

Abstract
Donald Straley Coffey completed his 85 year life’s journey on November 9, 2017. In his wake, he left a legion of inspired and loyal students, fellows, and faculty colleagues from all over the world to carry on his passion both for life in general and his 50 year quest to conquer cancer. Early in his career, Dr. Coffey developed a series of animal models to study how androgen regulates the growth of both normal and abnormal prostatic epithelium. As part of these early studies, Dr. Coffey uncovered a paradox in that anti-androgen treatment given at the “wrong” time paradoxically enhanced, not inhibited, normal prostate growth. Advances over the last several years concerning the paracrine-dependent stem cell organization of the prostate provide a mechanistic explanation for this “Coffey Paradox”. This is based upon the realization that the normal function of the Androgen Receptor (AR) in the paracrine-dependent stem cell organization of the prostate is to induce terminal differentiation of normal prostate epithelial cells while suppressing their growth, despite the presence of high levels of stromal cell-derived paracrine growth factors. Such growth suppression involves ligand-dependent AR binding to the Tcf-4/β-catenin 3’c-Myc enhancer in prostate epithelial cells, which inhibits c-Myc transcription needed for proliferation. Therefore, if anti-androgen is given at the wrong time, it prevents such AR-dependent c-Myc down regulation, and thus paradoxically enhances epithelial regrowth (i.e. the Coffey Paradox) induced by exogenous androgen replacement in the castration regressed prostate. In contrast to the normal prostate epithelium, in prostate cancer cells retaining AR expression, androgen-induced AR signaling no longer reduces c-Myc transcription but instead up-regulates c-Myc translation and protein stability to stimulate malignant growth. Thus, in these AR expressing prostate cancer cells, AR signaling is converted from a growth suppressor to an oncogene, which involves a gain of function to upregulate c-Myc protein expression. Such a gain of function “addicts” these prostate cancer cells to AR signaling for their proliferation and survival, which provides the rationale for therapy targeted at inhibiting such AR signaling. While therapies targeted at maximally decreasing the level of androgen ligand are the most commonly used, recent studies have documented that a subset of patients progressing on such androgen ablation (i.e. castration-resistant disease) due to their adaptive increase in AR protein expression respond positively to rapid cycling between pharmacologically high and castration low levels of circulating androgen. [i.e. Bipolar Androgen Therapy (BAT)].
Keywords: Prostate, castration, prostate cancer, androgen receptor, stem cells, andromedins
Introduction to the “Coffey Paradox”
In the late 1960s, Donald S. Coffey reported his seminal studies on the biochemical changes in prostate tissue associated with androgen-induced DNA synthesis [1]. These studies, using the androgen-induced proliferative regrowth of the 2 week castration regressed rat prostate as the experimental model, documented that proliferative regrowth of the prostatic epithelium occurred over a 10-14 day period with the maximal rate of proliferative DNA synthesis occurring between 2-3 days post-exogenous androgen replacement [1]. Dr. Coffey went on to document in 1972 that anti-androgens, like cyproteroneactetate or flutamide, could prevent such proliferative regrowth of the regressed prostate if given simultaneously with exogenous androgen replacement to the castrated animals (Detailed in Figure 1, replicated from reference [2]). Paradoxically, however, if the castrated rats were given 54 hours of treatment with exogenous androgen alone followed by 18 hours of co-treatment with exogenous androgen plus anti-androgen, the rate of proliferative prostatic DNA synthesis was significantly enhanced by~50% as compared to when the animals received 72 hours of androgen replacement alone (Detailed in Figure 2, replicated from reference [2]).
Figure 1.
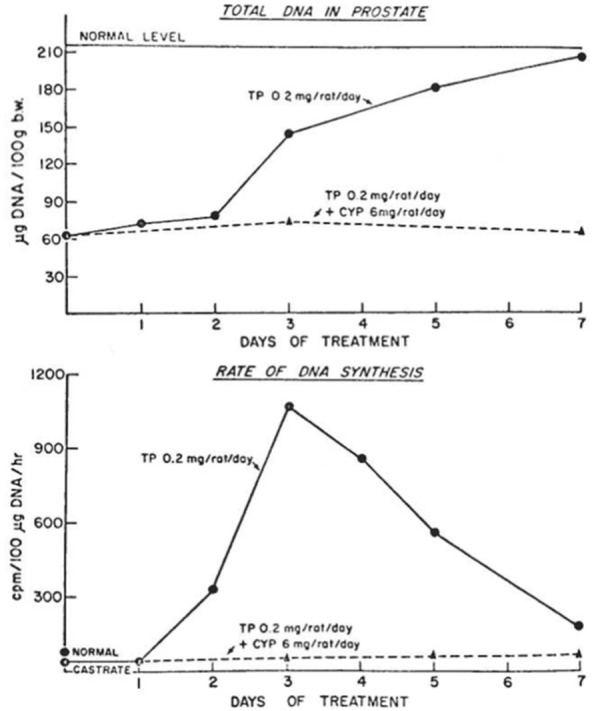
The effect of the anti-androgen cyproterone acetate (6 mg/rat/day) in blocking the exogenous androgen replacement (0.2 mg testosterone propionate/rat/day)-induced regrowth of the ventral prostate of a 2-week previously castrated rat based upon total DNA content expressed as µg DNA/100 grams of body weight (upper panel) and DNA synthesis expressed as tritiated thymidine incorporation/100 µg DNA/hr (lower panel). Figure replicated from reference [2], with permission.
Figure 2.
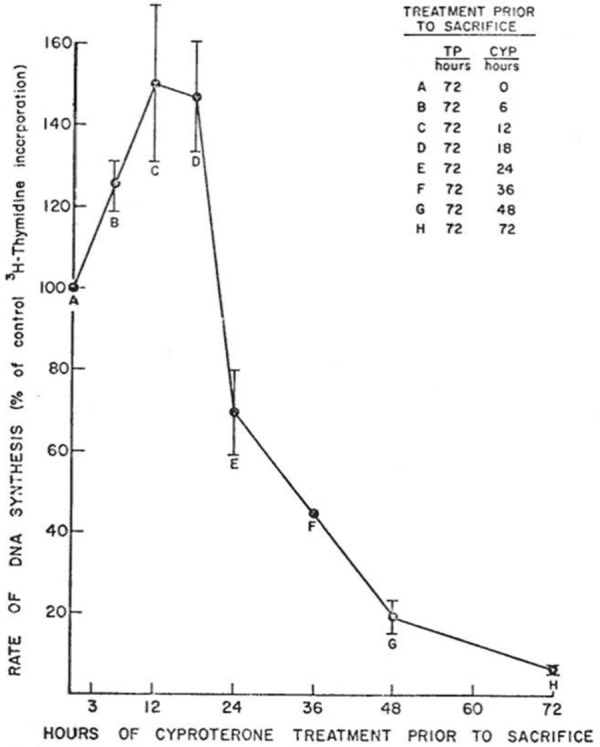
The effect of delaying treatment with anti-androgen (i.e. 6 mg cyproterone acetate) on its ability to block the stimulated rate of regenerative DNA synthesis in the 2-week regressed ventral prostate of rats after 3 days exogenous androgen replacement (i.e. 0.2 mg of testosterone propionate/day). Figure replicated from reference [2], with permission.
Androgen regulation of stem cell organization in normal prostate and the Coffey Paradox
The mechanism for this “Coffey Paradox” has only recently been resolved based upon several additional seminal observations. The first, discovered and championed by Gerry Cunha, is that in the normal prostate, epithelial cell number is dependent upon the level of ligand binding to the androgen receptor (AR) within prostate stromal cells, which stimulates their production and secretion of androgen-regulated stromal cell-derived paracrine peptide factors (i.e. andromedins) [3]. These andromedins then diffuse into the epithelial compartment and bind to cognate receptors to initiate signaling pathways that promote epithelial cell growth and survival. The second, discovered more than 30 years ago in collaboration with Dr. Coffey, is that the normal prostate can undergo successive cycles of androgen deprivation and replacement without diminishing its ability for continued regeneration [4]. Since then, a large number of independent groups have clarified how the human prostate is organized into prostate stromal and epithelial adult stem cell units, Figure 3, and how androgen-sensitive reciprocal paracrine interactions between these two compartments allows such profound cyclic regenerative growth capacity [4-9]. In the stromal compartment, adult mescenchymal stem cells (MSCs) respond to paracrine factors (e.g. SHH) secreted by prostate basal epithelial cells for their self-renewal and the generation of stromal progeny cells, which have a limited proliferative ability before differentiating into a variety of proliferatively quiescent stromal cell types (e.g. smooth muscle cells, adiptocytes, fibroblasts, pericytes, etc.), a subset of which express the AR [10-12]. When adequate levels of androgen are present to efficiently bind to and thus activate AR transcriptional signaling, these AR expressing stromal cells secrete andromedins that diffuse throughout the immediate microenvironment until binding their cognate receptors on specific cell types within the epithelial and stromal compartments to induce compartment-specific differentiation programs, Figure 3 [8].
Figure 3.
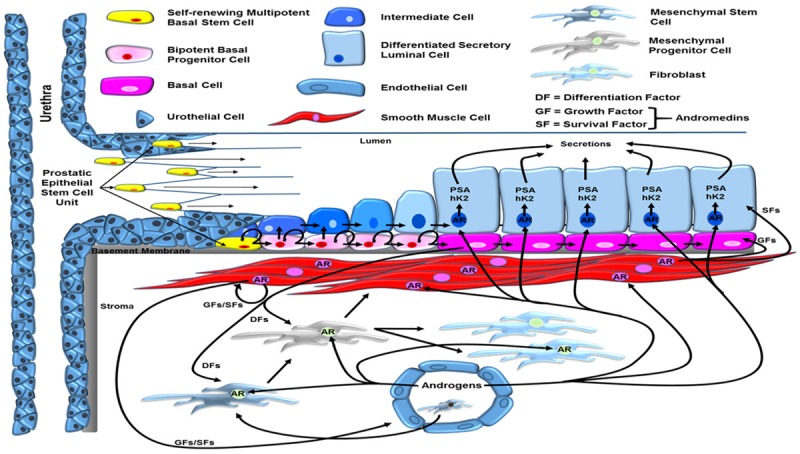
Reciprocal hierarchical expansion of epithelial and stromal adult stem cells in the human prostate. Prostate epithelial stem cells located in niches in the basal layer of the proximal ducts are induced to undergo hierarchical expansion in response to AR-dependent stromal-derived andromedins. This hierarchical expansion produces a stratified epithelium consisting of a continuous layer of ΔNp63-positive/basal cytokeratin-positive/AR-negative basal cells under a layer of ΔNp63-negative/luminal cytokeratin-positive/AR-positive/PSA-expressing secretory luminal cells. Adapted from Isaacs [8] and Moad, et al. [9].
The human prostate epithelial compartment is a tubuloalveolar structure containing a simple stratified epithelium composed of a continuous basal layer underneath a luminal layer. Functionally, this epithelial compartment is organized by epithelial adult stem cell units [8]. These prostate epithelial adult stem cells are located in niches in the basal layer of the proximal epithelial ducts, Figure 3 [7-9]. These prostate epithelial adult stem cells are androgen independent as documented by the classic tissue recombination studies of Gerry Cunha, which proved that epithelial morphogenesis and growth occurs even when AR protein is not expressed by any prostate epithelial cells, including adult stem cells, as long as there is ligand-dependent AR paracrine signaling in the supporting stromal cells [3,5]. The mechanism for this epithelial growth in the absence of epithelial AR expression is related to the hierarchical expansion and maturation of AR-negative epithelial adult stem cells and their progeny, Figure 3. E. Lynette Wilson et al. documented that these androgen insensitive epithelial stem cells are located in niches that control their survival and self-renewal, which are positioned in the basal epithelial layer at the opening of the proximal ducts as they enter the urethra [7,9]. AR-negative epithelial adult stem cells within these proximal duct basal niches undergo self-renewal division in which one daughter remains in the niche as a stem cell and the other daughter cell migrates out of the niche and undergoes differentiation. Though this migrating daughter occasionally differentiates into a non-proliferating, AR-negative neuroendocrine (NE) cell, the much more prevalent pathway is differentiation into a ΔNp63-positive/basal cytokeratin-positive/AR-negative proliferating progenitor cell. Using in situ lineage tracing on human prostate tissues, Rakesh Heer et al. documented these basal progenitor cells undergo a limited number of amplifying proliferations while they migrate in streams in the basal layer along the proximal-distal ductal axis, Figure 3 [9]. Such basal progenitor proliferation requires the androgen regulated production and secretion of diffusible stromal-derived paracrine andromedins [8]. These paracrine secreted andromedins diffuse from the stroma into the epithelial compartment where their binding to cognate receptors stimulates progenitor cell proliferation and subsequent maturation of a subset of these cells into ΔNp63-negativebasal and luminal cytokeratin-positive (i.e. intermediate) basal cells, initially documented by Schalken et al. [6]. These intermediate basal cells begin to express an increasing level of AR protein as they migrate from the basal to the luminal layer to become luminal intermediate cells during their flow to the more distal ducts. Ligand-dependent AR binding to response elements in the enhancer/promoter of AR target genes (e.g. PSA) induces differentiation of these luminal intermediate cells into ΔNp63-negativeluminal cytokeratin-positive/AR-positive/PSA-expressing secretory luminal cells [13].
While this adult stem cell organization provides a framework for understanding both the paracrine mechanism of androgen action in the normal prostate and how the gland maintains its ability to regenerate through multiple cycles of androgen deprivation and restoration, only recently has there been an understanding of what restricts the continuous overgrowth of normal adult prostate epithelial stem cell units in the continuous presence of high levels of andromedins in the prostate of non-castrated adult males. Immunocytochemical studies document that in the human prostate, the small fraction of proliferating normal prostate epithelial cells is located in the basal compartment and do not express AR protein, while the AR positive secretory-luminal cells are proliferative quiescent [14]. It has also been demonstrated experimentally that AR signaling activated by androgen binding in prostate epithelial cells induces their growth arrest and eventual differentiation into secretory-luminal cells [15,16]. Likewise, transgenic mouse studies have documented that when the AR gene is knocked out selectively in secretory-luminal cells within the prostate, then only these AR-deficient cells become hyper-proliferative and do not terminally differentiate [17-19]. These data document that AR-induced epithelial cell growth arrest limits the positive feed-forward proliferative stromal-driven paracrine loop. Thus, preventing continuous prostatic epithelial hyperplasia in the presence of high levels of stromal andromedins that are chronically maintained by physiological levels of androgen in an intact male [15,16]. The mechanism for such growth suppression is due to ligand occupied AR binding to the β-catenin/TCF-4 complex located at the c-Myc 3’ TCF-4 enhancer, which prevent stranscription of c-Myc [15,16].
When previously castrated hosts with a regressed prostate are given physiologic androgen replacement, adequate ligand occupancy of nuclear AR re-occurs within the prostatic stromal cells to re-stimulate their production and secretion of an adequate level of and romedins to enter the epithelial compartment and stimulate the proliferation of initially AR-negative/p63-positivebasal epithelial cells. The progeny of these initially AR-negative epithelial cells now express AR protein to which the replaced androgen binds. This ligand occupancy allows AR to bind to specific gene enhancers and promoters as a transcription factor inducing increased expression of NKX3.1, PSA, HK2, and luminal cytokeratins (e.g. CK18) coupled with down regulation of p63 and basal cytokeratins (e.g. CK14); thus differentiating these cells into intermediate cells. The ligand occupied AR in these intermediate cells also binds the β-catenin/TCF-4 complex located at the c-Myc 3’ TCF-4 enhancer preventing transcription of c-Myc and thus completing their terminal differentiation into non-proliferating secretory-luminal cells. On the other hand, if such castrated hosts are given androgen replacement alone for a sufficient amount of time to produce adequate levels of stromal andromedins to stimulate proliferation and differentiation of AR-negative basal cells into AR-positive intermediate progeny before initiating anti-androgen treatment, then anti-androgen prevents AR from binding to the β-catenin/TCF-4 complex at the c-Myc 3’ TCF-4 enhancer, which allows c-Myc expression and thus their continued proliferation. Thereby, explaining the Coffey Paradox.
AR acquires oncogenic functions during prostatic carcinogenesis
During prostatic carcinogenesis, stromal AR expression is not required for prostate cancer growth since there is a conversion from the normal AR-dependent stromal paracrine to a cell autonomous autocrine mechanism for AR-stimulated malignant growth [20]. Such a conversion is characterized by a lack of AR expression in the tumor stromal cells surrounding AR-positive human prostate primary cancer and their metastases [21]. During this molecular conversion from stromal dependent paracrine to cell autonomous autocrine AR signaling pathways, the prostate cancer cells become “addicted” to such cell autonomous AR oncogenic signaling [20,21]. Such cell autonomous AR-dependent growth stimulation involves secretion, extracellular binding, and signaling by autocrine growth factors that stimulate prostate cancer growth, not the andromedins secreted by normal prostate stromal cells [21]. Such cell autonomous extracellular autocrine signaling is necessary but not sufficient for the optimal growth of prostate cancer cells [21]. Thus, AR-induced growth stimulation of human prostate cancer also requires AR-dependent intracellular pathways to regulate the expression of critical transcription factors, like c-Myc [15,16,21]. For example, this oncogenic conversion involves a differential loss of AR suppression of c-Myc transcription coupled with a gain of AR up-regulation of c-Myc translation and protein stability that stimulates prostate cancer growth as documented by inhibition of both of these responses following treatment with an AR antagonist, such as bicalutamide [16,22].
Conclusions
In normal prostate epithelial cells, AR signaling induces both differentiation and growth suppression. During prostatic carcinogenesis, AR signaling loses its growth suppressive ability either by its lack of expression (i.e. in AR-negative PCa) or by molecular changes that prevent its suppressor function in cancer cells that continue to express AR [22-24]. Under this latter situation, continued AR expression can also undergo a gain of oncogenic function to stimulate prostate cancer cell growth via stimulating c-Myc expression [16,20-22]. Such a gain of function “addicts” prostate cancer cells to AR signaling for their proliferation and survival, providing the rationale for therapy targeted at inhibiting such AR signaling. While therapies targeted at maximally decreasing the level of androgen ligand are the most commonly used, recent studies have documented that a subset of patients progressing on such androgen ablation (i.e. castration resistance) due to their adaptive increase in AR protein expression respond positively to sequential cycles alternating rapidly between periods of acute supraphysiologic androgen followed by acute ablation to take advantage of a vulnerability produced by adaptive auto-regulation and binding of AR [25-29]. Resolving the mechanism (s) for the effectiveness of such Bipolar Androgen Therapy (BAT) is presently an active area of study with AR functioning in DNA replication and DNA damage repair being leading candidates.
Postscript
In a letter to Robert Hooke in 1675, Sir Isaac Newton stated that “If I have seen further, it is by standing on ye shoulders of Giants”. I have had the privilege and the joy to have stood on the shoulders of Donald Straley Coffey, Figure 4.
Figure 4.

The author standing on the shoulder of a giant (i.e. Donald Straley Coffey).
Acknowledgements
The author wouldlike to acknowledge the following sources of financial support: SKCCC Cancer Center Support Grant (P30 CA006973), Department of Defense Prostate Cancer Research Program (Award No W81XWH-16-1-0410), and NIH-Prostate SPORE Grant (P50 CA058236).
References
- 1.Coffey DS, Shimazaki J, Williams-Ashman HG. Polymerization of deoxyribonucleotides in relation to androgen-induced prostatic growth. Arch Biochem Biophys. 1968;124:184–198. doi: 10.1016/0003-9861(68)90319-6. [DOI] [PubMed] [Google Scholar]
- 2.Carter MF, Chung LWK, Coffey DS. The temporal requirements for androgen during the cell cycle of the prostate gland. In: King LR, Murphy GP, editors. Urologic Research. New York: Plenum Press; 1972. pp. 27–28. [Google Scholar]
- 3.Cunha GR, Chung LW, Shannon JM, Taguchi O, Fujii H. Hormone-induced morphogenesis and growth: role of mesenchymal-epithelial interactions. Recent Prog Horm Res. 1983;39:559–98. doi: 10.1016/b978-0-12-571139-5.50018-5. [DOI] [PubMed] [Google Scholar]
- 4.Isaacs J, Coffey DS. Etiology and disease process of benign prostatic hyperplasia. Prostate Suppl. 1989;2:33–50. doi: 10.1002/pros.2990150506. [DOI] [PubMed] [Google Scholar]
- 5.Wang Y, Hayward S, Cao M, Thayer K, Cunha G. Cell differentiation lineage in the prostate. Differentiation. 2001;68:270–279. doi: 10.1046/j.1432-0436.2001.680414.x. [DOI] [PubMed] [Google Scholar]
- 6.Schalken JA, van Leenders G. Cellular and molecular biology of the prostate: stem cell biology. Urology. 2003;62:11–20. doi: 10.1016/s0090-4295(03)00758-1. [DOI] [PubMed] [Google Scholar]
- 7.Goto K, Salm SN, Coetzee S, Xiong X, Burger PE, Shapiro E, Lepor H, Moscatelli D, Wilson EL. Proximal prostatic stem cells are program to regenerate a proximal-distal ductal axis. Stem Cells. 2006;24:1859–1868. doi: 10.1634/stemcells.2005-0585. [DOI] [PubMed] [Google Scholar]
- 8.Isaacs JT. Prostate stem cells and benign prostatic hyperplasia. Prostate. 2008;68:1025–1034. doi: 10.1002/pros.20763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moad M, Hannezo E, Buczacki SJ, Wilson L, El-Sherif A, Sims D, Pickard R, Wright NA, Williamson SC, Turnbull DM, Taylor RW, Greaves L, Robson CN, Simons BD, Heer R. Multipotent basal stem cells, maintained in localized proximal niches, support directed long-ranging epithelial flows in human prostates. Cell Rep. 2017;20:1609–1622. doi: 10.1016/j.celrep.2017.07.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lamm ML, Catbagan WS, Laciak RJ, Barnett DH, Hebner CM, Gaffield W, Walterhouse D, Iannaccone P, Bushman W. Sonic hedgehog activates mesenchymal Gli1 expression during prostate ductal bud formation. Dev Biol. 2002;249:349–366. doi: 10.1006/dbio.2002.0774. [DOI] [PubMed] [Google Scholar]
- 11.Karhadkar SS, Bova GS, Abdallah N, Dhara S, Gardner D, Maitra A, Isaacs JT, Berman DM, Beachy PA. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature. 2004;431:707–712. doi: 10.1038/nature02962. [DOI] [PubMed] [Google Scholar]
- 12.Peng YC, Levine CM, Zahid S, Wilson EL, Joyner AL. Sonic hedgehog signals to multiple prostate stromal stem cells that replenish distinct stromal subtypes during regeneration. Proc Natl Acad Sci U S A. 2013;110:20611–20616. doi: 10.1073/pnas.1315729110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vander Griend DJ, Karthaus WL, Dalrymple S, Meeker A, DeMarzo AM, Isaacs JT. The role of CD133 in normal human prostate stem cells and malignant cancer-initiating cells. Cancer Res. 2008;68:9703–9711. doi: 10.1158/0008-5472.CAN-08-3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Marzo AM, Meeker AK, Epstein JI, Coffey DS. Prostate stem cell compartments: expression of the cell cycle inhibitor p27Kip1 in normal, hyperplastic, and neoplastic cells. Am J Pathol. 1998;153:911–9. doi: 10.1016/S0002-9440(10)65632-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Antony L, van der Schoor F, Dalrymple SL, Isaacs JT. Androgen receptor (AR) suppresses normal human prostate epithelial cell proliferation via AR/beta-catenin/TCF-4 complex inhibition of c-MYC transcription. Prostate. 2014;74:1118–1131. doi: 10.1002/pros.22828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vander Griend DJ, Litvinov IV, Isaacs JT. Conversion of androgen receptor signaling from a growth suppressor in normal prostate epithelial cells to an oncogene in prostate cancer cells involves a gain of function in c-Myc regulation. Int J Biol Sci. 2014;10:627–42. doi: 10.7150/ijbs.8756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu CT, Altuwaijri S, Ricke WA, Huang SP, Yeh S, Zhang C, Niu Y, Tsai MY, Chang C. Increased prostate cell proliferation and loss of cell differentiation in mice lacking prostate epithelial androgen receptor. Proc Natl Acad Sci U S A. 2007;104:12679–84. doi: 10.1073/pnas.0704940104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simanainen U, Allan CM, Lim P, McPherson S, Jimenez M, Zajac JD, Davey RA, Handelsman DJ. Disruption of prostate epithelial androgen receptor impedes prostate lobe-specific growth and function. Endocrinology. 2007;148:2264–72. doi: 10.1210/en.2006-1223. [DOI] [PubMed] [Google Scholar]
- 19.Simanainen U, McNamara K, Gao YR, Handelsman DJ. Androgen sensitivity of prostate epithelium is enhanced by postnatal androgen receptor inactivation. Am J Physiol Endocrinol Metab. 2009;296:E1335–43. doi: 10.1152/ajpendo.00017.2009. [DOI] [PubMed] [Google Scholar]
- 20.Gao J, Arnold JT, Isaacs JT. Conversion from a paracrine to an autocrine mechanism of androgen-stimulated growth during malignant transformation of prostatic epithelial cells. Cancer Res. 2001;61:5038–5044. [PubMed] [Google Scholar]
- 21.Vander Griend DJ, D’Antonio J, Gurel B, Antony L, Demarzo AM, Isaacs JT. Cell-autonomous intracellular androgen receptor signaling drives the growth of human prostate cancer initiating cells. Prostate. 2010;70:90–9. doi: 10.1002/pros.21043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Litvinov IV, De Marzo AM, Isaacs JT. Is the Achilles’ heel for prostate cancer therapy a gain of function in androgen receptor signaling? J Clin Endocrinol Metab. 2003;88:2972–82. doi: 10.1210/jc.2002-022038. [DOI] [PubMed] [Google Scholar]
- 23.Brennen WN, Isaacs JT. Cellular origin of androgen receptor pathway-independent prostate cancer and implications for therapy. Cancer Cell. 2017;32:399–401. doi: 10.1016/j.ccell.2017.09.011. [DOI] [PubMed] [Google Scholar]
- 24.D’Antonio JM, Vander Griend DJ, Antony L, Ndikuyeze G, Dalrymple SL, Koochekpour S, Isaacs JT. Loss of androgen receptor-dependent growth suppression by prostate cancer cells can occur independently from acquiring oncogenic addiction to androgen receptor signaling. PLoS One. 2010;5:e11475. doi: 10.1371/journal.pone.0011475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Denmeade SR, Isaacs JT. Bipolar androgen therapy: the rationale for rapid cycling of supraphysiologic androgen/ablation in men with castration resistant prostate cancer. Prostate. 2010;70:1600–7. doi: 10.1002/pros.21196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Isaacs JT, D’Antonio JM, Chen S, Antony L, Dalrymple SP, Ndikuyeze GH, Luo J, Denmeade SR. Adaptive auto-regulation of androgen receptor provides a paradigm shifting rationale for bipolar androgen therapy (BAT) for castrate resistant human prostate cancer. Prostate. 2012;72:1491–505. doi: 10.1002/pros.22504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schweizer MT, Antonarakis ES, Wang H, Ajiboye AS, Spitz A, Cao H, Luo J, Haffner MC, Yegnasubramanian S, Carducci MA, Eisenberger MA, Isaacs JT, Denmeade SR. Effect of bipolar androgen therapy for asymptomatic men with castration-resistant prostate cancer: results from a pilot clinical study. Sci Transl Med. 2015;7:269ra2. doi: 10.1126/scitranslmed.3010563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Isaacs JT, Brennen WN, Denmeade SR. Rationale for bipolar androgen therapy (BAT) for metastatic prostate cancer. Cell Cycle. 2017;16:1639–1640. doi: 10.1080/15384101.2017.1360645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Teply BA, Wang H, Luber B, Sullivan R, Rifkind I, Bruns A, Spitz A, DeCarli M, Sinibaldi V, Pratz CF, Lu C, Silberstein JL, Luo J, Schweizer MT, Drake CG, Carducci MA, Paller CJ, Antonarakis ES, Eisenberger MA, Denmeade SR. Bipolar androgen therapy in men with metastatic castration-resistant prostate cancer after progression on enzalutamide: an open-label, phase 2, multicohort study. Lancet Oncol. 2018;19:76–86. doi: 10.1016/S1470-2045(17)30906-3. [DOI] [PMC free article] [PubMed] [Google Scholar]