

Abstract
Benign prostatic hyperplasia (BPH) is an enlargement of the prostate gland that is frequently found in aging men. Androgens are essential for the development and differentiated function of the prostate, as well as for proliferation and survival of prostatic cells. In man, dog and rodent, there are age-related decreases in serum testosterone. Despite the lower serum testosterone levels, benign prostatic hyperplasia increases with age in men and dogs, while age-dependent prostatic hyperplasia develops in the dorsal and lateral lobes of the rat prostate. The possible mechanisms that lead to prostate hyperplasia have been extensively studied over many years. It is clear that androgens, estrogens and growth factors contribute to the condition, but the exact etiology remains unknown. Prostate cancer (CaP) represents a significant cause of death among males worldwide. As is the case of BPH, it is clear that androgens (testosterone and dihydrotestosterone) and their metabolites play important roles in the disease, but cause-effect relationships have not been established. Androgen deprivation therapy has been used for decades, primarily in the metastatic stage, to inhibit androgen-dependent prostate cancer cell growth. Androgen deprivation, which can be achieved by targeting hormone biosynthesis or androgen receptor activation, results in symptom amelioration. However, most patients will develop hormone refractory cancer or castration-resistant prostate cancer (CRPC). Prostatic epithelial cells demonstrate enormous plasticity in response to androgen ablation. This characteristic of prostatic epithelial cells may give rise to different populations of cells, some of which may not be dependent on androgen. Consequently, androgen receptor positive and negative cells might co-exist within CRPC. A clear understanding of this possible cellular heterogeneity and plasticity of prostate epithelial cells is necessary to develop an optimal strategy to treat or prevent CRPC.
Keywords: Prostate, BPH, CRPC, androgens, aging, Leydig cell
Tribute to Donald S. Coffey, Ph.D.
This review is dedicated to the memory of Dr. Donald S. Coffey, whom we and countless others have been fortunate to have had as a dear friend and mentor. Much of our early interest in the role of testosterone in prostate function and disease came from interacting and working with Dr. Coffey (Don) in the context of a Program Project grant on benign prostatic hyperplasia (BPH) that he led for many years. Don freely shared his ideas and enthusiasm with all those involved with the grant, and particularly with young investigators just starting out. Working with and around Don was simply thrilling, always enriching, and never dull!! After the completion of the Program Project grant, our interest in testicular function and prostate health and disease, which had been stimulated by interactions with Don, continued for many years. We and many, many others will never forget the influence of this great man, or the consistent joy in interacting with him.
Introduction
Testicular androgens are essential for the formation and functioning of the prostate throughout life, and in particular for the proliferation and survival of cells within the gland. Testosterone is produced during the fetal and adult periods by two distinct populations of testicular cells, the fetal Leydig cells and the adult Leydig cells, respectively. The high levels of testosterone produced by the fetal Leydig cells decline postnatally, coincident with the loss in the numbers of these cells. Then, during postnatal weeks 2 and 3 in the rat, the fetal Leydig cells are gradually replaced by adult Leydig cells, and testosterone gradually increases to high levels with the pubertal transition to adulthood. During the pre-pubertal and pubertal periods, the conversion of testosterone (T) to dihydrotestosterone (DHT) within the prostate is considered by many investigators to stimulate the growth of the prostate to its adult size. Thereafter, a balance between prostatic cell proliferation and cell death is reached such that there is no further growth of the prostate. During aging, serum testosterone levels decrease in some species, including man, dog, and rat. Despite the lower serum testosterone levels, aging often is associated with increased prostatic cell proliferation relative to cell death, an imbalance that can lead to prostatic hyperplasia or cancer in men and dogs. The possible reason(s) for the imbalance is (are) uncertain. Clearly however, it is not only the serum testosterone concentration that determines whether or not there is abnormal prostate growth.
Our major objectives in this review are to discuss the role(s) considered to be played by androgens and androgen signaling in BPH and prostate cancer (CaP), with an emphasis on early work that led to current thinking.
Leydig cell development and steroidogenic function
Virilization of the male urogenital system depends upon the testosterone produced by the fetal Leydig cells. In the mouse, the fetal Leydig cells form from the differentiation of steroidogenic factor 1 (SF-1; NR5A1) - positive cells [1,2]. In the rat, the fetal Leydig cells begin to produce testosterone by gestational day 15.5, initially independent of luteinizing hormone (LH) and later in response to LH [3,4]. Late in fetal life, the fetal Leydig cells begin to regress, with only few persisting in the adult [5-8]. Early in the postnatal period, the fetal Leydig cells begin to be replaced by the forming adult Leydig cells [1,9]. The latter cells, which arise from stem cells that are present in the neonatal testis [10-13], produce high levels of testosterone in response to LH.
Age-related reductions in serum levels of testosterone [hypogonadism] can occur in both young and aging men. Indeed, significant decline in serum testosterone levels affects millions of American men [14,15] including 20-50% of men over age 60 and approximately 15% of men who are among the couples who seek infertility-related medical appointments [16-18]. There are many other men who also present with what is referred to as “low T”, including those with sickle cell disease and spinal cord injury [19]. In some men, reduced serum testosterone results from reduced serum LH and thus reduced stimulation of the Leydig cells (hypogonadotropic hypogonadism) [18]. In most hypogonadal men, however, serum LH either does not change or increases, indicative of primary testicular deficiency of testosterone biosynthesis (primary hypogonadism) [16,17]. Whether in aging or young men, reduced serum testosterone is associated with a number of metabolic and quality-of-life changes, including decreased lean body mass, bone mineral density, muscle mass, libido and sexual function, increased adiposity, osteoporosis and cardiovascular disorders, and altered mood [17,19].
Exogenous administered testosterone, known as testosterone replacement therapy (TRT), often is prescribed to reverse symptoms of low testosterone. A host of methods that are relatively easy to use and produce constant testosterone concentrations are available by which to do this. However, recent studies suggest that there may be increased risk of cardiovascular disease in older men after TRT [20-22]. There also are reports suggesting that exogenous testosterone treatment might increase the risk of CaP [23].
Benign prostatic hyperplasia
Of the hundreds of mammalian species, all of which have prostate glands, humans and dogs in particular develop BPH and CaP [24]. In the human, different anatomic regions within the prostate have different rates of BPH and carcinoma. Thus, the transition zone has a high incidence of BPH and a low incidence of carcinoma, whereas the peripheral zone has a high incidence of carcinoma and a low incidence of BPH [25,26]. BPH in humans is characterized primarily by stromal hyperplasia [27,28]. In dogs, BPH takes the form of overgrowth or hyperplasia of both the epithelial and stromal compartments throughout the gland. Despite the differences between dog and man, Coffey and others argued that there are sufficient similarities between the two to regard the dog as an appropriate model for the human [29-35]. This was a central tenant of the Coffey Program Project grant. We later discovered that prostatic hyperplasia also occurs in the dorsal and lateral lobes, but not the ventral lobe, of the aged Brown Norway rat prostate, with some similarities to both dog and man [36].
In both man and dog, the development of BPH requires functioning testes, advancing age, and the involvement of hormonal factors [37-39]. Walsh and Wilson [40] and others [41,42] demonstrated that BPH could be induced in young castrated dogs by administering androgens and estrogens concomitantly. In a long-term study conducted by Don Coffey, Larry Ewing and colleagues [29], it was reported that whereas 100% of intact control, aging beagles developed BPH, restoration of serum testosterone levels after castration of young dogs resulted in only 50% of the aging dogs developing BPH. This study made it clear that the testes, but not just the testosterone that they produce, are important for the development of canine BPH.
Androgens are essential for development and differentiated function of the prostate, as well as for proliferation and survival of cells within the gland. The issue of how BPH is initiated is made more complex by the fact that this condition occurs with aging as there is a gradual decline in the mean serum testosterone concentration in aging dogs and men. With the decline in testosterone, there is decline in the ratio of testosterone to 17β-estradiol in the serum, which may be critical in the pathogenesis of BPH [29]. There also may be altered sensitivity of the prostate to serum testosterone. Hyperplasia and/or hypertrophy of the prostate in the face of decreasing serum testosterone concentrations also might mean that the changes that are responsible for prostatic hyperplasia may be initiated early in adult canine life, and that testosterone later in life is permissive, not causative. Alternatively, as suggested by Coffey and others, there may be metabolic changes within the prostate that favor BPH. For example, it has been suggested that there may be increased production of the active androgen 5a-DHT [43,44]. Indeed, prostatic DHT levels have been reported in many studies to be several-fold greater in hyperplastic tissue compared with normal prostate in both man [45-49] and dog [50,51], providing evidence for such a metabolic shift. It should be noted however, that contrary to most studies, there are reports that there are no significant difference in prostatic DHT concentration between dogs with histologically normal prostates and those with spontaneous BPH [52], and that steroid hormone treatment regimens resulting in elevated prostatic DHT concentrations do not always result in high prostatic weight [53].
In the rat, as in dog and man, spontaneous as well as hormonally-induced hyperplasia can develop [36,54-56]. In both young and old Brown Norway rats, exogenously administered testosterone resulted in age- and lobe-specific overgrowth of the ventral, dorsal, and lateral lobes. In the case of old rats, both hyperplasia and hypertrophy were seen in the dorsal and lateral lobes of untreated control rats, as well as in rats treated with testosterone. Thus, despite the lower serum testosterone levels in old rats, age-dependent and spontaneous prostatic hyperplasia developed in the dorsal and lateral lobes of the prostate, though not in the ventral lobe [55]. The lobe-specific, age-dependent hyperplasia was enhanced by the administration of testosterone [54]. Castration led to decreased weight of all prostate lobes, but less rapidly and to a lesser magnitude in the dorsal and lateral lobes compared to the ventral lobe. Moreover, less cell death occurred in the prostates of old than young rats in response to castration. These studies revealed marked differences in cell death and survival among the different rat prostatic lobes in response to castration, and suggested an age-dependent response of apoptosis to reduced androgen. The lower rates of cell death observed for the dorsal and lateral lobes, and particularly so with increasing age, appear to be important components of the age-dependent and lobe-specific overgrowth observed for these lobes. Moreover, the age-dependent decline in apoptotic cell death observed in the prostates of old rats suggests that prostate cells develop an altered sensitivity to androgen as a function of age, and that survival of these cells is less dependent on androgen (Figure 1).
Figure 1.
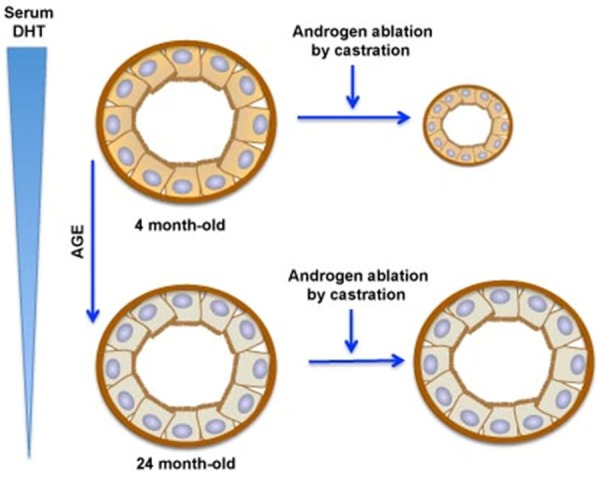
Age-dependent changes in androgen sensitivity in the dorsolateral lobe of Brown Norway rat prostatic acini. Prostatic epithelial cells of young adults (age 4 months) are androgen-dependent, and therefore castration reduces the size of the dorsolateral lobe. However, epithelial cells of the aging dorsolateral prostate become relatively insensitive to changes in androgens, and therefore are relatively unaffected by castration or by normally occurring androgen reductions.
What causes the imbalance of cell death and cell proliferation that leads to prostatic hyperplasia?
As indicated above, androgens are essential for the proliferation and survival of cells within the prostate gland. Despite the lower serum levels with aging, BPH increases with age in men and dogs, while age-dependent prostatic hyperplasia develops in the dorsal and lateral lobes of the rat prostate. These observations have proven difficult to explain. One possibility is age-related altered responsiveness of prostatic cells to androgen. For example, it has been reported that in response to castration, there are lower rates of cell death in the dorsal and lateral lobes of aged as compared to young rats, suggesting that prostatic cells may develop some measure of androgen independence with aging [57]. Isaacs and Coffey [58] reported that aging results in an increase in intra-prostatic DHT levels associated with BPH in both animal and human studies. However, there are studies in which this is disputed [59,60]. Given that testosterone functions via androgen receptors, the proliferative response of cells within the prostate to androgens presumably is dependent in some way upon the expression of androgen receptors. In a study that we conducted some years ago, we hypothesized that age-dependent hyperplasia in the dorsal and lateral lobes of Brown Norway rats might occur in relation to age-dependent and lobe-specific differences in androgen receptor expression [36,55]. Lobe-specific increased androgen receptor protein expression was seen that correlated with age-dependent hyperplasia in the dorsal and lateral lobes of the Brown Norway rat prostate. A series of studies by Prins et al. [61-63] of young adult Sprague Dawley rats also showed lobe-specific autoregulation of the androgen receptor. Evidence for increased androgen sensitivity of prostatic cells in the dorsal and lateral lobes of aging rats was also demonstrated by the observation of increases in cell proliferation and cell cycle markers in response to low levels of testosterone correlated with lobe-specific increases in androgen receptor levels of aged rats. These findings, taken together, provide evidence that the imbalance in cell death and cell proliferation that leads to age-dependent prostatic hyperplasia may be related to an altered sensitivity of the prostate to androgen, and suggest that this may be a function of nuclear androgen receptor expression changes with age.
The studies of the rat ventral, dorsal, and lateral prostatic lobes in response to altered hormonal environment, including differences in epithelial cell apoptosis [57] and androgen receptor expression after castration, suggest a role for factors in addition to androgen in the regulation of lobe-specific androgen receptor expression. In this regard, it has been suggested [55,64-66] that androgen receptor-dependent transcription of target genes may result in the production and secretion of peptide growth factors, including IGF-I, keratinocyte growth factor, fibroblast growth factor-related proteins such as keratinocyte growth factor, interleukin 6, and others. Suffice to say at this juncture that the exact etiology and pathophysiology of BPH remain unknown. The contributions of androgens and estrogens, growth factors, and chronic inflammation remain under investigation. Additionally, stromal-epithelial interactions are known to be very important for BPH pathogenesis and in the regulation of normal cell growth and differentiation [55].
In sum, androgens are essential for development and differentiated function, as well as proliferation and survival of cells within the prostate gland. In man, dog and rodent, there are age-related decreases in serum testosterone. Despite the lower serum testosterone levels, BPH and CaP increase with age in men while age-dependent prostatic hyperplasia develops in the dorsal and lateral lobes of the rat prostate. Clearly, it is not only the serum testosterone concentration that determines whether or not there is abnormal prostate growth.
Androgen/androgen receptor signaling and prostate cancer
Androgen/androgen receptor signaling also is considered to drive prostate malignancy [67], the second leading cause of cancer deaths in the US [68] and worldwide [69]. Given its critical role in the normal prostate, it is perhaps not surprising that the AR signaling axis is crucial for prostate carcinogenesis and subsequent phases of disease progression. For that reason, androgen-deprivation therapy has been the mainstay for CaP treatment for over seven decades, after Dr. Charles Huggins demonstrated that orchiectomy or treatment with high doses of estrogen led to regression of metastatic CaP [70]. Androgen deprivation therapy in the form of LHRH agonists/antagonists has typically been used as treatment for metastatic CaP. In addition, various anti-androgens, including flutamide, bicalutamide, and nilutamide, bind to the androgen receptor and inhibit its activity, and thus have been used along with/without chemotherapy [71-73]. However, many patients receiving androgen-deprivation therapy progress to metastatic castration-resistant CaP, characterized by cancer progression despite low serum testosterone levels (Figure 2). Although androgen-deprivation therapy is initially successful in most men, development of resistance is inevitable, normally occurring within a period of 18-24 months [74]. The resultant form of this disease is referred to as castration-resistant prostate cancer (CRPC), and is incurable and most often lethal [75,76]. To overcome this shortfall, taxane (docetaxel) has been used as the standard of care for metastatic CRPC. However, survival is not long-lasting [77,78].
Figure 2.
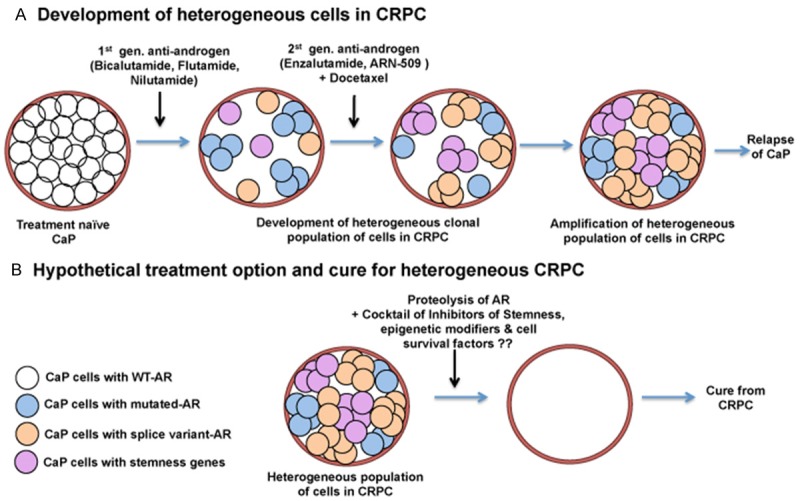
Model by which the treatment naïve prostate cancer (CaP) changes from complete androgen dependence to castration-resistant prostate cancer (CRPC). Treatment with 1st generation of anti-androgens kills a majority of wild-type, androgen receptor (AR)-expressing androgen sensitive cells. However, some of these cells acquire AR mutations, some express AR splice variants, and some trans-differentiate to cancer stem-like cells by expressing stemness genes. Second generation anti-androgens attacks only some of the mutated AR expressing cells; because of the plasticity of the CaP cells, some survive, repopulate heterogeneous cancer cells throughout the prostate, and there is cancer relapse. To target all mutated and splice variant AR positive-cells and cancer stem-like cells, total AR degradation together with inhibitors of stem-like cells will be necessary. Only this way will a lethal environment be created that will eradicate all CaP cells irrespective of their heterogeneity.
Given the importance of androgen receptor signaling during prostate carcinogenesis, and the vast body of work over the past 2-3 decades showing that most CRPC remain dependent on the AR signaling pathway, androgen receptor signaling has remained the major therapeutic target in CRPC even in the face of a very low androgenic environment. The dependence on androgen receptor signaling may be due to the increased AR gene copy number and expression of androgen receptors in the CRPC. Eighty percent of patients exhibit elevated androgen receptor gene copy number, and evidence for mRNA amplification has been observed in 30% of patients with CRPC [79-82]. Moreover, elevated levels of AR have been shown to hypersensitize cancer cells even to castrate levels of androgens [83-85], switch AR antagonists to agonists [86], and promote resistance to variety of AR-targeting agents [87].
In some patients with CRPC, AR mutations have been detected in primary CaP prior to androgen deprivation therapy, and it is generally believed that androgen deprivation therapy-mediated selection of such mutations can underlie resistance in patients with CaP [87-92]. However, much higher frequencies of AR mutations (5-30%) have been reported in CRPC tissue, circulating tumor cells, and circulating cell-free DNA samples compared to pre-treated tumor samples [80-82,89,93-98]. These AR mutations are clustered in domains responsible for ligand-binding (at the AR C-terminal domain) or transactivation activity (the AR N-terminal domain). Such alterations by mutation facilitate AR signaling in CRPC by conferring ligand promiscuity or ligand-independent transcriptional activity, thereby allowing AR to be activated even in the presence of low/absent levels of androgens. The best known AR mutation is the T878A mutant, first identified in the LNCaP cell line [99]. This is the archetypal promiscuous receptor, activated by estrogen, progesterone, and glucocorticoids [92,97,99-104]. AR mutations H875Y and L702H broaden ligand specificity by enabling AR activation by glucocorticoids [103-106]. AR mutations also confer agonistic properties to antiandrogens. For example, cancer cells with the T878A mutation are activated by flutamide and nilutamide, and those with the H875Y or W742C/L mutations are activated by nilutamide or bicalutamide, respectively [96,97,99,101,107-109].
Furthermore, increased expressions of constitutively active AR splice variants (AR-Vs) represent another layer of complexity that pertains to the molecular mechanism for disease progression after castration-resistance or androgen-deprivation [110]. Many alternatively spliced AR-Vs lack the C-terminal ligand-binding domain but retain the transactivating N-terminal domain, leading to constitutively active AR in the absence of ligands [111,112]. AR-Vs are either truncated versions of full-length AR (AR-V1 to AR-V11) or have missing/skipped exons (AR-V12 to AR-V14 and AR-V567es) [110]. Of the different AR isoforms identified in CaP, AR-V7 and ARv567es are the most common [110,113,114]. Both are upregulated in metastatic CRPC compared with hormone-naïve metastatic disease [114-116], although only V7 has been consistently described in human samples.
In addition to AR gene amplification, AR mutations and AR splice variants, AR activation in CRPC can occur in response to increased intratumoral synthesis of testosterone and dihydrotestosterone (DHT) from weak androgens produced by the adrenal glands as well as from de novo androgen synthesis from cholesterol [117,118]. A number of studies demonstrated that intratumoral levels of androgens in metastatic CRPC are elevated compared with untreated primary prostate cancers [119,120]. Increased levels of AKR1C3, HSD3B2 and CYP17A1, enzymes that are involved in androgen synthesis, have been detected in the intratumoral tissue from CRPC patients [119,121]. Therefore, abiraterone acetate, a steroidal antiandrogen that inhibits intratumoral androgen biosynthesis by blocking the hydroxylase and lyase activities of CYP17A, has been used to treat CRPC. However, the benefit of this treatment is also short-lived.
Why has targeting androgen receptor signaling not dramatically improved overall survival in individuals with metastatic castration-resistant prostate cancer?
We have enormous understanding about androgen receptor signaling, AR gene amplification, AR mutation, AR splice variants and intratumoral androgen synthesis in CaP tumor tissue before and after androgen deprivation therapy. However, overall survival has not improved beyond few months. Although clinical data indicate the benefits of second generation antiandrogens such as Enzalutamide (MDV3100) and ARN509, recent evidence also indicates the emergence of resistance. Enzalutamide is ineffective in preventing the growth of CRPC cell lines such as 22Rv1 unless the AR splice variant, AR-V7, is specifically knocked down [122]. Two independent groups have demonstrated the appearance of a specific missense mutation (F876L) in the AR ligand binding domain in cell lines, xenograft tumors, and clinical samples that have undergone prolonged treatment with enzalutamide and ARN509 [123,124]. Additionally, patients who are poor responders to enzalutamide treatment show increased expression of the glucocorticoid receptor in bone marrow biopsies [125]. Elevated glucocorticoid receptor expression has been implicated as a compensatory mechanism to overcome AR antagonism in vitro and in vivo. Therefore, it appears that the benefits of anti-androgen therapy are short-lived, and alternative approaches to combat emerging resistance are needed for the effective management of CRPC. One flaw of Enzalutamide and ARN509 is that these second generation antiandrogens, as well as earlier antiandrogens, target the AR-ligand binding domain. Now we know that CRPC also expresses AR splice variants that lack the ligand-binding domain, and therefore neither Enzalutamide or ARN509 alone will be effective in that scenario. More recent therapeutic modalities have attempted to target the AR-transactivation domain rather than the ligand-binding domain.
EPI-001, a small-molecule antagonist of AR N-terminal transactivation domain that inhibits protein-protein interactions necessary for AR transcriptional activity, has recently been developed. EPI-001 inhibited transcriptional activity of AR and its splice variants and reduced the growth of CRPC in a xenograft model [126-128]. These findings suggest that the development of small-molecule inhibitors that target the AR transactivation domain could be a promising strategy for CRPC. Currently, a stereoisomer of EPI-001, EPI-506, is under phase I/II clinical testing (NCT02606123) in post-abiraterone and post-enzalutamide settings. However, the outcomes of these clinical trials are not published yet.
Another alternative therapeutic approach is one that directly targets the degradation of the androgen receptor protein itself rather than its ligand binding and/or transcriptional activity. A plant-derived carbazole alkaloid, mahanine, showed inhibition of both ligand-dependent and ligand-independent AR transactivation, as well as AR protein degradation, leading to a decline in AR target gene expression [129]. Niclosamide, an FDA-approved antihelminthic drug, was identified as a potent inhibitor of the AR-V7 variant in prostate cancer cells. Niclosamide significantly down-regulated AR-V7 protein expression by enhancing protein degradation through a proteasome-dependent pathway [130]. It also inhibited AR-V7 transcription activity and reduced the recruitment of AR-V7 to the PSA promoter [130]. More importantly, this drug potentiates the effects of enzalutamide in vitro and in vivo, and resensitizes enzalutamide-resistant CaP cells [130]. This finding has elicited a phase I trial to assess the utility of niclosamide in combination with enzalutamide for treating AR-V7-positive CRPC (NCT02532114). The clinical outcome has not been published yet. There are additional selective agents that target AR degradation (SARDs; UT-69, UT-155, and (R)-UT-155) and markedly reduce the activity of wild-type and splice variant isoforms of AR at submicromolar doses [131]. However, the efficacy of these agents in clinical settings has yet to be determined.
It is important to emphasize that prostatic epithelial cells demonstrate enormous plasticity in response to androgen ablation. This innate characteristic of prostatic epithelial cells may give rise to different populations of cells, some of which are not dependent on androgen. Consequently, androgen receptor positive and negative cells might co-exist as important examples of cellular heterogeneity within CRPC.
Understanding CaP phenotypic and functional cellular heterogeneity is critical for the optimal treatment of CRPC [132-138]. To better understand this cellular heterogeneity, it is essential to identify the individual cell types within the tumor. The predominant histological subtypes found in prostatic adenocarcinoma [139] are luminal secretory cells, rare neuroendocrine cells, and some basal cells, with luminal epithelial cells accounting for the highest percentage within the gland. However, all these luminal epithelial cells are not the same. In fact, using multiple xenograft models and over 70 patient tumor samples, a comprehensive study was conducted recently to dissect the phenotypic, functional, and tumorigenic heterogeneities in human CaP. Four subtypes of prostate cancer cells, AR+PSA+, AR-PSA+, AR+PSA-, and AR-PSA, were seen in untreated human CaP tissue, reconfirming that Cap tissue is comprised of a heterogeneous pool of cells [140]. In another elegant study, the same group demonstrated that the PSA-/lo CaP cells possess unlimited tumor-propagating activity, whereas PSA+ CaP cells have limited activity [141]. Results from this study reinforce the intrinsic stem cell nature and castration-resistant properties of the PSA-/lo cancer cells. The heterogeneous properties of CaP cells present the ultimate challenge to effective therapy in this disease.
The revelations regarding heterogeneity of CaP cells bring us full circle to our earlier studies in which we observed a diversity of responses among cells in the aging Brown Norway rat prostate. We noted cellular heterogeneity in the normal rodent prostate gland, both during its development in various lobes (ventral, dorsal, lateral and anterior) and in response to androgen ablation by castration. To our surprise, we observed that although cell proliferation occurs in all lobes, the presence of physiological or exogenously administered androgen resulted in significant differences in cell death and androgen responsiveness in the ventral compared to the dorsal and lateral lobes [54,55,57,142-145]. While luminal epithelial cells in the ventral lobe are very sensitive to androgen ablation, indicated by significant loss of total DNA content and cell death, the dorsal and lateral lobes showed almost no change in total cell numbers or cell death. Sensitivity to androgen withdrawal also was found to depend on the age of the rat and the presence and absence of various survival factors (telomerase activity, TGF-alpha, Bcl2) [146-148]. These results suggest that although prostatic epithelial cells are dependent on androgen for their growth and survival, epithelial cells exhibit plasticity such that their function can be modified over time so as to form a heterogeneous pool of cells within the prostate gland, even before androgen withdrawal. These properties become even more apparent following androgen withdrawal (Figure 1).
It is interesting to note that approximately a quarter of CRPC patients who develop an aggressive phenotype have low to no AR expression, acquire neuroendocrine signatures, loss of function of tumor suppressor genes (PTEN, RB1, and TP53), overexpression of stemness markers (SOX2, Nanog, Oct4), and epigenetic reprogramming factors (EZH2, BMI) [149-161]. Therefore, unidirectional targeting of androgen receptor signaling will not result in a major benefit or cure from CRPC. A clear understanding about the cellular heterogeneity and plasticity of prostate epithelial cells is necessary to develop an optimal combinatorial strategy to treat or prevent CRPC. A hypothetical model is proposed in Figure 2.
Conclusions and future directions
It is clear that androgens and androgen receptor signaling are crucial for prostate growth and homeostasis, and for the development of BPH and CRPC. However, it is also evident that in the face of decreasing serum androgen, as during aging, prostatic cells adapt to survive in low concentrations of androgen. Thus, although androgens are essential, prostatic hyperplasia can occur in men, dogs and rats despite decreased androgen. Clearly, it is not only serum androgen concentration that determines whether or not there is abnormal prostate growth. This also is true of CRPC; heterogeneous cells evolve with various mutations and splice variants in AR after androgen deprivation. Therefore, traditional treatment with anti-androgen targeting to the ligand-binding domain of AR typically is not effective or curative for CRPC. Beside AR modifications, CRPC cells acquire various epigenetic changes and overexpression of stemness genes that make these cells extremely heterogeneous. Therefore, to treat CRPC effectively, anti-androgens are needed that target the AR transactivation domain or completely degrade ARs (wild-type, mutated and splice variants). The hope is that these, used in combination with other chemotherapy, will be able to create a lethal environment that kills all CaP cells irrespective of their heterogeneity. Until we understand the cellular heterogeneity in CRPC, it will remain difficult to cure this deadly disease.
References
- 1.Habert R, Lejeune H, Saez JM. Origin, differentiation and regulation of fetal and adult Leydig cells. Mol Cell Endocrinol. 2001;179:47–74. doi: 10.1016/s0303-7207(01)00461-0. [DOI] [PubMed] [Google Scholar]
- 2.Barsoum IB, Kaur J, Ge RS, Cooke PS, Yao HH. Dynamic changes in fetal Leydig cell populations influence adult Leydig cell populations in mice. FASEB J. 2013;27:2657–2666. doi: 10.1096/fj.12-225060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Shaughnessy PJ, Baker P, Sohnius U, Haavisto AM, Charlton HM, Huhtaniemi I. Fetal development of Leydig cell activity in the mouse is independent of pituitary gonadotroph function. Endocrinology. 1988;139:1141–1146. doi: 10.1210/endo.139.3.5788. [DOI] [PubMed] [Google Scholar]
- 4.Migrenne S, Pairault C, Racine C, Livera G, Géloso A, Habert R. Luteinizing hormone-dependent activity and luteinizing hormone-independent differentiation of rat fetal Leydig cells. Mol Cell Endocrinol. 2001;172:193–202. doi: 10.1016/s0303-7207(00)00339-7. [DOI] [PubMed] [Google Scholar]
- 5.Habert R, Picon R. Control of testicular steroidogenesis in foetal rat: effect of decapitation on testosterone and plasma luteinizing hormone-like activity. Acta Endocrinol. 1982;99:466–473. doi: 10.1530/acta.0.0990466. [DOI] [PubMed] [Google Scholar]
- 6.Habert R, Rouiller-Fabre V, Lecerf L, Levacher C, Saez JM. Developmental changes in testosterone production by the rat testis in vitro during late fetal life. Arch Androl. 1992;29:191–197. doi: 10.3109/01485019208987724. [DOI] [PubMed] [Google Scholar]
- 7.Huhtaniemi I, Pelliniemi LJ. Fetal Leydig cells. Cellular origin, morphology, life span, and special functional features. Proc Soc Exp Biol Med. 1992;201:125–140. doi: 10.3181/00379727-201-43493. [DOI] [PubMed] [Google Scholar]
- 8.Habert R. In vivo acute testicular testosterone response to injection of luteinizing hormone in the rat fetus. Acta Endocrinol. 1993;128:268–273. doi: 10.1530/acta.0.1280268. [DOI] [PubMed] [Google Scholar]
- 9.Haider SG. Cell biology of Leydig cells in the testis. Int Rev Cytol. 2004;233:181–241. doi: 10.1016/S0074-7696(04)33005-6. [DOI] [PubMed] [Google Scholar]
- 10.Mendis-Handagama SM, Ariyaratne HB. Differentiation of the adult Leydig cell population in the postnatal testis. Biol Reprod. 2001;65:660–671. doi: 10.1095/biolreprod65.3.660. [DOI] [PubMed] [Google Scholar]
- 11.Ge RS, Shan LX, Hardy MP. Pubertal development of Leydig cells. In: Payne AH, Hardy MP, Russell LD, editors. The Leydig Cell. Vienna, IL: Cache River Press; 1996. p. 159. [Google Scholar]
- 12.Lo KC, Lei Z, Rao ChV, Beck J, Lamb DJ. De novo testosterone production in luteinizing hormone receptor knockout mice after transplantation of leydig stem cells. Endocrinology. 2004;145:4011–4015. doi: 10.1210/en.2003-1729. [DOI] [PubMed] [Google Scholar]
- 13.Ge RS, Dong Q, Sottas CM, Papadopoulos V, Zirkin BR, Hardy MP. In search of rat stem Leydig cells: identification, isolation, and lineage-specific development. Proc Natl Acad Sci U S A. 2006;103:2719–2724. doi: 10.1073/pnas.0507692103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Surampudi PN, Wang C, Swerdloff R. Hypogonadism in the aging male diagnosis, potential benefits, and risks of testosterone replacement therapy. Int J Endocrinol. 2012;2012:1–20. doi: 10.1155/2012/625434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bhasin S, Huang G, Travison TG, Basaria S. Age-related changes in the male reproductive axis. In: De Groot LJ, Chrousos G, Dungan K, Feingold KR, Grossman A, Hershman JM, Koch C, Korbonits M, McLachlan R, New M, Purnell J, Rebar R, Singer F, Vinik A, editors. SourceEndotext [Internet] South Dartmouth (MA): MDText.com, Inc.; 2000-2014. [Google Scholar]
- 16.Kim HH, Schlegel PN. Endocrine manipulation in male infertility. Urol Clin N Am. 2008;35:303–318. doi: 10.1016/j.ucl.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 17.Hwang K, Walters RC, Lipshultz LI. Contemporary concepts in the evaluation and management of male infertility. Nat Rev Urol. 2011;8:86–94. doi: 10.1038/nrurol.2010.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sigman M, Howards SS. Male infertility. In: Walsh PC, Retik AB, Vaughan ED Jr, editors. Campbell’s Textbook of Urology. 7th ed. Philadelphia: WB Saunders Co.; 1998. p. 1287. [Google Scholar]
- 19.Bhasin S, Basaria S. Diagnosis and treatment of hypogonadism in men. Best Prac Res Clin Endocrinol Metab. 2011;25:251–270. doi: 10.1016/j.beem.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 20.Finkle WD, Greenland S, Ridgeway GK, Adams JL, Frasco MA, Cook MB, Fraumeni JF Jr, Hoover RN. Increased risk of non-fatal myocardial infarction following testosterone therapy prescription in men. PLoS One. 2014;9:e85805. doi: 10.1371/journal.pone.0085805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vigen R, O’Donnell CI, Barón AE, Grunwald GK, Maddox TM, Bradley SM, Barqawi A, Woning G, Wierman ME, Plomondon ME, Rumsfeld JS, Ho PM. Association of testosterone therapy with mortality, myocardial infarction, and stroke in men with low testosterone levels. JAMA. 2013;310:1829–1836. doi: 10.1001/jama.2013.280386. [DOI] [PubMed] [Google Scholar]
- 22.Xu L, Freeman G, Cowling BJ, Schooling CM. Testosterone therapy and cardiovascular events among men: a systematic review and meta-analysis of placebo-controlled randomized trials. BMC Med. 2013;11:108. doi: 10.1186/1741-7015-11-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bosland MC. Testosterone treatment is a potent tumor promoter for the rat prostate. Endocrinology. 2014;155:1–5. doi: 10.1210/en.2014-1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Marzo AM, Coffey DS, Nelson WG. New concepts in tissue specificity for prostate cancer and benign prostatic hyperplasia. Urology. 1999;53:29–40. doi: 10.1016/s0090-4295(98)00536-6. [DOI] [PubMed] [Google Scholar]
- 25.McNeal JE. Normal anatomy of the prostate and changes in benign prostatic hypertrophy and carcinoma. Semin Ultrasound CT MRI. 1988;9:329–334. [PubMed] [Google Scholar]
- 26.Qian J, Bostwick DG. The extent and zonal location of prostatic intraepithelial neoplasia and atypical adenomatous hyperplasia: relationship with carcinoma in radical prostatectomy specimens. Pathol Res Pract. 1995;191:860–867. doi: 10.1016/S0344-0338(11)80969-6. [DOI] [PubMed] [Google Scholar]
- 27.Moore RA. Benign hypertrophy of the prostate: a morphological study. J Urol. 1943;50:680–710. [Google Scholar]
- 28.McNeal JE. Origin and evolution of benign prostatic enlargement. Invest Urol. 1978;15:340–345. [PubMed] [Google Scholar]
- 29.Brendler CB, Berry SJ, Ewing LL, McCullough AR, Cochran RC, Strandberg JD, Zirkin BR, Coffey DS, Wheaton LG, Hiler ML, Bordy MJ, Niswender GD, Scott WW, Walsh PC. Spontaneous benign prostatic hyperplasia in the beagle. Age-associated changes in serum hormone levels, and the morphology and secretory function of the canine prostate. J Clin Invest. 1983;71:1114–1123. doi: 10.1172/JCI110861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berg OA. Parenchymatous hypertrophy of the canine prostate gland. Acta Endocrinol. 1958;27:140–154. doi: 10.1530/acta.0.0270140. [DOI] [PubMed] [Google Scholar]
- 31.Ofner P. Effects and metabolism of hormones in normal and neoplastic prostate tissue. Vitam Horm. 1968;26:237–291. doi: 10.1016/s0083-6729(08)60756-6. [DOI] [PubMed] [Google Scholar]
- 32.Zuckerman S, Groome JR. The aetiology of benign enlargement of the prostate in the dog. J Pathol Bacteriol. 1937;44:113–124. [Google Scholar]
- 33.Walsh PC. Experimental approaches to benign prostatic hypertrophy: animal models utilizing the dog, rat, and mouse. In: Grayhack JT, Wilson JD, Scherbenske MJ, editors. Benign Prostatic Hyperplasia, Department of Health, Education, and Welfare Publ No. 76-113, Wash. DC: 1976. pp. 215–222. [Google Scholar]
- 34.Brendler H. Benign prostatic hyperplasia: natural history. In: Grayhack JT, Wilson JD, Scherbenske MJ, editors. Benign Prostatic Hyperplasia, Department of Health, Education, and Welfare Publ No. 76-113, Wash. DC: 1976. pp. 101–103. [Google Scholar]
- 35.Wilson JD. The pathogenesis of benign prostatic hyperplasia. Am J Med. 1980;68:745–756. doi: 10.1016/0002-9343(80)90267-3. [DOI] [PubMed] [Google Scholar]
- 36.Banerjee PP, Banerjee S, Lai JM, Strandberg JD, Zirkin BR, Brown TR. Age-dependent and lobe-specific spontaneous hyperplasia in the brown Norway rat prostate. Biol Reprod. 1998;59:1163–1170. doi: 10.1095/biolreprod59.5.1163. [DOI] [PubMed] [Google Scholar]
- 37.Zuckerman S, McKeown T. The canine prostate in relation to normal and abnormal testicular changes. J Pathol Bacteriol. 1938;46:7–19. [Google Scholar]
- 38.Huggins C, Clark PJ. Quantitative studies of the prostatic secretion. II. The effect of castration and of estrogen injection on the normal and on the hyperplastic prostate glands of dogs. J Exp Med. 1940;72:747–761. doi: 10.1084/jem.72.6.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Juniewicz PE, Berry SJ, Coffey DS, Strandberg JD, Ewing LL. The requirement of the testis in establishing the sensitivity of the canine prostate to develop benign prostatic hyperplasia. J Urol. 1994;152:996–1001. doi: 10.1016/s0022-5347(17)32641-1. [DOI] [PubMed] [Google Scholar]
- 40.Walsh PC, Wilson JD. The induction of prostatic hypertrophy in the dog with androstanediol. J Clin Invest. 1976;57:1093–1097. doi: 10.1172/JCI108353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jacobi GH, Moore RJ, Wilson JD. Studies on the mechanism of 3a-androstanediol-induced growth of the dog prostate. Endocrinology. 1978;102:1748–1755. doi: 10.1210/endo-102-6-1748. [DOI] [PubMed] [Google Scholar]
- 42.DeKlerk DP, Coffey DS, Ewing LL, McDermott IR, Reiner WG, Robinson CH, Scott WW, Strandberg JD, Talalay P, Walsh PC, Wheaton LG, Zirkin BR. Comparison of spontaneous and experimentally induced canine prostatic hyperplasia. J Clin Invest. 1979;64:842–849. doi: 10.1172/JCI109532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shain SA, Nitchuk WM. Testosterone metabolism by the prostate of the aging canine. Mech Ageing Dev. 1979;11:23–35. doi: 10.1016/0047-6374(79)90061-7. [DOI] [PubMed] [Google Scholar]
- 44.Isaacs JT, Coffey DS. Changes in dihydrotestosterone metabolism associated with the development of canine benign prostatic hyperplasia. Endocrinology. 1981;108:445–453. doi: 10.1210/endo-108-2-445. [DOI] [PubMed] [Google Scholar]
- 45.Siiteri PK, Wilson JD. Dihydrotestosterone in prostatic hypertrophy. I. The formation and content of dihydrotestosterone in the hypertrophic prostate of man. J Clin Invest. 1970;49:1737–1745. doi: 10.1172/JCI106391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Albert J, Geller J, Geller S, Lopez D. Prostate concentrations of endogenous androgens by radioimmunoassay. J Steroid Biochem. 1976;7:301–307. doi: 10.1016/0022-4731(76)90131-x. [DOI] [PubMed] [Google Scholar]
- 47.Geller J, Albert J, Lopez D, Geller S, Niwayama G. Comparison of androgen metabolites in benign prostatic hypertrophy (BPH) and normal prostate. J Clin Endocrinol Metab. 1976;43:686–688. doi: 10.1210/jcem-43-3-686. [DOI] [PubMed] [Google Scholar]
- 48.Hammond GL. Endogenous steroid levels in the human prostate from birth to old age: a comparison of normal and diseased tissues. J Endocrinol. 1978;78:7–19. doi: 10.1677/joe.0.0780007. [DOI] [PubMed] [Google Scholar]
- 49.Kim EH, Brockman JA, Andriole GL. The use of 5-alpha reductase inhibitors in the treatment of benign prostatic hyperplasia. Asian J Urol. 2018;5:28–32. doi: 10.1016/j.ajur.2017.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gloyna RE, Siiteri PK, Wilson JD. Dihydrotestosterone in prostatic hypertrophy. II. The formation and content of dihydrotestosterone in the hypertrophic canine prostate and the effect of dihydrotestosterone on prostate growth in the dog. J Clin Invest. 1970;49:1746–1753. doi: 10.1172/JCI106392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lloyd JW, Thomas JA, Mawhinney MH. Androgens and estrogens in the plasma and prostatic tissue of normal dogs and dogs with benign prostatic hypertrophy. Invest Urol. 1975;13:220–222. [PubMed] [Google Scholar]
- 52.Ewing LL, Berry SJ, Higginbottom EG. Dihydrotestosterone concentration of beagle prostatic tissue: effect of age and hyperplasia. Endocrinology. 1983;113:2004–2009. doi: 10.1210/endo-113-6-2004. [DOI] [PubMed] [Google Scholar]
- 53.Herati AS, Kohn TP, Butler PR, Lipshultz LI. Effects of testosterone on benign and malignant conditions of the prostate. Curr Sex Health Rep. 2017;9:65–73. doi: 10.1007/s11930-017-0104-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Banerjee PP, Banerjee S, Dorsey R, Zirkin BR, Brown TR. Age- and lobe-specific responses of the Brown Norway rat prostate to androgen. Biol Reprod. 1994;51:675–684. doi: 10.1095/biolreprod51.4.675. [DOI] [PubMed] [Google Scholar]
- 55.Banerjee PP, Banerjee S, Brown TR. Increased androgen receptor expression correlates with development of age-dependent, lobe-specific spontaneous hyperplasia of the brown Norway rat prostate. Endocrinology. 2001;142:4066–4075. doi: 10.1210/endo.142.9.8376. [DOI] [PubMed] [Google Scholar]
- 56.Yan J, Brown TR. Cell proliferation and expression of cell cycle regulatory proteins that control the G1/S transition are age dependent and lobe specific in the Brown Norway rat model of prostatic hyperplasia. Endocrinology. 2008;149:193–207. doi: 10.1210/en.2007-1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Banerjee S, Banerjee PP, Brown TR. Castration-induced apoptotic cell death in the Brown Norway rat prostate decreases as a function of age. Endocrinology. 2000;141:821–832. doi: 10.1210/endo.141.2.7339. [DOI] [PubMed] [Google Scholar]
- 58.Isaacs JT, Coffey DS. Changes in dihydrotestosterone metabolism associated with the development of canine benign prostatic hyperplasia. Endocrinology. 1981;108:445–453. doi: 10.1210/endo-108-2-445. [DOI] [PubMed] [Google Scholar]
- 59.Marks LS, Mazer NA, Mostaghel E, Hess DL, Dorey FJ, Epstein Jl, Veltri RW, Makarov DV, Partin AW, Bostwick DC, Macairan ML, Nelson PS. Effect of testosterone replacement therapy on prostate tissue in men with late-onset hypogonadism: arandomized controlled trial. JAMA. 2006;296:2351–261. doi: 10.1001/jama.296.19.2351. [DOI] [PubMed] [Google Scholar]
- 60.Jarvis TR, Chughtai B, Kaplan SA. Testosterone and benign prostatic hyperplasia. Asian J Androl. 2015;17:212–216. doi: 10.4103/1008-682X.140966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Prins GS, Birch L. Immunocytochemical analysis of androgen receptor along the ducts of the separate rat prostate lobes after androgen withdrawal and replacement. Endocrinology. 1993;132:169–178. doi: 10.1210/endo.132.1.8419121. [DOI] [PubMed] [Google Scholar]
- 62.Prins GS, Cooke PS, Birch L, Donjacour AA, Yalcinkaya TM, Siiteri PK, Cunha GR. Androgen receptor expression and 5a-reductase activity along the prosimal-distal axis of the rat prostatic duct. Endocrinology. 1992;130:3066–3073. doi: 10.1210/endo.130.5.1572313. [DOI] [PubMed] [Google Scholar]
- 63.Prins GS, Woodham C. Autologous regulation of androgen receptor messenger ribonucleic acid in the separate lobes of the rat prostate gland. Biol Reprod. 1995;53:609–619. doi: 10.1095/biolreprod53.3.609. [DOI] [PubMed] [Google Scholar]
- 64.Culig Z, Hobisch A, Cronauer MV, Radmayr C, Trapman J, Hittmair A, Bartsch G, Klocker H. Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res. 1994;54:5474–5478. [PubMed] [Google Scholar]
- 65.Hobisch A, Eder IE, Putz T, Horniger W, Bartsch G, Klocker H, Culig Z. Interleukin-6 regulates prostate-specific protein expression in prostate carcinoma cells by activation of the androgen receptor. Cancer Res. 1998;58:4640–4645. [PubMed] [Google Scholar]
- 66.Klocker H, Culig Z, Eder IE, Nessler-Menardi C, Hobisch A, Putz T, Bartsch G, Peterziel H, Cato AC. Mechanism of androgen receptor activation and possible implications for chemoprevention trials. Eur Urol. 1999;35:413–419. doi: 10.1159/000019918. [DOI] [PubMed] [Google Scholar]
- 67.Lonergan PE, Tindall DJ. Androgen receptor signaling in prostate cancer development and progression. J Carcinog. 2011;10:20. doi: 10.4103/1477-3163.83937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 69.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–86. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 70.Huggins C, Stephens RC, Hodges CV. The effects of castration on advanced carcinoma of the prostate gland. Arch Surg. 1941;43:209. [Google Scholar]
- 71.Boccon-Gibod L, Fournier G, Bottet P, Marechal JM, Guiter J, Rischman P, Hubert J, Soret JY, Mangin P, Mallo C, Fraysse CE. Flutamide versus orchidectomy in the treatment of metastatic prostate carcinoma. Eur Urol. 1997;32:391–5. discussion 395-6. [PubMed] [Google Scholar]
- 72.Kolvenbag GJ, Nash A. Bicalutamide dosages used in the treatment of prostate cancer. Prostate. 1999;39:47–53. doi: 10.1002/(sici)1097-0045(19990401)39:1<47::aid-pros8>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 73.Todd NF, Lieberman R, Gulley JL, Dahut W, Arlen PM. Prolonged response to nilutamide in a patient with stage D0.5 prostate cancer who previously failed androgen deprivation therapy. Am J Ther. 2005;12:172–4. doi: 10.1097/01.mjt.0000144497.85706.e6. [DOI] [PubMed] [Google Scholar]
- 74.Asmane I, Céraline J, Duclos B, Rob L, Litique V, Barthélémy P, Bergerat JP, Dufour P, Kurtz JE. New strategies for medical management of castration-resistant prostate cancer. Oncology. 2011;80:1–11. doi: 10.1159/000323495. [DOI] [PubMed] [Google Scholar]
- 75.Scher HI, Buchanan G, Gerald W, Butler LM, Tilley WD. Targeting the androgen receptor: improving outcomes for castration-resistant prostate cancer. Endocr Relat Cancer. 2004;11:459–76. doi: 10.1677/erc.1.00525. [DOI] [PubMed] [Google Scholar]
- 76.Thoreson GR, Gayed BA, Chung PH, Raj GV. Emerging therapies in castration resistant prostate cancer. Can J Urol. 2014;21:98–105. [PubMed] [Google Scholar]
- 77.Gilbert DC, Parker C. Docetaxel for the treatment of prostate cancer. Future Oncol. 2005;1:307–14. doi: 10.1517/14796694.1.3.307. [DOI] [PubMed] [Google Scholar]
- 78.Arlen PM, Gulley JL. Docetaxel-based regimens, the standard of care for metastatic androgen-insensitive prostate cancer. Future Oncol. 2005;1:19–22. doi: 10.1517/14796694.1.1.19. [DOI] [PubMed] [Google Scholar]
- 79.Waltering KK, Urbanucci A, Visakorpi T. Androgen receptor (AR) aberrations in castration-resistant prostate cancer. Mol Cell Endocrinol. 2012;360:38–43. doi: 10.1016/j.mce.2011.12.019. [DOI] [PubMed] [Google Scholar]
- 80.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, Asangani IA, Ateeq B, Chun SY, Siddiqui J, Sam L, Anstett M, Mehra R, Prensner JR, Palanisamy N, Ryslik GA, Vandin F, Raphael BJ, Kunju LP, Rhodes DR, Pienta KJ, Chinnaiyan AM, Tomlins SA. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–43. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Beltran H, Yelensky R, Frampton GM, Park K, Downing SR, MacDonald TY, Jarosz M, Lipson D, Tagawa ST, Nanus DM, Stephens PJ, Mosquera JM, Cronin MT, Rubin MA. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur Urol. 2013;63:920–6. doi: 10.1016/j.eururo.2012.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, Beltran H, Abida W, Bradley RK, Vinson J, Cao X, Vats P, Kunju LP, Hussain M, Feng FY, Tomlins SA, Cooney KA, Smith DC, Brennan C, Siddiqui J, Mehra R, Chen Y, Rathkopf DE, Morris MJ, Solomon SB, Durack JC, Reuter VE, Gopalan A, Gao J, Loda M, Lis RT, Bowden M, Balk SP, Gaviola G, Sougnez C, Gupta M, Yu EY, Mostaghel EA, Cheng HH, Mulcahy H, True LD, Plymate SR, Dvinge H, Ferraldeschi R, Flohr P, Miranda S, Zafeiriou Z, Tunariu N, Mateo J, Perez-Lopez R, Demichelis F, Robinson BD, Schiffman M, Nanus DM, Tagawa ST, Sigaras A, Eng KW, Elemento O, Sboner A, Heath EI, Scher HI, Pienta KJ, Kantoff P, de Bono JS, Rubin MA, Nelson PS, Garraway LA, Sawyers CL, Chinnaiyan AM. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;162:454. doi: 10.1016/j.cell.2015.06.053. [DOI] [PubMed] [Google Scholar]
- 83.Kawata H, Ishikura N, Watanabe M, Nishimoto A, Tsunenari T, Aoki Y. Prolonged treatment with bicalutamide induces androgen receptor overexpression and androgen hypersensitivity. Prostate. 2010;70:745–54. doi: 10.1002/pros.21107. [DOI] [PubMed] [Google Scholar]
- 84.Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinänen R, Palmberg C, Palotie A, Tammela T, Isola J, Kallioniemi OP. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995;9:401–6. doi: 10.1038/ng0495-401. [DOI] [PubMed] [Google Scholar]
- 85.Koivisto P, Kononen J, Palmberg C, Tammela T, Hyytinen E, Isola J, Trapman J, Cleutjens K, Noordzij A, Visakorpi T, Kallioniemi OP. Androgen receptor gene amplification: a possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res. 1997;57:314–9. [PubMed] [Google Scholar]
- 86.Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–9. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 87.Carreira S, Romanel A, Goodall J, Grist E, Ferraldeschi R, Miranda S, Prandi D, Lorente D, Frenel JS, Pezaro C, Omlin A, Rodrigues DN, Flohr P, Tunariu N, S de Bono J, Demichelis F, Attard G. Tumor clone dynamics in lethal prostate cancer. Sci Transl Med. 2014;6:254ra125. doi: 10.1126/scitranslmed.3009448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tilley WD, Buchanan G, Hickey TE, Bentel JM. Mutations in the androgen receptor gene are associated with progression of human prostate cancer to androgen independence. Clin Cancer Res. 1996;2:277–85. [PubMed] [Google Scholar]
- 89.Taplin ME, Bubley GJ, Ko YJ, Small EJ, Upton M, Rajeshkumar B, Balk SP. Selection for androgen receptor mutations in prostate cancers treated with androgen antagonist. Cancer Res. 1999;59:2511–5. [PubMed] [Google Scholar]
- 90.Thompson J, Hyytinen ER, Haapala K, Rantala I, Helin HJ, Jänne OA, Palvimo JJ, Koivisto PA. Androgen receptor mutations in high-grade prostate cancer before hormonal therapy. Lab Invest. 2003;83:1709–13. doi: 10.1097/01.lab.0000107262.40402.44. [DOI] [PubMed] [Google Scholar]
- 91.Steinkamp MP, O’Mahony OA, Brogley M, Rehman H, Lapensee EW, Dhanasekaran S, Hofer MD, Kuefer R, Chinnaiyan A, Rubin MA, Pienta KJ, Robins DM. Treatment-dependent androgen receptor mutations in prostate cancer exploit multiple mechanisms to evade therapy. Cancer Res. 2009;69:4434–42. doi: 10.1158/0008-5472.CAN-08-3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chen EJ, Sowalsky AG, Gao S, Cai C, Voznesensky O, Schaefer R, Loda M, True LD, Ye H, Troncoso P, Lis RL, Kantoff PW, Montgomery RB, Nelson PS, Bubley GJ, Balk SP, Taplin ME. Abiraterone treatment in castration-resistant prostate cancer selects for progesterone responsive mutant androgen receptors. Clin Cancer Res. 2015;21:1273–80. doi: 10.1158/1078-0432.CCR-14-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wallén MJ, Linja M, Kaartinen K, Schleutker J, Visakorpi T. Androgen receptor gene mutations in hormone-refractory prostate cancer. J Pathol. 1999;189:559–63. doi: 10.1002/(SICI)1096-9896(199912)189:4<559::AID-PATH471>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 94.Kumar A, Coleman I, Morrissey C, Zhang X, True LD, Gulati R, Etzioni R, Bolouri H, Montgomery B, White T, Lucas JM, Brown LG, Dumpit RF, DeSarkar N, Higano C, Yu EY, Coleman R, Schultz N, Fang M, Lange PH, Shendure J, Vessella RL, Nelson PS. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat Med. 2016;22:369–78. doi: 10.1038/nm.4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jiang Y, Palma JF, Agus DB, Wang Y, Gross ME. Detection of androgen receptor mutations in circulating tumor cells in castration-resistant prostate cancer. Clin Chem. 2010;56:1492–5. doi: 10.1373/clinchem.2010.143297. [DOI] [PubMed] [Google Scholar]
- 96.Azad AA, Volik SV, Wyatt AW, Haegert A, Le Bihan S, Bell RH, Anderson SA, McConeghy B, Shukin R, Bazov J, Youngren J, Paris P, Thomas G, Small EJ, Wang Y, Gleave ME, Collins CC, Chi KN. Androgen receptor gene aberrations in circulating cell-free DNA: biomarkers of therapeutic resistance in castration-resistant prostate cancer. Clin Cancer Res. 2015;21:2315–24. doi: 10.1158/1078-0432.CCR-14-2666. [DOI] [PubMed] [Google Scholar]
- 97.Lallous N, Volik SV, Awrey S, Leblanc E, Tse R, Murillo J, Singh K, Azad AA, Wyatt AW, LeBihan S, Chi KN, Gleave ME, Rennie PS, Collins CC, Cherkasov A. Functional analysis of androgen receptor mutations that confer anti-androgen resistance identified in circulating cell-free DNA from prostate cancer patients. Genome Biol. 2016;17:10. doi: 10.1186/s13059-015-0864-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wyatt AW, Azad AA, Volik SV, Annala M, Beja K, McConeghy B, Haegert A, Warner EW, Mo F, Brahmbhatt S, Shukin R, Le Bihan S, Gleave ME, Nykter M, Collins CC, Chi KN. Genomic alterations in cell-free DNA and enzalutamide resistance in castration-resistant prostate cancer. JAMA Oncol. 2016;2:1598–1606. doi: 10.1001/jamaoncol.2016.0494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Veldscholte J, Ris-Stalpers C, Kuiper GG, Jenster G, Berrevoets C, Claassen E, van Rooij HC, Trapman J, Brinkmann AO, Mulder E. A mutation in the ligand binding domain of the androgen receptor of human LNCaP cells affects steroid binding characteristics and response to anti-androgens. Biochem Biophys Res Commun. 1990;173:534–40. doi: 10.1016/s0006-291x(05)80067-1. [DOI] [PubMed] [Google Scholar]
- 100.Berrevoets CA, Veldscholte J, Mulder E. Effects of antiandrogens on transformation and transcription activation of wild-type and mutated (LNCaP) androgen receptors. J Steroid Biochem Mol Biol. 1993;46:731–6. doi: 10.1016/0960-0760(93)90313-l. [DOI] [PubMed] [Google Scholar]
- 101.Suzuki H, Akakura K, Komiya A, Aida S, Akimoto S, Shimazaki J. Codon 877 mutation in the androgen receptor gene in advanced prostate cancer: relation to antiandrogen withdrawal syndrome. Prostate. 1996;29:153–8. doi: 10.1002/1097-0045(199609)29:3<153::aid-pros2990290303>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 102.Culig Z, Hobisch A, Cronauer MV, Cato AC, Hittmair A, Radmayr C, Eberle J, Bartsch G, Klocker H. Mutant androgen receptor detected in an advanced-stage prostatic carcinoma is activated by adrenal androgens and progesterone. Mol Endocrinol. 1993;7:1541–50. doi: 10.1210/mend.7.12.8145761. [DOI] [PubMed] [Google Scholar]
- 103.Zhao XY, Malloy PJ, Krishnan AV, Swami S, Navone NM, Peehl DM, Feldman D. Glucocorticoids can promote androgen-independent growth of prostate cancer cells through a mutated androgen receptor. Nat Med. 2000;6:939. doi: 10.1038/76287. [DOI] [PubMed] [Google Scholar]
- 104.Steketee K, Timmerman L, Ziel-van der Made AC, Doesburg P, Brinkmann AO, Trapman J. Broadened ligand responsiveness of androgen receptor mutants obtained by random amino acid substitution of H874 and mutation hot spot T877 in prostate cancer. Int J Cancer. 2002;100:309–17. doi: 10.1002/ijc.10495. [DOI] [PubMed] [Google Scholar]
- 105.Suzuki H, Sato N, Watabe Y, Masai M, Seino S, Shimazaki J. Androgen receptor gene mutations in human prostate cancer. J Steroid Biochem Mol Biol. 1993;46:759–65. doi: 10.1016/0960-0760(93)90316-o. [DOI] [PubMed] [Google Scholar]
- 106.Taplin ME, Bubley GJ, Shuster TD, Frantz ME, Spooner AE, Ogata GK, Keer HN, Balk SP. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N Engl J Med. 1995;332:1393–8. doi: 10.1056/NEJM199505253322101. [DOI] [PubMed] [Google Scholar]
- 107.Tan J, Sharief Y, Hamil KG, Gregory CW, Zang DY, Sar M, Gumerlock PH, deVere White RW, Pretlow TG, Harris SE, Wilson EM, Mohler JL, French FS. Dehydroepiandrosterone activates mutant androgen receptors expressed in the androgen-dependent human prostate cancer xenograft CWR22 and LNCaP cells. Mol Endocrinol. 1997;11:450–9. doi: 10.1210/mend.11.4.9906. [DOI] [PubMed] [Google Scholar]
- 108.Hara T, Miyazaki J, Araki H, Yamaoka M, Kanzaki N, Kusaka M, Miyamoto M. Novel mutations of androgen receptor: a possible mechanism of bicalutamide withdrawal syndrome. Cancer Res. 2003;63:149–53. [PubMed] [Google Scholar]
- 109.O’Neill D, Jones D, Wade M, Grey J, Nakjang S, Guo W, Cork D, Davies BR, Wedge SR, Robson CN, Gaughan L. Development and exploitation of a novel mutant androgen receptor modelling strategy to identify new targets for advanced prostate cancer therapy. Oncotarget. 2015;6:26029–40. doi: 10.18632/oncotarget.4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sprenger CC, Plymate SR. The link between androgen receptor splice variants and castration-resistant prostate cancer. Horm Cancer. 2014;5:207–17. doi: 10.1007/s12672-014-0177-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dehm SM, Tindall DJ. Alternatively spliced androgen receptor variants. Endocr Relat Cancer. 2011;18:R183–96. doi: 10.1530/ERC-11-0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mostaghel EA, Marck BT, Plymate SR, Vessella RL, Balk S, Matsumoto AM, Nelson PS, Montgomery RB. Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: induction of steroidogenesis and androgen receptor splice variants. Clin Cancer Res. 2011;17:5913–25. doi: 10.1158/1078-0432.CCR-11-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, Han M, Partin AW, Vessella RL, Isaacs WB, Bova GS, Luo J. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69:16–22. doi: 10.1158/0008-5472.CAN-08-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA, Page ST, Coleman IM, Nguyen HM, Sun H, Nelson PS, Plymate SR. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010;120:2715–30. doi: 10.1172/JCI41824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hörnberg E, Ylitalo EB, Crnalic S, Antti H, Stattin P, Widmark A, Bergh A, Wikström P. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS One. 2011;6:e19059. doi: 10.1371/journal.pone.0019059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Karantanos T, Evans CP, Tombal B, Thompson TC, Montironi R, Isaacs WB. Understanding the mechanisms of androgen deprivation resistance in prostate cancer at the molecular level. Eur Urol. 2015;67:470–9. doi: 10.1016/j.eururo.2014.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cai C, Balk SP. Intratumoral androgen biosynthesis in prostate cancer pathogenesis and response to therapy. Endocr Relat Cancer. 2011;18:R175–82. doi: 10.1530/ERC-10-0339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Logothetis CJ, Gallick GE, Maity SN, Kim J, Aparicio A, Efstathiou E, Lin SH. Molecular classification of prostate cancer progression: foundation for marker-driven treatment of prostate cancer. Cancer Discov. 2013;3:849–61. doi: 10.1158/2159-8290.CD-12-0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chang KH, Li R, Papari-Zareei M, Watumull L, Zhao YD, Auchus RJ, Sharifi N. Dihydrotestosterone synthesis bypasses testosterone to drive castration-resistant prostate cancer. Proc Natl Acad Sci U S A. 2011;108:13728–33. doi: 10.1073/pnas.1107898108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, True LD, Nelson PS. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 2008;68:4447–54. doi: 10.1158/0008-5472.CAN-08-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, Penning TM, Febbo PG, Balk SP. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66:2815–25. doi: 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]
- 122.Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA, Dehm SM. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013;73:483–9. doi: 10.1158/0008-5472.CAN-12-3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Joseph JD, Lu N, Qian J, Sensintaffar J, Shao G, Brigham D, Moon M, Maneval EC, Chen I, Darimont B, Hager JH. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013;3:1020–9. doi: 10.1158/2159-8290.CD-13-0226. [DOI] [PubMed] [Google Scholar]
- 124.Korpal M, Korn JM, Gao X, Rakiec DP, Ruddy DA, Doshi S, Yuan J, Kovats SG, Kim S, Cooke VG, Monahan JE, Stegmeier F, Roberts TM, Sellers WR, Zhou W, Zhu P. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide) Cancer Discov. 2013;3:1030–43. doi: 10.1158/2159-8290.CD-13-0142. [DOI] [PubMed] [Google Scholar]
- 125.Arora VK, Schenkein E, Murali R, Subudhi SK, Wongvipat J, Balbas MD, Shah N, Cai L, Efstathiou E, Logothetis C, Zheng D, Sawyers CL. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell. 2013;155:1309–22. doi: 10.1016/j.cell.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Andersen RJ, Mawji NR, Wang J, Wang G, Haile S, Myung JK, Watt K, Tam T, Yang YC, Bañuelos CA, Williams DE, McEwan IJ, Wang Y, Sadar MD. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell. 2010;17:535–46. doi: 10.1016/j.ccr.2010.04.027. [DOI] [PubMed] [Google Scholar]
- 127.Myung JK, Banuelos CA, Fernandez JG, Mawji NR, Wang J, Tien AH, Yang YC, Tavakoli I, Haile S, Watt K, McEwan IJ, Plymate S, Andersen RJ, Sadar MD. An androgen receptor N-terminal domain antagonist for treating prostate cancer. J Clin Invest. 2013;123:2948–60. doi: 10.1172/JCI66398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Brand LJ, Olson ME, Ravindranathan P, Guo H, Kempema AM, Andrews TE, Chen X, Raj GV, Harki DA, Dehm SM. EPI-001 is a selective peroxisome proliferator-activated receptor-gamma modulator with inhibitory effects on androgen receptor expression and activity in prostate cancer. Oncotarget. 2015;6:3811–24. doi: 10.18632/oncotarget.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Amin KS, Jagadeesh S, Baishya G, Rao PG, Barua NC, Bhattacharya S, Banerjee PP. A naturally derived small molecule disrupts ligand-dependent and ligand-independent androgen receptor signaling in human prostate cancer cells. Mol Cancer Ther. 2014;13:341–52. doi: 10.1158/1535-7163.MCT-13-0478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Liu C, Lou W, Zhu Y, Nadiminty N, Schwartz CT, Evans CP, Gao AC. Niclosamide inhibits androgen receptor variants expression and overcomes enzalutamide resistance in castration-resistant prostate cancer. Clin Cancer Res. 2017;23:323. doi: 10.1158/1078-0432.CCR-13-3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ponnusamy S, Coss CC, Thiyagarajan T, Watts K, Hwang DJ, He Y, Selth LA, McEwan IJ, Duke CB, Pagadala J, Singh G, Wake RW, Ledbetter C, Tilley WD, Moldoveanu T, Dalton JT, Miller DD, Narayanan R. Novel selective agents for the degradation of androgen receptor variants to treat castration-resistant prostate cancer. Cancer Res. 2017;77:6282–6298. doi: 10.1158/0008-5472.CAN-17-0976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Roudier MP, True LD, Higano CS, Vesselle H, Ellis W, Lange P, Vessella RL. Phenotypic heterogeneity of end-stage prostate carcinoma metastatic to bone. Hum Pathol. 2003;34:646–53. doi: 10.1016/s0046-8177(03)00190-4. [DOI] [PubMed] [Google Scholar]
- 133.Liu AY, Roudier MP, True LD. Heterogeneity in primary and metastatic prostate cancer as defined by cell surface CD profile. Am J Pathol. 2004;165:1543–56. doi: 10.1016/S0002-9440(10)63412-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kiskowski MA, Jackson RS 2nd, Banerjee J, Li X, Kang M, Iturregui JM, Franco OE, Hayward SW, Bhowmick NA. Role for stromal heterogeneity in prostate tumorigenesis. Cancer Res. 2011;71:3459–70. doi: 10.1158/0008-5472.CAN-10-2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Drake JM, Graham NA, Lee JK, Stoyanova T, Faltermeier CM, Sud S, Titz B, Huang J, Pienta KJ, Graeber TG, Witte ON. Metastatic castration-resistant prostate cancer reveals intrapatient similarity and interpatient heterogeneity of therapeutic kinase targets. Proc Natl Acad Sci U S A. 2013;110:E4762–9. doi: 10.1073/pnas.1319948110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Massard C, Oulhen M, Le Moulec S, Auger N, Foulon S, Abou-Lovergne A, Billiot F, Valent A, Marty V, Loriot Y, Fizazi K, Vielh P, Farace F. Phenotypic and genetic heterogeneity of tumor tissue and circulating tumor cells in patients with metastatic castration-resistant prostate cancer: a report from the PETRUS prospective study. Oncotarget. 2016;7:55069–55082. doi: 10.18632/oncotarget.10396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Scher HI, Graf RP, Schreiber NA, McLaughlin B, Jendrisak A, Wang Y, Lee J, Greene S, Krupa R, Lu D, Bamford P, Louw JE, Dugan L, Vargas HA, Fleisher M, Landers M, Heller G, Dittamore R. Phenotypic heterogeneity of circulating tumor cells informs clinical decisions between AR signaling inhibitors and taxanes in metastatic prostate cancer. Cancer Res. 2017;77:5687–5698. doi: 10.1158/0008-5472.CAN-17-1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Katoh M. Canonical and non-canonical WNT signaling in cancer stem cells and their niches: Cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity. Int J Oncol. 2017;51:1357–1369. doi: 10.3892/ijo.2017.4129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Humphrey PA. Histological variants of prostatic carcinoma and their significance. Histopathology. 2012;60:59–74. doi: 10.1111/j.1365-2559.2011.04039.x. [DOI] [PubMed] [Google Scholar]
- 140.Liu X, Chen X, Rycaj K, Chao HP, Deng Q, Jeter C, Liu C, Honorio S, Li H, Davis T, Suraneni M, Laffin B, Qin J, Li Q, Yang T, Whitney P, Shen J, Huang J, Tang DG. Systematic dissection of phenotypic, functional, and tumorigenic heterogeneity of human prostate cancer cells. Oncotarget. 2015;6:23959–86. doi: 10.18632/oncotarget.4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Qin J, Liu X, Laffin B, Chen X, Choy G, Jeter CR, Calhoun-Davis T, Li H, Palapattu GS, Pang S, Lin K, Huang J, Ivanov I, Li W, Suraneni MV, Tang DG. The PSA(-/lo) prostate cancer cell population harbors self-renewing long-term tumor-propagating cells that resist castration. Cell Stem Cell. 2012;10:556–69. doi: 10.1016/j.stem.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Banerjee PP, Banerjee S, Sprando RL, Zirkin BR. Regional cellular heterogeneity and DNA synthetic activity in rat ventral prostate during postnatal development. Biol Reprod. 1991;45:773–82. doi: 10.1095/biolreprod45.5.773. [DOI] [PubMed] [Google Scholar]
- 143.Banerjee PP, Banerjee S, Zirkin BR. DNA synthesis occurs throughout the rat ventral prostate during its postnatal development. Biol Reprod. 1993;48:248–51. doi: 10.1095/biolreprod48.2.248. [DOI] [PubMed] [Google Scholar]
- 144.Banerjee S, Banerjee PP, Zirkin BR. Cell proliferation in the dorsal and lateral lobes of the rat prostate during postnatal development. J Androl. 1993;14:310–8. [PubMed] [Google Scholar]
- 145.Banerjee PP, Banerjee S, Tilly KI, Tilly JL, Brown TR, Zirkin BR. Lobe-specific apoptotic cell death in rat prostate after androgen ablation by castration. Endocrinology. 1995;136:4368–76. doi: 10.1210/endo.136.10.7664656. [DOI] [PubMed] [Google Scholar]
- 146.Banerjee PP, Banerjee S, Zirkin BR, Brown TR. Lobe-specific telomerase activity in the intact adult brown Norway rat prostate and its regional distribution within the prostatic ducts. Endocrinology. 1998;139:513–9. doi: 10.1210/endo.139.2.5761. [DOI] [PubMed] [Google Scholar]
- 147.Banerjee S, Banerjee PP, Zirkin BR, Brown TR. Regional expression of transforming growth factor-alpha in rat ventral prostate during postnatal development, after androgen ablation, and after androgen replacement. Endocrinology. 1998;139:3005–13. doi: 10.1210/endo.139.6.6060. [DOI] [PubMed] [Google Scholar]
- 148.Banerjee PP, Banerjee S, Brown TR. Bcl-2 protein expression correlates with cell survival and androgen independence in rat prostatic lobes. Endocrinology. 2002;143:1825–32. doi: 10.1210/endo.143.5.8763. [DOI] [PubMed] [Google Scholar]
- 149.Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, Marotz C, Giannopoulou E, Chakravarthi BV, Varambally S, Tomlins SA, Nanus DM, Tagawa ST, Van Allen EM, Elemento O, Sboner A, Garraway LA, Rubin MA, Demichelis F. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med. 2016;22:298–305. doi: 10.1038/nm.4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Tan HL, Sood A, Rahimi HA, Wang W, Gupta N, Hicks J, Mosier S, Gocke CD, Epstein JI, Netto GJ, Liu W, Isaacs WB, De Marzo AM, Lotan TL. Rb loss is characteristic of prostatic small cell neuroendocrine carcinoma. Clin Cancer Res. 2014;20:890–903. doi: 10.1158/1078-0432.CCR-13-1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Aparicio AM, Shen L, Tapia EL, Lu JF, Chen HC, Zhang J, Wu G, Wang X, Troncoso P, Corn P, Thompson TC, Broom B, Baggerly K, Maity SN, Logothetis CJ. Combined tumor suppressor defects characterize clinically defined aggressive variant prostate cancers. Clin Cancer Res. 2016;22:1520–30. doi: 10.1158/1078-0432.CCR-15-1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, Goodrich MM, Labbé DP, Gomez EC, Wang J, Long HW, Xu B, Brown M, Loda M, Sawyers CL, Ellis L, Goodrich DW. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science. 2017;355:78–83. doi: 10.1126/science.aah4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen CC, Wongvipat J, Ku SY, Gao D, Cao Z, Shah N, Adams EJ, Abida W, Watson PA, Prandi D, Huang CH, de Stanchina E, Lowe SW, Ellis L, Beltran H, Rubin MA, Goodrich DW, Demichelis F, Sawyers CL. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science. 2017;355:84–88. doi: 10.1126/science.aah4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Dardenne E, Beltran H, Benelli M, Gayvert K, Berger A, Puca L, Cyrta J, Sboner A, Noorzad Z, MacDonald T, Cheung C, Yuen KS, Gao D, Chen Y, Eilers M, Mosquera JM, Robinson BD, Elemento O, Rubin MA, Demichelis F, Rickman DS. N-Myc induces an EZH2-mediated transcriptional program driving neuroendocrine prostate cancer. Cancer Cell. 2016;30:563–577. doi: 10.1016/j.ccell.2016.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Bizzarro V, Belvedere R, Milone MR, Pucci B, Lombardi R, Bruzzese F, Popolo A, Parente L, Budillon A, Petrella A. Annexin A1 is involved in the acquisition and maintenance of a stem cell-like/aggressive phenotype in prostate cancer cells with acquired resistance to zoledronic acid. Oncotarget. 2015;6:25076–92. doi: 10.18632/oncotarget.4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zhang K, Guo Y, Wang X, Zhao H, Ji Z, Cheng C, Li L, Fang Y, Xu D, Zhu HH, Gao WQ. WNT/β-catenin directs self-renewal symmetric cell division of hTERT(high) prostate cancer stem cells. Cancer Res. 2017;77:2534–2547. doi: 10.1158/0008-5472.CAN-16-1887. [DOI] [PubMed] [Google Scholar]
- 157.Smith BA, Sokolov A, Uzunangelov V, Baertsch R, Newton Y, Graim K, Mathis C, Cheng D, Stuart JM, Witte ON. A basal stem cell signature identifies aggressive prostate cancer phenotypes. Proc Natl Acad Sci U S A. 2015;112:E6544–52. doi: 10.1073/pnas.1518007112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Bae KM, Dai Y, Vieweg J, Siemann DW. Hypoxia regulates SOX2 expression to promote prostate cancer cell invasion and sphere formation. Am J Cancer Res. 2016;6:1078–88. [PMC free article] [PubMed] [Google Scholar]
- 159.Jeter CR, Liu B, Liu X, Chen X, Liu C, Calhoun-Davis T, Repass J, Zaehres H, Shen JJ, Tang DG. NANOG promotes cancer stem cell characteristics and prostate cancer resistance to androgen deprivation. Oncogene. 2011;30:3833–45. doi: 10.1038/onc.2011.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Russo MV, Esposito S, Tupone MG, Manzoli L, Airoldi I, Pompa P, Cindolo L, Schips L, Sorrentino C, Di Carlo E. SOX2 boosts major tumor progression genes in prostate cancer and is a functional biomarker of lymph node metastasis. Oncotarget. 2016;7:12372–85. doi: 10.18632/oncotarget.6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kerr CL, Hussain A. Regulators of prostate cancer stem cells. Curr Opin Oncol. 2014;26:328–33. doi: 10.1097/CCO.0000000000000080. [DOI] [PubMed] [Google Scholar]