

Summary
Heterotrophic plants provide excellent opportunities to study the effects of altered selective regimes on genome evolution. Plastid genome (plastome) studies in heterotrophic plants are often based on one or a few highly divergent species or sequences as representatives of an entire lineage, thus missing important evolutionary-transitory events.
Here we present the first infraspecific analysis of plastome evolution in any heterotrophic plant. By combining genome skimming and targeted sequence capture, we address hypotheses on the degree and rate of plastome degradation in a complex of leafless orchids (Corallorhiza striata) across its geographic range.
Plastomes provide strong support for relationships and evidence of reciprocal monophyly between C. involuta and the endangered C. bentleyi. Plastome degradation is extensive, occurring rapidly over a few million years, with evidence of differing rates of substitution among the two principal clades of the complex. Genome skimming and targeted sequence capture differ widely in coverage depth overall, with depth in targeted sequence capture datasets varying immensely across the plastome as a function of GC content.
These findings will help fill a knowledge gap in models of heterotrophic plastid genome evolution, and have implications for future studies in heterotrophs.
Keywords: Corallorhiza, hybrid capture, mycoheterotroph, parasite, plastid genome, pseudogenization
Introduction
Studies of plastid genome (plastome) evolution in heterotrophic plants often focus on one to a few taxa, yet none have focused on species-wide variation. In most studies to date, plastomes are sequenced and compared to those previously published. These have been of great importance in elucidating the large-scale patterns of plastome evolution due to relaxed purifying selective constraints on photosynthesis, revealing convergent patterns of gene degradation (Wolfe et al., 1992; Funk et al., 2007; McNeal et al., 2007; Wickett et al., 2008; Delannoy et al., 2011; Logacheva et al., 2011; Barrett et al., 2012; Logacheva et al., 2013; Li et al., 2013; Lam et al., 2015; Bellot & Renner, 2016; Lim et al., 2016; Naumann et al., 2016; Roquet et al., 2016; Samigullin et al., 2016). Recently, phylogenetic, comparative approaches have been taken across families, tribes, or genera containing parasites, representing a shift away from single-plastome studies (Wicke et al., 2013; Barrett et al., 2014; Feng et al., 2016; Braukmann et al., 2017). Such studies allow powerful phylogenetic comparisons of plastid genome evolution within related lineages. Researchers have expanded and refined models of plastome evolution to incorporate additional features of modification as a result of this rapidly accumulating body of data on the plastomes of heterotrophic plants. These include overall decreases in genome size, decreases in GC content, increasing frequency of rearrangements, accumulation of indels, losses of introns, etc. (Wicke et al., 2011, 2013, 2016; Barrett & Davis, 2012; Barrett et al., 2014; Naumann et al., 2016; reviewed in Graham et al., 2017).
Yet, no research has focused on the infraspecific level. Such studies would be highly informative on fine-scale mutational processes that ultimately result in such drastic changes observed at higher taxonomic levels, such as family or genus, and their timing (Wicke et al., 2013, 2016; Barrett et al., 2014; Feng et al., 2016; Braukmann et al., 2017). Comparisons of plastid genome evolution at higher taxonomic levels (e.g. across genera) may leave phylogenetic sampling ‘gaps’, characterized by drastic differences in gene content and genome size among the taxa sampled. Thus, key pieces of the process of plastome evolution may remain elusive. How much variation exists among plastomes within heterotrophic species and species complexes? At what timescale does degradation occur within and between closely related species – do these changes occur in rapid bursts or more gradually? Can substitution and insertion/deletion (indel) rate changes be observed at finer taxonomic levels, or are these only observable at larger scales?
Orchids have experienced a greater number of independent transitions to heterotrophy than any other group of land plants (>30; Freudenstein & Barrett, 2008; Freudenstein & Merckx, 2010; Merckx et al., 2013). They comprise a trophic spectrum from autotrophy (following initial heterotrophy during germination) and various degrees of partial heterotrophy (in which the plant obtains and utilizes carbon from its host on top of its own photosynthetic energy gain; Selosse & Martos, 2014; Gebauer et al., 2016), to complete reliance on host plants for nutrients. Among those are lineages with highly reduced plastomes (e.g., Epipogium – Schelkunov et al., 2015; Rhizanthella – Delannoy al., 2011), and groups with minimally reduced plastomes (e.g., some Neottieae; Logacheva et al., 2011; Feng et al., 2016).
Corallorhiza is an ideal system in which to address hypotheses of plastome evolution in heterotrophic lineages, containing both green (e.g. C. trifida, C. odontorhiza) and non-green members (e.g. members of the C. striata and C. maculata complexes). Here we focus on the leafless, North American C. striata species complex (Supporting Information Notes S1A) to address questions regarding patterns of intraspecific variation of plastomes in heterotrophs. Previous studies demonstrate evidence of accelerated plastome degradation, based on accumulation of pseudogenes and large deletions (Barrett & Freudenstein, 2010; Barrett & Davis, 2012; Barrett et al., 2014), thus making the C. striata complex an apt system in which to study the dynamics of plastid genome evolution in a phylogenetic context. The complex consists of three species: C. involuta (southern Mexico); the endangered C. bentleyi (Virginia and West Virginia, USA); and the more widespread C. striata ‘sensu stricto’ (Mexico, USA, and Canada; Fig. 1). The latter is composed of three varieties: C. striata var. striata (northern USA, Canada), var. vreelandii (southwestern USA, Mexico), and an undescribed, putative variety from the western Sierra Nevada (California, USA; Barrett & Freudenstein, 2011).
Fig. 1.
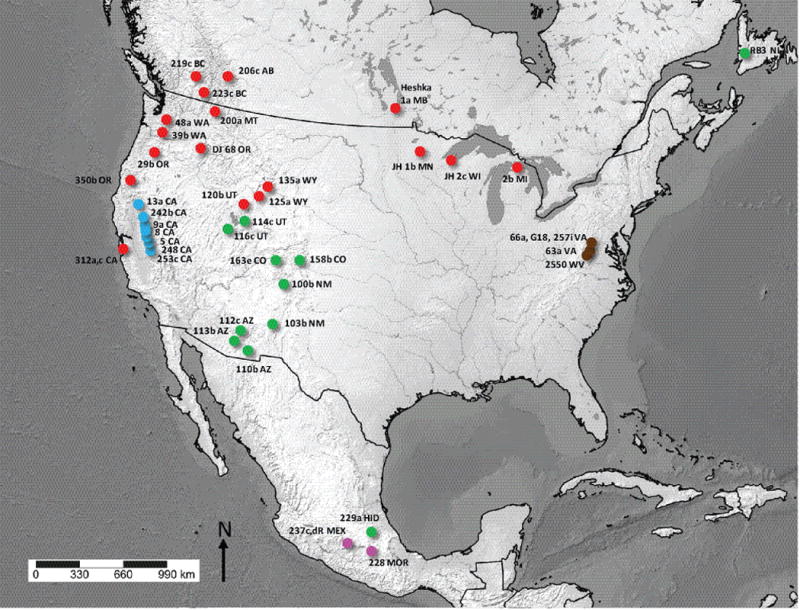
Sampling locations across North America for the Corallorhiza striata complex. Magenta, C. involuta; brown, C. bentleyi; blue, C. striata Sierra Nevadan accessions; green, C. striata var. vreelandii; red, C. striata var. striata.
We take a two-tiered approach to investigate infraspecific plastome evolution in the C. striata complex, incorporating genome skimming (GS) from shallow whole-genome shotgun sequencing and targeted sequence capture, with the objectives of: (1) resolving relationships among plastomes of the C. striata complex across its geographic range; (2) characterizing patterns of plastome variation among members of this complex; (3) quantifying the relative timing of gene loss and pseudogenization events in Corallorhiza broadly, and in the C. striata complex specifically; and (4) comparing patterns of coverage depth based on genome skimming versus sequence capture methods for assessing plastome evolution in heterotrophic plants.
Materials and Methods
We isolated genomic DNAs (gDNA) from 0.5 to 1 g of tissue using the CTAB method (Doyle & Doyle, 1987) with RNase digestion. We assessed DNA quality and quantity via electrophoresis and nucleic acid stain-based spectrophotometry. Subsequent plastid genome sequencing was performed via two popular methods: (1) Genome skimming (GS), or multiplexed, low-coverage sequencing of total genomic DNA (Cronn et al., 2008; Meyer & Kircher, 2010; Straub et al., 2011); and (2) targeted sequence capture (TSC), an alternative method that increases cost effectiveness by using specific probe hybridization based on reference genomes, reducing the fraction of non-target genomic DNA sequenced in genome skimming (e.g. Hodges et al., 2009; Bi et al., 2012; Sass et al., 2016). GS can be used to sequence nearly complete mitochondrial genomes and other high-copy elements (ribosomal DNA, transposable elements, etc.), while TSC further allows enrichment of the low copy and organellar fractions of gDNA.
Generating reference plastomes for the C. striata complex
GS sequencing procedures
We sequenced plastomes for representative individuals of each previously recognized taxonomic entity within the C. striata complex (including an individual from the Sierra Nevada), and one accession each of the green, photosynthetic C. trifida and Calypso bulbosa at Global Biologics, LLC (Columbia, MI, USA) following Illumina protocols (Notes S1B). Libraries were prepared using 1 μg of high molecular weight gDNA sheared to 300–400 bp via sonication. Paired-end sequencing was carried out on an Illumina NextSeq500 (150 bp) for a total of 13 samples (seven for this study and six for another). Total reads per library ranged from 6,015,539 for C. involuta (accession CFB 237c MEX) to 19,535,084 for C. striata var. striata (accession CFB 120b UT).
Plastid genome assembly of GS data
We processed reads following the quality control pipeline of Bi et al. (2012, 2013) and Singhal (2013). Briefly, we trimmed reads to remove adapters (CutAdapt, Martin, 2011), trimmed/filtered on quality (PHRED < 20, Trimmomatic v.0.32; Bolger et al., 2014), filtered for bacterial contaminants, and merged overlaps with Flash v.1.2.11 (Magoc & Salzberg, 2011). We assembled plastomes de novo with Velvet v.1.2.1 (Zerbino & Birney, 2009), and NOVOPlasty v.1.1 (Diercksens et al., 2016) over a range of kmer values. For NOVOPlasty assemblies we used the plastid gene matK from Corallorhiza striata var. vreelandii (Barrett & Davis, 2012) as a seed (insert range = 1.5, coverage cutoff = 30). We merged contigs (overlap 30 bp, similarity 95%, allowing gaps) in Sequencher (v.5.1, GeneCodes, USA). Contigs from both programs were merged in GENEIOUS (v.8.1, Biomatters Ltd, New Zealand) to build draft plastomes. We then mapped reads to draft plastomes in GENEIOUS to check for mis-assemblies and low-coverage regions (mismatch 5%, gap size 5 kb). We used UNIX grep searches against original reads to verify regions of ambiguity due to low coverage, and to validate inverted repeat boundaries via paired-end information. We annotated plastomes in DOGMA (Wyman et al., 2004) and SEQUIN (https://www.ncbi.nlm.nih.gov/Sequin; GenBank accession numbers: MG874034-MG874040).
Targeted sequence capture across the C. striata complex
Sampling and probe design
We sampled 48 individuals of C. striata and two of C. trifida from Mexico, USA, and Canada following Barrett & Freudenstein (2011; Notes S1B), to encompass geographic, taxonomic, and morphological diversity of the complex (gDNAs were isolated as above). We designed capture probes using annotated plastid genomes of C. striata vreelandii (Barrett & Davis, 2012) and C. trifida (Barrett et al., 2014). Both were randomly tiled as 60 bp fragments at 1-bp intervals following Sass et al. (2016) across an Agilent 1M microarray chip (Agilent Technologies, USA). Inclusion of both reference plastomes was to avoid bias; the plastome of C. striata vreelandii has experienced deletions (Barrett & Davis, 2012), and it is unknown whether other members of this complex have experienced similar deletions. Corallorhiza trifida was chosen as a backup probe, as it has a more intact plastome, and is phylogenetically close to the C. striata complex (Barrett et al., 2014).
Capture
We sheared gDNA to 200–300 bp fragments using a Qsonica 800R sonicator (Millard & Muriel Jacobs Genetics and Genomics Lab, CalTech; Qsonica, LLC., Newton, CT, USA). We chose this relatively shorter fragment size to maximize the number of on-target reads (Hodges et al., 2007). We prepared Illumina libraries following the protocol of Sass et al. (2016) at the Evolutionary Genetics Laboratory at UC, Berkeley. We quantified libraries using a Qubit Fluorometer and pooled them at equimolar ratios and checked fragment sizes with an Agilent Bioanalyzer (Agilent Technologies, USA). We carried out TSC of plastid genomes following Hodges et al. (2009). Briefly, we hybridized libraries at 65˚C for 65 h in an Agilent G2545A Hybridization Oven, eluted hybridized DNA, enriched the pool via PCR amplification (Phusion® High-Fidelity DNA Polymerase; ThermoFisher Scientific, Waltham, Massachusetts, USA), and then sequenced in a lane of 100 bp paired-end reads on an Illumina HiSeq2000 at the QB3 Vincent J Coates Genomic Sequencing Facility at UC, Berkeley (http://qb3.berkeley.edu/gsl). We cleaned the captured read data as above, with the only difference being that we skipped the removal step of low complexity reads, as plastid genomes often contain low-complexity, AT-rich intergenic regions.
Plastid DNA assembly from TSC data
As de novo assembly of captured reads resulted in numerous short contigs and low N50, we mapped captured reads to our annotated and closest GS reference sequences (according to Barrett & Freudenstein, 2009, 2011) using the native GENEIOUS mapper for 25 iterations under stringent conditions, or until no more reads were added. When assembled reads displayed unexpectedly high mismatches, we remapped reads to an alternative reference and chose the lowest overall distance between read consensus and reference. After trials under different parameter sets, the final mapping parameters using 5% maximum mismatch and a maximum gap size of 5 kb (all other settings at default). References also included contigs from a genome-skim de novo assembly of C. maculata var. occidentalis, in order to filter out plastid-like sequences from the mitochondrial and nuclear genomes.
We examined mapped read contigs visually along the plastome for misassemblies and low coverage. We corrected the few misassembled regions either by UNIX grep searches, or by iterative remapping in GENEIOUS using flanking sequences as seeds to extend through the region. Regions of consistently short stretches of low coverage depth due to low complexity were reassembled as above. Areas with coverage below 10x were automatically masked with ‘N’. We deposited all captured datasets in the NCBI Sequence Read Archive as ‘.bam’ alignments (Accession: SRP131512).
Alignment, phylogenetic analyses, and plastome sequence variation
Plastome alignment
We ‘sub-aligned’ consensus plastomes to each corresponding reference plastome using the MAFFT plugin for GENEIOUS (default settings). As necessary, we made minor adjustments manually by realigning those regions in GENEIOUS via the MUSCLE plugin (Edgar, 2004). We then transferred annotations from each closest GS reference, and adjusted manually. We again used MAFFT for profile alignment, and then used the same procedure to align orchid outgroup taxa. Alignments were deposited in DRYAD (doi:10.5061/dryad.g1d2s). We also aligned whole plastomes generated via GS with the MAUVE plugin for GENEIOUS (Darling et al., 2010) to investigate possible rearrangements.
Whole gene sets (i.e. coding regions plus those of Calypso and C. trifida) were aligned with the codon-based aligner MACSE (Ranwez et al., 2011), which allows for reading frame shifts. We extracted introns and spacers from GENEIOUS as individual alignments, refined in MUSCLE as above, and re-concatenated for downstream analyses. Indels were coded using the GapCoder module of SEQSTATE (Müller, 2005), under the ‘modified complex indel coding’ (mcic) method of Müller (2006). Indel content for each plastome was calculated as root-to-tip GTR distances using the ‘ape’ and ‘phytools’ packages in R (Paradis, 2004; Popescu et al., 2012; Revell, 2012).
Phylogenetic analyses
We generated trees with RAxML based on whole plastome alignments using ten independent searches under an unpartitioned GTR-GAMMA model with the default number of rate categories and 1,000 standard bootstrap pseudoreplicates. Following Barrett et al. (2014), non-triplet frame-shifts and premature stop codons were used as evidence of pseudogenes. We calculated the average numbers of nucleotide differences and nucleotide diversity (π) within and between each taxonomic entity for whole plastomes, and conducted a Mantel test for correlation between genetic and geographic distances between accessions (Notes S1).
We inferred divergence times with BEAST2 (Bouckaert et al., 2014) based on the nuclear internal transcribed Spacer (ITS) and the plastid genes matK, psaB, and rbcL, which have sufficient variation for resolving relationships at both high (e.g. orchid subfamilies) and low taxonomic levels (e.g. among and within species; Cameron, 2004; Freudenstein & Senyo, 2008; Barrett & Freudenstein, 2010). Our decision to use four genes is based on previous analyses using whole plastomes, which failed to converge due to the immense parameter space required to effectively model such a large dataset (Barrett et al., 2015; see Foster et al., 2017). We constructed the dataset based on sequences from Givnish et al. (2015), Cameron (2004), Freudenstein et al. (2017), the current study, and from GenBank (Notes S1C). We used an Uncorrelated Lognormal Relaxed Clock model with an unpartitioned GTR+GAMMA+I substitution model, with four rate categories, and parameters estimated from the data. We used a Yule speciation model, with lognormal distributed priors on nodes calibrated with minimum age fossil dates from Dendrobium (Conran et al., 2009) and a newly described genus from Baltic amber, Succinanthera (Poiret & Rasmusssen, 2017). We ran three analyses from random seeds in BEAST, each 2.0 × 108 generations, sampling every 10,000 generations. We tracked parameters in TRACER (Rambaut et al., 2014), and used effective sample sizes (ESS) of >200 for all parameters to assess stationarity and convergence. We combined runs in LogCombiner and TreeAnnotator (burn-in: 30%), and edited trees in FigTree (http://tree.bio.ed.ac.uk). Additional details are given in Notes S1. To roughly assess the timing of plastome degradation, we mapped gene losses on the chronogram, partitioned by functional gene classes (as in Wicke et al., 2013; Barrett et al., 2014).
We analyzed coevolution of sequence variation, changes in selection (dS, dN, dN/dS; indels measured as described above), and various parameters underlying the substitution process (transition:transversion ratio (ti/tv), GC content) with coevol v1.4 (Lartillot & Poujol, 2011), on a dataset of all protein-coding genes and one combined dataset of the non-coding regions. Coevol combines substitution models with multivariate Brownian Motion on the basis of Bayesian Inference and Markov Chain Monte Carlo methods in a phylogenetic framework. Coevol was run with two chains, each sampling every tenth generation. Convergence was checked by computing discrepancies (at most 0.01) and ESS of the posterior averages, tree lengths, and other summary statistics obtained from the independent runs. Posterior averages were computed from the merged chains, and the first 10% of samples per chain were excluded as burn-in.
We carried out all other statistical comparisons using Phylogenetically Independent Contrasts (PIC; Felsenstein, 1985; Garland, 1992) under Brownian Motion in the R packages ‘ape’, and ‘phytools’. Other statistical analyses were conducted in PAST v.3.8 (Hammer et al., 2001) or in R (R Core Team, 2014), including Pearson’s Correlation, Mann-Whitney U-tests (nonparametric, two-samples), and Kendall’s Tau (nonparametric correlation).
Plastome coverage depth and sequence capture efficiency
The percentage of reads mapping to GS reference plastomes was used as an estimate of capture efficiency across all TSC datasets, normalized by the total number of reads. Variation in coverage depth and GC content across the plastome was assessed using a sliding window analysis. Plastid read mappings were exported from GENEIOUS in ‘.bam’ format and converted to ‘.pileup’ in SAMTOOLS (Li et al., 2009). A PERL script was used to simultaneously calculate coverage depth and GC content across each reference mapping, using a sliding window size of 100 bp, in 100 bp increments (Nolte et al., 2013). PAST was used to test for spatial autocorrelation (Durbin & Watson, 1950); regions missing data were excluded.
Results
Plastome relationships and variation across the C. striata complex
Analysis of aligned plastomes yielded a highly resolved and supported tree for the C. striata complex (Fig. 2). Three low-coverage accessions were removed (C. involuta CFB 228aR; C. striata var. vreelandii CFB 229a and LR5). Plastomes of C. bentleyi and C. involuta occupy distinct sister clades, each receiving strong to medium bootstrap support (BS): 100 for C. involuta; 93 for C. bentleyi. Hereafter, strong support is ≥ 95%, medium support ≥ 85%, and weak support < 85%. Within C. bentleyi, one accession from Monroe Co., West Virginia, USA is sister to all others (from Allegheny and Bath Counties, Virginia, USA). The bentleyi-involuta clade is sister to a clade of all other C. striata (BS = 100). Within the latter clade are numerous accessions of C. striata vars. vreelandii and striata (each was respectively monophyletic), collectively sister to a clade of seven accessions from the western slope of the Sierra Nevada of California, USA; all of these relationships are supported at BS ≥ 99.
Fig. 2.
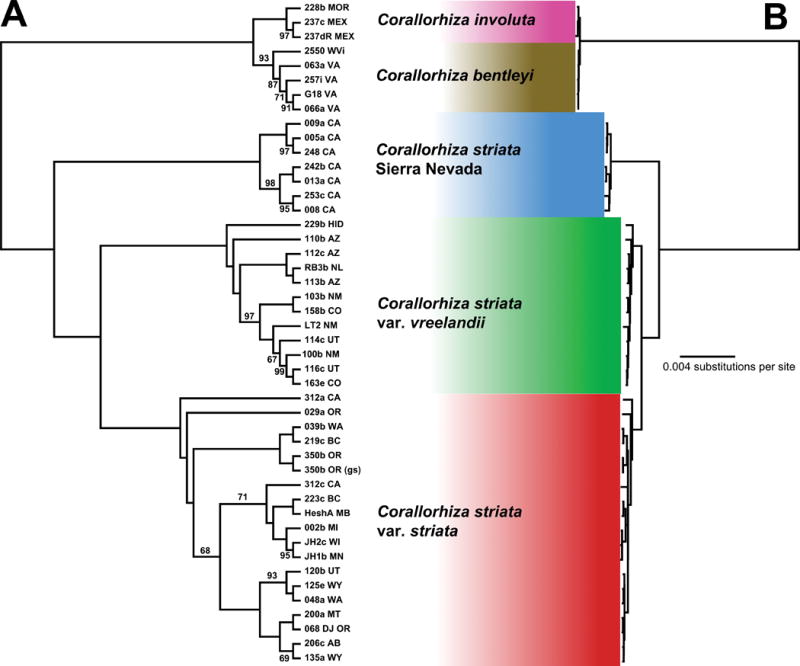
Relationships among members of the Corallorhiza striata species complex based on Maximum Likelihood analysis of whole aligned plastomes (unpartitioned GTR-GAMMA). Accession codes are followed by US, Canadian, or Mexican state/province. (a) Cladogram showing relationships and Maximum Likelihood Bootstrap Support (adjacent to branches; no number indicates 100%). (b) Phylogram showing branch lengths; scale bar units are substitutions per site. 350b OR (gs), genome skim dataset.
Within the ‘var. vreelandii clade’, all accessions are sister to an accession from Hidalgo, Mexico (CFB 229b), with the next split (moving from the root of the clade) including accessions from Arizona, USA (Fig. 2). This clade also includes one accession from Newfoundland, Canada, and a clade of accessions from Colorado, New Mexico, and Utah, USA. Within the C. striata var. striata clade, an accession from the Santa Cruz Mountains of California, USA (CFB 312a CA) is sister to all others, followed by an accession from the Cascade Range of Oregon (CFB 29a OR). This is successively followed by a clade composed of numerous accessions from the Rocky Mountains of the USA and Canada, with some from California and Oregon interspersed. Accession CFB 312c CA came from the same population as 312a CA (which diverges at the root of the ‘var. striata clade’), but the placement of the former has weak support (BS = 71).
Plastome divergence within the C. striata complex
Genome sizes for complete plastomes generated via GS range from 141,914 bp in C. striata var. striata to 124,420 bp in C. involuta (Notes S1B). The plastome of Corallorhiza bentleyi is similar in size to that of C. involuta at 124,481 bp, a difference of only 51 bp, while C. striata Sierra Nevada has a plastome of 137,068 bp, and is similar in size to the previously sequenced plastome of C. striata vreelandii (137,505 bp). The green, leafless C. trifida has a plastome of 149,384 bp, and thus is marginally longer than that of the leafy Calypso bulbosa (149,313 bp). Within the C. striata complex, GC content ranges from 36.3% in C. striata var. striata (accessions 120b UT and 350b OR) to 36.6 in C. bentleyi and C. involuta (here, including both IR copies). These two values show a negative correlation with plastome size (Spearman’s D = 67, P = 0.0121), even when corrected for phylogenetic relationships (FPIC = 11.93; P = 0.02594), though the range in variation is small for GC content overall. Total mean GC content was 35% and for all non-pseudogene, protein coding sequences it was 36.4%. For all genes and putative pseudogenes GC content was 37.4%, and for all non-coding DNA, GC content was 30.6%.
Polymorphisms ranged from 364 sites within C. striata var. striata (π = 0.00063; 18 haplotypes) to 2 in C. bentleyi (π = 0.00001; three haplotypes) (Notes S1E,G). Average pairwise polymorphisms ranged between 2405.0 sites between Sierra Nevadan C. striata and C. involuta. A Mantel correlation between genetic and geographic whole-plastome distances was significant (R = 0.54, P = 0.01). MAUVE detected no evidence of genomic rearrangements. Root-to-tip GTR distances indicated significantly different branch lengths among clades (Kruskal-Wallis H = 40.65, P < 0.0001; in all pairwise comparisons, Mann-Whitney P < 0.01 after Bonferroni correction). The number of putatively functional genes ranges from 78 to 80 (of a possible 116 based on C. trifida as a reference), in contrast to the range observed in plastome size across the C. striata complex (124,420–141,914 bp; (Fig. 3). Despite the total numbers being similar, the presence of putatively functional genes varies for many photosynthesis-related genes, including cytochrome (pet), photosystems I and II (psa, psb), photosystem assembly proteins (ycf3, 4), and one ‘housekeeping’ gene, trnTGGU (Fig. 3).
Fig. 3.
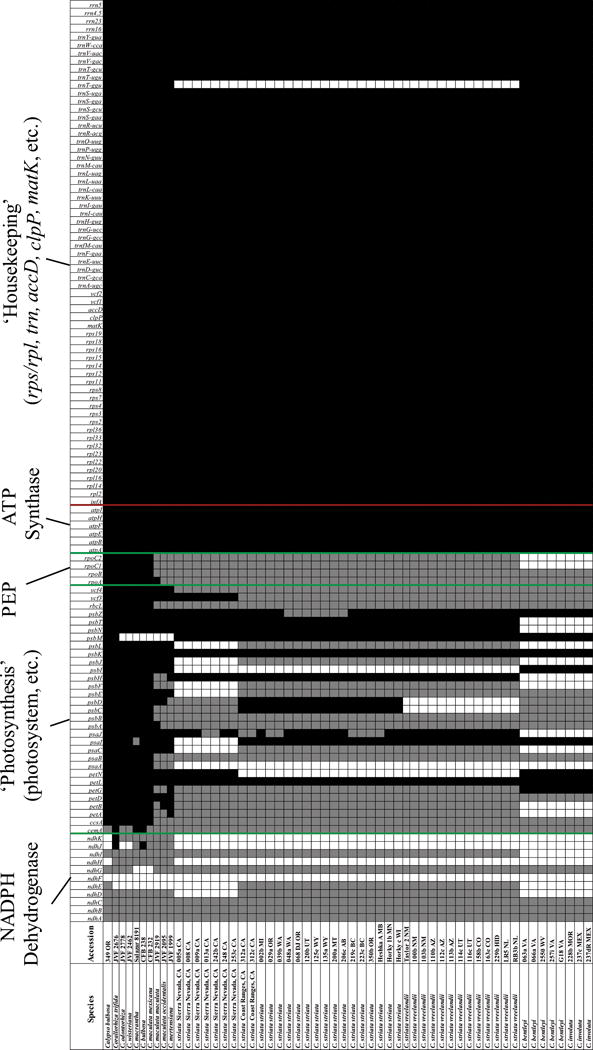
Summary of putatively functional genes (black), pseudogenes (gray), and gene losses (white) among Corallorhiza and the Corallorhiza striata complex. Collection numbers are in the second column, including US, Canadian, or Mexican state/province. Green lines indicate breaks between ndh, photosynthesis-related, rpo, and atp genes; red line marks ‘housekeeping’ genes.
Based on Bayesian molecular coevolutionary analysis, dS and dN are strongly correlated (posterior probabilities, pp: 1.00; pp – maximally controlled correlation, ppMC = 0.99). Neither dN nor dS apparently directly relate to changes of selective pressure as assessed by the ratio of dN/dS (pp = 0.40; ppMC = 0.49). However, we find a strong positive correlation between dN and the number of indels (pp = 0.95; ppMC = 0.7), and also a strong correlation between GC content and indels (pp = 0.93; ppMC = 0.76). A negative association exists between the indels and genome length. The indel/GC association is prominently pronounced in noncoding DNA (pp = 0.002, ppMC = 0.17) but co-correlates extensively with other genetic traits in protein-coding regions (pp < 0.001, ppMC = 0.54).
Timing of plastome degradation
Our analysis suggests that Corallorhiza has diversified c. 9.3 million yr ago (mya; 95% Highest Posterior Density (HPD) = 6.67–11.62 mya; Fig. 4a). The Corallorhiza striata complex likely originated c. 7.3 mya (95% HPD = 4.76–9.30), and the C. maculata complex arose c. 4.0 mya (HPD = 2.52–5.37 mya). Within the C. striata complex, C. bentleyi and C. involuta diverged relatively recently, c. 2.1 mya (0.97–3.53, mya), while the ancestor of the remaining accessions (C. striata sensu stricto) diverged c. 2.86 mya (1.36–4.33 mya). Varieties striata and vreelandii diverged c. 1.8 mya (0.70–2.95 mya).
Fig. 4.
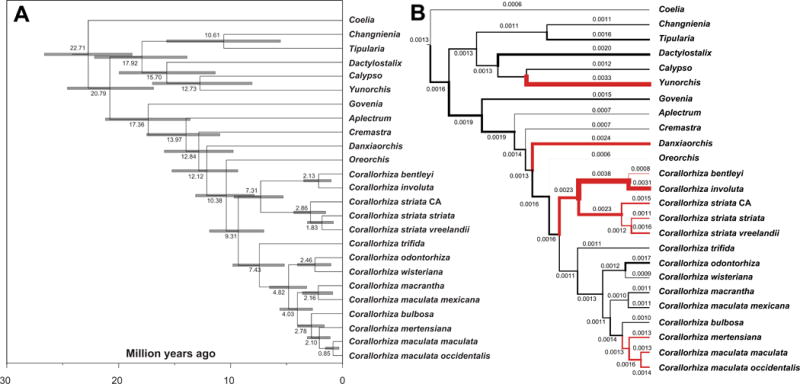
(a) Divergence time estimates from BEAST2 under an uncorrelated lognormal clock model (internal transcribed Spacer (ITS), matK, psaB, and rbcL), calibrated with fossils from Conran et al. (2009) and Poinar & Rasmussen (2017). Scale axis indicates millions of years before present. Node bars indicate the 95% Highest Posterior Density estimates. (b) Substitution rates per branch (substitutions·per site·per year) from BEAST2. Branch widths are scaled by substitution rate, and mean estimates are given for each branch. Red branches indicate putatively non-photosynthetic, fully mycoheterotrophic lineages.
We observe substitution rate increases (Fig. 4b) in our study group: at the origin of the C. striata complex; in the bentleyi-involuta clade; at the origin of fully mycoheterotrophic members of the C. maculata complex (a slight increase); and in the two other fully mycoheterotrophic lineages included here within Calypsoinae, Yunorchis (Yoania) and Danxiaorchis. Genes of the NADPH dehydrogenase complex (ndh) display evidence of degradation before the crown radiation of Corallorhiza, but these losses may either be shared or have occurred in parallel with other members of Calypsoinae for which sequenced plastomes are not yet available (Fig. 5). The relative timing of divergence events suggests that pseudogenization in photosynthesis-related complexes (psa/psb, rbcL, pet) and the RNA polymerase (rpo) began between 6.63–11.62 mya in the C. striata complex, and between 1.14 and 3.25 mya in the C. maculata complex (including only C. mertensiana, C. maculata var. maculata, and C. maculata var. occidentalis), with continued physical losses of the ndh, rpo, and photosynthesis-related complexes among the lineages diverging afterward. The loss of trnTGGU occurred after the split between the bentleyi-involuta clade and before the divergence of C. striata s.s.
Fig. 5.
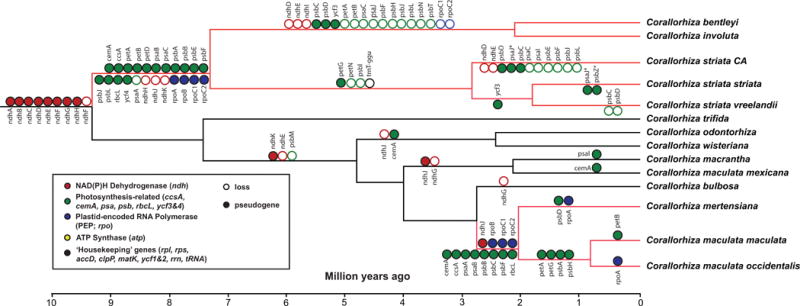
Putative pseudogenes and gene losses in Corallorhiza, based on the chronogram in Fig. 4. Genes are categorized roughly by functional class. Closed circles, pseudogenes; open circles, gene losses. *, pseudogene in some but not all accessions. Red branches correspond to fully mycoheterotrophic lineages.
Comparing genome skimming and sequence capture
The normalized percentage of mapped reads was higher for TSC than for GS (Notes S1). GS yielded a mean of 1.19 ± 0.85% reads mapped as opposed to TSC with 40.93 ± 10.90% (Mann-Whitney U, P < 0.0001). However, members of C. striata var. vreelandii and C. trifida (upon which probes were designed) had significantly higher capture efficiency, considered together (mean = 49.9%; Mann-Whitney, P < 0.001). Divergence (measured as patristic, or tip-to-tip GTR distances on the tree) from the C. striata vreelandii plastome used for probe design (accession LT2 NM) and capture efficiency were negatively correlated (Kendall’s tau = −0.314, P = 0.003).
Comparison of GS vs TSC mappings shows a striking contrast in terms of variation in coverage depth across the plastome (Fig. 6a). GS datasets give even coverage across the plastome, while TSC data vary immensely. GC content and coverage depth show a strong positive correlation across the plastome (Fig. 6b; Pearson R = 0.634; P < 0.0001), and further, values for sliding windows are not autocorrelated (Durbin-Watson, P = 0.48). Some spacers and introns have short stretches of low coverage depth, consistently across samples, in which coverage drops below 10× or sometimes to zero (rps16-trnQ, trnT-trnL, atpB-rbcL, psbB-psbT, ycf4-cemA, rpl16 intron, and ycf1 (partial copy)-rpl32 (ndhF has been lost in all accessions)). Zero-coverage spots occurred in one or more of the above long spacers or introns (mean length = 760.3 bp, vs total mean length of 422.4 bp for all introns/spacers) with relatively low GC content (mean = 24.0%, vs of 30.6% for all spacers/introns) and AT-rich microsatellites or homopolymer runs.
Fig. 6.
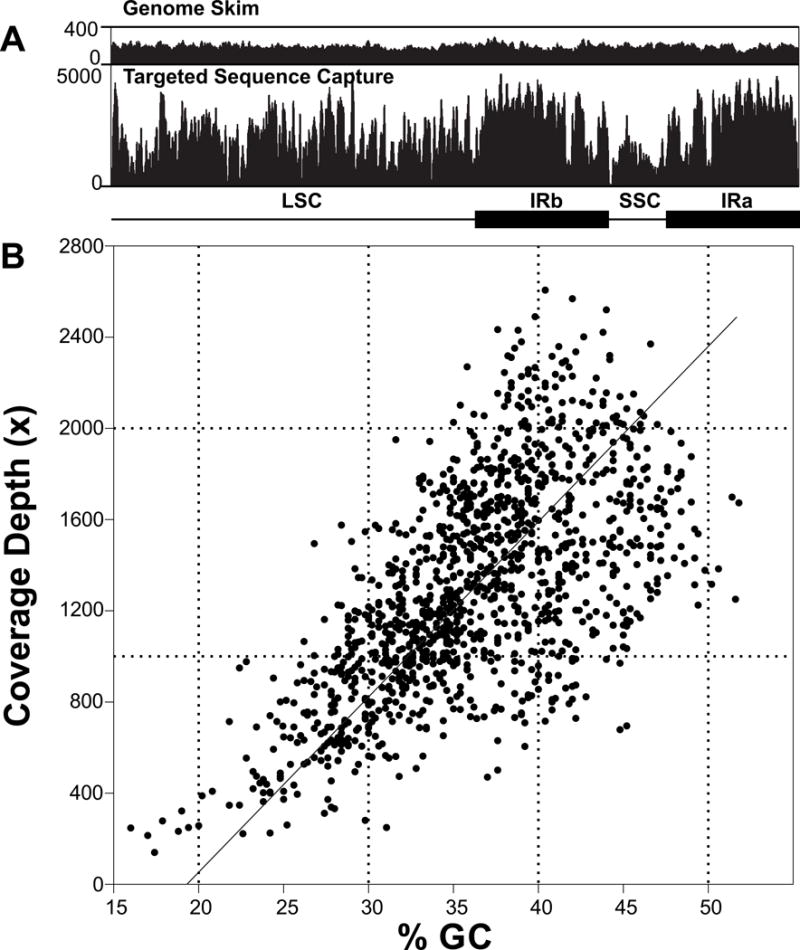
(a) Comparison of read coverage depth distribution across the entire plastome as a result of genome skimming (above) and targeted sequence capture (below), for Corallorhiza striata var. striata accession 350b OR. LSC, large single copy; SSC, small single copy; IR, inverted repeat. (b) The relationship between mean GC content and coverage depth across the plastomes of the C. striata complex for target capture data, based on a sliding window analysis. Slope of the best-fit line indicates the Pearson Correlation Coefficient (R = 0.634, P < 0.0001).
Discussion
This study represents to our knowledge the finest-scale investigation of plastome evolution in any heterotrophic plant clade. We find: strongly supported plastid relationships; drastic variation in plastome size at an infraspecific scale; rapid plastome degradation; heterogeneity in substitution rates among lineages; and contrasting distributions of coverage between GS and TSC methods.
Plastome relationships
Plastid relationships are strongly supported among members of the C. striata complex. Reciprocal monophyly is observed among plastomes of C. bentleyi and C. involuta (Fig. 1), providing evidence for recognition of separate species (Greenman 1898; Freudenstein, 1997; Barrett & Freudenstein, 2011). It is unknown whether these two species are the result of a long-distance dispersal event or isolation via recent vicariance. Sister to C. striata vars. vreelandii and striata is a Sierra Nevadan clade that excludes coastal California and Oregon. This suggests barriers to pollen/seed flow to the east and west of the Sierra Nevada, a situation observed in other taxa (Raven & Axelrod, 1974; Forister et al., 2004; Gugger et al., 2010). Alpine and arid lowlands to the east and the Central Valley to the west likely prevent gene flow between the Coast Ranges, Sierras, and Rocky Mountains (e.g. Calsbeek et al., 2003). Thus, Sierran populations – from a plastid perspective – might have evolved in isolation from the more widespread varieties to the north and southeast. Accessions from coastal CA and western OR diverge near the base of the var. striata clade (Fig. 1), suggesting the possibility of coastal refugia. Additional sampling at the nexus of the Sierras, Coast, and Cascade Ranges will allow fine-scale determination of this putative phylogeographic break.
Patterns of plastome variation
‘Leaflessness’ per se does not appear to be related to drastic changes in the plastome, as evidenced by the leafy Calypso and the leafless, green species of Corallorhiza having highly similar plastomes in terms of size and gene content, despite having experienced degradation of the ndh complex (Fig. 3; Barrett et al., 2014). Weakening of selective pressure already evident in plastomes of leafless, green species (e.g. losses of ndh) may already be in the early stages in many leafy orchids, though this deserves more comprehensive study. Alternatively, the idiosyncratic losses of these genes in some leafy taxa but not in others may be explained by their inhabiting low- or variable-light environments. It is clear that mycoheterotrophy can be implicated in relaxed selection on this gene complex (Wicke et al., 2013; Barrett et al., 2014; Kim et al., 2015; Kim & Chase, 2017; Lin et al., 2017).
A promising system in which to address these questions is the orchid tribe Neottieae, including both photosynthetic and non-photosynthetic species (Pridgeon et al., 2005; Chase et al., 2015; Feng et al., 2016). Cephalanthera damasonium is polymorphic for green and albino individuals, representing a potentially transitional stage from partial to full mycoheterotrophy (Julou et al., 2005; Abadie et al., 2006; Roy et al., 2013). Albino individuals suffer fitness disadvantages relative to green individuals, from water retention to lower seed output; thus, any successful transition to full mycoheterotrophy would require a concerted suite of morphological adaptations (Roy et al., 2013). While selection likely has consequences for nuclear genes encoding morphological structures, it may also drive changes in plastid gene content. Analogous to the potentially deleterious retention of leaves and stomata in non-photosynthetic Cephalanthera damasonium, retention of ndh and other photosynthesis-associated genes may incur similar fitness costs if retained and expressed. It will be informative to see whether species exhibiting polymorphic albinism (e.g. C. damasonium, Epipactis helleborine; Salmia, 1986) display evidence of ndh loss.
Our study demonstrates for the first time that plastome size and gene content vary substantially within a single complex of closely related heterotrophic lineages. This is significant because while many studies have focused on the ‘endpoint’ of plastome degradation, few have focused on the beginning of the process in an infraspecific context. The most pronounced differences span the deepest split in the complex, between the clades comprising C. bentleyi/involuta and C. striata sensu stricto (Figs 3, 5). These clades have 19 pseudogenes in common yet they share only four genes that have been deleted (ndh, ‘photosynthesis-related’, and rpo). No variation is observed in protein-coding gene content within var. vreelandii. The photosystem gene psaJ has become a pseudogene in two sister accessions from the Sierra Nevada (13a CA and 242b CA from Tehama and Placer Counties, California, respectively), representing the two northernmost localities in the Sierra Nevada. Both psaJ and psbZ have become pseudogenes in some accessions of var. striata. For psaJ these accessions are mostly from coastal regions in California, Oregon, and Washington (USA) but some are from British Columbia, and Manitoba, Canada. The psaJ pseudogene is the result of a variable poly-T mononucleotide repeat, and thus has likely evolved independently in multiple lineages. Accessions with psbZ pseudogenes occupy a northern Rocky Mountain clade comprising accessions from Alberta, Montana, Oregon, Utah, and Wyoming.
The rapid and extensive plastome divergence observed here within a single species complex may have implications for reproductive isolation among members with different plastome types, in reference to plastid-nuclear incompatibilities (e.g. Coyne & Orr, 2004; Greiner et al., 2011). It is unclear whether pollen- or seed-mediated gene flow would ultimately cause a population-level decrease in fitness, say, if pollen or seed from C. striata var. striata regularly reaches individuals of C. striata var. vreelandii or vice versa (e.g. in Utah or South Dakota, USA where they grow in regional proximity; Fig. 1). Multiple nuclear loci from across the genome and additional population-level sampling would allow the detection of putative hybrids, suggesting a lack of such barriers. Crossing studies between taxa with different plastome types would ultimately be informative on potential plastid-nuclear or other incompatibilities, but the inability to culture Corallorhiza necessitates field-based approaches. In any case, such drastic divergence in genome size/gene content in mycoheterotrophs represents a potentially informative, yet unexplored, study system for rapid post-zygotic isolation.
Results from Bayesian coevolutionary analyses identify elevated rates of dN and dS together in fully mycoheterotrophic taxa relative to green taxa, suggesting overall elevated mutation rates in the plastomes of the C. striata complex. Correlation between dN, GC content, and indels indicates relaxation of purifying selection in protein sequences and possibly structure, though dN/dS ratios were not significantly correlated with any of these (largely due to co-elevated dN and dS rates). The nature of these co-correlates in coding regions remains elusive though, mainly because several of the examined genetic traits produce weak positive or negative correlations with molecular evolutionary rates or selective pressure itself.
The variation observed among the two principal clades is astounding in that a difference of 17,494 bp in length occurs within a single species complex. Schelkunov et al. (2015) reported a difference of 11,063 bp in the plastomes of congeners Epipogium aphyllum (30,650 bp) and E. roseum (19,047 bp), explained by the loss of one copy of the inverted repeat. Comparable length variation is observed among species in a fully mycoheterotrophic clade of Neottia, with plastomes of 110,246 bp in N. listeroides to 83,190 bp in N. acuminata (Feng et al., 2016). Extensive variation is observed also among genera of fully mycoheterotrophic Ericaceae (Gruzdev et al., 2016, Logacheva et al., 2016, Ravin et al., 2016, Braukmann et al., 2017) and parasitic broomrapes (Cusimano & Wicke, 2016). The range in plastome length observed here is not due to the loss of an inverted repeat copy, but to differential rates of deletion in bentleyi-involuta clade versus C. striata sensu stricto.
Timing of plastome degradation
Corallorhiza has experienced rapid plastid genome degradation, with initial diversification of the genus occurring c. 9.3 mya (HPD = 6.67–11.62; Figs 4a, 5). Conservatively, this suggests that degradation within the fully mycoheterotrophic C. striata and C. maculata complexes began less than 11.62 and 4.02 mya, respectively, taking into account the upper 95%HPD limit for the stem node of each (Fig. 4A). Thus, C. bentleyi and C. involuta have lost 1/8 of their plastome in a few million years, and collectively, members of the C. striata complex have physically or functionally lost most photosynthetic genes.
Divergence times have been incorporated into two recent studies of plastome degradation in Orobanchaceae (Cusimano & Wicke, 2016; Wicke et al., 2016) and in the orchid tribe Neottieae (Feng et al., 2016). A conservative estimate of the photosynthetic loss in holoparasitic Orobanchaceae places the start of this process at < 50 mya, with many genes hypothesized to have been lost within the first 5–10 million yr following the transition to holoparasitism (Cusimano & Wicke, 2016). Estimates within Neottieae are < 28 mya for Aphyllorchis, and < 21 mya for fully mycoheterotrophic Neottia (Feng et al., 2016). Both lineages are in more advanced stages of degradation relative to Corallorhiza, with losses of function in the atp complex, and additionally several ‘housekeeping genes’ (trn/rps/rpl/rrn) in Orobanchaceae.
There is an apparent correlation between the degree of loss per clade and their respective times of divergence. Older clades show extreme plastome degradation, likely because they had time to accumulate mutations resulting in pseudogenes and losses (Wicke & Naumann, 2017). It remains to be explored whether there is a linear relationship between time and degree of degradation, or if this has accelerated over time, as a positive feedback. One hypothesis is that rapid loss of function occurs as selection pressure weakens on photosynthesis. Assuming that many changes observed in the plastid genome are mirrored in the nuclear genome, and further on the hypothesis that evolutionary rates are elevated overall in nonphotosynthetic parasites (e.g. Duff & Nickrent, 1997; Bromham et al., 2013), the relationship between functional gene loss and substitution rate might instead be nonlinear, and have a sort of ratcheting effect on mutation rate itself, as suggested by Wicke et al. (2016). Our findings are similar to those of Bromham et al. (2013) and Wicke et al. (2016) in that dN and dS but not dN/dS have increased in the C. striata complex, suggesting that substitution rates are accelerated, and not only due to relaxed selection on photosynthesis. The alternative would be punctuated bursts of losses; such a scenario could take place soon after the loss of photosynthesis until a new genomic equilibrium is achieved; i.e., only genes with essential functions remain in the plastome (i.e. ‘stationary phase’ sensu Naumann et al., 2016). These two scenarios could happen together: ‘punctuational’ stages of gene loss, with each stage of stasis representing a new, temporary equilibrium with higher mutation rates.
Within the C. striata complex we see rapid pseudogenization (Fig. 5), presumably following initial loss of photosynthesis in the clade, followed by physical losses of many pseudogenes. Members of the C. striata complex may be in a stationary phase, evidenced by functional atp genes with intact reading frames, and loss of only a single tRNA in C. striata sensu stricto (i.e. excluding the bentleyi-involuta clade), against a ‘background’ of massive ndh and photosynthesis gene degradation. However, total length reduction within the complex occurred over only a few million years, suggesting the bentleyi-involuta ancestor may have entered a new phase, having experienced a higher total amount of deletions than the rest of the complex. Thus, the pattern here seems to partially fit both aforementioned scenarios. It will be important to test whether similar patterns are observed in nuclear-encoded photosynthetic machinery, and whether there is evidence to support divergent evolutionary trajectories based on gene expression and substitution patterns in genes involved in DNA repair and recombination.
Comparing genome skimming and sequence capture
Deep coverage of the plastome for TSC allowed many-fold more accessions to be sequenced in a single Illumina run than with GS alone (see Stull et al., 2013), suggesting TSC is a viable option in rapidly evolving plastomes. Coverage was even across the plastome in GS datasets (Fig. 6A), but extremely uneven in TSC datasets; in the latter, areas with higher GC content had deeper coverage (Fig. 6b). Very high and very low GC content can be problematic for capture-based methods (Asan et al., 2011; Clark et al., 2011; Samorodnitsky et al., 2015). Particularly troubling are regions with < 20% GC, which here result in very low coverage (Fig. 6b; Samorodnitsky et al., 2015).
Genome skimming may be a better choice if spacer/intron regions are of primary interest, while TSC is more cost effective for coding regions (and recent pseudogenes), given the same number of samples to be sequenced. Pseudogenes may over evolutionary time end up becoming deleted before enough mutations accumulate to significantly reduce their GC content to the point where TSC becomes ineffective (Wicke et al., 2013; Barrett et al., 2014; Graham et al., 2017). Targeted sequence capture may be particularly useful in heterotrophic taxa, as the possibility exists for reduced cellular plastid populations—especially of chloroplasts—but the latter hypothesis has not been tested explicitly (e.g. see observations from Molina et al., 2014; Wicke et al., 2013; Feng et al., 2016). Regions with the lowest GC content (long stretches of AT, repeats, etc.) had the lowest capture efficiency (Fig. 6b; Zhou & Holliday, 2012), but ironically these are often the regions of highest interest with the most SNP information in fine-scale population genetic studies using organellar DNA in plants (e.g. Shaw et al., 2007, 2014).
Conclusion
We have conducted the first explicit investigation of plastid genome evolution at the infraspecific level for any heterotrophic plant, and have demonstrated that the changes observed at higher levels can be detected at lower levels as well. We demonstrate that in only a few million years, drastic changes in plastome size and functional gene content can occur due to relaxed selective constraints on photosynthesis. Such studies will be important in the future for refining models of genomic change in organisms with radically altered selective regimes. As technology advances, it will be informative to include elements of the nuclear genome, either via capture-based methods, transcriptomes, or even proteomes in such studies. Similar studies in other groups are needed for comparison to the patterns observed here, to allow phylogenetically independent estimates of the scale and rate of genome modification across clades that display convergent, radical changes in nutritional lifestyle.
Supplementary Material
Notes S1 Photographs, accession information (including vouchers and coverage depth of the plastme), divergence time estimation, and polymorphism information for the Corallorhiza striata complex.
Acknowledgments
We thank Lydia Smith (UC, Berkeley Evolutionary Genetics Lab), Ke Bi (UC, Berkeley Computational Genomics Resource Lab), and the Vincent J. Coates Genomics Sequencing Laboratory (supported by NIH S10 Instrumentation Grants S10RR029668 and S10RR027303) for expert assistance with capture experiments. We thank Sean Blake (Global Biologics, LLC), Igor Antoneschkin (CalTech), and Laragen, Inc. for assistance with library and sequencing services. We thank Will Iles for expert advice in divergent time estimation, and Ian Tindel for assistance with initial analyses. We thank five anonymous reviewers and Marc-Andre Selosse for helpful comments. Research was supported by NSF DEB 0816661 Research Opportunity Award Supplement to CFB and Chelsea Specht (UC, Berkeley), and the California State University Program for Education & Research in Biotechnology (CSUPERB).
Footnotes
Supporting Information
Additional Supporting Information may be found online in the Supporting Information tab for this article:
Please note: Wiley Blackwell are not responsible for the content or functionality of any supporting information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Author contributions
C.F.B. conceived of and designed the experiment, collected material, carried out lab work and analyses, wrote the paper and figures; S.W. conducted analyses and revised drafts of the paper; C.S. conducted lab work and revised drafts of the paper.
References
- Abadie JC, Puttsepp U, Gebauer G, Faccio A, Bonfante P, Selosse MA. Cephalanthera longifolia (Neottieae, Orchidaceae) is mixotrophic: a comparative study between green and nonphotosynthetic individuals. Canadian Journal of Botany-Revue Canadienne De Botanique. 2006;84:1462–1477. [Google Scholar]
- Asan, Xu Y, Jiang H, Tyler-Smith C, Xue YL, Jiang T, Wang JW, Wu MZ, Liu X, Tian G, et al. Comprehensive comparison of three commercial human whole-exome capture platforms. Genome Biology. 2011;12:R95. doi: 10.1186/gb-2011-12-9-r95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett CF, Baker WJ, Comer JR, Conran JG, Lahmeyer SC, Leebens-Mack JH, Li J, Lim GS, Mayfield-Jones DR, Perez L, et al. Plastid genomes reveal support for deep phylogenetic relationships and extensive rate variation among palms and other commelinid monocots. New Phytologist. 2015;209:855–870. doi: 10.1111/nph.13617. [DOI] [PubMed] [Google Scholar]
- Barrett CF, Davis JI. The plastid genome of the mycoheterotrophic Corallorhiza striata (Orchidaceae) is in the relatively early stages of degradation. American Journal of Botany. 2012;99:1513–1523. doi: 10.3732/ajb.1200256. [DOI] [PubMed] [Google Scholar]
- Barrett CF, Freudenstein JV. Patterns of morphological and plastid DNA variation in the Corallorhiza striata species complex (Orchidaceae) Systematic Botany. 2009;34:496–504. [Google Scholar]
- Barrett CF, Freudenstein JV. An integrative approach to delimiting species in a rare but widespread mycoheterotrophic orchid. Molecular Ecology. 2011;20:2771–2786. doi: 10.1111/j.1365-294X.2011.05124.x. [DOI] [PubMed] [Google Scholar]
- Barrett CF, Freudenstein JV, Taylor DL, Kõljalg U. Rangewide analysis of fungal associations in the fully mycoheterotrophic Corallorhiza striata complex (Orchidaceae) reveals extreme specificity on ectomycorrhizal Tomentella (Thelephoraceae) across North America. American Journal of Botany. 2010;97:628–643. doi: 10.3732/ajb.0900230. [DOI] [PubMed] [Google Scholar]
- Barrett CF, Freudenstein JV, Li J, Mayfield-Jones DR, Perez L, Pires JC, Santos C. Investigating the path of plastid genome degradation in an early-transitional clade of heterotrophic orchids, and implications for heterotrophic angiosperms. Molecular Biology and Evolution. 2014;31:3095–3112. doi: 10.1093/molbev/msu252. [DOI] [PubMed] [Google Scholar]
- Bellot S, Renner SS. The plastomes of two species in the endoparasite genus Pilostyles (Apodanthaceae) each retain just five or six possibly functional genes. Genome Biology and Evolution. 2016;8:189–201. doi: 10.1093/gbe/evv251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi K, Vanderpool D, Singhal S, Linderoth T, Moritz C, Good JM. Transcriptome-based exon capture enables highly cost-effective comparative genomic data collection at moderate evolutionary scales. BMC Genomics. 2012;13:403. doi: 10.1186/1471-2164-13-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi K, Linderoth T, Vanderpool D, Good JM, Nielsen R, Moritz C. Unlocking the vault: next-generation museum population genomics. Molecular Ecology. 2013;22:6018–6032. doi: 10.1111/mec.12516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouckaert R, Heled J, Kuhnert D, Vaughan T, Wu CH, Xie D, Suchard MA, Rambaut A, Drummond AJ. BEAST 2: A Software platform for Bayesian evolutionary analysis. PLoS Computational Biology. 2014;10:e1003537. doi: 10.1371/journal.pcbi.1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braukmann TWA, Broe MB, Stefanovic S, Freudenstein JV. On the brink: the highly reduced plastomes of nonphotosynthetic Ericaceae. New Phytologist. 2017;216:254–266. doi: 10.1111/nph.14681. [DOI] [PubMed] [Google Scholar]
- Bromham L, Cowman PF, Lanfear R. Parasitic plants have increased rates of molecular evolution across all three genomes. BMC Evolutionary Biology. 2013;13:126. doi: 10.1186/1471-2148-13-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calsbeek R, Thompson JN, Richardson JE. Patterns of molecular evolution and diversification in a biodiversity hotspot: the California Floristic Province. Molecular Ecology. 2003;12:1021–1029. doi: 10.1046/j.1365-294x.2003.01794.x. [DOI] [PubMed] [Google Scholar]
- Cameron KM. Utility of plastid psaB gene sequences for investigating intrafamilial relationships within Orchidaceae. Molecular Phylogenetics and Evolution. 2004;31:1157–1180. doi: 10.1016/j.ympev.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Chase MW, Cameron KM, Freudenstein JV, Pridgeon AM, Salazar G, Van den Berg C, Schuiteman A. An updated classification of Orchidaceae. Botanical Journal of the Linnean Society. 2015;177:151–174. [Google Scholar]
- Clark MJ, Chen R, Lam HYK, Karczewski KJ, Chen R, Euskirchen G, Butte AJ, Snyder M. Performance comparison of exome DNA sequencing technologies. Nature Biotechnology. 2011;29:908–U206. doi: 10.1038/nbt.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conran JG, Bannister JM, Lee DE. Earliest orchid macrofossils: Early Miocene Dendrobium and Earina (Orchidaceae: Epidendroideae) from New Zealand. American Journal of Botany. 2009;96:466–474. doi: 10.3732/ajb.0800269. [DOI] [PubMed] [Google Scholar]
- Coyne JA, Orr HA. Speciation. Sunderland, MA, USA: Sinauer Associates; 2004. [Google Scholar]
- Cronn R, Liston A, Parks M, Gernandt DS, Shen R, Mockler T. Multiplex sequencing of plant chloroplast genomes using Solexa sequencing-by-synthesis technology. Nucleic Acids Research. 2008;36:e122. doi: 10.1093/nar/gkn502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusimano N, Wicke S. Massive intracellular gene transfer during plastid genome reduction in nongreen Orobanchaceae. New Phytologist. 2016;210:680–693. doi: 10.1111/nph.13784. [DOI] [PubMed] [Google Scholar]
- Darling AE, Mau B, Perna NT. progressiveMauve: Multiple Genome Alignment with Gene Gain, Loss and Rearrangement. PLoS ONE. 2010;5:e11147. doi: 10.1371/journal.pone.0011147. https://doi.org/10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delannoy E, Fujii S, Colas des Francs-Small C, Brundrett M, Small I. Rampant gene loss in the underground orchid Rhizanthella gardneri highlights evolutionary constraints on plastid genomes. Molecular Biology and Evolution. 2011;28:2077–2086. doi: 10.1093/molbev/msr028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dierckxsens N, Mardulyn P, Smits G. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Research. 2017;45:e18. doi: 10.1093/nar/gkw955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle JJ, Doyle JL. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemical Bulletin. 1987;19:11–15. [Google Scholar]
- Duff RJ, Nickrent DL. Characterization of mitochondrial small-subunit ribosomal RNAs from holoparasitic plants. Journal of Molecular Evolution. 1997;45:631–639. doi: 10.1007/pl00006267. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. Phylogenies and the comparative method. American Naturalist. 1985;125:1–15. doi: 10.1086/703055. [DOI] [PubMed] [Google Scholar]
- Feng YL, Wicke S, Li JW, Han Y, Lin CS, Li DZ, Zhou TT, Huang WC, Huang LQ, Jin XH. Lineage-specific reductions of plastid genomes in an orchid tribe with partially and fully mycoheterotrophic species. Genome Biology and Evolution. 2016;8:2164–2175. doi: 10.1093/gbe/evw144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forister ML, Fordyce JA, Shapiro AM. Geological barriers and restricted gene flow in the holarctic skipper Hesperia comma (Hesperiidae) Molecular Ecology. 2004;13:3489–3499. doi: 10.1111/j.1365-294X.2004.02349.x. [DOI] [PubMed] [Google Scholar]
- Foster CSP, Sauquet H, Van der Merwe M, McPherson H, Rossetto M, Ho SYW. Evaluating the impact of genomic data and priors on Bayesian estimates of the angiosperm evolutionary timescale. Systematic Biology. 2017;66:338–351. doi: 10.1093/sysbio/syw086. [DOI] [PubMed] [Google Scholar]
- Freudenstein JV. A monograph of Corallorhiza (Orchidaceae) Harvard Papers in Botany. 1997;10:5–51. [Google Scholar]
- Freudenstein JV, Senyo DM. Relationships and evolution of matK in a group of leafless orchids (Corallorhiza and Corallorhizinae; Orchidaceae: Epidendroideae) American Journal of Botany. 2008;95:498–505. doi: 10.3732/ajb.95.4.498. [DOI] [PubMed] [Google Scholar]
- Freudenstein JV, Yukawa T, Luo YB. A reanalysis of relationships among Calypsoinae (Orchidaceae: Epidendroideae): floral and vegetative evolution and the placement of Yoania. Systematic Botany. 2017;42:17–25. [Google Scholar]
- Funk HT, Berg S, Krupinska K, Maier UG, Krause K. Complete DNA sequences of the plastid genomes of two parasitic flowering plant species, Cuscuta reflexa and Cuscuta gronovii. BMC Plant Biology. 2007;7:45. doi: 10.1186/1471-2229-7-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garland T, Harvey PH, Ives AR. Procedures for the analysis of comparative data using phylogenetically independent contrasts. Systematic Biology. 1992;41:18–32. [Google Scholar]
- Gebauer G, Preiss K, Gebauer AC. Partial mycoheterotrophy is more widespread among orchids than previously assumed. New Phytologist. 2016;211:11–15. doi: 10.1111/nph.13865. [DOI] [PubMed] [Google Scholar]
- Givnish TJ, Spalink D, Ames M, Lyon SP, Hunter SJ, Zuluaga A, Iles WJ, Clements MA, Arroyo MT, Leebens-Mack J, et al. Orchid phylogenomics and multiple drivers of their extraordinary diversification. Proceeding of the Royal Society B. 2015;282:20151553. doi: 10.1098/rspb.2015.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham SW, Lam VKY, Merckx VSFT. Plastomes on the edge: the evolutionary breakdown of mycoheterotroph plastid genomes. New Phytologist. 2017;214:48–55. doi: 10.1111/nph.14398. [DOI] [PubMed] [Google Scholar]
- Greenman JM. Diagnoses of new and critical Mexican phanerogams (Corallorhiza involuta) Proceedings of the American Academy of Arts. 1898;33:474. [Google Scholar]
- Greiner S, Rauwolf U, Meurer J, Herrmann RG. The role of plastids in plant speciation. Molecular Ecology. 2011;20:671–691. doi: 10.1111/j.1365-294X.2010.04984.x. [DOI] [PubMed] [Google Scholar]
- Gruzdev E, Mardanov A, Beletsky A, Kadnikov V, Kochieva E, Ravin N, Skryabin K. Reduction of the chloroplast genome and the loss of photosynthetic pathways in the mycoheterotrophic plant Monotropa hypopitys, as revealed by genome and transcriptome sequencing. FEBS Journal. 2016;283:341–342. [Google Scholar]
- Gugger PF, Sugita S, Cavender-Bares J. Phylogeography of Douglas-fir based on mitochondrial and chloroplast DNA sequences: testing hypotheses from the fossil record. Molecular Ecology. 2010;19:1877–1897. doi: 10.1111/j.1365-294X.2010.04622.x. [DOI] [PubMed] [Google Scholar]
- Hammer Ø, Harper DAT, Ryan PD. PAST: Paleontological statistics software package for education and data analysis. Palaeontologia Electronica. 2001;4 http://palaeo-electronica.org/2001_1/past/issue1_01.htm. [Google Scholar]
- Hodges E, Rooks M, Xuan ZY, Bhattacharjee A, Gordon DB, Brizuela L, McCombie WR, Hannon GJ. Hybrid selection of discrete genomic intervals on custom-designed microarrays for massively parallel sequencing. Nature Protocols. 2009;4:960–974. doi: 10.1038/nprot.2009.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges E, Xuan Z, Balija V, Kramer M, Molla MN, Smith SW, Middle CM, Rodesch MJ, Albert TJ, Hannon GJ, et al. Genome-wide in situ exon capture for selective resequencing. Nature Genetics. 2007;39:1522–1527. doi: 10.1038/ng.2007.42. [DOI] [PubMed] [Google Scholar]
- Julou T, Burghardt B, Gebauer G, Berveiller D, Damesin C, Selosse MA. Mixotrophy in orchids: insights from a comparative study of green individuals and nonphotosynthetic individuals of Cephalanthera damasonium. New Phytologist. 2005;166:639–653. doi: 10.1111/j.1469-8137.2005.01364.x. [DOI] [PubMed] [Google Scholar]
- Kim HT, Chase MW. Independent degradation in genes of the plastid ndh gene family in species of the orchid genus Cymbidium (Orchidaceae; Epidendroideae) PLoS ONE. 2017;12:e0187318. doi: 10.1371/journal.pone.0187318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HT, Kim JS, Moore MJ, Neubig KM, Williams NH, Whitten WM, Kim JH. Seven new complete plastome sequences reveal rampant independent loss of the ndh gene family across orchids and associated instability of the Inverted Repeat/Small Single-Copy Region boundaries. PLoS ONE. 2015;10(11):e0142215. doi: 10.1371/journal.pone.0142215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam VK, Soto Gomez M, Graham SW. The highly reduced plastome of mycoheterotrophic Sciaphila (Triuridaceae) is colinear with its green relatives and is under strong purifying selection. Genome Biology and Evolution. 2015;7:2220–2236. doi: 10.1093/gbe/evv134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lartillot N, Poujol R. A phylogenetic model for investigating correlated evolution of substitution rates and continuous phenotypic characters. Molecular Biology and Evolution. 2011;28:729–744. doi: 10.1093/molbev/msq244. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zhang TC, Qiao Q, Ren ZM, Zhao JY, Yonezawa T, Hasegawa M, Crabbe MJC, Li JQ, Zhong Y. Complete chloroplast genome sequence of holoparasite Cistanche deserticola (Orobanchaceae) reveals gene loss and horizontal gene transfer from its host Haloxylon ammodendron (Chenopodiaceae) PLoS ONE. 2013;8:e58747. doi: 10.1371/journal.pone.0058747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim GS, Barrett CF, Pang CC, Davis JI. Drastic reduction of plastome size in the mycoheterotrophic Thismia tentaculata relative to that of its autotrophic relative Tacca chantrieri. American Journal of Botany. 2016;103:1129–1137. doi: 10.3732/ajb.1600042. [DOI] [PubMed] [Google Scholar]
- Lin CS, Chen JJW, Chiu CC, Hsiao HCW, Yang CJ, Jin XH, Leebens-Mack J, de Pamphilis CW, Huang YT, Yang LH, et al. Concomitant loss of NDH complex-related genes within chloroplast and nuclear genomes in some orchids. Plant Journal. 2017;90:994–1006. doi: 10.1111/tpj.13525. [DOI] [PubMed] [Google Scholar]
- Logacheva MD, Schelkunov MI, Penin AA. Sequencing and analysis of plastid genome in mycoheterotrophic orchid Neottia nidus-avis. Genome Biology and Evolution. 2011;3:1296–1303. doi: 10.1093/gbe/evr102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logacheva MD, Schelkunov MI, Shtratnikova VY, Matveeva MV, Penin AA. Comparative analysis of plastid genomes of non-photosynthetic Ericaceae and their photosynthetic relatives. Scientific Reports. 2016;6:30042. doi: 10.1038/srep30042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magoc T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeal JR, Arumugunathan K, Kuehl JV, Boore JL, dePamphilis CW. Systematics and plastid genome evolution of the cryptically photosynthetic parasitic plant genus Cuscuta (Convolvulaceae) BMC Biology. 2007;5:55–73. doi: 10.1186/1741-7007-5-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merckx V, Freudenstein JV. Evolution of mycoheterotrophy in plants: a phylogenetic perspective. New Phytologist. 2010;185:605–609. doi: 10.1111/j.1469-8137.2009.03155.x. [DOI] [PubMed] [Google Scholar]
- Merckx V. Mycoheterotrophy: the biology of plants living on fungi. New York, USA: Springer; 2013. [Google Scholar]
- Meyer M, Kircher M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harbor Protocols. 2010;2010:pdb.prot5448. doi: 10.1101/pdb.prot5448. [DOI] [PubMed] [Google Scholar]
- Molina J, Hazzouri KM, Nickrent D, Geisler M, Meyer RS, Pentony MM, Flowers JM, Pelser P, Barcelona J, Inovejas SA, et al. Possible loss of the chloroplast genome in the parasitic flowering plant Rafflesia lagascae (Rafflesiaceae) Molecular Biology and Evolution. 2014;31:793–803. doi: 10.1093/molbev/msu051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller K. SeqState – primer design and sequence statistics for phylogenetic DNA data sets. Applied Bioinformatics. 2005;4:65–69. doi: 10.2165/00822942-200504010-00008. [DOI] [PubMed] [Google Scholar]
- Müller K. Incorporating information from length-mutational events into phylogenetic analysis. Molecular Phylogenetics and Evolution. 2006;38:667–676. doi: 10.1016/j.ympev.2005.07.011. [DOI] [PubMed] [Google Scholar]
- Naumann J, Der JP, Wafula EK, Jones SS, Wagner ST, Honaas LA, Ralph PE, Bolin JF, Maass E, Neinhuis C, et al. Detecting and characterizing the highly divergent plastid genome of the nonphotosynthetic parasitic plant Hydnora visseri (Hydnoraceae) Genome Biology and Evolution. 2016;8:345–363. doi: 10.1093/gbe/evv256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolte V, Pandey RV, Kofler R, Schlötterer C. Genome-wide patterns of natural variation reveal strong selective sweeps and ongoing genomic conflict in Drosophila mauritiana. Genome Research. 2013;23:99–110. doi: 10.1101/gr.139873.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis E, Claude J, Strimmer K. APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics. 2004;20:289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- Poinar G, Rasmussen FN. Orchids from the past, with a new species in Baltic amber. Botanical Journal of the Linnean Society. 2017;183:327–333. [Google Scholar]
- Popescu AA, Huber KT, Paradis E. APE 3.0: New tools for distance-based phylogenetics and evolutionary analysis in R. Bioinformatics. 2012;28:1536–1537. doi: 10.1093/bioinformatics/bts184. [DOI] [PubMed] [Google Scholar]
- Pridgeon AM, Cribb PJ, Chase MW, Rasmussen FN, editors. Genera Orchidacearum. Volume 4 Epidendroideae (Part 1) Oxford, UK: Oxford University Press; 2005. [Google Scholar]
- Ranwez V, Harispe S, Delsuc F, Douzery EJP. MACSE: Multiple Alignment of Coding Sequences: accounting for frameshifts and stop codons. PLoS ONE. 2011;6:e22594. doi: 10.1371/journal.pone.0022594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2014. [Google Scholar]
- Rambaut A, Suchard MA, Xie D, Drummond AJ. Tracer v1.6. 2014 Available from http://tree.bio.ed.ac.uk/software/tracer/
- Raven PH, Axelrod DI. Angiosperm biogeography and past continental movements. Annals of the Missouri Botanical Garden. 1974;61:539–673. [Google Scholar]
- Ravin NV, Gruzdev EV, Beletsky AV, Mazur AM, Prokhortchouk EB, Filyushin MA, Kochieva EZ, Kadnikov VV, Mardanov AV, Skryabin KG. The loss of photosynthetic pathways in the plastid and nuclear genomes of the non-photosynthetic mycoheterotrophic eudicot Monotropa hypopitys. BMC Plant Biology. 2016;16:153–161. doi: 10.1186/s12870-016-0929-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revell LJ. PHYTOOLS: an R package for phylogenetic comparative biology (and other things) Methods in Ecology and Evolution. 2012;3:217–223. [Google Scholar]
- Roquet C, Coissac E, Cruaud C, Boleda M, Boyer F, Alberti A, Gielly L, Taberlet P, Thuiller W, Van Es J, et al. Understanding the evolution of holoparasitic plants: the complete plastid genome of the holoparasite Cytinus hypocistis (Cytinaceae) Annals of Botany. 2016;118:885–896. doi: 10.1093/aob/mcw135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samigullin TH, Logacheva MD, Penin AA, Vallejo-Roman CM. Complete plastid genome of the recent holoparasite Lathraea squamaria reveals earliest stages of plastome reduction in Orobanchaceae. PLoS ONE. 2016;11:e0150718. doi: 10.1371/journal.pone.0150718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samorodnitsky E, Jewell BM, Hagopian R, Miya J, Wing MR, Lyon E, Damodaran S, Bhatt D, Reeser JW, Datta J, et al. Evaluation of hybridization capture versus amplicon-based methods for whole-exome sequencing. Human Mutation. 2015;36:903–914. doi: 10.1002/humu.22825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sass C, Iles WJD, Barrett CF, Smith SY, Specht CD. Revisiting the Zingiberales: using multiplexed exon capture to resolve ancient and recent phylogenetic splits in a charismatic plant lineage. PeerJ. 2016;4:e1584. doi: 10.7717/peerj.1584. https://doi.org/10.7717/peerj.1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schelkunov MI, Shtratnikova VY, Nuraliev MS, Selosse MA, Penin AA, Logacheva MD. Exploring the limits for reduction of plastid genomes: a case study of the mycoheterotrophic orchids Epipogium aphyllum and Epipogium roseum. Genome Biology and Evolution. 2015;7:1179–1191. doi: 10.1093/gbe/evv019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selosse MA, Martos F. Do chlorophyllous orchids heterotrophically use mycorrhizal fungal carbon? Trends in Plant Science. 2014;19:683–685. doi: 10.1016/j.tplants.2014.09.005. [DOI] [PubMed] [Google Scholar]
- Shaw J, Lickey EB, Schilling EE, Small RL. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: The tortoise and the hare III. American Journal of Botany. 2007;94:275–288. doi: 10.3732/ajb.94.3.275. [DOI] [PubMed] [Google Scholar]
- Shaw J, Shafer HL, Leonard OR, Kovach MJ, Schorr M, Morris AB. Chloroplast dna sequence utility for the lowest phylogenetic and phylogeographic inferences in angiosperms: the tortoise and the hare IV. American Journal of Botany. 2014;101:1987–2004. doi: 10.3732/ajb.1400398. [DOI] [PubMed] [Google Scholar]
- Singhal S, Moritz C. Testing hypotheses for genealogical discordance in a rainforest lizard. Molecular Ecology. 2012;21:5059–5072. doi: 10.1111/j.1365-294X.2012.05747.x. [DOI] [PubMed] [Google Scholar]
- Straub SCK, Fishbein M, Livshultz T, Foster Z, Parks M, Weitemier K, Cronn RC, Liston A. Building a model: developing genomic resources for common milkweed (Asclepias syriaca) with low coverage genome sequencing. BMC Genomics. 2011;12:211. doi: 10.1186/1471-2164-12-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stull GW, Moore MJ, Mandala VS, Douglas NA, Kates HR, Qi XS, Brockington SF, Soltis PS, Soltis DE, Gitzendanner MA. A targeted enrichment strategy for massively parallel sequencing of angiosperm plastid genomes. Applications in Plant Sciences. 2013;1:1200497. doi: 10.3732/apps.1200497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicke S, Naumann J. Molecular evolution of plastid genomes in parasitic flowering plants. In: Chaw S-M, Jansen RK, editors. Advances in botanical research. Cambridge, MA, USA: Elsevier; 2017. https://doi.org/10.1016/bs.abr.2017.11.014. [Author, if possible, please update the doi with the page numbers.] [Google Scholar]
- Wicke S, Müller KF, de Pamphilis CW, Quandt D, Wickett NJ, Zhang Y, Renner SS, Schneeweiss GM. Mechanisms of functional and physical genome reduction in photosynthetic and nonphotosynthetic parasitic plants of the broomrape family. Plant Cell. 2013;25:3711–3725. doi: 10.1105/tpc.113.113373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicke S, Müller KF, dePamphilis CW, Quandt D, Bellot S, Schneeweiss GM. Mechanistic model of evolutionary rate variation en route to a nonphotosynthetic lifestyle in plants. Proceedings of the National Academy of Sciences, USA. 2016;113:9045–9050. doi: 10.1073/pnas.1607576113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicke S, Schneeweiss GM, dePamphilis CW, Müller KF, Quandt D. The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Molecular Biology. 2011;76:273–297. doi: 10.1007/s11103-011-9762-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickett NJ, Fan Y, Lewis PO, Goffinet B. Distribution and evolution of pseudogenes, gene losses, and a gene rearrangement in the plastid genome of the nonphotosynthetic liverwort, Aneura mirabilis (Metzgeriales, Jungermanniopsida) Journal of Molecular Evolution. 2008;67:111–122. doi: 10.1007/s00239-008-9133-1. [DOI] [PubMed] [Google Scholar]
- Wolfe KH, Morden CW, Palmer JD. Function and evolution of a minimal plastid genome from a nonphotosynthetic parasitic plant. Proceedings of the National Academy of Sciences, USA. 1992;89:10648–10652. doi: 10.1073/pnas.89.22.10648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyman SK, Jansen RK, Boore JL. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 2004;20:3252–3255. doi: 10.1093/bioinformatics/bth352. [DOI] [PubMed] [Google Scholar]
- Zhou LC, Holliday JA. Targeted enrichment of the black cottonwood (Populus trichocarpa) gene space using sequence capture. BMC Genomics. 2012;13:703. doi: 10.1186/1471-2164-13-703. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Notes S1 Photographs, accession information (including vouchers and coverage depth of the plastme), divergence time estimation, and polymorphism information for the Corallorhiza striata complex.