

Abstract
Improvements in the function, quality of life, and longevity of patients with Duchenne muscular dystrophy (DMD) have been achieved through a multidisciplinary approach to management across a range of health-care specialties. In part 3 of this update of the DMD care considerations, we focus on primary care, emergency management, psychosocial care, and transitions of care across the lifespan. Many primary care and emergency medicine clinicians are inexperienced at managing the complications of DMD. We provide a guide to the acute and chronic medical conditions that these first-line providers are likely to encounter. With prolonged survival, individuals with DMD face a unique set of challenges related to psychosocial issues and transitions of care. We discuss assessments and interventions that are designed to improve mental health and independence, functionality, and quality of life in critical domains of living, including health care, education, employment, interpersonal relationships, and intimacy.
Introduction
This paper is part 3 of an update of the 2010 Duchenne muscular dystrophy (DMD) care considerations.1,2 In parts 1 and 2 of this Review, we provide updated guidance on the management of this severe, progressive neuro-muscular disease across a range of specialties. In part 3, we present considerations for primary care, emergency management, psychosocial care, and transitions of care across the lifespan. With the exception of psychosocial care, these are new additions to the care considerations, the inclusion of which was motivated by feedback from patients, their families, and DMD advocacy groups.
The first point of medical contact for people with DMD is often their primary care provider or, in acute medical situations, the local emergency department. The rarity of the disease means that clinicians in these settings often have little experience with the complications of DMD, making it difficult for them to provide optimum management. The need for improved primary care and emergency management is a key concern for people with DMD.
Many individuals with DMD are now living into their 30s and beyond.3 With prolonged survival, an entirely new set of issues needs to be addressed with regard to mental health, psychosocial care, and transitions to adulthood. These issues cover some of the basic components of human fulfilment and happiness, and span the domains of education, vocation, maturation, independence, personal relationships, emotional health, and intimacy. The importance of these issues reflects a fundamental change in DMD care: recognition that in addition to increasing longevity, helping individuals to achieve their best possible quality of life is an essential part of health care.
The RAND Corporation–University of California Los Angeles Appropriateness Method (RAM), described in part 1, involves the step-wise rating of clinical scenarios for appropriateness, and then for necessity, by a group of experts. However, owing to the paucity of the relevant scientific literature, the RAM process was deemed to be inappropriate for three of the topics in part 3: primary care, emergency management, and transitions of care across the lifespan. For the fourth topic, psychosocial care, a modified RAM method was used, either because the evidence in the scientific literature had not changed significantly since 2010 or because the panel of experts reached timely unanimous agreement on the subtopic under discussion. Therefore, the care considerations in part 3 largely reflect the consensus opinion of expert panellists, based on their translation of the scarce scientific literature into clinical practice. A complete description of the methods is provided in part 1 and the appendix. Figure 1 in part 1 of this Review provides a brief overview of assessments and interventions across all topics, organised by stage of disease, and is intended to serve as a stand-alone pocket guide to care.
Figure 1. Considerations for psychosocial care of individuals with Duchenne muscular dystrophy.

DMD=Duchenne muscular dystrophy.
Primary care
Primary care providers are typically physicians, nurse practitioners, or physician assistants who specialise in paediatrics, family medicine, or internal medicine, and provide a so-called medical home for their patients. The American Academy of Pediatrics describes the medical home as accessible, family centred, continuous, comprehensive, coordinated, compassionate, and culturally effective.4 All patients, particularly those with special needs, should benefit from a medical home characterised by accessibility and continuity of care.5 The expert panel on primary care and emergency management (committee members listed in part 1 of this Review) strongly endorses the concept of a medical home for individuals with DMD, or equivalent models of primary care such as the UK general practitioner model. These providers should work in partnership with the individual’s neuromuscular specialist.
The primary care provider is often the first medical professional to hear a family’s concern about their child’s muscle weakness, initiating the process that leads to a diagnosis of DMD. A guide to the evaluation of a child with neuromotor delay can be found in part 1 of this Review; the ChildMuscleWeakness.org website provides additional resources. A child with hypotonia and an elevated serum creatine kinase level should be referred to a neuromuscular specialist; however, the primary care provider will often be the person to inform the family that DMD is suspected. The possibility of a diagnosis of DMD should be delivered directly but compassionately, bearing in mind that these early medical discussions tend to set the tone for how patients and their families react to the flow of information throughout the diagnostic process. The primary care provider should make the most of the opportunity to establish a strong and trusting relationship with an affected family, which can provide a much-needed source of stability and support as patients interact with a host of specialised medical providers. The primary care provider should be aware of the significant improvements in survival that have been achieved with contemporary DMD management so that the family is not presented with an overly pessimistic view of the child’s prognosis.6
Panel 1 outlines considerations for primary care, summarising the roles of the primary care provider as part of the care team. The goals are for the primary care provider to provide first-line care for acute and chronic medical issues, coordinate care with appropriate specialists, provide trusted advice and continuity of care across the lifespan, and optimise the wellbeing and quality of life of patients and their family members. The recommended structure endorsed by this approach to DMD primary care might be challenged in the context of international variations in health-care delivery. In such circumstances, adaptation of the goals and objectives of the recommended interventions to the local health-care environment is beneficial.
Panel 1. Considerations for primary care of individuals with Duchenne muscular dystrophy.
Immunisation
Administer all non-live-virus vaccinations recommended by the US Centers for Disease Control and Prevention (CDC)7
Aim to give live-virus vaccines before the initiation of steroid treatment; live-virus vaccines are contraindicated in individuals with Duchenne muscular dystrophy (DMD) on high-dose daily corticosteroids (>20 mg per day or >2 mg/kg per day prednisone or equivalent)
Annually administer injectable influenza vaccine to individuals with DMD and all close contacts (do not give the live-virus nasal vaccine, which is contraindicated)
Follow the CDC pneumococcal vaccination schedule,8 integrating PCV13 with PPSV23
Nutrition (see part 1 of this Review)
Ensure that individuals with DMD receive nutritional counselling to prevent obesity and malnutrition
Encourage adequate nutrient intake (especially calcium and vitamin D)
Refer to registered dieticians for nutritional counselling
Dental care
Ensure that individuals with DMD have regular dental care
Ensure that the primary care provider or dentist applies varnish as per protocol9
Ensure that fluoride is supplemented for patients with unfluoridated water
Safety counselling
Raise awareness that individuals with DMD are prone to falls as muscle weakness progresses, especially when they begin to lose ambulation
Consult the multidisciplinary clinical team (including occupational or physical therapist) for appropriate safety practices (eg, use of wheelchair and advice on safety devices) to minimise risk of falls
Emphasise that seatbelts should be worn in motor vehicles at all times; individuals with DMD with poor trunk control might require special positioning devices
Ensure that individuals with DMD who sit in their wheelchairs in motor vehicles are aware that they should secure the wheelchair according to manufacturer guidelines
Monitoring for adrenal insufficiency (see part 1 of this Review)
For individuals with DMD taking corticosteroids:
Educate the family not to miss any doses of prescribed corticosteroid and to be vigilant for signs of adrenal insufficiency (such as lethargy) in association with febrile illnesses, vomiting, surgeries, and other physiological stresses
Supply the patient or family with a stress dose of steroids at home for symptoms of adrenal insufficiency and ensure that they seek immediate medical attention when a stress dose is needed
Psychosocial care for patients and family members
Monitor physical and developmental milestones and be aware of DMD-specific neurodevelopmental and neuropsychological issues, such as the increased prevalence of intellectual disability, attention-deficit hyperactivity disorder, and autism spectrum disorder
Refer to a psychologist for psychological and neuropsychological assessments and interventions when appropriate (see main text)
Refer to a speech-language pathologist for suspected delays
Help the family with special educational needs (eg, in the USA, plans include the Individualized Education Programs and 504 plans)
Identify community resources that might enhance individual and family functioning and coping, such as local social service agencies and patient advocacy organisations
Help to initiate discussions about transitions of care
Ensure that adults with DMD have completed advance directives, when appropriate, and that they have appointed a health-care power of attorney
Other screening
Do standardised screenings such as hearing and vision screening, and screening for mood disorders and substance abuse, on the usual schedule
Screen for cardiovascular risk factors, such as hypertension and hypercholesterolaemia
Emergency management
The local emergency department might be inexperienced in caring for individuals with DMD. To optimise patient outcomes, medical providers need a good background in the issues uniquely relevant to the emergency management of individuals with DMD. Panel 2 provides an overview of key issues related to the emergency care of patients with DMD, and is intended to be a useful pocket guide for emergency medicine providers, primary care clinicians, and neuromuscular specialists. The panel is also a resource for patients, who can take this guide with them when they seek emergency care.
Panel 2. Key issues related to emergency care of patients with Duchenne muscular dystrophy*.
Advance directives, history, and contacts
Determine whether there are restrictions on resuscitation
Ask for the patient’s emergency card and baseline test results, including electrocardiogram results
Obtain a brief history with a focus on baseline respiratory and cardiac status, including use of relevant devices and medications
Determine whether the patient is treated with chronic steroid therapy
Contact the patient’s neuromuscular specialist
Breathing problems
Ask about respiratory symptoms and home equipment
Monitor blood oxygen saturation (SpO2) levels via pulse oximetry; even mild hypoxaemia (SpO2 <95% in room air) is a concern; do a blood gas analysis if necessary
Treat with non-invasive ventilation and frequent application of a cough assistance device (or manual assisted coughing if device is unavailable); use the patient’s home equipment when available
Obtain a portable chest radiograph
Obtain early consultation with a respiratory therapist and respiratory physician
Cardiac problems
Ask about cardiac symptoms
Monitor heart rate and rhythm
Obtain an electrocardiogram (this is typically abnormal and Q waves might be expected) and portable chest radiograph
Measure blood levels of B-type natriuretic peptide or troponin I, or both, as indicated
Consider worsening cardiomyopathy, congestive heart failure, and arrhythmias
Obtain an echocardiogram when necessary
Obtain early consultation with a cardiologist
Endocrine problems
Determine whether stress steroid dosing is necessary
For critical adrenal insufficiency, administer intravenous or intramuscular hydrocortisone: 50 mg for children <2 years old; 100 mg for children ≥2 years and adults
In less critical situations, consult the PJ Nicholoff Steroid Protocol10
Obtain early consultation with an endocrinologist
Orthopaedic problems
Assess for long-bone or vertebral fractures as indicated
Review critical precautions related to sedation and anaesthesia if applicable (see text)
Consider fat embolism syndrome if individual has dyspnoea or altered mental status
Obtain consultation with an orthopaedic specialist early in the process
Disposition after discharge from emergency care
Be aware that most patients will need hospital admission (eg, to initiate or intensify respiratory or cardiac therapy or to manage fractures)
Early in the process, initiate emergency transport by skilled personnel to a centre specialising in the care of patients with Duchenne muscular dystrophy, in cooperation with the individual’s neuromuscular specialist
See appendix for more details.
A detailed discussion of emergency management is presented in the appendix. Additionally, details on respiratory, cardiac, and orthopaedic and surgical management (including fractures, anaesthesia, and sedation) can be found in part 2 of this Review. A detailed discussion of the use of glucocorticoids and steroid-induced adrenal insufficiency in DMD are discussed in part 1; we recommend the PJ Nicholoff Steroid Protocol for stress steroid dosing.10
Psychosocial care
Comprehensive care of individuals with DMD should include surveillance and management of the psychosocial effect of the disease across the lifespan, which now extends well into adulthood for many patients. Many people with DMD are well adjusted to their conditions and the world around them, and lead independent, productive lives. Nevertheless, living with a chronic illness and adjusting to the changes that define the disease require coping strategies and skills, and ongoing emotional support. People with DMD are increasingly recognised to be at risk of delays in cognitive and social development. Comprehensive psychosocial care should therefore address social and cognitive development, as well as quality of life and factors that affect patient and family functioning across environments, including home, school, and work.
Since the 2010 care considerations were published,1,2 dystrophin expression in the brain has been shown to vary depending on the genetic mutation in patients with DMD.11,12 Moreover, smaller total brain volume, smaller grey matter volume, lower white matter fractional anisotropy, and higher white matter mean and radial diffusivity have been shown in an MRI study of people with DMD compared with healthy controls.13 In this study, the subgroup of DMD patients lacking isoform Dp140 made the largest contribution to the difference in grey matter volume. From a functional perspective, the severity of a patient’s cognitive delay seems to vary with the location of the dystrophin gene mutation and with the effect of that mutation on dystrophin isoforms in the patient’s CNS.14–17 A well recognised pattern of cognitive strengths and weaknesses can be observed in the neuropsychological profiles of patients with DMD,18–20 which might warrant diagnosis of a comorbid neurodevelopmental or neuropsychiatric disorder. High rates of intellectual disability (17–27%), learning disabilities (26%), autism spectrum disorder (15%), attention-deficit hyperactivity disorder (32%), and anxiety (27%) have been reported in people with DMD.17,21–23 Learning difficulties involving language-based skills, such as reading, are also more common than other learning difficulties in this population.24 Furthermore, increased attention has been paid to the course of emotional and social functioning in individuals with DMD over the lifetime.25 The care considerations have been updated to emphasise appropriate screening and assessment of these conditions, and to discuss interventions that might be necessary for individuals with DMD.
Psychosocial support should be developed and implemented across the lifespan in a manner that promotes thinking about the future and sets expectations that individuals will actively participate in their care and daily activities. The key components needed to promote psychosocial health of patients and their families are outlined in panel 3 and described in more detail below. Additionally, the panel’s care considerations for psychosocial care by stage of disease are presented in figure 1.
Panel 3. Components of clinic-based psychosocial care of patients with Duchenne muscular dystrophy.
Care coordination
The care coordinator is a point of contact for patients with Duchenne muscular dystrophy (DMD) and families; they should be health professionals with sufficient training or experience in the clinical care of patients with DMD
The role of the care coordinator is to provide information, coordinate (and possibly schedule) appointments, and facilitate communication with clinicians across disciplines
Routine mental health screening
At each neuromuscular clinic visit, mental health and quality of life should be screened
Screening can be informal and does not require comprehensive assessment
An appropriate tool for paediatric patients is the Strengths and Difficulties Questionnaire;26 for adult patients, the Patient Health Questionnaire 9-item depression scale (PHQ-9)27 and the Generalized Anxiety Disorder 7-item scale (GAD-7)28 are appropriate; for parents of patients aged 5–17 years, the Personal Adjustment and Role Skills Scale (PARSIII) is suitable29,30 (scale and scoring programme is available on the Parent Project Muscular Dystrophy website)
Screening can be conducted by a social worker or mental health professional or by other clinic staff with sufficient training or experience in this area (eg, a nurse or attending physician)
If screening is positive, a referral should be made to a psychologist and psychiatrist for further assessment or treatment
Every clinic should have a plan to assess and address suicidal ideation or other acute safety concerns
Caregiver emotional adjustment should be monitored and intervention or support offered as needed
Siblings of a person with DMD should be provided with opportunities to connect with other siblings of patients with DMD and with access to mental health services as needed
Psychological care
All individuals with DMD should be expected to live rich, fulfilling lives, and those without additional neurodevelopmental or psychological disorders may achieve a high level of independence in managing their disease; however, all patients and their families might need psychosocial support
The neuromuscular care team should include a mental health professional (ie, psychologist or psychiatrist) with training and experience in assessing and treating psychiatric conditions in the context of chronic medical or neurodevelopmental conditions
When mental health concerns are identified, the mental health professional should provide further evaluation of individuals with DMD and their family members, and provide cognitive or behavioural interventions to treat psychiatric conditions
Standard, evidence-based practices should be used for those who need more formal mental health treatment
Neuropsychological evaluations should be done when cognitive delays, difficulties with emotional and behavioural regulation, or concerns about social skills exist; re-evaluations should be done every 2–3 years to monitor developmental progress and response to interventions
Neuropsychological evaluations should be considered within the first year of diagnosis to establish a baseline, or when transitioning to adulthood if government-based assistance might be necessary to promote functional independence
Pharmacological interventions
The neuromuscular team should include a psychiatrist or other physician with training and experience in providing medication to treat behavioural or emotional disorders in the context of chronic medical or neurodevelopmental conditions
Standard prescribing practices should be followed
Selective serotonin-reuptake inhibitors should be prescribed for depression, anxiety, and obsessive–compulsive disorder
α-Adrenoceptor agonists (first choice) or atypical antipsychotics (second choice) should be prescribed for aggression and anger or emotional dysregulation
Stimulants or α-adrenoceptor agonists should be prescribed for attention-deficit hyperactivity disorder
Psychological care
Patients and their family members are at increased risk of depression and anxiety, particularly at major care transition points in the progression of the disease. The neuromuscular care team should include a mental health clinician (ie, psychologist, psychiatrist, social worker, or psychiatric nurse) who has specialised training and experience in assisting families and individuals with chronic medical or neurodevelopmental conditions. At each neuromuscular clinic visit, mental health and quality of life should be screened. An appropriate tool for paediatric patients is the Strengths and Difficulties Questionnaire,26 which is available in multiple languages. For adult patients, the Patient Health Questionnaire 9-item depression scale27 and the Generalized Anxiety Disorder 7-item scale28 are recommended. Finally, for parents of patients aged 5–17 years, the Personal Adjustment and Role Skills Scale is recommended (scale and scoring programme is available on the Parent Project Muscular Dystrophy website).29,30 If screening is positive, the mental health clinician should be engaged for further assessment. At a minimum, each clinic should have a plan to address suicidality. Formal annual evaluation by the mental health clinician is advised to assess the psychological wellbeing of the individual, parents, and siblings. When formal treatment of psychological disorders is needed, standard evidence-based practices should be used, ideally those applied in populations with chronic medical conditions.
Psychopharmacological interventions should be considered for treatment of moderate-to-severe psychiatric symptoms or for milder symptoms when nonpharmacological interventions are not effective. Standard prescribing practices and guidelines should apply, with additional care considerations focused on the individual’s general medical condition. Depending on the patient’s age and disease status, special attention to cardiac status, medication interactions, and side-effects when combined with existing medications (eg, weight gain) might be needed. Comprehensive assessment is essential and should generally include neuropsychological testing, given the prevalence of learning issues and the frequency of comorbidities that can complicate diagnosis (eg, attention-deficit hyperactivity disorder in combination with anxiety and learning disabilities).
Close monitoring with routine follow-up is advised, because both psychiatric presentation and relevant medical issues change over time (eg, emerging inattention as educational demands increase, or development of cardiac disease). Progression of cardiac disease, in particular, might affect decisions related to use of stimulants, atypical antipsychotics, and some antidepressants. Close collaboration with medical specialists (cardiology and neurology) is essential in helping individuals and families weigh the risks and benefits of therapy. For paediatric patients (ages 2–18 years), clinics might consider using the Pediatric Outcomes Questionnaire to track health-related quality of life over time (available through the American Academy of Orthopaedic Surgeons website).
Neuropsychological evaluations
Referral for neuropsychological evaluations should be considered at diagnosis, but is essential when concerns about developmental progress arise. A neuropsychologist is a clinical psychologist who has specialised training and experience in the formal assessment of cognitive and psychological functions. By observation and implementation of standardised tests, a neuropsychologist can assess a child’s cognitive development, academic skills (or school readiness for younger individuals), social functioning, emotional adjustment, and behavioural regulation. In the USA, school districts often do similar testing, but the scope of such testing is usually narrower than that done by neuropsychologists and results might not be interpreted with appropriate reference to the medical context. A comprehensive assessment allows targeted recommendations, and neuropsychologists should work with families to develop intervention plans that can be implemented at home and at school. Re-evaluations can be done every 2–3 years to monitor a child’s developmental progress and response to interventions, but are often warranted when acute change in functioning occurs, new concerns arise, or a major transition involving home or school occurs.
Educational support
DMD will affect a student’s ability to consistently access his educational environment. Accommodations should be provided to maximise a child’s ability to function normally with his peers. With the support of occupational and physical therapy, educational activities and services might need modification on the basis of a child’s physical functioning. Accommodations should also be made for frequent medically related absences. In children who have cognitive, behavioural, or learning needs, additional services should be considered, such as speech and language therapy, applied behaviour analysis, or specialised academic instruction. Delivery and goals of these services should be formalised. In the USA, access to special education accommodations and services is mandated by federal legislation under the Individuals with Disabilities Education Act, which requires a formal educational plan. The plan should be updated annually to reflect a child’s changing needs and developmental progress.
Transitions of care across the lifespan
People with DMD need education and support to make transitions throughout the lifespan. In particular, strategies are needed to help people with DMD to navigate the transition from adolescence to adult life. At ages when adolescents and young adults generally have a desire for greater independence, those with DMD often have increasing health-care needs and physical reliance on others for activities of daily living, which can create challenges in making a successful transition to adult lifestyles. Resources, such as funding, equipment, and care personnel to meet physical needs, can be hard to identify, limited in availability, or difficult to acquire. Some young adults with DMD might experience psychosocial hardships or neurocognitive impairments that affect autonomy and independence. However, for most individuals, full participation in future planning and decision making should be an expectation once proper support and resources are identified and in place.
The 2010 care considerations did not specifically address care transitions. Several conference and workshop summaries have reported on transitions into adult life with DMD, and surveys on experiences of individuals with DMD have indicated deficits in transition guidance and planning.31–35 However, research regarding successful transition interventions and outcomes is scarce. In developing the current DMD care considerations, the expert panel on transitions of care reviewed the following: published position papers on transition; literature regarding adolescents and young adults with DMD; and transition tools published by advocacy groups, private organisations, and governmental agencies.36–40 An overview of transition planning is shown in figure 2.
Figure 2. Components of young adulthood to be addressed during transition planning for individuals with Duchenne muscular dystrophy.
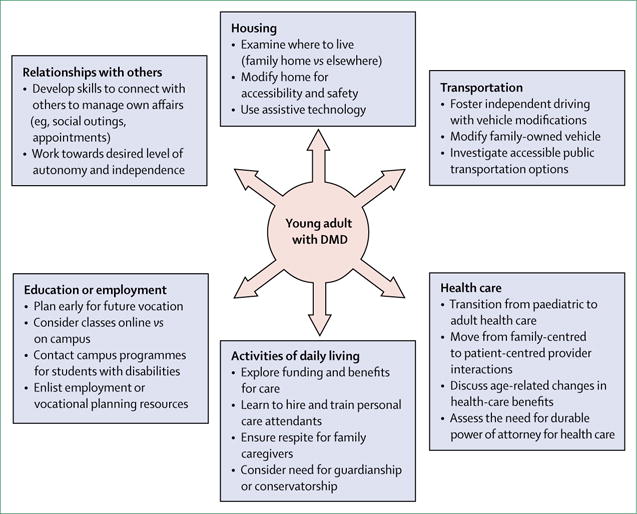
DMD=Duchenne muscular dystrophy.
Timing of transition planning
Beginning in early childhood, providers, educators, and parents should strive to engage young people with DMD in their care and future planning. In alignment with recommendations of the American Academy of Pediatrics, the American Academy of Family Physicians, and the American College of Physicians, patients and their families should be made aware of plans for health-care transition by the time the patient is 12 years old, with initiation of transition discussions and planning by age 13–14 years.36 This plan should include services that need to be provided, providers of these services, and details of how they will be financed. The plan should be based on the needs, wants, and values of the individual with DMD and his family. The plan should be reviewed and updated annually.
In England, Education, Health and Care (EHC) plans have been in use since 2014 for young people (0–25 years old) who have special educational needs or disabilities. EHC plans are produced by local authorities, with input from affected individuals and their family members, and include outcomes in education or employment, independent living, health, and social inclusion.
Care coordination
A care coordinator or social worker should be responsible for transition planning, without which care can become fragmented and recommendations can go unheeded, such that an individual’s needs are not met.29 The care coordinator can serve as a central resource for questions; facilitate communication between providers and the family; navigate insurance benefit eligibility; assist with acquisition of prescribed equipment; identify community care agencies; advocate for students in their schools; ensure transmission of medical records; identify and protect financial resources; and enlist the help of social care systems, such as human service organisations, specialty clinics, and advocacy groups (eg, Parent Project Muscular Dystrophy, the Muscular Dystrophy Association [MDA], Action Duchenne, Muscular Dystrophy UK, and DMD Pathfinders).
Health care
Transition planning should include a plan for continuity of health care with paediatric providers (primary care and subspecialists) until adult care is established. On the basis of the availability of providers in the community, the make-up of the health-care team might vary, but multidisciplinary care should continue throughout adult life.41 The transition coordinator should facilitate the adolescent’s self-management of his health care, facilitate referrals to appropriate adult providers, and ensure transfer of medical records.
From a young age, people with DMD know that they are living with a progressive disease and they should be included in health-care discussions. When an individual shows an interest and ability to self-advocate in a variety of settings and when he participates in discussions about his care and needs, it is appropriate to move from family-centred to patient-centred interactions.38,42 At least annually, the young person with DMD, his medical providers, and his caregivers should engage in developmentally appropriate discussions about long-term care goals. Time alone with at least one provider should begin in early adolescence. Relevant tools include the MDA Roadmap to Independence,43 which offers anticipatory guidance about health-related concepts in the later stages of DMD.
Adults with DMD have voiced concerns that issues of prognosis, longevity, and preparations for medical decision making are not discussed early and often enough.35 People who know the expected course of DMD can provide insight into health-care decisions that are typically part of this progression, such as those related to intensive care for illness, ventilation options, and management of heart failure. Major physical changes—including loss of ambulation, development of scoliosis, and deterioration in respiratory or cardiac function—should prompt re-evaluation of care goals. Providers should prepare patients and their families for anticipated complications, so that potential interventions are considered in advance of acute medical developments and crises. Anticipatory discussions can be empowering for patients, and can promote self-determination based on personal values and goals of care.
Palliative care consultation can be useful at various times throughout the lifespan of a person with DMD: at diagnosis, at the time of major treatment decisions, during life-threatening events, and at the terminal phase of care.44 The Vision of Hope curriculum45 integrates the principles of paediatric palliative care into the care of individuals with DMD. When the goal of care is comfort and death with dignity, hospice services should be offered to provide emotional support and physical care for patients and their families.
Advance care planning involves decisions about the care one would want to receive if unable to speak for oneself, based on personal values and preferences.46 This planning is particularly important in DMD, given that the clinical course can be characterised by periods of stability interspersed with episodes of medical crisis.46 Advance care planning tools, such as “Voicing my choices”,47 allow individuals with DMD to express how they prefer to be comforted, supported, treated, and remembered. Laws vary, but in many places, minors can participate in formulating advance directives. Anyone aged 18 years or older should have a designated health-care power of attorney. Providers should offer a format for these discussions, either in their clinics or by referral to advance care planning experts.
Education, employment, and pursuit of vocations
Education planning meetings should start when a person with DMD is around 13 years old and occur at least annually; these meetings should include a focus on the student’s needs and goals, assessment of personal strengths and interests, and assessment of physical challenges in the school environment. Challenges typically include acquisition of necessary equipment and technology, modifications for accessibility and transportation, and establishment of a plan that balances medical needs with practical issues associated with school attendance.
Examples of additional resources for education-related transition planning are provided in the appendix. These resources include government agencies and services accessed in educational settings or through employers. Additionally, several advocacy groups (eg, the Muscular Dystrophy Association Young Adult Programs48) offer planning strategies, peer-to-peer advice, moderated online discussion forums, support networks, and web pages.
Not all people with DMD seek education beyond high school, and generally accepted aims of transition programming towards adult roles need to be considered for each individual.49 The particular strengths and talents of individuals should be catalogued to create a programme of meaningful, rewarding daily activities. Any viable plan should take into account the necessary support structure, including availability of relevant financial, community, educational or vocational, technological, and caregiving resources. For patients with intellectual disabilities, government-funded or private-funded employment opportunities might exist. For a young person who is not ready or able to participate fully in adult activities, families might need guidance on guardianship or conservatorship.
Housing, transportation, and assistance with activities of daily living
As teenagers with DMD enter young adulthood, they should be encouraged to explore options for independent living. They will need accessible housing and, probably, assistance with activities of daily living. Transportation issues affect an individual’s autonomy and independence, opportunities for employment and education, and social activities. Health-care providers should discuss options for safe transportation, including independent driving with vehicle modifications, modifications to family-owned vehicles, and use of public transportation. Considerations for housing, transportation, and care are outlined in the appendix.
Although a family member is often the most trusted source of caregiving, caregiver fatigue, ageing of the caregiver, and altered family dynamics are reasons to hire personal care attendants, and to establish a respite plan that starts in the teenage years.50 Without respite, family caregivers can experience loss in their personal life, leisure activities, and connections to the community.51,52 Alternative providers should be instructed on daily routines and proper transfer techniques in anticipation of respite needs and emergency support.53
Because of the fragmented nature of disability benefits systems, adults with DMD need to have considerable financial aptitude. Families of teenagers and young adults with DMD should learn details of their private insurance, state or regional programmes, and national social support systems. Adult health care and services are often scarce, and a number of organisations are involved in initiatives to address the resource gaps within the adult DMD community.
Relationships with others
Social connections are crucial for health, wellbeing, recreation, and quality of life. As young people graduate from high school, established peer groups often dissolve, and physical mobility becomes more difficult for people with DMD. Opportunities for personal contact and social engagement happen less naturally and become a rarer commodity for young adults with DMD.53 Suggested steps to create and maintain community connections through social groups and leisure activities are outlined in the appendix. Although participation is common, it is our consensus opinion that excessive time devoted to computer games can become socially isolating because young men with DMD remain at home, limiting their interactions with family and friends.
Dating, intimacy, and sexuality are high-priority topics among men with DMD. Health-care providers should initiate discussions about these topics, as developmentally appropriate (appendix). At-home discussions about relationships, dating, sexual orientation, and marriage are complex and potentially awkward for young adults with DMD, in part due to their dependence on family members for all aspects of physical care. Thus, providers should encourage and facilitate such discussions, including at the time of medical appointments. Potential topics for questions and answers include masturbation, having sex with a partner, and fathering children. It is common for individuals with more advanced cardiorespiratory disease, or with a history of fragile bones, to be concerned about changes in heart rate, blood pressure, respiration, and injury in the context of acts of intimacy. Referrals might be needed outside the clinic for more appropriate counselling and follow-up, since not all medical settings offer a secure, comfortable environment for such discussions.
Conclusions and future directions
Primary care, emergency management, and transitions of care across the lifespan are new additions to the DMD care considerations. Primary care and emergency medicine clinicians with a special interest in DMD are rare, and these areas have been understudied. As a result, our care considerations are based primarily on the consensus opinion of expert panellists. Elucidation of best practices in primary care and emergency management would improve quality of care and greatly benefit patients with DMD. More resources will need to be devoted to clinician education and to the support of relevant clinical networks, initiatives, and research to achieve this goal.
Neurodevelopmental and neuropsychiatric conditions are increasingly recognised to be common in DMD; however, the prevalence and optimum treatment of these disorders are not well characterised. We hope that by formally integrating psychosocial care within the neuromuscular clinic, these issues will be more effectively addressed and better understood by clinical providers. Continued study on the development, validation, and application of psychosocial measurement tools and interventions for DMD is needed to further establish evidence-based best practices. Improved psychosocial care could help to reduce disease complications, improve patient quality of life, and reduce caregiver burden.
More emphasis is needed on measures to help people with DMD to attain appropriate levels of education, find employment, and achieve autonomous, fulfilling lifestyles and full societal inclusion. As with many other disabling chronic conditions, adolescents and adults with DMD face obstacles to full participation in many aspects of life. The issue of transitions of care requires substantially more study and the allocation of more medical, psychosocial, and societal resources. Once best practices for transition are established, relevant resources should be made accessible so that patients and their families can derive maximum benefit.
Supplementary Material
Acknowledgments
We thank Sharon Barrell and Danielle Hennis (RTI International) for editorial and graphical support, respectively. We thank Julie Bolen (US Centers for Disease Control and Prevention [CDC]) and Elizabeth Pinsky (Massachusetts General Hospital and Harvard Medical School) for their contribution to the design and conduct of the project and review of the manuscript. This work was supported by CDC contract 200-2007-22644-023. The CDC provided honoraria and travel expenses for committee members to attend an in-person meeting. Funding was provided under the Muscular Dystrophy Care and Treatment Act, legislation that calls for cooperation of the CDC with professional organisations and the patient community in the development, issuance, and periodic review and update of care considerations for Duchenne muscular dystrophy. The findings and conclusions presented in this paper are those of the authors and do not necessarily represent the official position of the CDC.
Footnotes
Contributors
DJB, KB, CMB, SDA, AB, MKC, LC, ARH, AK, KK, JN, GN, JP, NS, CJT, DRW, and LMW provided intellectual expertise in the study design, generation and interpretation of data, review of the literature, writing of the article, and the decision to publish. DJB, aided by CMB and NS, drafted and edited the article and approved the final version.
Declaration of interests
DJB was a paid consultant for Hill-Rom Corporation and has US patents (8651107, 8844530, and 9795752) for respiratory devices, as well as related international patents and patent applications. KB was a consultant for Solid Ventures, Catabasis, LGC Ltd, Bristol Myers Squibb, PTC Therapeutics, GLC Research, Eli Lilly, and Publicis Life Brands Resolute; she has received grant support from PTC Therapeutics. SDA is a principal investigator for multicentre clinical trials sponsored by PTC Therapeutics and Sarepta Pharmaceuticals. MKC received an honorarium from Parent Project Muscular Dystrophy. DRW is a paid consultant for Health Research Inc and Marathon Pharmaceuticals. LMW has received grant support and honoraria from Novartis and Amgen. All other authors declare no competing interests.
Contributor Information
David J Birnkrant, Department of Pediatrics, MetroHealth Medical Center, Case Western Reserve University, Cleveland, OH, USA.
Katharine Bushby, John Walton Muscular Dystrophy Research Centre, Institute of Genetic Medicine, Newcastle University, Newcastle upon Tyne, UK.
Carla M Bann, RTI International, Research Triangle Park, NC, USA.
Susan D Apkon, Department of Rehabilitation Medicine, Seattle Children’s Hospital, Seattle, WA, USA.
Angela Blackwell, RTI International, Research Triangle Park, NC, USA.
Mary K Colvin, Massachusetts General Hospital and Harvard Medical School, Boston, MA, USA.
Linda Cripe, Department of Pediatrics, Nationwide Children’s Hospital, The Ohio State University, Columbus, OH, USA.
Adrienne R Herron, Rare Disorders and Health Outcomes Team, National Center on Birth Defects and Developmental Disabilities, Centers for Disease Control and Prevention, Atlanta, GA, USA; Oak Ridge Institute for Science and Education, Oak Ridge, TN, USA.
Annie Kennedy, Parent Project Muscular Dystrophy, Hackensack, NJ, USA.
Kathi Kinnett, Parent Project Muscular Dystrophy, Hackensack, NJ, USA.
James Naprawa, Emergency Department, University of California San Francisco Benioff Children’s Hospital, Oakland, CA, USA.
Garey Noritz, Department of Pediatrics, Nationwide Children’s Hospital, The Ohio State University, Columbus, OH, USA.
James Poysky, Baylor College of Medicine, Houston, TX, USA.
Natalie Street, Rare Disorders and Health Outcomes Team, National Center on Birth Defects and Developmental Disabilities, Centers for Disease Control and Prevention, Atlanta, GA, USA.
Christina J Trout, Stead Family Department of Pediatrics, University of Iowa, Iowa City, IA, USA.
David R Weber, Division of Endocrinology and Diabetes, Golisano Children’s Hospital, University of Rochester Medical Center, Rochester, NY, USA.
Leanne M Ward, Division of Endocrinology and Metabolism, Children’s Hospital of Eastern Ontario, and University of Ottawa, Ottawa, ON, Canada.
References
- 1.Bushby K, Finkel R, Birnkrant DJ, et al. for the DMD Care Considerations Working Group Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9:77–93. doi: 10.1016/S1474-4422(09)70271-6. [DOI] [PubMed] [Google Scholar]
- 2.Bushby K, Finkel R, Birnkrant DJ, et al. for the DMD Care Considerations Working Group Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol. 2010;9:177–89. doi: 10.1016/S1474-4422(09)70272-8. [DOI] [PubMed] [Google Scholar]
- 3.Saito T, Kawai M, Kimura E, et al. Study of Duchenne muscular dystrophy long-term survivors aged 40 years and older living in specialized institutions in Japan. Neuromuscul Disord. 2017;27:107–14. doi: 10.1016/j.nmd.2016.11.012. [DOI] [PubMed] [Google Scholar]
- 4.Medical Home Initiatives for Children With Special Needs Project Advisory Committee, American Academy of Pediatrics. The medical home. Pediatrics. 2002;110(1 Pt 1):184–86. [Google Scholar]
- 5.Homer CJ, Klatka K, Romm D, et al. A review of the evidence for the medical home for children with special health care needs. Pediatrics. 2008;122:e922–37. doi: 10.1542/peds.2007-3762. [DOI] [PubMed] [Google Scholar]
- 6.Passamano L, Taglia A, Palladino A, et al. Improvement of survival in Duchenne muscular dystrophy: retrospective analysis of 835 patients. Acta Myol. 2012;31:121–25. [PMC free article] [PubMed] [Google Scholar]
- 7.Centers for Disease Control and Prevention Immunization schedules. 2017 https://www.cdc.gov/vaccines/schedules/ (accessed Jan 12, 2018).
- 8.Immunization Action Coalition. Ask the experts: diseases and vaccines. 2017 http://www.immunize.org/askexperts/experts_pneumococcal_vaccines.asp#ppsv23_rec (accessed Jan 12, 2018).
- 9.Clark MB, Slayton RL, Section on Oral Health Fluoride use in caries prevention in the primary care setting. Pediatrics. 2014;134:626–33. doi: 10.1542/peds.2014-1699. [DOI] [PubMed] [Google Scholar]
- 10.Kinnett K, Noritz G. The PJ Nicholoff Steroid Protocol for Duchenne and Becker muscular dystrophy and adrenal suppression. PLoS Curr. 2017;9 doi: 10.1371/currents.md.d18deef7dac96ed135e0dc8739917b6e. ecurrents.md. d18deef7dac96ed135e0dc8739917b6e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ricotti V, Mandy WP, Scoto M, et al. Neurodevelopmental, emotional, and behavioural problems in Duchenne muscular dystrophy in relation to underlying dystrophin gene mutations. Dev Med Child Neurol. 2016;58:77–84. doi: 10.1111/dmcn.12922. [DOI] [PubMed] [Google Scholar]
- 12.Young HK, Barton BA, Waisbren S, et al. Cognitive and psychological profile of males with Becker muscular dystrophy. J Child Neurol. 2008;23:155–62. doi: 10.1177/0883073807307975. [DOI] [PubMed] [Google Scholar]
- 13.Doorenweerd N, Straathof CS, Dumas EM, et al. Reduced cerebral gray matter and altered white matter in boys with Duchenne muscular dystrophy. Ann Neurol. 2014;76:403–11. doi: 10.1002/ana.24222. [DOI] [PubMed] [Google Scholar]
- 14.D’Angelo MG, Lorusso ML, Civati F, et al. Neurocognitive profiles in Duchenne muscular dystrophy and gene mutation site. Pediatr Neurol. 2011;45:292–99. doi: 10.1016/j.pediatrneurol.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taylor PJ, Betts GA, Maroulis S, et al. Dystrophin gene mutation location and the risk of cognitive impairment in Duchenne muscular dystrophy. PLoS One. 2010;5:e8803. doi: 10.1371/journal.pone.0008803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wingeier K, Giger E, Strozzi S, et al. Neuropsychological impairments and the impact of dystrophin mutations on general cognitive functioning of patients with Duchenne muscular dystrophy. J Clin Neurosci. 2011;18:90–95. doi: 10.1016/j.jocn.2010.07.118. [DOI] [PubMed] [Google Scholar]
- 17.Sarrazin E, von der Hagen M, Schara U, von Au K, Kaindl AM. Growth and psychomotor development of patients with Duchenne muscular dystrophy. Eur J Paediatr Neurol. 2014;18:38–44. doi: 10.1016/j.ejpn.2013.08.008. [DOI] [PubMed] [Google Scholar]
- 18.Cotton S, Crowe SF, Voudouris N. Neuropsychological profile of Duchenne muscular dystrophy. Child Neuropsychol. 1998;4:110–17. [Google Scholar]
- 19.Perumal AR, Rajeswaran J, Nalini A. Neuropsychological profile of Duchenne muscular dystrophy. Appl Neuropsychol Child. 2015;4:49–57. doi: 10.1080/21622965.2013.802649. [DOI] [PubMed] [Google Scholar]
- 20.Hinton VJ, De Vivo DC, Nereo NE, Goldstein E, Stern Y. Poor verbal working memory across intellectual level in boys with Duchenne dystrophy. Neurology. 2000;54:2127–32. doi: 10.1212/wnl.54.11.2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pane M, Lombardo ME, Alfieri P, et al. Attention deficit hyperactivity disorder and cognitive function in Duchenne muscular dystrophy: phenotype–genotype correlation. J Pediatr. 2012;161:705–09.e1. doi: 10.1016/j.jpeds.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 22.Hendriksen JG, Vles JS. Neuropsychiatric disorders in males with Duchenne muscular dystrophy: frequency rate of attention-deficit hyperactivity disorder (ADHD), autism spectrum disorder, and obsessive–compulsive disorder. J Child Neurol. 2008;23:477–81. doi: 10.1177/0883073807309775. [DOI] [PubMed] [Google Scholar]
- 23.Banihani R, Smile S, Yoon G, et al. Cognitive and neurobehavioral profile in boys with Duchenne muscular dystrophy. J Child Neurol. 2015;30:1472–82. doi: 10.1177/0883073815570154. [DOI] [PubMed] [Google Scholar]
- 24.Hendriksen JG, Vles JS. Are males with Duchenne muscular dystrophy at risk for reading disabilities? Pediatr Neurol. 2006;34:296–300. doi: 10.1016/j.pediatrneurol.2005.08.029. [DOI] [PubMed] [Google Scholar]
- 25.Snow WM, Anderson JE, Jakobson LS. Neuropsychological and neurobehavioral functioning in Duchenne muscular dystrophy: a review. Neurosci Biobehav Rev. 2013;37:743–52. doi: 10.1016/j.neubiorev.2013.03.016. [DOI] [PubMed] [Google Scholar]
- 26.Goodman R. The strengths and difficulties questionnaire: a research note. J Child Psychol Psychiatry. 1997;38:581–86. doi: 10.1111/j.1469-7610.1997.tb01545.x. [DOI] [PubMed] [Google Scholar]
- 27.Kroenke K, Spitzer RL, Williams JB. The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med. 2001;16:606–13. doi: 10.1046/j.1525-1497.2001.016009606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spitzer RL, Kroenke K, Williams JB, Lowe B. A brief measure for assessing generalized anxiety disorder: the GAD-7. Arch Intern Med. 2006;166:1092–97. doi: 10.1001/archinte.166.10.1092. [DOI] [PubMed] [Google Scholar]
- 29.Hendriksen JG, Poysky JT, Schrans DG, Schouten EG, Aldenkamp AP, Vles JS. Psychosocial adjustment in males with Duchenne muscular dystrophy: psychometric properties and clinical utility of a parent-report questionnaire. J Pediatr Psychol. 2009;34:69–78. doi: 10.1093/jpepsy/jsn067. [DOI] [PubMed] [Google Scholar]
- 30.Stein RE, Jessop DJ. Functional status II(R). A measure of child health status. Med Care. 1990;28:1041–55. doi: 10.1097/00005650-199011000-00006. [DOI] [PubMed] [Google Scholar]
- 31.Rahbek J, Steffensen BF, Bushby K, de Groot IJ. 206th ENMC International Workshop: Care for a novel group of patients—adults with Duchenne muscular dystrophy. Naarden, The Netherlands, 23–25 May 2014. Neuromuscul Disord. 2015;25:727–38. doi: 10.1016/j.nmd.2015.05.005. [DOI] [PubMed] [Google Scholar]
- 32.Gibson BE, Young NL, Upshur RE, McKeever P. Men on the margin: a Bourdieusian examination of living into adulthood with muscular dystrophy. Soc Sci Med. 2007;65:505–17. doi: 10.1016/j.socscimed.2007.03.043. [DOI] [PubMed] [Google Scholar]
- 33.Abbott D, Carpenter J, Bushby K. Transition to adulthood for young men with Duchenne muscular dystrophy: research from the UK. Neuromuscul Disord. 2012;22:445–46. doi: 10.1016/j.nmd.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 34.Rahbek J, Werge B, Madsen A, Marquardt J, Steffensen BF, Jeppesen J. Adult life with Duchenne muscular dystrophy: observations among an emerging and unforeseen patient population. Pediatr Rehabil. 2005;8:17–28. doi: 10.1080/13638490400010191. [DOI] [PubMed] [Google Scholar]
- 35.Schrans DGM, Abbott D, Peay HL, et al. Transition in Duchenne muscular dystrophy: an expert meeting report and description of transition needs in an emergent patient population: (Parent Project Muscular Dystrophy Transition Expert Meeting 17–18 June 2011, Amsterdam, The Netherlands) Neuromuscul Disord. 2013;23:283–86. doi: 10.1016/j.nmd.2012.08.009. [DOI] [PubMed] [Google Scholar]
- 36.American Academy of Pediatrics, American Academy of Family Physicians, American College of Physicians-American Society of Internal Medicine. A consensus statement on health care transitions for young adults with special health care needs. Pediatrics. 2002;110(6 Pt 2):1304–06. [PubMed] [Google Scholar]
- 37.Rosen DS, Blum RW, Britto M, Sawyer SM, Siegel DM, Society for Adolescent Medicine Transition to adult health care for adolescents and young adults with chronic conditions: position paper of the Society for Adolescent Medicine. J Adolesc Health. 2003;33:309–11. doi: 10.1016/s1054-139x(03)00208-8. [DOI] [PubMed] [Google Scholar]
- 38.Got Transition. 2014–2017 http://www.gottransition.org/ (accessed Aug 30, 2017).
- 39.Muscular Dystrophy Campaign. Guide to transition for 13-25 year olds with muscle disease. 2010;(1) http://www.musculardystrophyuk.org/app/uploads/2015/02/transition-guide-factsheets-web.pdf (accessed Aug 30, 2017).
- 40.Muscular Dystrophy Association. Young adult programs. https://www.mda.org/young-adults (accessed Aug 30, 2017).
- 41.Wagner KR, Lechtzin N, Judge DP. Current treatment of adult Duchenne muscular dystrophy. Biochim Biophys Acta. 2007;1772:229–37. doi: 10.1016/j.bbadis.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 42.Sawicki GS, Lukens-Bull K, Yin X, et al. Measuring the transition readiness of youth with special healthcare needs: validation of the TRAQ—Transition Readiness Assessment Questionnaire. J Pediatr Psychol. 2011;36:160–71. doi: 10.1093/jpepsy/jsp128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muscular Dystrophy Association. MDA road map to independence for young adults: a guide to becoming your own advocate and living successfully with neuromuscular disease. https://www.mda.org/sites/default/files/publications/Road_Map_to_Independence.pdf (accessed Aug 30, 2017).
- 44.National Hospice and Palliative Care Organization. Advance care planning. 2017 http://www.nhpco.org/advance-care-planning (accessed Aug 30, 2017).
- 45.Johns Hopkins Berman Institute of Bioethics. Vision of hope: integration of palliative care in chronic pediatric disease. http://www.bioethicsinstitute.org/research/projects-2/hope?doing_wp_cron=1447434377.8737630844116210937500 (accessed Aug 30, 2017).
- 46.Ruston C, Erby L, Cohn R, Geller G. Integrating palliative care in life-limiting pediatric neuromuscular conditions: the case of SMA-type 1 and Duchenne muscular dystrophy. J Palliat Care Med. 2012;2:1. [Google Scholar]
- 47.Aging with dignity. Voicing my choices. 2017 https://agingwithdignity.org/shop/product-details/voicing-my-choices (accessed Aug 30, 2017)
- 48.Muscular Dystrophy Association. Young adult programs. 2017 https://www.mda.org/young-adults (accessed Jan 19, 2018).
- 49.Hamdani Y, Mistry B, Gibson BE. Transitioning to adulthood with a progressive condition: best practice assumptions and individual experiences of young men with Duchenne muscular dystrophy. Disabil Rehabil. 2015;37:1144–51. doi: 10.3109/09638288.2014.956187. [DOI] [PubMed] [Google Scholar]
- 50.Yamaguchi M, Suzuki M. Becoming a back-up carer: parenting sons with Duchenne muscular dystrophy transitioning into adulthood. Neuromuscul Disord. 2015;25:85–93. doi: 10.1016/j.nmd.2014.09.001. [DOI] [PubMed] [Google Scholar]
- 51.Magliano L, D’Angelo MG, Vita G, et al. Psychological and practical difficulties among parents and healthy siblings of children with Duchenne vs. Becker muscular dystrophy: an Italian comparative study. Acta Myol. 2014;33:136–43. [PMC free article] [PubMed] [Google Scholar]
- 52.Magliano L, Patalano M, Sagliocchi A, et al. Burden, professional support, and social network in families of children and young adults with muscular dystrophies. Muscle Nerve. 2015;52:13–21. doi: 10.1002/mus.24503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pangalila RF, van den Bos GA, Stam HJ, van Exel NJ, Brouwer WB, Roebroeck ME. Subjective caregiver burden of parents of adults with Duchenne muscular dystrophy. Disabil Rehabil. 2012;34:988–96. doi: 10.3109/09638288.2011.628738. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.