

Abstract
AIM
To explore the state of autophagy and related mechanisms in the murine retinal microvascular endothelial cells (RMECs) under hypoxia stimulation.
METHODS
The murine RMECs were primarily cultured and randomly divided into three groups: hypoxia group (cultured in 1% O2 environment), hypoxia+autophagy inhibition group [pretreated with 5 mmol/L 3-methyladenine (3-MA) for 4h followed by incubation in 1% O2] and control group (cultured under normoxic condition). The state of autophagy in RMECs was examined by assaying the turnover of light chain 3B (LC3BB) and expression of Beclin-1, Atg3 and Atg5 proteins with Western blotting, by detecting formation of autophagosomes with transmission electron microscopy (TEM) and by counting the number of GFP+ puncta in RMECs. The protein levels of AMPK, P-AMPK, Akt, P-Akt, m-TOR and P-mTOR were also assayed by Western blotting.
RESULTS
Primary murine RMECs were successfully cultured. Under hypoxic conditions, the ratio of LC3BB-II/I and the expression of Beclin-1, Atg3 and Atg5 proteins were increased when compared with the control group. In addition, the numbers of autophagosome and the GFP+ puncta were also increased under hypoxia. However, pre-treatment with 3-MA obviously attenuated these changes in autophagy in RMECs under hypoxia. Protein expression of P-Akt and P-AMPK was increased but P-mTOR level was decreased in cells exposed to hypoxia.
CONCLUSION
In murine RMECs autophagy is activated under hypoxia possibly through activation of the AMPK/mTOR signaling pathway.
Keywords: autophagy, retinal microvascular endothelial cells, hypoxia
INTRODUCTION
Hypoxia is an important pathophysiological stress generated during blood vessel damages and other disease conditions to cause the release of many pathogenic factors and result in a series of pathological changes. The retina is one of the most metabolically active human tissues, so it is highly sensitive to hypoxia and consequent oxidative stress. Therefore, a variety of ischemic retinopathies are closely related to hypoxia, such as retinopathy of prematurity, proliferative diabetic retinopathy and retinal venous occlusion[1]–[3]. Retinal microvascular endothelial cell (RMEC), located in the branched microvascular structures, are responsible for nutrient supply and excretion of metabolites of neurosensory retina. The anatomy and physiology of these endothelial cells are consistent with nutritional demand and protection of the key tissues that maintain vision. On the one hand, endothelial cells must ensure supply of oxygen and other nutrients to the metabolically active retina. On the other hand, endothelial cells participate in the formation of the blood-retinal barrier to protect the retina from damages by the molecular toxins, microorganisms and inflammatory cells in circulation. However, the characteristics of RMEC implementing these functions can also easily induce many pathological processes, such as retinal vascular leakage, angiogenesis, transport of microorganisms and inflammatory cells, etc[4]. Therefore, RMECs is a key participant in these retinal ischemic diseases and its dysfunction plays very important role in the development and progress of these diseases.
Autophagy is a self-degradation system through an autophagosomal-lysosomal pathway widely existing in eukaryotic cells, for the removal of misfolded proteins, polymerized macromolecules and damaged organelles to realize the function of material recycle and synthesis of new proteins, thus maintaining cellular survival and homeostasis in response to various environment. Autophagy maintains at a basic level under normal condition and it is an adaptive response of the cells to the metabolic stress and exogenous stimulation (e.g. lack of intracellular nutrition, anoxia and infection). At the same time, it induces initiative cell death as a way of programmed cell death. In recent years, autophagy has been reported to be involved in multiple pathological processes[5]. If the activity of autophagy is abnormal, a variety of diseases would take place, such as malignant tumors, neurodegenerative diseases and cardiovascular diseases[6]. Our previous study suggested that CoCl2-induced hypoxia could activate autophagy in a rhesus macaque choroid-retinal endothelial cell line (RF/6A) and this effect was effectively inhibited by an autophagy inhibitor 3-methyladenine (3-MA)[7]. However, despite the wide use of CoCl2 to mimic cellular hypoxia, it is different from hypoxia in the body which is due to the decrease of oxygen concentration actually. In addition, the RF/6A cell line is of mixed origin (choroidal and retinal) and these two types of cells have been reported to present molecular diversity and respond quite differently to external stimuli[8]–[9]. Given this, we used murine RMECs (from a single origin) to investigate the state of autophagy stimulated by low oxygen concentration and explored the underlying molecular mechanisms in the present study.
MATERIALS AND METHODS
Isolation of Retinal Microvascular Endothelial Cells
We isolated primary mouse RMECs from the retina of C57BL/6J mice as previously described[10]. Briefly, we harvest the eyeballs of mice and remove the residual retinal pigment epithelial cells and the visible main blood vessels. In a new culture dish, the separated retinal tissue was minced into pieces as small as possible. The minced retinal tissues were then digested with collagenase until cells were separated under an inverted phase contrast microscope (IX51, Olympus, Japan). The dissociated cells were then filtered through a 100 µm cell strainer and spin at 1000 rpm for 5min. The isolated cells were cultured in the endothelial cell basal medium (M199, Gibco, USA) supplemented with 10% fetal bovine serum, vascular endothelial growth factor, epidermal growth factor, basic fibroblast growth factor and insulin-like growth factors-1. Cells were cultured on the plate until visible clones were formed from microvascular endothelial cells that migrated from vascular fragments. Cells other than the clones were scratched off using a cell scraper. The remaining clones were then picked and expanded in culture plates for further experiments. All animals-based experiments were approved by the Institutional Animal Care and Use Committee of Xi'an Medical University and conformed to the Statement for the Use of Animals in Ophthalmic and Vision Research.
Immunofluorescent Staining of CD34
Isolated cells were grown on collagen I-coated coverslips on 6-well plates and when cells reached 60%-70% confluence they were immunofluorescently stained for CD34 expression. Briefly, cells were fixed with 4% paraformaldehyde for 15min at room temperature (RT) and then permeabilized with 0.5% Triton X-100 for 20min. The cells were blocked in normal goat serum for 30min at RT and then stained with primary CD34 antibody (1:200, Abcam, USA) at 4°C for overnight in a wet box. After washed with 1×PBS for 3 times, the coverslips were incubated with Cy3-conjugated goat anti-rabbit secondary antibody (1:100, Wuhan Boster, China) for 1h at RT in dark. For the control cells, a mouse IgG isotype control (1:100, Sigma, USA) was used instead of CD34 primary antibody. The coverslips were washed for 3 times with 1×PBS and mounted to slides with ProLong Gold Antifade Mountant with DAPI (Thermo Fisher Scientific, USA). Images were then collected by using a fluorescence microscope (Olympus BX53, Olympus, Japan).
Hypoxia-induced Autophagy
To investigate the hypoxia-induced autophagy, RMECs were randomly classified into the control group, hypoxia group and 3-MA+hypoxia group. Briefly, 5×105 RMECs cells were seeded on each well of 6-well plates in serum-free endothelial cell basal medium. The hypoxic group were incubated in a hypoxic incubator (1% O2/5% CO2/94% N2, BioSpherix, USA) for 24h as previously described[11]. For the 3-MA+hypoxia group, RMECs were pre-treated with 5 mmol/L 3-MA (Sigma, USA) for 4h and then incubated under hypoxic condition for 24h. In the control group, cells were kept in a normoxic incubator throughout the experiment.
Transmission Electron Microscopy
We then investigated the formation of autophagosome in RMECs by transmission electron microscopy (TEM). Briefly, cells were treated as mentioned above and then harvested by trypsinization. The cells were fixed with 2.5% glutaraldehyde in 0.1 mol/L phosphate buffer at 4°C for 2h, followed by incubation with 1% osmic acid at 20°C for 2h. Cells were then dehydrated with serial gradients of ethanol and embedded in Embed-812 medium for 48h. The blocks were sliced into sections of 60-80 nm using an Ultrotome (Leica, Germany) on uncoated copper grids. The slices were subjected to staining with the solution of 0.2% lead citrate and 1% uranyl acetate. The slices were imaged with a TEM (HT7700-SS, HITACHI, Japan) according to manufacturer's protocol.
Immunoblotting for Autophagic Markers
RMECs were treated as mentioned above. The whole cell lysates were harvested with the RIPA buffer (Beyotime, China), and the protein concentration was assayed by using a BCA Protein Assay kit (Beyotime, China). A total of 40 µg cell lysate per lane was loaded onto the 10% sodium dodecyl sulfate polyacrylamide gel (SDS-PAGE) and separated by electrophoresis at 120 V. The proteins were transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, USA) at 200 mA for 90min and then blocked with 5% non-fat dry milk in TBST for 1h at RT. The membranes were then incubated with anti-Beclin-1 (1:250, Abcam, USA), anti-Atg3 (1:1000, Proteintech Group, Inc., China), anti-Atg5 (1:1000, Proteintech Group, Inc., China), anti-microtubule associated protein 1 light chain 3B (LC3BB) (1:1000, Cell Signaling, USA), anti-AMPK α1+AMPKα 2 (1:800, Abcam, USA), anti-phospho-AMPKα (Thr172) (1:1000, Cell Signaling, USA), anti-Akt (1:1000, Cell Signaling, USA), anti-phospho-Akt (1:2000, Cell Signaling, USA), anti-mTOR (mammalian target of rapamycin) (1:1000, Proteintech Group, Inc., China), anti-phospho-mTOR (s2448) (1:500, Bioworld, USA) and anti-GADPH (1:1000, Hangzhou Xianzhi biology Co., Ltd., China) primary antibodies for overnight at 4°C, followed by incubation with horseradish peroxidase (HRP)-conjugated goat anti-rabbit or goat anti-mouse secondary antibodies (1:50000, Boster, China) for 1h at RT. Then the membranes were incubated with ECL Western blotting Substrate (Thermo Fisher scientific, USA) and imaged using a ChemiDoc XRS imaging system (Bio-Rad, USA). The protein expression level of each gene was quantified by measuring the gray scale intensity of the corresponding bands with the QuantityOne software (Bio-Rad, USA). The expression levels of target genes were normalized by GAPDH.
Stable Cell Line Expressing the GFP-LC3BB Chimeric Protein
The PUC19-mus-LC3BB plasmid containing the full-length encoding sequence for mouse LC3BB was obtained from Sino Biological Inc., China. The LC3BB encoding sequence was amplified by polymerase chain reaction (PCR) with the following primers: Forward GGACTCAGATCTCGA GATGCCGTCCGAGAAGACCTT, Reverse: TGGTGGCGAT GGATCCCACAGCCATTGCTGTCCCGA. The PCR product of LC3BB was extracted from 1.0% agarose gel with the QIAquick Gel Extraction Kit (Qiagen, USA). The purified PCR product was digested with XhoI and BamHI and then inserted into the pEGFP-N3 vector between these two sites (referred as pEGFP-LC3BB). The RMECs were transiently transfected with 1 µg of pEGFP-LC3BB using lipofectamine 2000 (Thermo Fisher Scientific, USA) according to the manufactures instructions. The cells transfected with 1 µg of the empty pEGFP-N3 vector were used as a control. After transfected for 24h, RMECs were treated with either DMSO or 5 mmol/L 3-MA for 4h and then incubated in a hypoxic incubator for 24h. The cells were imaged with a fluorescence microscope and the number of cells with GFP puncta was counted in 150 cells from 3 individual experiments. The ratio of cells with GFP puncta to GFP-positive cells was calculated.
Statistical Analysis
All data analyses were performed by using the SPSS 19.0 software (IBM, USA). Data were presented as mean±standard deviation (SD) of at least 3 experiments. The means of different treatment groups were compared using the One-way analysis of variance (ANOVA) and the LSD post hoc test. A P<0.05 was considered as significantly different.
RESULTS
Isolation and Identification of Murine RMECs
We have successfully isolated and characterized mouse RMEC in this study, although its isolation and primary culture has been proven to be very difficult. The morphology of RMECs prepared from C57BL/6J mice was shown in Figure 1. These cells exhibited a similar polygonal morphology with close cell-cell apposition. To confirm that these cells were endothelial cells, we examined the expression of an endothelial cell-specific marker, CD34, with immunofluorescence. Nearly 100% of the cultured cells expressed CD34 with high level on their surface (Figure 2).
Figure 1. The cobblestone-shaped murine RMECs.

Bar=20 µm.
Figure 2. Representative images of primary murine RMECs by immunofluorescence.
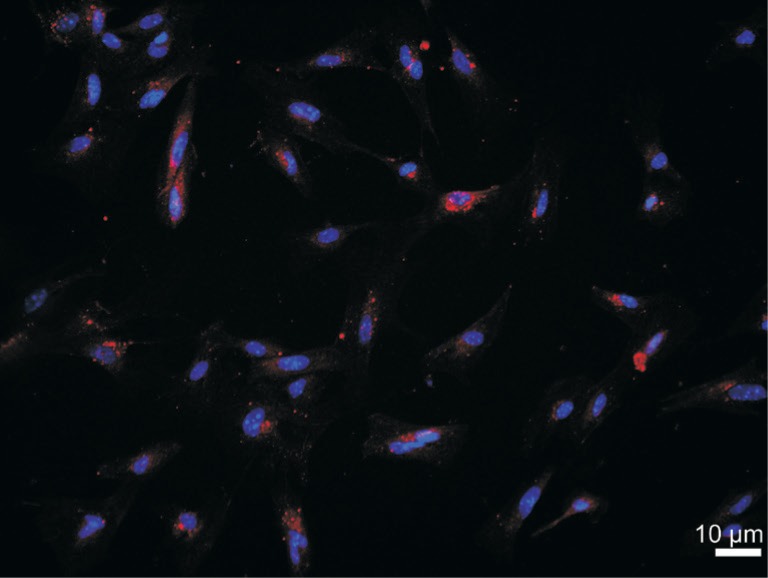
CD34 (red) was mainly located on the surface of cells. The nucleus (blue) was marked by DAPI. Bar=10 µm.
Autophagosome Formation in RMECs by Transmission Electron Microscopy
Activation of autophagy in RMECs in different groups was monitored by TEM. As shown in Figure 3, formation of autophagosome was significantly increased in the hypoxia group compared to the control group, and this effect was blocked by pre-treatment with 3-MA.
Figure 3. Representative TEM micrographs demonstrating increased autophagosome formation in RMECs in the hypoxia group compared to the control and hypoxia+3-MA group.

The black arrows indicated the double-membrane bounded autophagosomes digesting organelles or cytosolic contents.
Expression of Autophagic Markers, LC3BB, Beclin-1, Atg3 and Atg5 in RMECs
The protein expression of LC3BB, Beclin-1, Atg3 and Atg5 was further observed by Western blotting in RMECs (Figure 4A). The ratio of LC3BB-II/I was significantly higher in the hypoxia group than that of the control group (0.24±0.03 vs 0.66±0.03, P<0.01), while pre-treatment with 3-MA significantly decreased it (0.40±0.10 vs 0.66±0.03, P<0.01) (Figure 4B). The results of other autophagic markers, Beclin-1, Atg3 and Atg5 and the variation tendency in different groups were similar to LC3BB. The protein expression level of Beclin-1, Atg3 and Atg5 in the control, hypoxia and hypoxia+3-MA groups were 0.26±0.04, 0.65±0.07, 0.46±0.09; 0.28±0.04, 0.54±0.07, 0.39±0.02 and 0.25±0.03, 0.72±0.01, 0.45±0.07, respectively (Figure 4C-4E). In together, these results suggested that the expression of autophagic markers of RMECs was elevated in hypoxia and induction of cellular autophagy by hypoxia could be partially inhibited by 3-MA.
Figure 4. Expression of autophagic markers, LC3B, Beclin-1, Atg3 and Atg5 in RMECs.

A: Representative immunoblot bands of LC3B-I, LC3B-II, Beclin-1, Atg3 and Atg5 in RMECs in three groups. B, C, D and E: Quantification of the Western blots data. The levels of LC3B, Beclin-1, Atg3 and Atg5 were normalized by GAPDH. aP<0.01 vs control; bP<0.01 vs hypoxia; cP<0.05 vs control; dP<0.05 vs hypoxia.
GFP-LC3BB Chimeric Protein Expression in RMECs
We constructed RMECs ectopically expressing a GFP-LC3BB chimeric protein, and the number of the GFP+ puncta under a fluorescence microscope represents an approximation of the formation autophagosome in cells. As shown in Figure 5, in the hypoxia group, the number of GFP+ puncta was significantly higher than that of the control group, and it was decreased in the hypoxia+3-MA group compared to the hypoxia group but still higher than that of the control group.
Figure 5. GFP+ puncta in RMECs.

A: Representative images of endogenous GFP+ puncta in the three groups visualized by fluorescence microscopy (Bar=50 µm); B: The quantification of GFP+ puncta. aP<0.01 vs control; bP<0.05 vs hypoxia.
Detecting Key Components of Akt/mTOR and AMPK/mTOR Signaling Pathways
To investigate the main signaling pathways involved in regulation of autophagy in RMECs, several key factors of the PI3K/Akt/mTOR (inhibitory role) and AMPK/mTOR (activating role) signaling pathways, P-Akt, P-AMPK and P-mTOR were assayed by Western blotting (Figure 6A). In the hypoxia group, expression of P-Akt and P-AMPK was significantly increased compared to that of the control cells (0.50±0.05 vs 0.15±0.02, P<0.01; 0.45±0.04 vs 0.12±0.05, P<0.01 respectively). Expression of P-mTOR was decreased when compared with the control group (0.18±0.04 vs 0.69±0.09, P<0.01). In cells pre-treated with 3-MA, expression of P-Akt and P-AMPK was decreased compared to the hypoxia group (0.36±0.00 vs 0.50±0.05, P<0.05; 0.27±0.04 vs 0.45±0.04, P<0.01) but both were still higher than control group (P<0.01; Figure 6B-6C). On the contrary, expression of P-mTOR in the 3-MA pre-treated cells was up-regulated compared to the hypoxia group (0.58±0.09 vs 0.18±0.04, P<0.01) but it was still lower than control group (0.58±0.09 vs 0.69±0.09, P>0.05; Figure 6D). The protein levels of total Akt, AMPK and mTOR were not obviously different among the three groups.
Figure 6. Detecting key components of the Akt/mTOR and AMPK/mTOR signaling pathways.

A: Representative immunoblots for Akt, P-Akt, AMPK, P-AMPK, mTOR and P-mTOR in RMECs in three treatment groups; B, C and D: Quantification of p-Akt (B), p-AMPK (C) and p-mTOR (D) expression in different groups. Bars represtented the resutls of 3 independent experiments. The levels of P-Akt, P-AMPK and P-mTOR were normalized by GAPDH. aP<0.01 vs Control; bP<0.01 vs Hypoxia; cP<0.05 vs Control; dP<0.05 vs Hypoxia.
DISCUSSION
In view of the similar pattern of vascular development between mouse and human retina[12], murine RMECs were adopted in this study to investigate the role of autophagy. With the progress of biotechnology, different versions of detection methods of autophagy are available at present, such as static and dynamic tests, direct and indirect detection. TEM is a kind of qualitative detection method to inspect autophagy, and this method is direct and objective but with more stringent requirements. Because of its direct presentation of the morphology of autophagosome and other related subcellular structures, it is the gold standard to detect autophagy[13]. In this study, we found autophagy in RMECs was activated under hypoxic environment, showing as formation of more autophagosomes and this effect was partially inhibited by pretreatment with 3-MA, an inhibitor of the enzyme necessary for autophagosome formation[14].
Autophagy process is strictly regulated by the Atg and more than 30 specific Atgs have been found and their coding proteins are involved collaboratively in various stages of autophagosome formation. Among them, LC3BB and Beclin-1, playing an important role in the formation and maturation of autophagosome, have been widely used as specific autophagic markers. Liang et al[15] cloned the first mammalian autophagy gene, named Beclin-1 in 1998. Beclin-1, the homologous gene of yeast Atg6, is associated with formation of early autophagosome, and it is also the essential component for the involvement of other autophagy genes in the process of autophagosome formation[16]. LC3B, existing in the two forms, LC3B-I and LC3B-II, is the homologous protein of Atg8 found in mammalian cells. In the process of autophagy, the cytosolic form of LC3B (LC3B-I) combines with phosphatidylethanolamine (PE) after processing and modification by the ubiquitin system to form LC3B-II, existing in the inner and outer membrane of autophagosome[13]. LC3B-II can combine with green fluorescent protein (GFP) to form GFP-LC3B and detecting its content can reflect the activity of autophagy[17]. LC3B-II protein or the number of GFP-LC3B can be detected by Western blotting or immunofluorescent assay for GFP-LC3B. Because of the possible false-positive result caused by a higher affinity of LC3B antibody to LC3B-II and the subjective factors during counting the number of GFP-LC3B, these two techniques need to be combined to observe the formation of autophagosome[18]. Thus, we detected the expression of these proteins in RMECs by western blotting combined with GFP-LC3B here. In consistence with our previous findings[7] and results of TEM, we found LC3BB-II/I and Beclin-1 expression was obviously elevated the number of GFP+puncta in cells under hypoxia compared to the control cells which were partially inhibited by pretreatment with 3-MA.
The formation of autophagosome is critical to autophagy, relying on two ubiquitin system, Atg12-Atg5 conjugated system and Atg3, Atg4, Atg7, Atg8 grease system. The two systems are not independent, but are closely related to each other. Atg3 and Atg5 play a key role in the formation of autophagosome. Atg5 (located at 6q21 on human chromosome 6), regulats the process of autophagy and combines the conservative protein Atg12 to form Atg12-Atg5 protein system, which is conducive to the extension of autophagosome membrane. Atg3 (located at 3q13 on human chromosome 3), plays as E2 enzyme catalytic role in the whole process of autophagy and promotes the combination of Atg8 and phosphatidyl inositol to participate in every stage of autophagosome formation[19]–[20]. Thus, testing the expression of Atg3 and Atg5 in RMECs to deduce the role of autophagy in this cell may provide a new direction and strategy for retinal diseases. In consistence with LC3B and Beclin-1, we also found expression of Atg3 and Atg5 was obviously elevated in RMECs under hypoxia to further confirm the activation of autophagy.
These results above were similar to the research findings on the state of autophagy under hypoxia or ischemia[7],[21]–[23]. Under hypoxia, autophagy can protect cells and reduce cell damage. Recent studies have found that hypoxia can induce autophagy activity to promote survival of cells under stress[24]–[26]. This protective effects of autophagy on the hypoxia stress of the body are mainly realized through two ways: 1) direct removal of reactive oxygen species (ROS) or degradation of damaged mitochondria to indirectly remove ROS to avoid the oxidative damage of cells[24]; 2) removal of other oxidative damaged organelles or proteins to maintain homeostasis in cells[27]–[28].
The molecular mechanism of autophagy is very complex and may be involved multiple signals. In hypoxic conditions, autophagy related proteins were increased in RMECs and expression of P-Akt and P-AMPK was up-regulated and P-mTOR was down-regulated. However, there was no significant difference in expression of Akt, AMPK and mTOR between the hypoxia group and the control group. This result suggested that changes in phosphorylation levels of Akt, AMPK and mTOR induced by hypoxia led to activation of autophagy. mTOR (mammalian target of rapamycin) is a key regulatory factor in the initialization stage of autophagy and its activation (phosphorylation) can inhibit autophagy in cells[29]. The signaling pathways depending on mTOR include PI3K/Akt/mTOR and AMPK/TSC1/2/mTOR pathways. PI3K/AKT plays an inhibitory role in autophagy through activating mTOR. In addition, PI3K/Akt/mTOR signaling pathway has become a research hotspot in tumor and this pathway can induce tumorigenesis through a variety of mechanisms, including participating in autophagy[30]. AMPK is involved in activating autophagy and the main mechanism is to directly inhibit mTOR through phosphorylation and AMPK can also activate TSC to inhibit mTOR[31]. It is known that hypoxia stress can damage the structure and function of mitochondria, resulting in the imbalance of cell energy and the increase of AMP/ATP ratio can promote the formation of AMPK. Here, up-regulated of P-AMPK and down-regulated P-mTOR indicated that AMPK/mTOR pathway may play a main role in activation of autophagy in RMECs under hypoxia and the PI3K/Akt/mTOR pathway was weak of action. Our results were consistent with previous reports that AMPK and mTOR were the key regulating factors of autophagy under ischemia-hypoxia conditions and the inhibitory effects of mTOR on autophagy was attenuated by decreased P-mTOR level resulting in activation of autophagy[32]. Furthermore, P-AMPK can inhibit the activity of mTORC1, which is one of mTOR and the major factor in regulating autophagy, to eventually enhance autophagy[33]–[34].
Hypoxia has been recognized as the most important initiating factor for many retinal diseases. This study identified the activation of autophagy in RMECs under hypoxia mainly through the AMPK/mTOR signaling pathway. However, the underlying mechanisms for autophagy changes and their effects on cell survival or death as well as retinal vascular diseases need further investigation. Besides, this experimental set-up had some limitations. Different autophagy inhibitors act on different stages of autophagy activation with various mechanisms. In this study, we only adopted an early inhibitor 3-MA, which acts on the autophagy induction stage. Thus, several inhibitors with different mechanisms of action should be used for mutual verification of the experimental results. Furthermore, the murine RMECs are different from human RMECs. These results obtained in the present study cannot fully reflect the actual condition in human retinas. Therefore, it is recommended to adopt human retinal cells for further researches in vitro and then test acquired results in vivo.
Acknowledgments
Foundation: Supported by the National Natural Science Foundation of China (No.81500726).
Conflicts of Interest: Li R, None; Wang LZ, None; Du JH, None; Zhao L, None; Yao Y, None.
REFERENCES
- 1.Grammas P, Riden M. Retinal endothelial cells are more susceptible to oxidative stress and increased permeability than brain-derived endothelial cells. Microvasc Res. 2003;65(1):18–23. doi: 10.1016/s0026-2862(02)00016-x. [DOI] [PubMed] [Google Scholar]
- 2.Li SY, Fu ZJ, Lo AC. Hypoxia-induced oxidative stress in ischemic retinopathy. Oxid Med Cell Longev. 2012;2012:426769. doi: 10.1155/2012/426769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kermorvant-Duchemin E, Sapieha P, Sirinyan M, Beauchamp M, Checchin D, Hardy P, Sennlaub F, Lachapelle P, Chemtob S. Understanding ischemic retinopathies: emerging concepts from oxygen-induced retinopathy. Doc Ophthalmol. 2010;120(1):51–60. doi: 10.1007/s10633-009-9201-x. [DOI] [PubMed] [Google Scholar]
- 4.Bharadwaj AS, Appukuttan B, Wilmarth PA, Pan Y, Stempel AJ, Chipps TJ, Benedetti EE, Zamora DO, Choi D, David LL, Smith JR. Role of the retinal vascular endothelial cell in ocular disease. Prog Retin Eye Res. 2013;32:102–180. doi: 10.1016/j.preteyeres.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mehrpour M, Esclatine A, Beau I, Codogno P. Overview of macroautophagy regulation in mammalian cells. Cell Res. 2010;20(7):748–762. doi: 10.1038/cr.2010.82. [DOI] [PubMed] [Google Scholar]
- 6.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368(7):651–662. doi: 10.1056/NEJMra1205406. [DOI] [PubMed] [Google Scholar]
- 7.Li R, Du J, Chang Y. Role of autophagy in hypoxia-induced angiogenesis of RF/6A cells in vitro. Curr Eye Res. 2016;41(12):1566–1570. doi: 10.3109/02713683.2016.1145234. [DOI] [PubMed] [Google Scholar]
- 8.Zamora DO, Riviere M, Choi D, Pan Y, Planck SR, Rosenbaum JT, David LL, Smith JR. Proteomic profiling of human retinal and choroidal endothelial cells reveals molecular heterogeneity related to tissue of origin. Mol Vis. 2007;13:2058–2065. [PubMed] [Google Scholar]
- 9.Stewart EA, Samaranayake GJ, Browning AC, Hopkinson A, Amoaku WM. Comparison of choroidal and retinal endothelial cells: characteristics and response to VEGF isoforms and anti-VEGF treatments. Exp Eye Res. 2011;93(5):761–766. doi: 10.1016/j.exer.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 10.Su X, Sorenson CM, Sheibani N. Isolation and characterization of murine retinal endothelial cells. Mol Vis. 2003;9:171–178. [PubMed] [Google Scholar]
- 11.You JJ, Yang CM, Chen MS, Yang CH. Regulation of Cyr61/CCN1 expression by hypoxia through cooperation of c-Jun/AP-1 and HIF-1α in retinal vascular endothelial cells. Exp Eye Res. 2010;91(6):825–836. doi: 10.1016/j.exer.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 12.Fruttiger M. Development of the mouse retinal vasculature: angiogenesis versus vasculogenesis. Invest Ophthalmol Vis Sci. 2002;43(2):522–527. [PubMed] [Google Scholar]
- 13.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu YT, Tan HL, Shui G, Bauvy C, Huang Q, Wenk MR, Ong CN, Codogno P, Shen HM. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem. 2010;285(14):10850–10861. doi: 10.1074/jbc.M109.080796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, Herman B, Levine B. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol. 1998;72(11):8586–8596. doi: 10.1128/jvi.72.11.8586-8596.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K, Tavernarakis N, Hickman JA, Geneste O, Kroemer G. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. Embo J. 2007;26(10):2527–2539. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jin Y, Chen C, Meng L, Chen J, Li M, Zhu Z, Lin J. A CE-LIF method to monitor autophagy by directly detecting LC3B proteins in HeLa cells. Analyst. 2012;137(23):5571–5575. doi: 10.1039/c2an36011j. [DOI] [PubMed] [Google Scholar]
- 18.Barth S, Glick D, Macleod KF. Autophagy: assays and artifacts. J Pathol. 2010;221(2):117–124. doi: 10.1002/path.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pyo JO, Nah J, Jung YK. Molecules and their functions in autophagy. Exp Mol Med. 2012;44(2):73–80. doi: 10.3858/emm.2012.44.2.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu ZQ, Ni T, Hong B, Wang HY, Jiang FJ, Zou S, Chen Y, Zheng XL, Klionsky DJ, Liang Y, Xie Z. Dual roles of Atg8-PE deconjugation by Atg4 in autophagy. Autophagy. 2012;8(6):883–892. doi: 10.4161/auto.19652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abounit K, Scarabelli TM, McCauley RB. Autophagy in mammalian cells. World J Biol Chem. 2012;3(1):1–6. doi: 10.4331/wjbc.v3.i1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chu CT. Eaten alive: autophagy and neuronal cell death after hypoxia-ischemia. Am J Pathol. 2008;172(2):284–287. doi: 10.2353/ajpath.2008.071064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sciarretta S, Hariharan N, Monden Y, Zablocki D, Sadoshima J. Is autophagy in response to ischemia and reperfusion protective or detrimental for the heart? Pediatr Cardiol. 2011;32(3):275–281. doi: 10.1007/s00246-010-9855-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen L, Liao B, Qi H, Xie LJ, Huang L, Tan WJ, Zhai N, Yuan LB, Zhou Y, Yu LJ, Chen QF, Shu W, Xiao S. Autophagy contributes to regulation of the hypoxia response during submergence in Arabidopsis thaliana. Autophagy. 2015;11(12):2233–2246. doi: 10.1080/15548627.2015.1112483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Samokhvalov V, Scott BA, Crowder CM. Autophagy protects against hypoxic injury in C. elegans. Autophagy. 2008;4(8):1034–1041. doi: 10.4161/auto.6994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu YL, Jahangiri A, De Lay M, Aghi MK. Hypoxia-induced tumor cell autophagy mediates resistance to anti-angiogenic therapy. Autophagy. 2012;8(6):979–981. doi: 10.4161/auto.20232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40(2):280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Granato M, Santarelli R, Filardi M, Gonnella R, Farina A, Torrisi MR, Faggioni A, Cirone M. The activation of KSHV lytic cycle blocks autophagy in PEL cells. Autophagy. 2015;11(11):1978–1986. doi: 10.1080/15548627.2015.1091911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brech A, Ahlquist T, Lothe RA, Stenmark H. Autophagy in tumour suppression and promotion. Mol Oncol. 2009;3(4):366–375. doi: 10.1016/j.molonc.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nazio F, Strappazzon F, Antonioli M, Bielli P, Cianfanelli V, Bordi M, Gretzmeier C, Dengjel J, Piacentini M, Fimia GM, Cecconi F. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat Cell Biol. 2013;15(4):406–416. doi: 10.1038/ncb2708. [DOI] [PubMed] [Google Scholar]
- 31.Yefimova MG, Messaddeq N, Harnois T, Meunier AC, Clarhaut J, Noblanc A, Weickert JL, Cantereau A, Philippe M, Bourmeyster N, Benzakour O. A chimerical phagocytosis model reveals the recruitment by Sertoli cells of autophagy for the degradation of ingested illegitimate substrates. Autophagy. 2013;9(5):653–666. doi: 10.4161/auto.23839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arsham AM, Howell JJ, Simon MC. A novel hypoxia-inducible factor-independent hypoxic response regulating mammalian target of rapamycin and its targets. J Biol Chem. 2003;278(32):29655–29660. doi: 10.1074/jbc.M212770200. [DOI] [PubMed] [Google Scholar]
- 33.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30(2):214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]