

Abstract
Background
Blepharophimosis‐ptosis‐epicanthus inversus syndrome (BPES) is a malformation of the eyelids. Forkhead Box L2 (FOXL2) is the only gene known to be associated with BPES.
Methods
We identified two Han Chinese BPES families with premature ovarian insufficiency (POI). Sanger sequencing and in vitro functional analysis were performed to identify the genetic cause.
Results
Sanger sequencing identified two novel mutations (c.462_468del, c.988_989insG) in FOXL2, one in each family. The in vitro functional analysis confirmed that both novel mutations were associated with impaired transactivation of downstream genes. Specifically, the single‐base insertion, c.988_989insG, led to subcellular mislocalization and aggregation of the encoded protein, which validated the hypothesis that the two novel FOXL2 mutations are deleterious and associated with POI in the two BPES families.
Conclusion
The novel mutations identified in the present study will enhance the present knowledge of the mutation spectrum of FOXL2. The in vitro experiments provide further insights into the molecular mechanism by which the two new variants mediate disease pathogenesis and may contribute to elucidating the genotype‐phenotype correlation between the two novel FOXL2 mutations and POI.
Keywords: blepharophimosis‐ptosis‐epicanthus inversus syndrome, FOXL2, novel mutation, premature ovarian insufficiency
1. INTRODUCTION
Blepharophimosis‐ptosis‐epicanthus inversus syndrome (BPES, OMIM 110100) is an autosomal dominant condition characterized by eyelid malformations, involving ptosis, a narrowed horizontal palpebral aperture, epicanthus inversus, and telecanthus. The incidence of BPES is approximately 1 per 50,000 in the general population (Oley & Baraitser, 1988). BPES may be categorized into two clinical subsets based on the additional presence or absence of premature ovarian insufficiency (POI). BPES type I manifests with palpebral malformations associated with POI, whereas patients with BPES type II exhibit isolated eyelid malformations (Zlotogora, Sagi, & Cohen, 1983). Both types of BPES are caused by mutations in the Forkhead Box L2 gene (FOXL2, OMIM 605597).
FOXL2, which encodes a forkhead transcription factor, is the only gene known to be associated with BPES. Apart from the mesenchyme of developing eyelids, FOXL2 is highly expressed in granulosa cells of the ovary and gonadotropic cells of the anterior pituitary (Batista, Vaiman, Dausset, Fellous, & Veitia, 2007; Cocquet et al., 2003; Ellsworth et al., 2006). The pattern of its expression suggests that FOXL2 might play a pivotal role in regulating the development of the ovaries and maintaining the female gonads in vertebrate species (Cocquet et al., 2003; Leung, Fuller, & Chu, 2016). The vast majority of variations that lead to truncated FOXL2 are generally related to BPES type I, while the variations that result in full‐length proteins are often associated with preserved ovarian function and BPES type II (Crisponi et al., 2001). To date, numerous germline mutations in FOXL2 have been identified to be responsible for BPES with POI (Kaur, Hussain, Naik, Murthy, & Honavar, 2011), whereas the genotype‐phenotype correlation between FOXL2 mutation and the type of BPES developed has not yet been fully delineated.
In this study, we identified two novel mutations of FOXL2 in two Han Chinese families with BPES type I by screening FOXL2. The aim of the present work was to comparatively evaluate the effect of the novel mutations on FOXL2 bioactivity via in vitro functional analysis and delineate the correlation between the two novel FOXL2 mutations and the types of BPES. The present insights into these novel mutations might contribute to improving diagnostics, genetic counselling, as well as fertility guidance in clinical settings. The results of the endocrine examination, FOXL2 sequencing, and in vitro functional analysis contributed to elucidating the genotype‐phenotype correlation between the two novel FOXL2 mutations and POI.
2. MATERIALS AND METHODS
2.1. Patients
Two probands from two families were admitted to the Reproductive and Genetic Hospital of CITIC‐Xiangya due to eyelid malformations and primary infertility (Figure 1a). The probands received detailed ophthalmic examinations by ophthalmologists and were diagnosed with BPES based on the following criteria: blepharophimosis, ptosis, epicanthus inversus, and telecanthus. Photographs of the probands’ eyelids were taken before or after surgery for assessment of BPES‐related features (Figure 1a).
Figure 1.
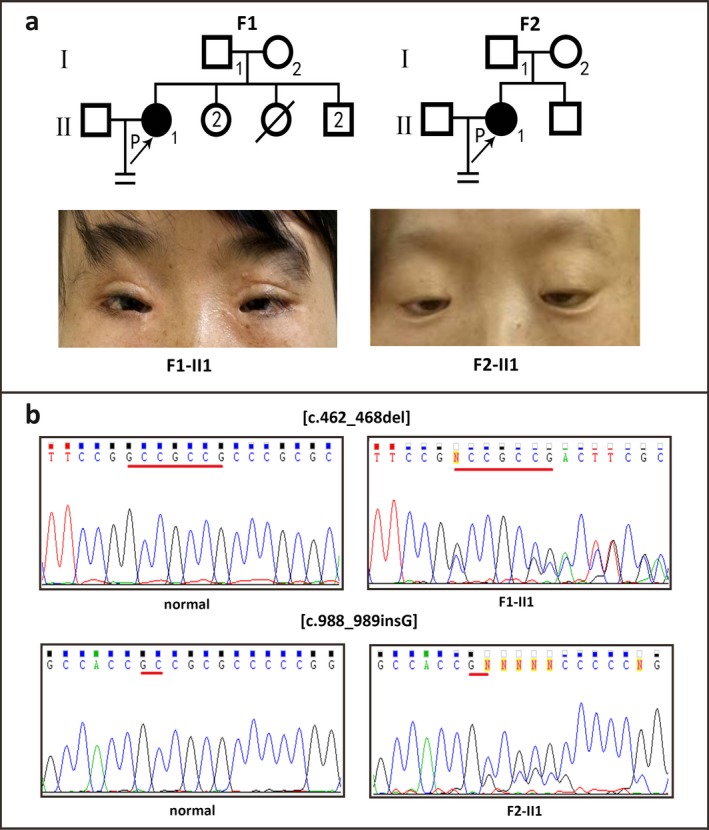
Pedigree and Sanger sequencing analysis of the probands from the two Han Chinese families. Panel a: Pedigree and eyelid photographs of the probands from two families in this study before or after surgery. Affected members are indicated by filled symbols; unaffected relatives are indicated by open symbols. The number of siblings is indicated in the symbol. The arrow indicates the proband. Numbers are allotted to family members whose DNA samples were used in this study. Panel b: Sanger sequencing analysis of the two families in this study.
Premature ovarian insufficiency, which is defined as the depletion or loss of normal ovarian function, is characterized by the acquisition of hypergonadotropic hypogonadism in women before the age of 40 years (Caburet et al., 2014; The ESHRE Guideline Group on POI et al., 2016). All affected female participants in this study presented with elevated serum gonadotrophin concentrations and primary infertility and had received the diagnosis of POI in the locality according to the latest criteria (The ESHRE Guideline Group on POI et al., 2016). The probands of family F1 and F2 experienced irregular menstrual cycles; the proband from family F2 presented with a decline in antral follicle count, indicating decreased ovarian reserve. Since they received traditional Chinese medicine treatments after their irregular cycles and we could not obtain the hormonal characteristics and clinical features of the probands before treatment, the information on these features after the treatment is provided in Table 1.
Table 1.
Hormonal characteristics and clinical features of two probands after the reatment of trational Chinese medicine
| Patient | Age/years | LH (mIU/ml) | FSH (mIU/ml) | Prolactin (ng/ml) | Estradiol (pg/ml) | Progesterone (ng/ml) | Testosterone (ng/ml) | Follicle numbers | Clinical information |
|---|---|---|---|---|---|---|---|---|---|
| F1‐II1 | 34 | 6.58 | 17.49↑ | 19.55 | 40 | 0.15 | 0.85 | Left: 5, Right: 10 | BPES; Menarche 16 years; irregular cycles |
| F2‐II1 | 30 | 10.33 | 17.84↑ | 7.16 | 42 | 0.2 | 0.59 | Left: 2, Right: 3 | BPES; Menarche 15 years; irregular cycles |
Ref. value of female follicle period: LH: 1.80–11.78 mIU/ml, FSH: 3.03–8.08 mIU/ml, Prolactin: 5.18–26.53 ng/ml, Estradiol: 21–251 pg/ml, Progesterone: <0.1–0.3 ng/ml, Testosterone: 0.38–1.97 ng/ml.
All affected women were found to have a normal 46, XX karyotype, and FMR1 CGG repeats in the normal polymorphic range. No associated endocrinopathies or autoimmune disorders were found. Informed consent was obtained from all participants in this study, which was approved by the ethics committee of the Reproductive and Genetic Hospital of CITIC‐Xiangya.
2.2. Sequencing and variant analysis
Genomic DNA was extracted from whole‐blood leucocytes of patients using a QIAamp Blood DNA Mini kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. The entire coding region and intron‐exon boundaries of FOXL2 were amplified from genomic DNA using polymerase chain reaction (PCR) as previously described (Bell, Murday, Patton, & Jeffery, 2001) and directly sequenced using the Applied Biosystems 3130 genetic analyser (ABI, USA). Subsequently, bioinformatics analysis of all identified mutations was performed using MutationTaster (http://www.mutationtaster.org/) and Combined Annotation Dependent Depletion (CADD, http://cadd.gs.washington.edu/).
2.3. Plasmid constructs
Since FOXL2 is a single‐exon gene, its full‐length open reading frame was amplified from the genomic DNA of patients using primers incorporating restriction enzyme sites (EcoRI‐KpnI). The amplified gDNA products were purified, digested, and then cloned into the digested plasmids of phosphorylated enhanced green fluorescent protein (EGFP)‐N1 (Clontech, CA, USA), leading to the production of fusion proteins with the EGFP on the C terminus of FOXL2 (the former codon of termination codon in the wild‐type FOXL2 or predicted termination codon in mutant FOXL2 followed by the open reading frame of EGFP, which were designated as FOXL2‐WT‐EGFP, FOXL2‐Pro156Argfs‐EGFP, and FOXL2‐ Ala330Glyfs‐EGFP, respectively). All expression constructs were sequenced to confirm the presence of the desired mutations and exclude PCR‐induced mutations.
2.4. Cell culture and transient transfection
CHO cells were seeded at a density of 2.5 × 105 cells in a 12‐well plate and allowed to grow to 70%–80% confluence prior to transfection under standard conditions in complete media (DMEM/F12 [Gibco, USA] and 10% FBS at 37°C with 5% CO2). CHO cells were transiently transfected with expression vectors containing wild‐type or mutated proteins, using Lipofectamine® 2000 (Thermo Fisher Scientific, Carlsbad, CA, USA) according to the manufacturer's instructions. Cells transfected with an empty vector (without FOXL2) served as the negative control. The transfected cells were incubated for 36 hr prior to their preparation for immunofluorescence and RNA extraction.
2.5. Immunofluorescence and confocal microscopy
Immunofluorescence was performed as previously described (Caburet et al., 2001) with transfected CHO cells grown on coverslips. Fluorescent images were captured by confocal microscopy (Olympus FV1000, Tokyo, Japan).
2.6. Quantitative real‐time PCR
It has been reported that FOXL2 represses the promoter activity of StAR, whereas it directly increases sirtuin 1 (SIRT1) transcription (Benayoun et al., 2009; Pisarska, Bae, Klein, & Hsueh, 2004). In order to confirm that mutant proteins alter the expression of target genes, the expression of endogenous StAR and SIRT1 mRNA was measured by Quantitative real‐time PCR (qPCR). Transfected cells were incubated for 36 hr, and then total mRNA was extracted using Trizol (Invitrogen, Carlsbad, CA, USA). RNA (1.5 μg) was reverse transcribed to synthesize cDNA in a 20‐μl reaction mixture. Quantitative SYBR Green real‐time PCR was performed on an Applied Biosystems 7500 system (ABI) using the 20‐μl mixture. The housekeeping gene GAPDH was used as a control. The primer sequences were as follows: StAR, forward 5′‐ACATGAAAGGACTGAGGCACC‐3′ and reverse 5′‐CTCCTTCTTCCAGCCTTCCTG‐3′; and SIRT1, forward 5′‐AGCTGGGGTTTCTGTTAGCTG‐3′ and reverse 5′‐GACACAGAGATGGCTGGAACT‐3′.
2.7. Statistical analysis
Statistical analysis was performed via Student's t test and one‐way ANOVA using SPSS software, version 19.0 (SPSS, Chicago, IL, USA). The differences were considered significant at p‐values < .05. The results of the real‐time PCR are expressed as bar graphs.
3. RESULTS
3.1. Sequencing and in silico analysis
The proband from Family F1 (II1) was found to harbor a heterozygous deletion mutation of FOXL2, c.462_468del, leading to a frameshift at codon 156. This mutation was predicted to result in the formation of a truncated FOXL2 protein of 267 amino acids (p.Pro156Argfs*113, Figure 1b). The proband from Family F2 (II1) carried a heterozygous insertion mutation (c.988_989insG), which was predicted to result in an extended protein of 532 amino acids (p.Ala330Glyfs*204, Figure 1b). Pedigree analysis revealed that their parents are all free of the mutations, which indicated that the two novel mutations are de novo in the probands.
The two mutations, c.462_468del and c.988_989insG, were not found in the dbSNP, 1000 Genomes Project, ExAC, and HGMD public databases, suggesting they were novel. All mutations identified in the present study were predicted as pathogenic mutations by bioinformatics analysis. Schematic representation of the FOXL2 gene and locations of the mutations identified in this study are shown in Figure 2c.
Figure 2.
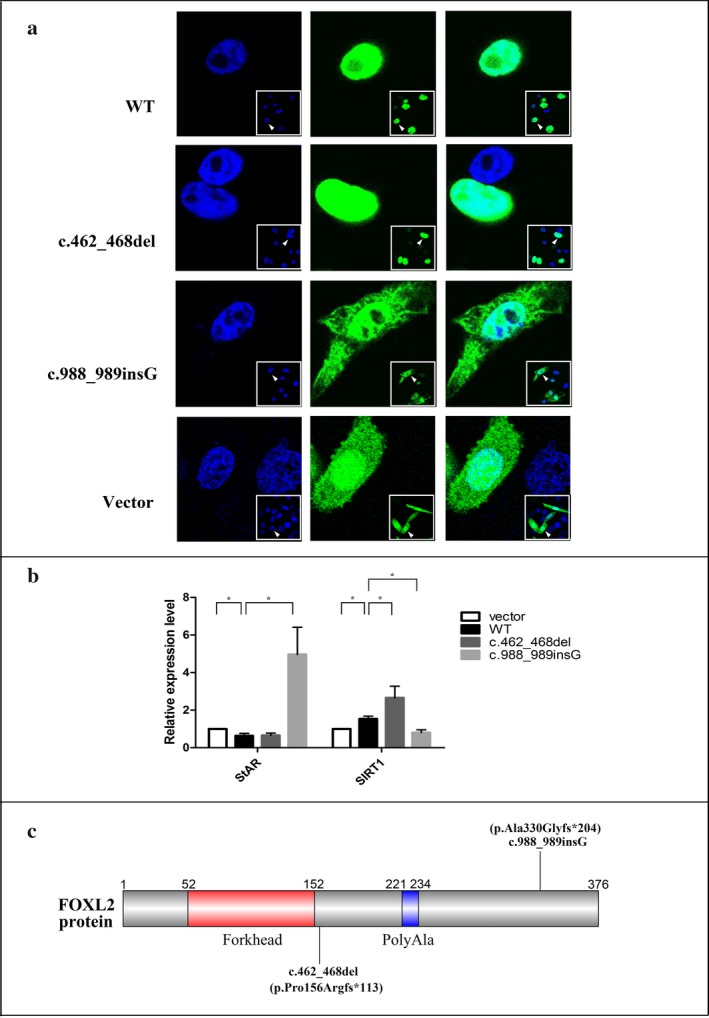
In vitro functional analysis of the two novel mutations and the schematic structure of FOXL2. Panel a: Subcellular localization of wild‐type and mutant FOXL2 proteins. The left panel shows nuclear staining with DAPI. The middle panel corresponds to the subcellular localization of FOXL2 as a fusion protein with GFP. The right panel is a merged image of the previous two images. Panel b: The expression level of endogenous StAR and SIRT mRNA in CHO cells transiently transfected with empty vector, wild‐type FOXL2, and mutant FOXL2 respectively. Statistical significance of the departure of the observed ratio from the expected ratio is represented by *p < .05. Panel c: The schematic structure of FOXL2 and the location of two mutations identified in this study
3.2. Subcellular location of FOXL2 protein
As expected, the wild‐type FOXL2 protein localizes exclusively in the nucleus in a diffused manner and cells transfected with the empty vector displayed significant cytoplasmic mislocalization. Cells transfected with the mutant construct (FOXL2‐Pro156Argfs‐EGFP) displayed the same pattern as cells transfected with the wild‐type construct, whereas cells transfected with mutant construct containing the FOXL2‐Ala330Glyfs‐EGFP displayed significant cytoplasmic mislocalization as well as nuclear and cytoplasmic aggregation (Figure 2a).
3.3. Real‐time qPCR
To assess the effects of the novel FOXL2 mutations (c.462_468del and c.988_989insG) on transactivation capacity, we performed real‐time qPCR to measure the expression of endogenous StAR and SIRT1 mRNA. When cells were transfected with wild‐type FOXL2, the expression level of endogenous StAR mRNA was the lowest, which was consistent with the wild‐type FOXL2 being a repressor of StAR expression. The expression level of endogenous StAR mRNA in cells transfected with the truncated FOXL2 was similar to that in cells transfected with the wild‐type FOXL2. However, the levels were dramatically elevated in cells transfected with the FOXL2 mutant encoding the extended protein, suggesting a dominant negative effect over endogenous wild‐type FOXL2 (Figure 2b). The expression level of endogenous SIRT1 mRNA in cells transfected with the wild‐type FOXL2 was higher than that in cells transfected with the empty vector, which was consistent with the view that SIRT1 transcription is directly upregulated by the FOXL2 protein. Furthermore, the mRNA expression level of endogenous SIRT1 in cells transfected with the extended FOXL2‐encoding mutant gene was decreased compared with cells transfected with wild‐type FOXL2. In contrast, the levels were remarkably increased in cells transfected with the truncated FOXL2, suggesting the truncated FOXL2 behaved in a hypermorphic manner on SIRT1 (Figure 2b).
4. DISCUSSION
In the present study, we analysed two Han Chinese families with BPES type I and identified two novel mutations (c.462_468del and c.988_989insG). Immunofluorescence and confocal microscopy revealed that the extended FOXL2, p.Ala330Glyfs*204, induced significant mislocalization and aggregation. Furthermore, we demonstrated that both novel mutations had a strong impact on FOXL2 transactivation abilities. Our data suggest that the novel mutations in FOXL2 are deleterious and represent disease‐associated mutations in the present probands with BPES and POI.
The forkhead nuclear transcription factor encoded by human FOXL2 contains two domains: the DNA‐binding forkhead domain (FHD) and the 14‐residue polyAla tract, which are strictly conserved in mammals (Adell & Muller, 2004; Baron et al., 2005; Crisponi et al., 2001;). Investigations of other forkhead transcription factors, such as FOXP2 and FOXP3, have illustrated that the FHD is pivotal for correct nuclear localization (Mizutani et al., 2007; Schubert, Jeffery, Zhang, Ramsdell, & Ziegler, 2001). A frameshift mutation or nonsense mutation is commonly thought to result in truncated or elongated protein and consequent loss of function; different loss‐of‐function proteins may show varying levels of bioactivity (Dipietromaria et al., 2009; Moumné, Fellous, & Veitia, 2005). The novel truncated FOXL2 (p.Pro156Argfs*113) with a complete FHD localizes exclusively in the nucleus in a diffuse manner, which is consistent with its function. The single base insertion (c.988_989insG) is predicted to result in the formation of an elongated FOXL2 protein containing a complete FHD. This phenomenon may induce misfolding that hides the nuclear localization signals or disrupts interactions with nuclear transporters, ultimately resulting in mislocalization, and similar results were observed in our functional study.
Normal bioactivity of FOXL2 is important for ovarian function (Georges et al., 2013). As a nuclear transcription factor, FOXL2 directly represses StAR transcription but upregulates SIRT1 transcription (Benayoun et al., 2009; Pisarska et al., 2004). The StAR gene encodes a protein involved in the rate‐limiting step in steroid hormone synthesis that plays an important role in folliculogenesis (Pisarska et al., 2004). SIRT1 has been found to play a crucial role in the cell stress response, and SIRT1‐knockout mice were found to be infertile (Di Emidio et al., 2014; McBurney et al., 2003). The novel mutations (c.462_468del and c.988_989insG) profoundly impaired the transactivation abilities of FOXL2, which was corroborated by the quantification of endogenous StAR and SIRT1 mRNA levels in CHO cells via qPCR. Therefore, the altered transactivation of StAR and SIRT1 promoters may explain the ovarian phenotype in the present patients. The phenotype of patient from family II was more severe than that of patient from family I, which may be due to a dominant negative effect (Caburet et al., 2001; Méduri et al., 2010). It has reported that a dominant negative effect may be caused by the interaction between mutated FOXL2 protein and the wild‐type protein in the aggregates (Caburet et al., 2001). This effect showed extensive nuclear and cytoplasmic protein aggregation for such a dominant negative effect presented in extended protein and was reported previously (Caburet et al., 2001; Méduri et al., 2010). This is consistent with the view that the mutant FOXL2 proteins implicated in BPES type I strongly decrease the transactivating effect on the promoters of target genes, whereas the mutant FOXL2 proteins implicated in BPES type II are still (at least partially) active (Dipietromaria et al., 2009). Although we did not perform whole‐exome sequencing to eliminate other putative disease‐causative mutations, two mutations identified in this study have been found to have a strong impact on FOXL2 transactivation abilities. Therefore, we suggest that the two mutations of FOXL2 identified in the present study are pathogenic mutations implicated in BPES type I in the two Han Chinese probands.
Differential diagnosis of BPES types I and II is pivotal for female patients carrying FOXL2 mutations, as it may allow the patients to predict fertility and plan appropriate therapy. Unfortunately, the genotype‐phenotype correlation between FOXL2 mutation and the type of BPES developed has not yet been fully delineated. It has been reported that mutations predicted to result in truncated proteins without the FHD are associated with BPES type I, whereas the phenotype associated with mutations leading to truncated or extended proteins with an intact forkhead and poly‐Ala tract were impossible to predict (De Baere et al., 2003). In this study, two novel mutations (c.462_468del and c.988_989insG) resulting in a truncated protein with complete FHD and an extended protein with an intact forkhead and poly‐Ala tract, respectively, were both responsible for BPES type I. Therefore, we demonstrated that a female patient who harbors the novel mutations might have a high risk of infertility, and females with BPES whether onset of POF should be examined by both a clinical geneticist and an endocrinologist for assessment of ovarian function, rather than relying only on molecular testing as a predictor for POI risk.
In conclusion, we identified two novel mutations of FOXL2 in two Han Chinese families. Our experimental findings suggest that the two novel mutations in the FOXL2 gene are pathogenic mutations and implicated in BPES type I in the two families, which was confirmed by the in vitro functional analysis. Our findings also contribute to the delineation of genotype‐phenotype correlation between the two novel FOXL2 mutations and POI, and enrich knowledge of FOXL2 gene mutation spectrum.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
ACKNOWLEDGMENTS
We thank the patients and their family members for their support and participation in this research. We also thank the genetic counselling team at the Reproductive and Genetic Hospital of CITIC‐Xiangya and the clinicians who referred the patients for clinical study. This work was supported by a grant from the National Natural Science Fund of China (No. 81471432).
Yang X‐W, He W‐B, Gong F, et al. Novel FOXL2 mutations cause blepharophimosis‐ptosis‐epicanthus inversus syndrome with premature ovarian insufficiency. Mol Genet Genomic Med. 2018;6:261–267. https://doi.org/10.1002/mgg3.366
National Natural Science Fund of China (No. 81471432).
Contributor Information
Juan Du, Email: tandujuan@csu.edu.cn.
Yue‐Qiu Tan, Email: tanyueqiu@csu.edu.cn.
REFERENCES
- Adell, T. , & Muller, W. E. (2004). Isolation and characterization of five Fox (Forkhead) genes from the sponge Suberites domuncula . Gene, 334, 35–46. [DOI] [PubMed] [Google Scholar]
- Baron, D. , Batista, F. , Chaffaux, S. , Cocquet, J. , Cotinot, C. , Cribiu, E. , … Fellous, M. (2005). Foxl2 gene and the development of the ovary: A story about goat, mouse, fish and woman. Reproduction Nutrition, 45, 377–382. [DOI] [PubMed] [Google Scholar]
- Batista, F. , Vaiman, D. , Dausset, J. , Fellous, M. , & Veitia, R. A. (2007). Potential targets of FOXL2, a transcription factor involved in craniofacial and follicular development, identified by transcriptomics. Proceedings of the National Academy of Sciences of the United States of America, 104, 3330–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell, R. , Murday, V. A. , Patton, M. A. , & Jeffery, S. (2001). Two families with blepharophimosis/ptosis/epicanthus inversus syndrome have mutations in the putative forkhead transcription factor FOXL2. Genet Test, 5, 335–338. [DOI] [PubMed] [Google Scholar]
- Benayoun, B. A. , Batista, F. , Auer, J. , Dipietromaria, A. , L'Hote, D. , De Baere, E. , & Veitia, R. (2009). A. Positive and negative feedback regulates the transcription factor FOXL2 in response to cell stress: Evidence for a regulatory imbalance induced by disease‐causing mutations. Human Molecular Genetics, 18, 632–644. [DOI] [PubMed] [Google Scholar]
- Caburet, S. , Arboleda, V. A. , Llano, E. , Overbeek, P. A. , Barbero, J. L. , Oka, K. , … Vilain, E. (2014). Mutant cohesin in premature ovarian failure. New England Journal of Medicine, 370, 943–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caburet, S. , Demarez, A. , Moumné, L. , Fellous, M. , De Baere, E. , & Veitia, R. A. (2001). A recurrent polyalanine expansion in the transcription factor FOXL2 induces extensive nuclear and cytoplasmic protein aggregation. Journal of Medical Genetics, 41, 932–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocquet, J. , De Baere, E. , Gareil, M. , Pannetier, M. , Xia, X. , Fellous, M. , & Veitia, R. A. (2003). Structure, evolution and expression of FOXL2 transcription unit. Cytogenet Genome Research, 101, 206–211. [DOI] [PubMed] [Google Scholar]
- Crisponi, L. , Deiana, M. , Loi, A. , Chiappe, F. , Uda, M. , Amati, P. , … Pilia, G. (2001). The putative forkhead transcription factor FOXL2 is mutated in blepharophimosis/ptosis/epicanthus inversus syndrome. Nature Genetics, 27, 159–166. [DOI] [PubMed] [Google Scholar]
- De Baere, E. , Beysen, D. , Oley, C. , Lorenz, B. , Cocquet, J. , De Sutter, P. , … Messiaen, L. (2003). FOXL2 and BPES: Mutational hotspots, phenotypic variability, and revision of the genotype‐phenotype correlation. American Journal of Human Genetics, 72, 478–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Emidio, G. , Falone, S. , Vitti, M. , D'Alessandro, A. M. , Vento, M. , Di Pietro, C. , … Tatone, C. (2014). SIRT1 signalling protects mouse oocytes against oxidative stress and is deregulated during aging. Human Reproduction, 29, 2006–2017. [DOI] [PubMed] [Google Scholar]
- Dipietromaria, A. , Benayoun, B. A. , Todeschini, A. L. , Rivals, I. , Bazin, C. , & Veitia, R. A. (2009). Towards a functional classification of pathogenic FOXL2 mutations using transactivation reporter systems. Human Molecular Genetics, 18, 3324–3333. [DOI] [PubMed] [Google Scholar]
- Ellsworth, B. S. , Egashira, N. , Haller, J. L. , Butts, D. L. , Cocquet, J. , Clay, C. M. , … Camper, S. A. (2006). FOXL2 in the pituitary: Molecular, genetic, and developmental analysis. Molecular Endocrinology, 20, 2796–2805. [DOI] [PubMed] [Google Scholar]
- Georges, A. , Auguste, A. , Bessière, L. , Vanet, A. , Todeschini, A. L. , & Veitia, R. A. (2013). FOXL2: A central transcription factor of the ovary. Journal of Molecular Endocrinology, 52, R17–R33. [DOI] [PubMed] [Google Scholar]
- Kaur, I. , Hussain, A. , Naik, M. N. , Murthy, R. , & Honavar, S. G. (2011). Mutation spectrum of Fork‐Head Transcriptional Factor Gene (FOXL2) in Indian Blepharophimosis Ptosis Epicanthus Inversus Syndrome (BPES) patients. British Journal of Ophthalmology, 95, 881–886. [DOI] [PubMed] [Google Scholar]
- Leung, D. T. , Fuller, P. J. , & Chu, S. (2016). Impact of FOXL2 mutations on signaling in ovarian granulosa cell tumors. International Journal of Biochemistry & Cell Biology, 72, 51–54. [DOI] [PubMed] [Google Scholar]
- McBurney, M. W. , Yang, X. , Jardine, K. , Hixon, M. , Boekelheide, K. , Webb, J. R. , … Lemieux, M. (2003). The mammalian SIR2alpha protein has a role in embryogenesis and gametogenesis. Molecular and Cellular Biology, 23, 38–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méduri, G. , Bachelot, A. , Duflos, C. , Bständig, B. , Poirot, C. , Genestie, C. , … Touraine, P. (2010). FOXL2 mutations lead to different ovarian phenotypes in BPES patients: Case report. Human Reproduction, 25, 235–243. [DOI] [PubMed] [Google Scholar]
- Mizutani, A. , Matsuzaki, A. , Momoi, M. Y. , Fujita, E. , Tanabe, Y. , & Momoi, T. (2007). Intracellular distribution of a speech/language disorder associated FOXP2 mutant. Biochemical and Biophysical Research Communications, 353, 869–874. [DOI] [PubMed] [Google Scholar]
- Moumné, L. , Fellous, M. , & Veitia, R. A. (2005). Deletions in the polyAlanine‐containing transcription factor FOXL2 lead to intranuclear aggregation. Human Molecular Genetics, 14, 3557–3564. [DOI] [PubMed] [Google Scholar]
- Oley, C. , & Baraitser, M. (1988). Blepharophimosis, ptosis, epicanthus inversus syndrome (BPES syndrome). Journal of Medical Genetics, 25, 47–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisarska, M. D. , Bae, J. , Klein, C. , & Hsueh, A. J. (2004). Forkhead L2 is expressed in the ovary and represses the promoter activity of the steroidogenic acute regulatory gene. Endocrinology, 145, 3424–3433. [DOI] [PubMed] [Google Scholar]
- Schubert, L. A. , Jeffery, E. , Zhang, Y. , Ramsdell, F. , & Ziegler, S. F. (2001). Scurfin (FOXP3) acts as a repressor of transcription and regulates T cell activation. Journal of Biological Chemistry, 276, 37672–37679. [DOI] [PubMed] [Google Scholar]
- The ESHRE Guideline Group on POI , Webber, L. , Davies, M. , Anderson, R. , Bartlett, J. , Braat, D. , … Vermeulen, N. (2016). ESHRE Guideline: Management of women with premature ovarian insufficiency. Human Reproduction, 31, 926–937. [DOI] [PubMed] [Google Scholar]
- Zlotogora, J. , Sagi, M. , & Cohen, T. (1983). The blepharophimosis, ptosis and epicanthus inversus syndrome: Delineation of two types. American Journal of Human Genetics, 35, 1020–1027. [PMC free article] [PubMed] [Google Scholar]