

Abstract
Mapping the protein-protein interaction (PPi) landscape is of critical importance to furthering our understanding how cells and organisms function. Optogenetic methods, that is, approaches that utilize genetically encoded fluorophores or fluorogenic enzyme reactions, uniquely enable the visualization of biochemical phenomena in live cells with high spatial and temporal accuracy. Applying optogenetic methods to the detection of PPis requires the engineering of protein-based systems in which an optical signal undergoes a substantial change when the two proteins of interest interact. In recent years, researchers have developed a number of creative and effective optogenetic methods that achieve this goal, and used them to further elaborate our map of the PPi landscape. In this review we provide an introduction to the general principles of optogenetic PPi detection, and then provide a number of representative examples of how these principles have been applied. We have organized this review by categorizing methods based on whether the signal generated is reversible or irreversible in nature, and whether the signal is localized or non-localized at the subcellular site of the PPi. We discuss these techniques giving both their benefits and drawbacks to enable rational choices about their potential use.
INTRODUCTION
Much like satellite imaging provides us with detailed maps of the roads and buildings of our cities, optical microscopy provides us with maps of the inner structure of cells. Extending this metaphor, a cell contains roads of actin and tubulin (the cytoskeleton), and specialized communities walled in by membranes (organelles). The mitochondrial reticulum is the power plant and the nucleus is the government headquarters. For this large-scale cellular infrastructure, diffraction-limited fluorescence microscopy can provide us with generally adequate maps of the cell. However, standard optical imaging methods fall short when it comes to learning about the daily life of the residents of the cell/city: the many thousands of different proteins. From basic biochemical studies, we know the general picture of what the life of a protein is like. That is, new proteins are constantly synthesized in ribosomes (birth), folded by chaperones (training), carry out their specific functions (work), and are eventually recycled by proteasomes (retired). All proteins play a role in the intricate flow of matter and energy that keeps a cell functioning, but none can perform in isolation. To fully map and understand a cell, we require molecular tools to visualize this dynamic dance of proteins interacting with each other.
Protein-protein interactions (PPis) occur on a length scale that is well below the diffraction limit for visible light, and so they cannot be visualized directly using standard optical imaging methods. Super-resolution imaging methods can provide the resolution necessary to conclude that two proteins are interacting, or at least in very close proximity to each other.1–3 This review focuses on molecular approaches to experimentally visualizing PPis in live cells. PPi interactions that demonstrably occur in the intracellular milieu (as opposed to in vitro) are, by definition, of biologically relevance. The common theme of these molecular approaches is that the experimentalist must insert a protein “spy” into the cell that will reliably report back on the interactions of two or more specific proteins.
Our focus will be on emerging technologies and so we refer the reader to previous reviews for detailed descriptions of more established technologies.4–10 To organize the various technologies, we categorize them with respect to whether the signal generated is reversible or irreversible in nature, and whether the signal is localized at the site of interaction or is non-localized and appears throughout the whole cell or subcellular compartment.
GENERAL PRINCIPLES
Resonance Energy Transfer
In order to detect a PPi a detectable signal change must occur. This can be achieved optically through the use of absorbance, fluorescence, and bioluminescence (Figure 1(a–c)) changes that occur as a result of the PPi. Förster resonance energy transfer (FRET) is a process where either an excited fluorescent molecule or bioluminescent molecule excites another fluorescent molecule through the non-radiative transfer of energy. FRET occurs when an excited fluorophore called the donor has an emission profile that overlaps with the absorbance profile of another chromophore, called the acceptor. The donor and acceptor together are called a FRET pair. The efficiency of FRET depends on the overlap of the donor emission and the acceptor absorbance spectra, the quantum yield of the donor, and the distance and orientation between the donor and acceptor.11 The donor can be a fluorophore or the bioluminescent product of a luciferase (Figure 1(d) and (e)). In the case of the bioluminescent product being the donor, the process is also known as bioluminescence resonance energy transfer (BRET).
FIGURE 1.
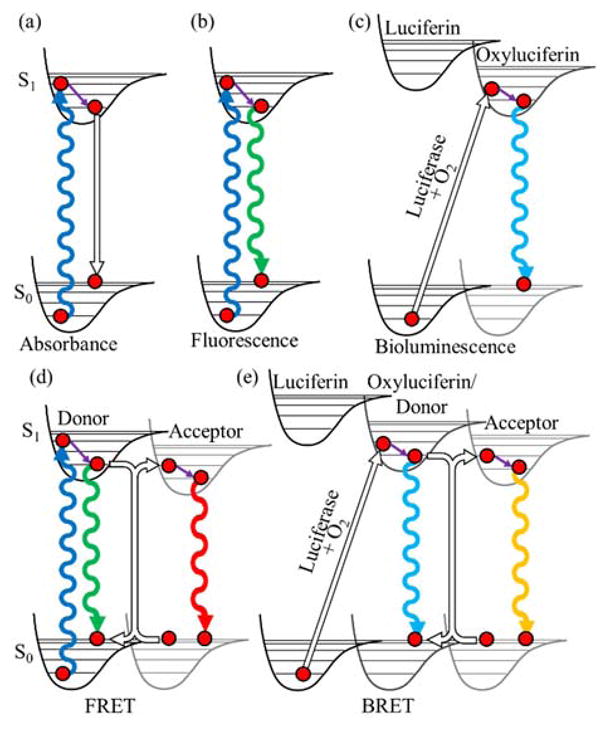
Jablonski diagrams to represent various optical imaging modes. (a) Absorbance. (b) Fluorescence. (c) Bioluminescence. (d) FRET. (e) BRET. Undulating lines represent transitions involving photons with up and down arrows representing absorbance or emission, respectively. Hollow arrows represent non-absorbing or non-radiative transitions. Purple arrows represent relaxation by internal conversion. All excited states have a probability to relax non-radiatively, as shown in (a).
The distance dependence of FRET is 1/r6 where r is the distance between the donor and acceptor.11 Each FRET pair is defined by a Förster radius which describes the distance at which 50% of the excited donor molecules will transfer their energy via FRET. Measurable FRET changes occur between 1–10 nm, creating an optical ruler that can report the distance between the donor and acceptor, assuming that the orientation is known or random. This strong distance dependence makes FRET one of the most useful photophysical phenomenon for detection of PPis.
Reporter genes
Reporter genes are genetic elements that code for proteins that are capable of producing a characteristic detectable signal. For examples, reporter genes could encode for proteins that can create fluorescent, luminescent, colorimetric, or radioactive signals. The reporter gene could allow survival, induce death, or create a phenotypic change. They are generally used by putting the transcription of the reporter gene under the control of a PPi.
When a PPi leads to expression of a reporter protein, these proteins will freely diffuse throughout a cell. Accordingly, the location of the reporter protein or detectable product does not contain information about the location of the PPi. Similarly, the resulting signal from the reporter gene is the result of a chain of processes that each serve to amplify the signal: transcription, translation, and possibly enzymatic production. This amplification can be beneficial as it can allow for the detection of rarer PPi events, but it is more susceptible to false positives as low background levels of non-specific interactions can potentially result in a large signal.
Protein complementation
Protein complementation is the underlying principle for many PPi detection technologies. When a protein is divided into two or more polypeptide chains, it is sometimes possible to reconstitute the complete folded protein structure simply by bringing the fragment polypeptides into close proximity. If the split protein is one that normally creates a detectable signal (i.e., a reporter protein), the proximity-dependent reconstitution can be used as a PPi sensor. To use a split reporter protein to detect a putative PPi between proteins X and Y, protein X must be genetically fused to one half of the split protein and protein Y to the second half of the split protein. If X and Y interact, the two halves of the split protein are brought together allowing them to reconstitute the intact reporter protein, leading to the creation of a detectable signal. Creating such protein fragment complementation assays requires genetically splitting a reporter protein, such as a fluorescent protein (FP), into two polypeptides. Each fragment should be soluble and partially fold in such a way that it remains possible to regain function when brought into the proximity of its other half.
In the case of FP complementation, this approach is commonly referred to as bimolecular fluorescence complementation (BiFC). For luminescence this approach is known as bimolecular luminescence complementation (BiLC).
REPRESENTATIVE EXAMPLES
Irreversible non-local PPi detection
Gene-level control
In this context, gene-level control is the use of a PPi to control the expression of a reporter gene. Among the many examples of such systems is the yeast 2-hybrid system which relies on reconstitution of a split transcription factor, Gal4, to activate transcription (Figure 2(a)).12 Controlling a transcription factor’s activity can also be achieved by sequestering the transcription factor, by adding a genetic tag that prevents the transcription factor from functioning normally. The sequestration can be turned off by cleaving the genetic tag with the use of a specific protease. For example, PPi-dependent reconstitution of a split protease could lead to cleavage of the genetic tag, thus releasing the transcription factor. Split TEV protease has been used to cleave a linker to release a transcription factor, Gal4, from the membrane allowing it to diffuse throughout the cell and induce transcription (Figure 2(b)).13 Similarly, PPi-induced reconstitution of split ubiquitin creates the cleavage site for endogenous ubiquitin proteases.14 This system has been used in yeast for detection of interactions between membrane proteins. Proteolytic cleavage releases a transcription factor that enters the nucleus and drives expression of a reporter gene.15 Pu et al. have reported a split T7 RNA polymerase system16 in which a PPi reconstitutes the polymerase which then transcribes a reporter gene (Figure 2(c)). Split Cre recombinase, developed by Hirrlinger et al.,17 can be used to flip the orientation of a reversed reporter gene in response to a PPi, or activate a gene by removal of an inserted stop codon (Figure 2(d)). Split Cre recombinase is an all-or-none detection system as, once the DNA orientation has been modified, the split Cre-PPi has no further control over the reporter gene. For such all-or-none systems, special care must be taken to control for false positives.
FIGURE 2.

Gene-level control PPi detection systems. (a) Yeast 2-hybrid based on split Gal4.12 The Gal4 transcription factor is split into two parts, the DNA binding domain (DBD) and the activation domain (AD). When fused proteins of interest (X and Y) interact, transcription is initiated and the reporter gene is expressed. (b) Split TEV protease system.13 A transcription factor is tethered to the membrane with a linker containing a TEV protease cut site. The interaction of proteins X and Y leads to reconstitution of TEV protease which cleaves the linker releasing the transcription factor which then activates expression of the reporter gene. (c) Split RNA polymerase.16 The PPi between X and Y brings the two parts of T7 RNA polymerase together allowing transcription of the reporter gene to occur. (d) Split Cre recombinase. Initially the reporter gene is inactive, due to an inverted orientation or an insertion that disables the gene. The interaction of X and Y reconstitutes Cre recombinase which flips the gene, or excises part of the DNA, to enable proper expression of the reporter gene.
Survival controllers
Survival control tethers cell survival to the PPi of interest. One method to achieve survival control is through the production of a critical metabolite. Remy et al. created a protein complementation-based survival assay based on split dihydrofolate reductase (DHFR).18 DHFR is required in the biosynthetic pathways for serine, methionine, purines, and thymidylate.19,20 The split DHFR systems are designed such that complementation provides resistance to an antibiotic that inhibits DHFR. The antibiotic trimethoprim specifically inhibits bacterial DHFR, leading to cell death if this is the only DHFR enzyme present. Cells in which there has been a PPi-dependent reconstitution of split mammalian DHFR, which is resistant to trimethoprim, will survive.21 This approach is limited to bacterial systems or eukaryote DHFR knock-outs in which the bacterial enzyme is expressed. This limitation was circumvented by creating a mutant DHFR which provides resistance to a different antibiotic, methotrexate.20 Methotrexate inhibits the endogenous DHFR in yeast and mammalian cells, normally leading to cell death. PPi-induced complementation of the split mutant DHFR allows cells to survive the addition of methotrexate.22
An alternate method of survival control is through antibiotic resistance genes. Antibiotic resistance genes provide the ability to survive an antibiotic treatment, but otherwise are unnecessary for survival. β-lactamase is a commonly used antibiotic resistance protein that catalyzes the hydrolysis of the β-lactam ring of antibiotics such as penicillins and cephalosporins. Wehrman et al. developed a split β-lactamase that only degrades its target antibiotic when reconstituted.23 Accordingly, cell survival can be linked to split β-lactamase reconstitution via a PPi. As these antibiotics are specifically designed to affect bacteria and not mammalian cells, this system is only applicable to protein interactions in bacteria.
Split enzymes
Split enzyme PPi detection systems catalyze a reaction that converts a substrate into a product, such that a conveniently detectable signal is produced (Figure 3). Split β-lactamase and split β-galactosidase both have a series of substrates that have been developed which result in colourful,24 fluorescent,8,25–27 or luminescent28 products throughout the cell.8 Split horseradish peroxidase similarly produces colourful, fluorescent and luminescent products but also requires a heme cofactor to function.29 Massoud et al. have developed a split version of herpes simplex virus type 1 thymidine kinase (TK).30 As a kinase it doesn’t produce an optically detectable product, instead it catalyzes the intracellular accumulation of an externally supplied positron emitting radionuclide, which can be imaged with positron emission tomography (PET). This is achieved by treating cells with the membrane permeable biomolecule, 9-(4-[18F]-fluoro-3-hydroxymethylbutyl)-guanine (FHBG). FHBG that has been phosphorylated by reconstituted TK becomes impermeable to the cell membrane and therefore accumulates inside the cell. This technique is most useful for deep tissue imaging as the detected gamma ray photons from 18F decay are detectable through 1 cm of mouse tissue.30 This split TK system has reversible association-dissociation, however the 110 minute half-life of 18F means that this system is not amenable to dynamic monitoring of PPi events.30
FIGURE 3.
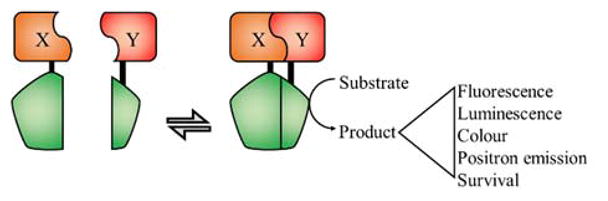
General strategy for PPi detection with a split reporter enzyme. The interaction of X and Y reconstitutes the function of the split reporter enzyme which, in the presence of an appropriate substrate, can produce a product with a detectable phenotype.
To develop an alternative method for PPi detection, Wu et al.31 used an intein protein that naturally occurs as a ‘split’ enzyme that catalyzes an auto-splicing reaction. DnaE is a set of two proteins, Int-n and Int-c, that work together to function as the intein. The N-terminal amino acids of Int-n and the C-terminal amino acids of Int-c are spliced together to form a new polypeptide called the extein as well as truncated Int-n and Int-c proteins.31 The extein and the remainders of Int-n and Int-c are not attached and diffuse apart. This system is set up such that a PPi interaction brings Int-n and Int-c together such that they form the active complex which splices together the two halves of a split reporter protein (Figure 4(f)).32 This system has the potential to allow the use of reporter proteins that are not otherwise amenable to genetic splitting, as the splicing reaction can reconstitute the native sequence of the split reporter protein.
FIGURE 4.
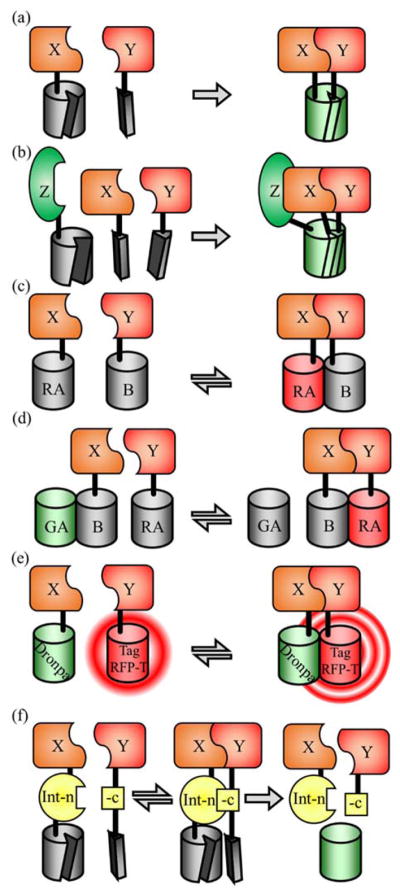
FP-based PPi detection systems. (a) Split FP. When the interaction of X and Y occurs, the two halves of the split FP associate to form the intact and functional FP. This process is essentially irreversible, so proteins X and Y remain associated. (b) Tripartite split FP. An FP is split into three parts that are only capable of forming their chromophore when all three are brought together by a three way PPi, schematically represented here with proteins X, Y, and Z. (c) Dimerization dependent FPs (ddFP). The red fluorescence of RA is increased when the X and Y PPi brings the RA and B copies together to allow its reversible dimerization to occur. (d) FP exchange (FPX). The B copy from (c) is capable of activating the fluorescence of both RA and GA, a green version of RA, but only one at a time. The interaction of X and Y serves to shift the equilibrium between the GA-B complex and the RA-B complex, resulting in a red-green ratiometric change in fluorescence hue. (e) FLINC. The interaction of X and Y induces an interaction between Dronpa and TagRFP-T, resulting in increased fluorescence fluctuations from TagRFP-T. (f) Split intein-based PPi detection. When X and Y interact, the Int-n and Int-c protein fragments associate to form a functional intein protein which catalyzes a splicing reaction to form an intact reporter protein. The spliced reporter protein is not attached to the now truncated X-Int-n and Y-Int-c proteins and will diffuse away.
Irreversible localized PPi detection
Split fluorescent proteins (FPs)
The catalog of FP variants, many of which have recently been quantitatively evaluated by Cranfill et al.,33 is extensive and provides seemingly limitless opportunities for creating systems for detection of PPis. As FPs are only fluorescent after folding and the subsequent maturation of the chromophore, an FP that has been split into two polypeptides will be non-fluorescent until the two fragments are brought into close proximity. Once in close proximity, the β-barrel structure can fold and the chromophore can autogenically mature to its fluorescent state. The first FP to be divided into two fragments was enhanced GFP (EGFP). In the original implementation, the smaller fragment contained the last four β-strands of the β-barrel and the larger fragment contained the first seven β-strands and the central helix. When these two fragments were brought together using an antiparallel leucine zipper PPi, the intact EGFP was reconstituted and became fluorescent.34 Since this first version was reported, many more split FPs have been engineered (Table 1). A few split FPs share the original split location, with others splitting off the last three β-strands.6 In 2008 a third split site that only splits off one β-strand (the 11th) was developed using an improved EGFP called superfolderGFP (sfGFP) (Figure 4(a)).35 A tripartite split where sfGFP was split into three parts (β-strands 1–9, β-strand 10, and β-strand 11) has also been reported.36 Reconstitution of the FP requires that all three parts be brought into proximity (Figure 4(b)).37,38
TABLE 1.
Split FPs
| Fluorescent protein | Excitation (nm) | Emission (nm) | N- and C- terminal cut sites | References |
|---|---|---|---|---|
| SCFP3A | 433 | 474 | 173,155 | 45 |
| Cerulean | 439 | 479 | 154,155 | 96 |
| 172,173 | 96 | |||
| ECFP | 452 | 478 | 154,155 | 46,97 |
| 172,173 | 46 | |||
| EGFP | 488 | 507 | 157,158 | 34,98 |
| 158,159 | 99 | |||
| sfGFP | 485 | 507 | 214,215 | 35 |
| sfGFP (tripartite) | 485 | 507 | 193,194–212,213 | 36 |
| frGFP | 485 | 507 | 157,158 | 100 |
| mKG | 494 | 507 | 168,169 | 101 |
| mKG2 | 494 | 507 | 169,170 | 42 |
| Dronpa | 503 | 518 | 164,165 | 102 |
| EYFP | 515 | 527 | 154,155 | 44,46 |
| 155,156 | 47 | |||
| 172,173 | 46,96 | |||
| Venus | 515 | 528 | 154,155 | 96 |
| 157,158 | 41 | |||
| 172,173 | 96 | |||
| sfYFP | 515 | 527 | 154,155 | 48 |
| Citrine | 516 | 529 | 154,155 | 96 |
| mRFP (Q66T) | 549 | 570 | 168,169 | 103 |
| dsRED monomer | 558 | 583 | 168,169 | 104 |
| mEOS3.2a | 507 [572] | 516 [580] | 164,165 | 3 |
| mCherry | 587 | 610 | 159,160 | 105 |
| mLumin | 587 | 621 | 151,152 | 106 |
| FusionRed | 575 | 605 | 189,188 | 107 |
| UnaGb | 497 | 527 | 84,85 | 81 |
| IFP1.4b | 684 | 710 | 132,133 | 79 |
| iSplit (iRFP)b | 690 | 713 | 120,119 | 50 |
mEOS3.2 photoconverts from green to red upon illumination with 405 nm light. The wavelengths in square brackets are the red state excitation and emission wavelengths.
Exogenous chromophore FPs.
Split versions of β-barrel-based FPs have a few limitations on how they can be used. The first limitation is a tendency to self-assemble creating a false positive signal that reduces their ability to identify weak or rare PPis. A second limitation is the irreversible nature of split FP reconstitution. Once a split FP has been reconstituted, it will not dissociate and the proteins involved in the PPis are permanently held in close proximity.39 A third limitation is the lag time between the binding event, the protein folding, and subsequent maturation of the chromophore. For split FPs this limits the time resolution for detection of PPi to 10s of minutes, at best.6 Despite these limitations, split FPs have allowed for the identification of many PPis and even have been used in high throughput screening for inhibitors of specific PPis.40–43 It should be noted that there is inconsistent naming for these split FPs with many groups renaming constructs or reusing a name. One split EYFP is simultaneously called YN and YC,44,45 YN155 and YC15546 and, YFPN and YFPC.47 YN and YC are reused with a split superfolder YFP.48 The extensive list of existing split FPs reflects the broad utility of these tools, however, there is always room for improvement and future efforts should focus on engineering variants that overcome the limitations discussed above.
FPs with an exogenous chromophore
Filonov et al. developed a split FP system, iSplit, which is based on a near-infrared biliverdin-binding protein that is not homologous to the β-barrel FPs discussed above. The iSplit design is based on a bacterial phytochrome RpBphP2 which was truncated to create iRFP49 which binds the exogenous chromophore biliverdin and fluoresces in the near-infrared region.50 iRFP is a truncated version of RpBphP2 which only retains two of the four parental domains, GAF and PAS. iSplit splits the remaining GAF and PAS domains which renders the protein non-fluorescent until the two domains are brought together due to interaction of the tethered proteins of interest (Figure 5(a)).
FIGURE 5.

PPi detection systems based on split FPs that use exogenous chromophores. (a) iSplit50 is a split version of the iRFP protein.49 Reconstitution of iRFP due to the interaction of X and Y activates the fluorescence of the bound biliverdin molecule. (b) IFP PCA79 is a split version of IFP1.4 that is reported to be reversible.78 (c) uPPI81 is based on split UnaG and depends on the X and Y PPi to activate fluorescence of a bound bilirubin molecule.
Reversible local PPi detection
Dimerization dependent fluorescence
Dimerization dependent FPs (ddFPs), developed by Alford et al., are a class of FPs that have been modified to enable detection of dynamic and reversible PPis.51 The goal in developing ddFPs was to create a reversible version of split FPs by creating an FP pair that change fluorescence intensity upon dimerization. The red fluorescent ddRFP pair increases in brightness by 10-fold upon dimerization and decreases back to basal fluorescence once dissociated (Figure 4(c)). The ddFP design was later extended to create green and yellow pairs.52 As these proteins undergo rapid and reversible changes in fluorescence, they are suitable for imaging of dynamic PPis. Compared to FRET, ddFPs sacrifice the molecular ruler aspect of FRET pairs in order to achieve larger intensiometric changes.
The two parts of a ddFP are generally referred to as the A and B copies. The A copy is a very dim red FP containing a mature chromophore that is quenched. The B copy was engineered such that it no longer forms a chromophore but still folds into the β-barrel structure. When A and B dimerize their interface interaction causes a subtle change in the conformation of the A protein that reduces the quenching and the fluorescence of A increases by 10-fold. It was later found that the B copy could activate the A copies associated with all three colours (i.e., red, green, and yellow). Use of the green and red A copies with a single B copy allows the exchange of the B copy to switch between bright red or green fluorescence creating a ratiometric assay for a dynamic PPi such as calmodulin and M13 binding Ca2+, in a technique termed FP exchange (FPX) (Figure 4(d)).53
FLINC, developed by Mo et al., is another example of dimerization-dependent changes in fluorescence (Figure 4(e)).54 When TagRFP-T55 and Dronpa56 come into contact, the inherent fluorescence fluctuations of TagRFP-T increase in a detectable manner, as determined using photochromic super-resolution optical fluctuation imaging (pcSOFI).57,58 This phenomenon was used to construct an intermolecular version of the protein kinase A (PKA) activity reporter (AKAR1),59–61 by co-expression of Dronpa fused to a phosphopeptide-binding domain and TagRFP-T bound to PKA substrate peptide. This approach enables detection of PPis in micro domains smaller than the diffraction limit and thereby opens new doors into probing how the interacting proteins and their local environment control PPis.
Split luciferases
Similar to FPs, luciferases have been split and engineered to make protein fragment complementation assays in a general strategy known as bimolecular luminescence complementation (BiLC).5 There are currently four classes of research-ready split luciferases that can be categorized based on their protein structure and substrate. The first class of split luciferases are the D-luciferin-based beetle luciferases which are 542–550 amino acids in length and include the firefly, Photinus pyralis,62 and two click beetle (Pyrophorus plagiophthalamus63 and Pyrearinus termitilluminans)64 luciferases (Figure 6(a)). These luciferases are homologous and each contains a large N-terminal domain and a smaller C-terminal domain. They have been split such that the N-terminal and C-terminal fragments overlap and both contain the last α-helix of the N-terminal domain.65 The homology is strong enough that an attempt to improve the CBR(395–542) fragment resulted in a C-terminal fragment, McLuc1, that is capable of activating all three N-terminal fragments.63 This could potentially enable FPX-like assays to be performed using these split luciferases.
FIGURE 6.

The four types of research-ready split luciferases. (a) Split D-luciferin luciferases include Firefly (shown),62 click beetle red,63 and Emerald luciferases.64 (b) Renilla luciferase using the substrate coelenterazine.66 (c) Gaussia luciferase using the substrate coelenterazine.68 (d) Oplophorus luciferases (NanoLuc shown) using the substrate furimazine.70 In all four types shown the interaction of X and Y initiates complementation and the subsequent enzymatic creation of photons. In the absence of the PPi, no light is produced.
The second class is the Renilla luciferase which is a coelenterazine-based luciferase that is 311 amino acids in length and has a globular protein structure (Figure 6(b)).66 It has been split to create two non-overlapping fragments of 1–229 and 230–311 amino acids.
The third class is Gaussia luciferase which is another coelenterazine-based luciferase. It is much shorter at 185 amino acids and is the brightest luciferase that uses the coelenterazine substrate (Figure 6(c)).67 Its structure has yet to be determined but it has low sequence homology to Renilla luciferase. However, unlike Renilla it is excreted from the cell implying it is optimized for an extracellular environment.68 Gaussia luciferase has been split into two non-overlapping fragments composed of the residues 1–93 and 94–185.
The fourth class is based on the Oplophorus luciferase which is a large 109 kDa complex which is composed of two 35 kDa proteins and two 19 kDa proteins.69 Promega’s NanoLuc™ (Ref. 70) is an optimized variant of the 19 kDa luciferase enzyme that no longer forms a complex (Figure 6(d)). This optimization effort also included the development of an improved substrate, furimazine, which is more stable and provides higher luminescent signal intensity than coelenterazine. NanoLuc has an 10-stranded β-barrel structure with “lid” domain composed of three α-helices (PDB ID: 5IBO).71 Promega has also developed a split version of NanoLuc (called NanoBiT) splitting the protein between the 9th and 10th β-strands. The two polypeptides of NanoBiT were subsequently evolved for improved function and solubility.72 The NanoBiT’s larger N-terminal fragment is named 11S and the C-terminal fragment is named 114. Zhao et al. developed a second split location for NanoLuc to create a 1–65 N-terminal fragment and 66–171 C-terminal fragment with both parts of the split protein containing parts of the β-barrel and the α-helical lid.73
In addition to the variants discussed above, many other luciferases have been used for research applications.74 In addition, some modified versions of the split luciferases discussed above have been reported.32,75,76
FlimPIA
The firefly luminescent intermediate-based protein-protein interaction assay (FlimPIA) takes advantage of the fact that two enzymatic steps are required to produce light using firefly luciferase (Figure 7).77 Kurihara et al. split firefly luciferase such that the two catalytic actions of the enzyme (i.e., adenylation of luciferin with ATP to form luciferyl adenylate and pyrophosphate; and the oxidation of luciferyl adenylate into light, oxyluciferin, and AMP) are encoded by separate polypeptides. The presence of both polypeptides is required to yield luminescence and, of particular relevance to the detection of PPis, the overall transformation from luciferin to oxyluciferin plus light is more efficient when the enzymes are in close spatial proximity. Bringing these two enzymes together creates up to a 40-fold increase in the signal to background ratio. The signal increase is due to the increased local concentration of luciferyl adenylate when the acceptor is in close proximity to the donor. Though this technique is yet in the early stages and not yet confirmed in cellulo, it may ultimately enable detection of PPis that could not be detected with any of the other techniques discussed. FlimPIA has the potential to detect PPis where the interacting proteins are on distant parts of a large complex that are 10s of nanometers apart. In contrast, the other techniques discussed in this review require the reporter proteins to come into direct physical contact and in the correct orientation, or to come within a few nanometers of each other in the case of FRET.
FIGURE 7.

FlimPIA PPi detection system.77 In the FlimPIA system, the two enzymatic functions of Firefly luciferase have been split into separate polypeptides. One polypeptide is the donor capable of the adenylation step, and the second polypeptide is the acceptor which is only capable of the oxidation step. This system is potentially useful for detecting the formation of large protein complexes with dimensions that exceed the working range of FRET.
FPs with an exogenous chromophore
While none of the split β-barrel FPs have been reported to undergo a reversible interaction, there are two examples of split FPs with exogenous chromophores that have been reported to undergo reversible complementation. IFP1.4 is a bacteriophytochrome (DrBphP) biliverdin-binding FP developed by Shu et al.78 This protein was split to make IFP PCA79 which fortuitously proved to be reversible, unlike iSplit (Figure 5(b)). Similarly, the green fluorescent bilirubin-binding UnaG FP80 was split into two fragments nUnaG and cUnaG which are non-fluorescent unless brought into contact.81 This system, which was designated uPPI, is fully reversible, as the authors demonstrated by fusing the two fragments to of FKBP and FRB and inducing association with rapamycin and dissociation with FK506 (Figure 5(c)). Notably, a common issue shared by all systems based on FPs with an exogenous chromophore (e.g., IFP1.4, iSplit, and UnaG) is that the intracellular concentration of the biliverdin or bilirubin in the cell type of interest could potentially be a limiting factor for the fluorescent signal.
FRET and BRET
FRET and BRET are both reversible PPi detection techniques that preserve information about the subcellular location of the interaction. These techniques have been the subject of recent reviews and articles discussing FP FRET pairs82,83 and BRET pairs.84 FRET and BRET and have number of inherent advantages, including the fact that energy transfer is fast (nanosecond time scale) and the phenomenon lends itself to quantitative determination of interprotein distances with nanometer precision. FRET and BRET typically require measuring two emission wavelengths and analyzing the ratio between them. Both techniques can also provide an intensiometric signal when used in conjunction with a non-fluorescent acceptor. It is important to note that, while FP-based FRET pairs have been shown to exhibit 20-fold ratio changes in vitro,85 it is rare to achieve more than a 2-fold change in FRET ratio in cellulo. However, due to the self-correcting nature of ratiometric signals, significant results have been reported with ratio changes of less than 10%.86,87
Reversible non-localized PPi detection
Fluoppi is a recently reported FP-based technology for detecting protein interactions that does not depend on changes in fluorescence intensity, but rather on changes on the distribution of fluorescence in a cell. Specifically, the Fluoppi system is designed such that a PPi results in the creation of condensed liquid phase aggregates (Figure 8). In the absence of the PPi, the Azami green FP88 fused to one protein of interest, X, will form tetramers that evenly distributed throughout a cell. The second component, the PB1 domain fused to the second protein of interest, Y, forms linear oligomers in the cell.89,90 Interaction of X and Y induces the FP tetramers and PB1 oligomers to clump together creating an aggregate that transitions into a condensed liquid phase. These aggregates themselves fuse to create bright puncta throughout the cytoplasm of a cell. If the interaction of X and Y is disrupted, the aggregates dissociate and fluorescence distributes evenly throughout the cell, illustrating that the system is reversible.91 The formation of large aggregates necessarily erases any information on the subcellular location of the PPi.
FIGURE 8.
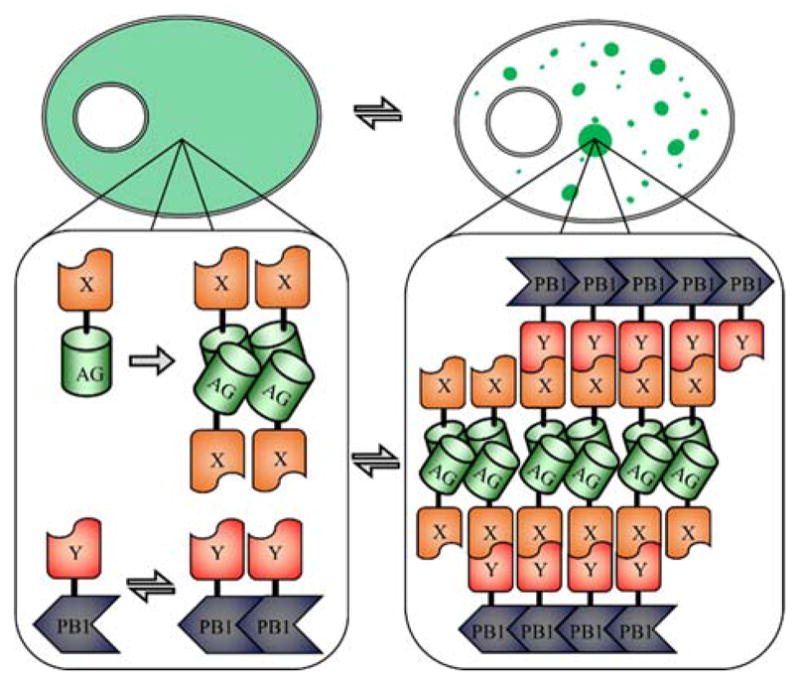
Fluoppi PPi detection system.91 Shown at the top is a schematic representation of a cell before and after the PPi. Shown at the bottom is the protein aggregate formed upon interaction of proteins X and Y, leading to the formation of large fluorescent puncta throughout the cell. These aggregates are stabilized by the tetramerization of the Azami green FP (AG) and linear oligomerization of PB1.
Reversible non-localized PPi detection has also been performed using several approaches in which one protein (the bait) is induced to form clusters using either light or a small molecule. If a second protein (the prey) undergoes a change in localization that is identical to that of the bait protein, it is taken as evidence that the two proteins are involved in a PPi. Examples include the LINC assay based on light-induced clustering of cryptochrome 2 (CRY2),92 the bisarsenical dye-induced aggregation of tetracysteine-labeled proteins,93 and the ECLIPSE assay based on rapamycin-induced recruitment to endosomes.94
CONCLUSION
Detecting PPis in cells and organisms is a major challenge that must be overcome if we are to ever gain a complete understanding of the complex mechanisms of life. In this review we have cataloged some of the exciting recent progress towards addressing this challenge using a diverse range of approaches. However, this review also demonstrates the limitations of our current technologies, particularly when it comes to multiplexed detection of PPis in single cells. As an illustrative example, Mitogen-activated protein kinase 1 and the CREB-binding protein are each estimated to participate in more 100 different PPis.95 None of the techniques described in this review would enable us to detect more than a few of these interactions in a single experiment. This limitation means that single cell imaging of the complex interplay of the myriad of binding partners for a target protein is likely to remain well beyond our reach for the foreseeable future. Inevitably, this and other limitations are certain to be overcome, as researchers use their ingenuity and creativity to develop ever more clever ways to “spy” on PPis as they occur in live cells.
TABLE 2.
Split luciferases.
| Organism | Species | Luciferase name | Length (amino acids) | Size kDa | Split name | N-terminal split | Split name | C-terminal split | Substrate |
|---|---|---|---|---|---|---|---|---|---|
| Firefly | Photinus pyralis | Firefly Luciferase | 550 | 61 | NLuc | 2–416 | CLuc | 398–550 | D-luciferin |
| Click beetle | Pyrophorus plagiophthalamus | Click Beetle Red | 542 | 64 | CBR (1–414) | 1–414 | CBR (395–542) | 395–542 | D-luciferin |
| Click beetle | Pyrearinus termitilluminans | Emerald Luciferase | 542 | 64 | ELucN | 1–415 | ELucC | 394–542 | D-luciferin |
| Sea pansy | Renilla reniformis | Renilla Luciferase | 311 | 36 | N-Rluc | 1–229 | C-Rluc | 230–311 | Coelenterazine |
| Copepod | Gaussia princeps | Gaussia Luciferase | 185 | 20 | Gluc(1) | 1–93 | Gluc(2) | 94–185 | Coelenterazine |
| Shrimp | Oplophorus gracilirostris | Nanoluc | 171 | 19 | N65 | 1–65 | 66C | 66–171 | Furimazine |
| NanoBiT | 171 | 19 | 11S | 1–156 | 114 | 158–168 | Furimazine |
Acknowledgments
Work in the Campbell lab is supported by grants from CIHR (MOP-123514), NSERC (RGPIN 288338-2010), Brain Canada, and NIH (U01 NS094246 and UO1 NS090565). MDW was supported, in part, by scholarships from the University of Alberta.
References
- 1.Bates M, Huang B, Dempsey GT, Zhuang X. Multicolor super-resolution imaging with photo-switchable fluorescent probes. Science. 2007;317:1749–1753. doi: 10.1126/science.1146598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shroff H, Galbraith CG, Galbraith JA, White H, Gillette J, Olenych S, et al. Dual-color superresolution imaging of genetically. Proc Natl Acad Sci U S A. 2007;104:20308–20313. doi: 10.1073/pnas.0710517105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu Z, Xing D, Su QP, Zhu Y, Zhang J, Kong X, et al. Super-resolution imaging and tracking of protein–protein interactions in sub-diffraction cellular space. Nat Commun. 2014;5:1–8. doi: 10.1038/ncomms5443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buntru A, Trepte P, Klockmeier K, Schnoegl S, Wanker EE. Current approaches toward quantitative mapping of the interactome. Front Genet. 2016;7:1–9. doi: 10.3389/fgene.2016.00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vidi P-A, Watts VJ. Fluorescent and bioluminescent protein-fragment complementation assays in the study of G protein-coupled receptor oligomerization and signaling. Mol Pharmacol. 2009;75:733–739. doi: 10.1124/mol.108.053819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shyu YJ, Hu CD. Fluorescence complementation: an emerging tool for biological research. Trends Biotechnol. 2008;26:622–630. doi: 10.1016/j.tibtech.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 7.Rao VS, Srinivas K, Sujini GN, Kumar GNS. Protein-protein interaction detection: Methods and analysis. Int J Proteomics. 2014;2014:1–12. doi: 10.1155/2014/147648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Galarneau A, Primeau M, Trudeau L-E, Michnick SW. Beta-lactamase protein fragment complementation assays as in vivo and in vitro sensors of protein protein interactions. Nat Biotechnol. 2002;20:619–622. doi: 10.1038/nbt0602-619. [DOI] [PubMed] [Google Scholar]
- 9.Porterfield WB, Prescher JA. Tools for visualizing cell-cell ‘interactomes’. Curr Opin Chem Biol. 2015;24:121–130. doi: 10.1016/j.cbpa.2014.11.006. [DOI] [PubMed] [Google Scholar]
- 10.Waadt R, Schlücking K, Schroeder JI, Kudla J. Protein fragment bimolecular fluorescence complementation analyses for the in vivo study of protein-protein interactions and cellular protein complex localizations. In: Sanchez-Serrano JJ, Salinas Julio, editors. Methods in Molecular Biology. Humana Press; 2014. pp. 629–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lakowicz JR, editor. Principles of Fluorescence Spectroscopy. Springer US; Boston, MA: 2006. [DOI] [Google Scholar]
- 12.Fields S, Song O. A novel genetic system to detect protein-protein interactions. Nature. 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- 13.Wehr MC, Laage R, Bolz U, Fischer TM, Grünewald S, Scheek S, et al. Monitoring regulated protein-protein interactions using split TEV. Nat Methods. 2006;3:985–93. doi: 10.1038/nmeth967. [DOI] [PubMed] [Google Scholar]
- 14.Johnsson N, Varshavsky A. Split ubiquitin as a sensor of protein interactions in vivo. Proc Natl Acad Sci U S A. 1994;91:10340–4. doi: 10.1073/pnas.91.22.10340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stagljar I, Korostensky C, Johnsson N, te Heesen S. A genetic system based on split-ubiquitin for the analysis of interactions between membrane proteins in vivo. Proc Natl Acad Sci U S A. 1998;95:5187–92. doi: 10.1073/pnas.95.9.5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pu J, Zinkus-Boltz J, Dickinson BC. Evolution of a split RNA polymerase as a versatile biosensor platform. Nat Publ Gr. 2017;13:1–27. doi: 10.1038/nchembio.2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirrlinger J, Scheller A, Hirrlinger PG, Kellert B, Tang W, Wehr MC, et al. Split-Cre complementation indicates coincident activity of different genes in vivo. PLoS One. 2009;4:1–10. doi: 10.1371/journal.pone.0004286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Remy I, Michnick SW. Clonal selection and in vivo quantitation of protein interactions with protein-fragment complementation assays. Proc Natl Acad Sci U S A. 1999;96:5394–9. doi: 10.1073/pnas.96.10.5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bertino JR, Hillcoat BL. Regulation of dihydrofolate reductase and other folate-requiring enzymes. Adv Enzyme Regul. 1968;6:335–49. doi: 10.1016/0065-2571(68)90021-6. [DOI] [PubMed] [Google Scholar]
- 20.Remy I, Campbell-Valois FX, Michnick SW. Detection of protein-protein interactions using a simple survival protein-fragment complementation assay based on the enzyme dihydrofolate reductase. Nat Protoc. 2007;2:2120–2125. doi: 10.1038/nprot.2007.266. [DOI] [PubMed] [Google Scholar]
- 21.Pelletier JN, Campbell-Valois FX, Michnick SW. Oligomerization domain-directed reassembly of active dihydrofolate reductase from rationally designed fragments. Proc Natl Acad Sci USA. 1998;95:12141–12146. doi: 10.1073/pnas.95.21.12141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chumakov SP, Kravchenko YE, Chumakov PM. Protein complementation as tool for studying protein-protein interactions in living cells. Mol Biol. 2012;46:627–638. [PubMed] [Google Scholar]
- 23.Wehrman T, Kleaveland B, Her J-H, Balint RF, Blau HM. Protein-protein interactions monitored in mammalian cells via complementation of beta -lactamase enzyme fragments. Proc Natl Acad Sci U S A. 2002;99:3469–3474. doi: 10.1073/pnas.062043699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Broome AM, Bhavsar N, Ramamurthy G, Newton G, Basilion JP. Expanding the utility of β-galactosidase complementation: Piece by piece. Mol Pharm. 2010;7:60–74. doi: 10.1021/mp900188e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gong H, Zhang B, Little G, Kovar J, Chen H, Xie W, et al. β-Galactosidase activity assay using far-red-shifted fluorescent substrate DDAOG. Anal Biochem. 2009;386:59–64. doi: 10.1016/j.ab.2008.11.031. [DOI] [PubMed] [Google Scholar]
- 26.Ho NH, Weissleder R, Tung CH. A self-immolative reporter for β-galactosidase sensing. ChemBioChem. 2007;8:560–566. doi: 10.1002/cbic.200600386. [DOI] [PubMed] [Google Scholar]
- 27.Spotts JM, Dolmetsch RE, Greenberg ME. Time-lapse imaging of a dynamic phosphorylation-dependent protein-protein interaction in mammalian cells. Proc Natl Acad Sci USA. 2002;99:15142–15147. doi: 10.1073/pnas.232565699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bronstein I, Martin CS, Fortin JJ, Olesen CEM, Voyta JC. Chemiluminescence: Sensitive detection technology for reporter gene assays. Clin Chem. 1996;42:1542–1546. [PubMed] [Google Scholar]
- 29.Martell JD, Yamagata M, Deerinck TJ, Phan S, Kwa CG, Ellisman MH, et al. A split horseradish peroxidase for the detection of intercellular protein–protein interactions and sensitive visualization of synapses. Nat Biotechnol. 2016;34:774–780. doi: 10.1038/nbt.3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Massoud TF, Paulmurugan R, Gambhir SS. A molecularly engineered split reporter for imaging protein-protein interactions with positron emission tomography. Nat Med. 2010;16:921–926. doi: 10.1038/nm.2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu H, Hu Z, Liu XQ. Protein trans-splicing by a split intein encoded in a split DnaE gene of Synechocystis sp. PCC6803. Proc Natl Acad Sci USA. 1998;95:9226–9231. doi: 10.1073/pnas.95.16.9226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ozawa T, Kaihara A, Sato M, Tachihara K, Umezawa Y. Split luciferase as an optical probe for detecting protein-protein interactions in mammalian cells based on protein splicing. Anal Chem. 2001;73:2516–21. doi: 10.1021/ac0013296. [DOI] [PubMed] [Google Scholar]
- 33.Cranfill PJ, Sell BR, Baird MA, Allen JR, Lavagnino Z, de Gruiter HM, et al. Quantitative assessment of fluorescent proteins. Nat Methods. 2016:1–7. doi: 10.1038/nmeth.3891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ghosh I, Hamilton AD, Regan L. Antiparallel leucine zipper-directed protein reassembly: application to the green fluorescent protein. J Am Chem Soc. 2000;122:5658–5659. [Google Scholar]
- 35.Cabantous S, Terwilliger TC, Waldo GS. Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat Biotechnol. 2005;23:102–107. doi: 10.1038/nbt1044. [DOI] [PubMed] [Google Scholar]
- 36.Cabantous S, Nguyen HB, Pedelacq J-D, Koraïchi F, Chaudhary A, Ganguly K, et al. A new protein-protein interaction sensor based on tripartite split-GFP association. Sci Rep. 2013;3:2854. doi: 10.1038/srep02854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kellermann SJ, Rath AK, Rentmeister A. Tetramolecular fluorescence complementation for detection of specific RNAs in vitro. ChemBioChem. 2013;14:200–204. doi: 10.1002/cbic.201200734. [DOI] [PubMed] [Google Scholar]
- 38.Waldo GS, Cabantous S. Protein- protein interaction detection system using fluorescent protein.pdf. 2010 https://www.google.com/patents/US7666606.
- 39.Magliery TJ, Wilson CGM, Pan W, Mishler D, Ghosh I, Hamilton AD, et al. Detecting protein-protein interactions with a green fluorescent protein fragment reassembly trap: Scope and mechanism. J Am Chem Soc. 2005;127:146–157. doi: 10.1021/ja046699g. [DOI] [PubMed] [Google Scholar]
- 40.Zych C, Domling A, Ayyavoo V. Development of a robust cell-based high-throughput screening assay to identify targets of HIV-1 viral protein R dimerization. Drug Des Devel Ther. 2013;7:403–412. doi: 10.2147/DDDT.S44139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ding Z, Liang J, Lu Y, Yu Q, Songyang Z, Lin S-Y, et al. A retrovirus-based protein complementation assay screen reveals functional AKT1-binding partners. Proc Natl Acad Sci U S A. 2006;103:15014–15019. doi: 10.1073/pnas.0606917103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kojima T, Karasawa S, Miyawaki A, Tsumuraya T, Fujii I. Novel screening system for protein-protein interactions by bimolecular fluorescence complementation in Saccharomyces cerevisiae. J Biosci Bioeng. 2011;111:397–401. doi: 10.1016/j.jbiosc.2010.12.013. [DOI] [PubMed] [Google Scholar]
- 43.Offenborn JN, Waadt R, Kudla J. Visualization and translocation of ternary Calcineurin-A/Calcineurin-B/Calmodulin-2 protein complexes by dual-color trimolecular fluorescence complementation. New Phytol. 2015;208:269–279. doi: 10.1111/nph.13439. [DOI] [PubMed] [Google Scholar]
- 44.Hu CD, Chinenov Y, Kerppola TK. Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol Cell. 2002;9:789–798. doi: 10.1016/s1097-2765(02)00496-3. [DOI] [PubMed] [Google Scholar]
- 45.Waadt R, Schmidt LK, Lohse M, Hashimoto K, Bock R, Kudla J. Multicolor bimolecular fluorescence complementation reveals simultaneous formation of alternative CBL/CIPK complexes in planta. Plant J. 2008;56:505–516. doi: 10.1111/j.1365-313X.2008.03612.x. [DOI] [PubMed] [Google Scholar]
- 46.Hu C-D, Kerppola TK. Simultaneous visualization of multiple protein interactions in living cells using multicolor fluorescence complementation analysis. Nat Biotechnol. 2003;21:539–545. doi: 10.1038/nbt816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walter M, Chaban C, Schütze K, Batistic O, Weckermann K, Näke C, et al. Visualization of protein interactions in living plant cells using bimolecular fluorescence complementation. Plant J. 2004;40:428–438. doi: 10.1111/j.1365-313X.2004.02219.x. [DOI] [PubMed] [Google Scholar]
- 48.Ottmann C, Weyand M, Wolf A, Kuhlmann J, Ottmann C. Applicability of superfolder YFP bimolecular fluorescence complementation in vitro. Biol Chem. 2009;390:81–90. doi: 10.1515/BC.2009.008. [DOI] [PubMed] [Google Scholar]
- 49.Filonov GS, Piatkevich KD, Ting LM, Zhang J, Kim K, Verkhusha VV. Bright and stable near-infrared fluorescent protein for in vivo imaging. Nat Biotechnol. 2011;29:757–761. doi: 10.1038/nbt.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Filonov GS, Verkhusha VV. A near-infrared bifc reporter for in vivo imaging of protein-protein interactions. Chem Biol. 2013;20:1078–1086. doi: 10.1016/j.chembiol.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alford SC, Abdelfattah AS, Ding Y, Campbell RE. A fluorogenic red fluorescent protein heterodimer. Chem Biol. 2012;19:353–360. doi: 10.1016/j.chembiol.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alford SC, Ding Y, Simmen T, Campbell RE. Dimerization-dependent green and yellow fluorescent proteins. ACS Synth Biol. 2012;1:569–575. doi: 10.1021/sb300050j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ding Y, Li J, Enterina JR, Shen Y, Zhang I, Tewson PH, et al. Ratiometric biosensors based on dimerization-dependent fluorescent protein exchange. Nat Methods. 2015;12:195–198. doi: 10.1038/nmeth.3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mo GCH, Ross B, Hertel F, Manna P, Yang X, Greenwald E, et al. Genetically encoded biosensors for visualizing live-cell biochemical activity at super-resolution. Nat Publ Gr. 2017;14:427–434. doi: 10.1038/nmeth.4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shaner NC, Lin MZ, McKeown MR, Steinbach PA, Hazelwood KL, Davidson MW, et al. Improving the photostability of bright monomeric orange and red fluorescent proteins. Nat Methods. 2008;5:545–551. doi: 10.1038/nmeth.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ando R, Mizuno H, Miyawaki A. Regulated fast nucleocytoplasmic shuttling observed by reversible protein highlighting. Science. 2004;306:1370–1373. doi: 10.1126/science.1102506. [DOI] [PubMed] [Google Scholar]
- 57.Dedecker P, Mo GCH, Dertinger T, Zhang J. Widely accessible method for superresolution fluorescence imaging of living systems. Proc Natl Acad Sci. 2012;109:10909–10914. doi: 10.1073/pnas.1204917109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dertinger T, Colyer R, Iyer G, Weiss S, Enderlein J. Fast, background-free, 3D super-resolution optical fluctuation imaging (SOFI) Proc Natl Acad Sci. 2009;106:22287–22292. doi: 10.1073/pnas.0907866106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang J, Ma Y, Taylor SS, Tsien RY. Genetically encoded reporters of protein kinase A activity reveal impact of substrate tethering. Proc Natl Acad Sci U S A. 2001;98:14997–15002. doi: 10.1073/pnas.211566798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang J, Hupfeld CJ, Taylor SS, Olefsky JM, Tsien RY. Insulin disrupts beta-adrenergic signalling to protein kinase A in adipocytes. Nature. 2005;437:569–573. doi: 10.1038/nature04140. [DOI] [PubMed] [Google Scholar]
- 61.Komatsu N, Aoki K, Yamada M, Yukinaga H, Fujita Y, Kamioka Y, et al. Development of an optimized backbone of FRET biosensors for kinases and GTPases. Mol Biol Cell. 2011;22:4647–56. doi: 10.1091/mbc.E11-01-0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Luker KE, Smith MCP, Luker GD, Gammon ST, Piwnica-Worms H, Piwnica-Worms D. Kinetics of regulated protein-protein interactions revealed with firefly luciferase complementation imaging in cells and living animals. Proc Natl Acad Sci U S A. 2004;101:12288–12293. doi: 10.1073/pnas.0404041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hida N, Awais M, Takeuchi M, Ueno N, Tashiro M, Takagi C, et al. High-sensitivity real-time imaging of dual protein-protein interactions in living subjects using multicolor luciferases. PLoS One. 2009:4. doi: 10.1371/journal.pone.0005868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Misawa N, Kafi AKM, Hattori M, Miura K, Masuda K, Ozawa T. Rapid and high-sensitivity cell-based assays of protein–protein interactions using split Click beetle luciferase complementation: An approach to the study of G-protein-coupled receptors. Anal Chem. 2010;82:2552–2560. doi: 10.1021/ac100104q. [DOI] [PubMed] [Google Scholar]
- 65.Conti E, Franks NP, Brick P. Crystal structure of firefly luciferase throws light on a superfamily of adenylate-forming enzymes. Structure. 1996;4:287–298. doi: 10.1016/s0969-2126(96)00033-0. [DOI] [PubMed] [Google Scholar]
- 66.Loening AM, Fenn TD, Gambhir SS. Crystal structures of the luciferase and green fluorescent protein from Renilla reniformis. J Mol Biol. 2007;374:1017–1028. doi: 10.1016/j.jmb.2007.09.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tannous BA, Kim DE, Fernandez JL, Weissleder R, Breakefield XO. Codon-optimized gaussia luciferase cDNA for mammalian gene expression in culture and in vivo. Mol Ther. 2005;11:435–443. doi: 10.1016/j.ymthe.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 68.Remy I, Michnick SW. A highly sensitive protein-protein interaction assay based on Gaussia luciferase. Nat Methods. 2006;3:977–979. doi: 10.1038/nmeth979. [DOI] [PubMed] [Google Scholar]
- 69.Inouye S, Watanabe K, Nakamura H, Shimomura O. Secretional luciferase of the luminous shrimp Oplophorus gracilirostris: cDNA cloning of a novel imidazopyrazinone luciferase. FEBS Lett. 2000;481:19–25. doi: 10.1016/s0014-5793(00)01963-3. [DOI] [PubMed] [Google Scholar]
- 70.Hall MP, Unch J, Binkowski BF, Valley MP, Butler BL, Wood MG, et al. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem Biol. 2012;7:1848–1857. doi: 10.1021/cb3002478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lovell S, Mehzabeen N, Battaile KP, Wood MG, Encell LP, Wood KV. PDB ID: 5IBO 1.95A resolution structure of NanoLuc luciferase. 2016 doi: 10.2210/pdb5ibo/pdb. [DOI] [Google Scholar]
- 72.Dixon AS, Schwinn MK, Hall MP, Zimmerman K, Otto P, Lubben TH, et al. NanoLuc complementation reporter optimized for accurate measurement of protein interactions in cells. ACS Chem Biol. 2016;11:400–408. doi: 10.1021/acschembio.5b00753. [DOI] [PubMed] [Google Scholar]
- 73.Zhao J, Nelson TJ, Vu Q, Truong T, Stains CI. Self-assembling NanoLuc luciferase fragments as probes for protein aggregation in living cells. ACS Chem Biol. 2016;11:132–138. doi: 10.1021/acschembio.5b00758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xu T, Close D, Handagama W, Marr E, Sayler G, Ripp S. The expanding toolbox of in vivo bioluminescent imaging. Front Oncol. 2016;6:1–8. doi: 10.3389/fonc.2016.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Endoh T, Mie M, Funabashi H, Sawasaki T, Endo Y, Kobatake E. Construction of intramolecular luciferase complementation probe for detecting specific RNA. Bioconjug Chem. 2007;18:956–962. doi: 10.1021/bc060351o. [DOI] [PubMed] [Google Scholar]
- 76.Kim SB, Kanno A, Ozawa T, Tao H, Umezawa Y. Nongenomic activity of ligands in the association of androgen receptor with Src. ACS Chem Biol. 2007;2:484–492. doi: 10.1021/cb7000439. [DOI] [PubMed] [Google Scholar]
- 77.Kurihara M, Ohmuro-Matsuyama Y, Ayabe K, Yamashita T, Yamaji H, Ueda H. Ultra sensitive firefly luciferase-based protein-protein interaction assay (FlimPIA) attained by hinge region engineering and optimized reaction conditions. Biotechnol J. 2016;11:91–99. doi: 10.1002/biot.201500189. [DOI] [PubMed] [Google Scholar]
- 78.Shu X, Royant A, Lin MZ, Aguilera TA, Lev-Ram V, Steinbach Pa, et al. Mammalian expression of infrared fluorescent proteins engineered from a bacterial phytochrome. Science. 2009;324:804–807. doi: 10.1126/science.1168683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tchekanda E, Sivanesan D, Michnick SW. An infrared reporter to detect spatiotemporal dynamics of protein-protein interactions. Nat Methods. 2014;11:641–644. doi: 10.1038/nmeth.2934. [DOI] [PubMed] [Google Scholar]
- 80.Kumagai A, Ando R, Miyatake H, Greimel P, Kobayashi T, Hirabayashi Y, et al. A bilirubin-inducible fluorescent protein from eel muscle. Cell. 2013;153:1602–1611. doi: 10.1016/j.cell.2013.05.038. [DOI] [PubMed] [Google Scholar]
- 81.To TL, Zhang Q, Shu X. Structure-guided design of a reversible fluorogenic reporter of protein-protein interactions. Protein Sci. 2016;25:748–753. doi: 10.1002/pro.2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bajar BT, Wang ES, Zhang S, Lin MZ, Chu J. A guide to fluorescent protein FRET pairs. Sensors (Switzerland) 2016;16:1–24. doi: 10.3390/s16091488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lam AJ, St-Pierre F, Gong Y, Marshall JD, Cranfill PJ, Baird MA, et al. Improving FRET dynamic range with bright green and red fluorescent proteins. Nat Methods. 2012;9:1005–1012. doi: 10.1038/nmeth.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.De A, Jasani A, Arora R, Gambhir SS. Evolution of BRET biosensors from live cell to tissue-scale in vivo imaging. Front Endocrinol (Lausanne) 2013;4:1–6. doi: 10.3389/fendo.2013.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nguyen AW, Daugherty PS. Evolutionary optimization of fluorescent proteins for intracellular FRET. Nat Biotechnol. 2005;23:355–360. doi: 10.1038/nbt1066. [DOI] [PubMed] [Google Scholar]
- 86.San Martín A, Ceballo S, Ruminot I, Lerchundi R, Frommer WB, Barros LF. A Genetically Encoded FRET Lactate Sensor and Its Use To Detect the Warburg Effect in Single Cancer Cells. PLoS One. 2013:8. doi: 10.1371/journal.pone.0057712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jones AM, Danielson JAh, Manojkumar SN, Lanquar V, Grossmann G, Frommer WB. Abscisic acid dynamics in roots detected with genetically encoded FRET sensors. Elife. 2014;3:e01741. doi: 10.7554/eLife.01741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Karasawa S, Araki T, Yamamoto-Hino M, Miyawaki A. A green-emitting fluorescent protein from Galaxeidae coral and its monomeric version for use in fluorescent labeling. J Biol Chem. 2003;278:34167–34171. doi: 10.1074/jbc.M304063200. [DOI] [PubMed] [Google Scholar]
- 89.Ren J, Wang J, Wang ZX, Wu JW. Structural and biochemical insights into the homotypic PB1-PB1 complex between PKCζ and p62. Sci China Life Sci. 2014;57:69–80. doi: 10.1007/s11427-013-4592-z. [DOI] [PubMed] [Google Scholar]
- 90.Saio T, Yokochi M, Kumeta H, Inagaki F. PCS-based structure determination of protein-protein complexes. J Biomol NMR. 2010;46:271–280. doi: 10.1007/s10858-010-9401-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Watanabe T, Seki T, Fukano T, Sakaue-Sawano A, Karasawa S, Kubota M, et al. Genetic visualization of protein interactions harnessing liquid phase transitions. Sci Rep. 2017;7:46380. doi: 10.1038/srep46380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Taslimi A, Vrana JD, Chen D, Borinskaya S, Mayer BJ, Kennedy MJ, et al. An optimized optogenetic clustering tool for probing protein interaction and function. Nat Commun. 2014;5:4925. doi: 10.1038/ncomms5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Martin BR, Deerinck TJ, Ellisman MH, Taylor SS, Tsien RY. Isoform-Specific PKA Dynamics Revealed by Dye-Triggered Aggregation and DAKAP1α-Mediated Localization in Living Cells. Chem Biol. 2007;14:1031–1042. doi: 10.1016/j.chembiol.2007.07.017. [DOI] [PubMed] [Google Scholar]
- 94.Lee KH, Lee S, Lee WY, Yang HW, Heo WD. Visualizing dynamic interaction between calmodulin and calmodulin-related kinases via a monitoring method in live mammalian cells. Proc Natl Acad Sci. 2010;107:3412–3417. doi: 10.1073/pnas.0911262107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ota M, Gonja H, Koike R, Fukuchi S. Multiple-localization and hub proteins. PLoS One. 2016;11:1–18. doi: 10.1371/journal.pone.0156455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shyu YJ, Liu H, Deng X, Hu C-D. Identification of new fluorescent protein fragments for bimolecular fluorescence complementation analysis under physiological conditions. Biotechniques. 2006;40:61–66. doi: 10.2144/000112036. [DOI] [PubMed] [Google Scholar]
- 97.Zhang S, Ma C, Chalfie M. Combinatorial marking of cells and organelles with reconstituted fluorescent proteins. Cell. 2004;119:137–144. doi: 10.1016/j.cell.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 98.Demidov VV, Dokholyan NV, Witte-Hoffmann C, Chalasani P, Yiu H-W, Ding F, et al. Fast complementation of split fluorescent protein triggered by DNA hybridization. Proc Natl Acad Sci U S A. 2006;103:2052–2056. doi: 10.1073/pnas.0511078103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Remy I, Michnick SW. A cDNA library functional screening strategy based on fluorescent protein complementation assays to identify novel components of signaling pathways. Methods. 2004;32:381–388. doi: 10.1016/j.ymeth.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 100.Sarkar M, Magliery TJ. Re-engineering a split-GFP reassembly screen to examine RING-domain interactions between BARD1 and BRCA1 mutants observed in cancer patients. Mol Biosyst. 2008;4:599–605. doi: 10.1039/b802481b. [DOI] [PubMed] [Google Scholar]
- 101.Ueyama T, Kusakabe T, Karasawa S, Kawasaki T, Shimizu A, Son J, et al. Sequential binding of cytosolic phox complex to phagosomes through regulated adaptor proteins: Evaluation using the novel monomeric Kusabira-green system and live imaging of phagocytosis. J Immunol. 2008;181:629–640. doi: 10.4049/jimmunol.181.1.629. [DOI] [PubMed] [Google Scholar]
- 102.Lee YR, Park JH, Hahm SH, Kang LW, Chung JH, Nam KH, et al. Development of bimolecular fluorescence complementation using dronpa for visualization of protein-protein interactions in cells. Mol Imaging Biol. 2010;12:468–478. doi: 10.1007/s11307-010-0312-2. [DOI] [PubMed] [Google Scholar]
- 103.Jach G, Pesch M, Richter K, Frings S, Uhrig JF. An improved mRFP1 adds red to bimolecular fluorescence complementation. Nat Methods. 2006;3:597–600. doi: 10.1038/nmeth901. [DOI] [PubMed] [Google Scholar]
- 104.Kodama Y, Wada M. Simultaneous visualization of two protein complexes in a single plant cell using multicolor fluorescence complementation analysis. Plant Mol Biol. 2009;70:211–217. doi: 10.1007/s11103-009-9467-0. [DOI] [PubMed] [Google Scholar]
- 105.Fan JY, Cui ZQ, Wei HP, Zhang ZP, Zhou YF, Wang YP, et al. Split mCherry as a new red bimolecular fluorescence complementation system for visualizing protein-protein interactions in living cells. Biochem Biophys Res Commun. 2008;367:47–53. doi: 10.1016/j.bbrc.2007.12.101. [DOI] [PubMed] [Google Scholar]
- 106.Chu J, Zhang Z, Zheng Y, Yang J, Qin L, Lu J, et al. A novel far-red bimolecular fluorescence complementation system that allows for efficient visualization of protein interactions under physiological conditions. Biosens Bioelectron. 2009;25:234–239. doi: 10.1016/j.bios.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 107.Kost LA, Putintseva EV, Pereverzeva AR, Chudakov DM, Lukyanov KA, Bogdanov AM. Bimolecular fluorescence complementation based on the red fluorescent protein FusionRed. Russ J Bioorganic Chem. 2016;42:619–623. [Google Scholar]