thin filament mutants. Biophys J. 2004;86:1618–1624. doi: 10.1016/S0006-3495(04)74229-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cammarato A, Dambacher CM, Knowles AF, Kronert WA, Bodmer R, Ocorr K, Bernstein SI. Myosin transducer mutations differentially affect motor function, myofibril structure, and the performance of skeletal and cardiac muscles. Mol Biol Cell. 2008;19:553–562. doi: 10.1091/mbc.E07-09-0890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cammarato A, Ahrens CH, Alayari NN, Qeli E, Rucker J, Reedy MC, Zmasek CM, Gucek M, Cole RN, Van Eyk JE, et al. A mighty small heart: the cardiac proteome of adult Drosophila melanogaster. PLoS ONE. 2011a;6:e18497. doi: 10.1371/journal.pone.0018497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cammarato A, Li XE, Reedy MC, Lee CF, Lehman W, Bernstein SI. Structural basis for myopathic defects engendered by alterations in the myosin rod. J Mol Biol. 2011b;414:477–484. doi: 10.1016/j.jmb.2011.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cammarato A, Ocorr S, Ocorr K. Enhanced assessment of contractile dynamics in Drosophila hearts. Biotechniques. 2015;58:77–80. doi: 10.2144/000114255. [DOI] [PubMed] [Google Scholar]
- Campbell KS, Sorrell VL. Cell- and molecular-level mechanisms contributing to diastolic dysfunction in HFpEF. J Appl Physiol. 2015;119:1228–1232. doi: 10.1152/japplphysiol.01168.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahari M, Dawson JF. Do cardiac actin mutations lead to altered actomyosin interactions? Biochem Cell Biol. 2015;93:330–334. doi: 10.1139/bcb-2014-0156. [DOI] [PubMed] [Google Scholar]
- Davis J, Davis LC, Correll RN, Makarewich CA, Schwanekamp JA, Moussavi-Harami F, Wang D, York AJ, Wu HD, Houser SR, et al. A tension-based model distinguishes hypertrophic versus dilated cardiomyopathy. Cell. 2016;165:1147–1159. doi: 10.1016/j.cell.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Winter JM, Joureau B, Lee EJ, Kiss B, Yuen M, Gupta VA, Pappas CT, Gregorio CC, Stienen GJM, Edvardson S, et al. Mutation-specific effects on thin filament length in thin filament myopathy. Ann Neurol. 2016;79:959–969. doi: 10.1002/ana.24654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorov VV, Lozinsky IT, Sosunov EA, Anyukhovsky EP, Rosen MR, Balke CW, Efimov IR. Application of blebbistatin as an excitation-contraction uncoupler for electrophysiologic study of rat and rabbit hearts. Heart Rhythm. 2007;4:619–626. doi: 10.1016/j.hrthm.2006.12.047. [DOI] [PubMed] [Google Scholar]
- Green EM, Wakimoto H, Anderson RL, Evanchik MJ, Gorham JM, Harrison BC, Henze M, Kawas R, Oslob JD, Rodriguez HM, et al. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science. 2016;351:617–621. doi: 10.1126/science.aad3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groth AC, Fish M, Nusse R, Calos MP. Construction of transgenic Drosophila by using the site-specific integrase from phage phiC31. Genetics. 2004;166:1775–1782. doi: 10.1534/genetics.166.4.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunning PW, Ghoshdastider U, Whitaker S, Popp D, Robinson RC. The evolution of compositionally and functionally distinct actin filaments. J Cell Sci. 2015;128:2009–2019. doi: 10.1242/jcs.165563. [DOI] [PubMed] [Google Scholar]
- Haigh SE, Salvi SS, Sevdali M, Stark M, Goulding D, Clayton JD, Bullard B, Sparrow JC, Nongthomba U. Drosophila indirect flight muscle specific Act88F actin mutants as a model system for studying congenital myopathies of the human ACTA1 skeletal muscle actin gene. Neuromuscul Disord. 2010;20:363–374. doi: 10.1016/j.nmd.2010.03.008. [DOI] [PubMed] [Google Scholar]
- Hinken AC, Solaro RJ. A dominant role of cardiac molecular motors in the intrinsic regulation of ventricular ejection and relaxation. Physiology (Bethesda) 2007;22:73–80. doi: 10.1152/physiol.00043.2006. [DOI] [PubMed] [Google Scholar]
- Hiromi Y, Hotta Y. Actin gene mutations in Drosophila; heat shock activation in the indirect flight muscles. EMBO J. 1985;4:1681–1687. doi: 10.1002/j.1460-2075.1985.tb03837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitchcock-DeGregori SE. Tropomyosin: function follows structure. Adv Exp Med Biol. 2008;644:60–72. doi: 10.1007/978-0-387-85766-4_5. [DOI] [PubMed] [Google Scholar]
- Jaskolski F, Mulle C, Manzoni OJ. An automated method to quantify and visualize colocalized fluorescent signals. J Neurosci Methods. 2005;146:42–49. doi: 10.1016/j.jneumeth.2005.01.012. [DOI] [PubMed] [Google Scholar]
- Kass DA, Bronzwaer JG, Paulus WJ. What mechanisms underlie diastolic dysfunction in heart failure? Circ Res. 2004;94:1533–1542. doi: 10.1161/01.RES.0000129254.25507.d6. [DOI] [PubMed] [Google Scholar]
- Katz J, Sheng J. Applications of holography in fluid mechanics and particle dynamics. Annu Rev Fluid Mech. 2010;42:531–555. [Google Scholar]
- Kaushik G, Spenlehauer A, Sessions AO, Trujillo AS, Fuhrmann A, Fu Z, Venkatraman V, Pohl D, Tuler J, Wang M, et al. Vinculin network-mediated cytoskeletal remodeling regulates contractile function in the aging heart. Sci Transl Med. 2015;7:292ra299. doi: 10.1126/scitranslmed.aaa5843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kron SJ, Spudich JA. Fluorescent actin-filaments move on myosin fixed to a glass-surface. Proc Natl Acad Sci USA. 1986;83:6272–6276. doi: 10.1073/pnas.83.17.6272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo T, Gimeno JR, Bahl A, Steffensen U, Steffensen M, Osman E, Thaman R, Mogensen J, Elliott PM, Doi Y, et al. Prevalence, clinical significance, and genetic basis of hypertrophic cardiomyopathy with restrictive phenotype. J Am Coll Cardiol. 2007;49:2419–2426. doi: 10.1016/j.jacc.2007.02.061. [DOI] [PubMed] [Google Scholar]
- Lehman W. Thin filament structure and the steric blocking model. Compr Physiol. 2016;6:1043–1069. doi: 10.1002/cphy.c150030. [DOI] [PubMed] [Google Scholar]
- Lehman W, Craig R, Vibert P. Ca2+-induced tropomyosin movement in Limulus thin filaments revealed by three-dimensional reconstruction. Nature. 1994;368:65–67. doi: 10.1038/368065a0. [DOI] [PubMed] [Google Scholar]
- Lehman W, Hatch V, Korman V, Rosol M, Thomas L, Maytum R, Geeves MA, Van Eyk JE, Tobacman LS, Craig R. Tropomyosin and actin isoforms modulate the localization of tropomyosin strands on actin filaments. J Mol Biol. 2000;302:593–606. doi: 10.1006/jmbi.2000.4080. [DOI] [PubMed] [Google Scholar]
- Lehman W, Galinska-Rakoczy A, Hatch V, Tobacman LS, Craig R. Structural basis for the activation of muscle contraction by troponin and tropomyosin. J Mol Biol. 2009;388:673–681. doi: 10.1016/j.jmb.2009.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman W, Orzechowski M, Li XE, Fischer S, Raunser S. Gestalt-binding of tropomyosin on actin during thin filament activation. J Muscle Res Cell Motil. 2013;34:155–163. doi: 10.1007/s10974-013-9342-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XE, Tobacman LS, Mun JY, Craig R, Fischer S, Lehman W. Tropomyosin position on F-actin revealed by EM reconstruction and computational chemistry. Biophys J. 2011;100:1005–1013. doi: 10.1016/j.bpj.2010.12.3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang B, Chen Y, Wang CK, Luo Z, Regnier M, Gordon AM, Chase PB. Ca2+ regulation of rabbit skeletal muscle thin filament sliding: role of cross-bridge number. Biophys J. 2003;85:1775–1786. doi: 10.1016/S0006-3495(03)74607-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA. 2002;287:1308–1320. doi: 10.1001/jama.287.10.1308. [DOI] [PubMed] [Google Scholar]
- Maytum R, Westerdorf B, Jaquet K, Geeves MA. Differential regulation of the actomyosin interaction by skeletal and cardiac troponin isoforms. J Biol Chem. 2003;278:6696–6701. doi: 10.1074/jbc.M210690200. [DOI] [PubMed] [Google Scholar]
- McKillop DF, Geeves MA. Regulation of the interaction between actin and myosin subfragment 1: evidence for three states of the thin filament. Biophys J. 1993;65:693–701. doi: 10.1016/S0006-3495(93)81110-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller A, editor. The Internal Anatomy and Histology of the Imago of Drosophila melanogaster. Wiley; 1950. [Google Scholar]
- Mogensen J, Klausen IC, Pedersen AK, Egeblad H, Bross P, Kruse TA, Gregersen N, Hansen PS, Baandrup U, Borglum AD. Alpha-cardiac actin is a novel disease gene in familial hypertrophic cardiomyopathy. J Clin Invest. 1999;103:R39–R43. doi: 10.1172/JCI6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss RL, Razumova M, Fitzsimons DP. Myosin crossbridge activation of cardiac thin filaments: implications for myocardial function in health and disease. Circ Res. 2004;94:1290–1300. doi: 10.1161/01.RES.0000127125.61647.4F. [DOI] [PubMed] [Google Scholar]
- Muller M, Mazur AJ, Behrmann E, Diensthuber RP, Radke MB, Qu Z, Littwitz C, Raunser S, Schoenenberger CA, Manstein DJ, et al. Functional characterization of the human alpha-cardiac actin mutations Y166C and M305L involved in hypertrophic cardiomyopathy. Cell Mol Life Sci. 2012;69:3457–3479. doi: 10.1007/s00018-012-1030-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundia MM, Demers RW, Chow ML, Perieteanu AA, Dawson JF. Subdomain location of mutations in cardiac actin correlate with type of functional change. PLoS ONE. 2012;7:e36821. doi: 10.1371/journal.pone.0036821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson TM, Doan TP, Kishimoto NY, Whitby FG, Ackerman MJ, Fananapazir L. Inherited and de novo mutations in the cardiac actin gene cause hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2000;32:1687–1694. doi: 10.1006/jmcc.2000.1204. [DOI] [PubMed] [Google Scholar]
- Orzechowski M, Moore JR, Fischer S, Lehman W. Tropomyosin movement on F-actin during muscle activation explained by energy landscapes. Arch Biochem Biophys. 2014;545:63–68. doi: 10.1016/j.abb.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Pirani A, Xu C, Hatch V, Craig R, Tobacman LS, Lehman W. Single particle analysis of relaxed and activated muscle thin filaments. J Mol Biol. 2005;346:761–772. doi: 10.1016/j.jmb.2004.12.013. [DOI] [PubMed] [Google Scholar]
- Pollard TD. Actin and actin-binding proteins. Cold Spring Harb Perspect Biol. 2016;8:8. doi: 10.1101/cshperspect.a018226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razzaq A, Schmitz S, Veigel C, Molloy JE, Geeves MA, Sparrow JC. Actin residue glu(93) is identified as an amino acid affecting myosin binding. J Biol Chem. 1999;274:28321–28328. doi: 10.1074/jbc.274.40.28321. [DOI] [PubMed] [Google Scholar]
- Rynkiewicz MJ, Schott V, Orzechowski M, Lehman W, Fischer S. Electrostatic interaction map reveals a new binding position for tropomyosin on F-actin. J Muscle Res Cell Motil. 2015;36:525–533. doi: 10.1007/s10974-015-9419-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah AP, Nongthomba U, Kelly Tanaka KK, Denton ML, Meadows SM, Bancroft N, Molina MR, Cripps RM. Cardiac remodeling in Drosophila arises from changes in actin gene expression and from a contribution of lymph gland-like cells to the heart musculature. Mech Dev. 2011;128:222–233. doi: 10.1016/j.mod.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardiff JC. Thin filament mutations: developing an integrative approach to a complex disorder. Circ Res. 2011;108:765–782. doi: 10.1161/CIRCRESAHA.110.224170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobacman LS. Thin filament-mediated regulation of cardiac contraction. Annu Rev Physiol. 1996;58:447–481. doi: 10.1146/annurev.ph.58.030196.002311. [DOI] [PubMed] [Google Scholar]
- Toko H, Takahashi H, Kayama Y, Oka T, Minamino T, Okada S, Morimoto S, Zhan DY, Terasaki F, Anderson ME, et al. Ca2+/calmodulin-dependent kinase II delta causes heart failure by accumulation of p53 in dilated cardiomyopathy. Circulation. 2010;122:891–899. doi: 10.1161/CIRCULATIONAHA.109.935296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vang S, Corydon TJ, Borglum AD, Scott MD, Frydman J, Mogensen J, Gregersen N, Bross P. Actin mutations in hypertrophic and dilated cardiomyopathy cause inefficient protein folding and perturbed filament formation. FEBS J. 2005;272:2037–2049. doi: 10.1111/j.1742-4658.2005.04630.x. [DOI] [PubMed] [Google Scholar]
- Viswanathan MC, Kaushik G, Engler AJ, Lehman W, Cammarato A. A Drosophila melanogaster model of diastolic dysfunction and cardiomyopathy based on impaired troponin-T function. Circ Res. 2014;114:e6–e17. doi: 10.1161/CIRCRESAHA.114.302028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan MC, Blice-Baum AC, Schmidt W, Foster DB, Cammarato A. Pseudo-acetylation of K326 and K328 of actin disrupts Drosophila melanogaster indirect flight muscle structure and performance. Front Physiol. 2015;6:116. doi: 10.3389/fphys.2015.00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogler G, Ocorr K. Visualizing the beating heart in Drosophila. J Vis Exp. 2009;31:1425. doi: 10.3791/1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Ecken J, Muller M, Lehman W, Manstein DJ, Penczek PA, Raunser S. Structure of the F-actin-tropomyosin complex. Nature. 2015;519:114–117. doi: 10.1038/nature14033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie HB, Cammarato A, Rajasekaran NS, Zhang H, Suggs JA, Lin HC, Bernstein SI, Benjamin IJ, Golic KG. The NADPH metabolic network regulates human αB-crystallin cardiomyopathy and reductive sress in Drosophila melanogaster. PLoS Genet. 2013;9:e1003544. doi: 10.1371/journal.pgen.1003544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Daniels J, Glaser AE, Wolf MJ. Raf-mediated cardiac hypertrophy in adult Drosophila. Dis Model Mech. 2013;6:964–976. doi: 10.1242/dmm.011361. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.