

Abstract
This mini-review article is part of a special issue dedicated to Donald S. Coffey, a pioneer translational research scientist, exemplary mentor, and leader in urologic and urologic oncology research. This article first briefly reflects on life and scientific lessons from Don Coffey. It then reviews the development of two prostate cancer targeting RNA aptamers, xPSM-A9 and xPSM-A10, through in vitro selection for aptamers that bind to the extracellular domain of the Prostate Specific Membrane Antigen (PSMA). These 2’-fluorpyrimidine RNA aptamers selectively bind PSMA on the surface of prostate cancer cells, inhibit PSMA glutamate carboxypeptidase activity, and internalize into PSMA-expressing cancer cells. The truncation of both aptamers, through experimentation as well as logical design, has produced smaller isoforms including A10-3, A10-3.2, A9g and A9L. The larger aptamer isoforms xPSM-A9 and xPSM-A10 are limited to production by in vitro transcription and polyacrylamide gel purification, while smaller isoforms can be generated by chemically synthesis. A series of aptamer conjugates have been developed through chemical crosslinking, complementary annealing strategies, or a combination of both, for the targeting of experimental therapeutics to and into prostate cancer cells. The resulting aptamer conjugates, including nanoparticles and siRNA conjugates, selectively target PSMA-positive prostate cancer cells and xenograft tumors, and demonstrate potent cytotoxic and tumoricidal activity. These experimental therapeutic agents provide a platform for realizing and optimizing the potential of tumor-selective targeting and drug delivery.
Keywords: Aptamers, PSMA, nanoparticles, RNAi therapy, siRNA therapy, prostate cancer
Introduction - a tribute to Dr. Coffey and his lessons on apple pies and scientific research
Most everyone can pinpoint a simple event, or choice, that forever changed life for the better. One day, as a first year graduate student at the Johns Hopkins School of Medicine, I chose to attend a small student group discussion on cancer led by the legendary Donald S. Coffey. Honestly, I did not know much about Don at the time, beyond his biography in the graduate program and some of his lab’s work on telomerase. The moment I walked into the classroom, I could see that he took a different approach to teaching. There, sitting on the edge of a desk, he used a soda can, a slinky, and a large tensegrity toy to explain the complexities of cancer, the nucleus, gene regulation, and cancer biology. No slides. No chalk. No whiteboard. His excitement and curiosity were infectious. I stayed after the session, just to meet with him for a few minutes. The next few hours were a blur. He whisked me across the campus, visiting the labs of his collaborators and colleagues. At one point, we ate dinner. We discussed molecular and cellular biology, cancer, physics, family, and sports. I fell for every one of his magic tricks and never could snatch that quarter out of his hand, which I later learned was one of his favorite tricks. I think I caught the last bus home that night. If you were fortunate enough to know Don Coffey, you have probably had a similar experience. His energy, compassion, curiosity, joy, and knowledge were boundless, and he was always ready and willing to share this with anyone.
If you did not get the chance to meet Don Coffey, I hope that this article and others in this issue can give you a sense of his unique and selfless personality, his dedication to students, science, teaching, learning, creativity, and innovation. While we could fill several issues with stories and lessons from Don Coffey, I have chosen to summarize one lesson he often taught his students and fellows. Then, as he would say, I will get to the science. When I officially joined the Coffey lab, there were four ongoing projects. Alan Meeker was studying telomeres and telomerase, Natsuki Takaha was studying HMGI(Y) and tumor cell heterogeneity, Kathy Bell-McGuinn was studying cancer cell plasticity and histone deacetylase inhibitors, and Dan Krovich was studying the cancer cell nuclear matrix. This diversity of projects echoes a theme that Don kept in his lab: everyone had their own highly independent project. He strongly advocated that each trainee needed ownership of his or her own project, and most importantly, he or she had to have tachycardia (i.e. - excitement) for the science. Don instructed me to avoid experiments for few months, to read as much as possible, and to meet with him regularly to plan and develop my own project from scratch. It was during one of these meetings that I asked him how he, as a mentor and professor, could maintain knowledge and expertise in such a diversity of projects. His humble answer was that he was not an expert in any of these fields. Rather, it was his job was to help each student and fellow become the expert. In this discussion, he made an analogy between scientific training and learning how to bake award-winning apple pies.
Scientific training and apple pies
If you want to become a world-renowned expert in apple pies, here are Don’s suggestions. First, read everything you can on apple pies. Use every available resource. Through this reading, identify the top three apple pie chefs in the world. Read everything that they ever have written. Find out whom they have trained, and read everything that they have written too. Finally, make your first apple pie. Share it with your family and friends and receive their feedback. Next, travel to restaurants and trade shows where you may find these experts, their trainees, and their pies. Talk to the experts and those who have baked with them. Try their pies. Learn the common spices, tastes, and techniques used by these experts. Importantly, also study what makes each chef’s pie unique. Get back to the kitchen and start making your own pies again. Incorporate what you have learned from all experts, and what you have learned within your own kitchen. Enter your pies into baking contests. Value feedback from judges and from your peers. Get back to the kitchen and repeat this process. In a short time period, similar to the time required for a Ph.D. project or fellowship, you will become one of the best apple pie chefs in the world. If not the best, certainly among the best. This is precisely the approach that I implemented with Don Coffey, from the beginning of my research starting, where we worked to develop prostate-targeted RNA aptamers through in vitro selection.
In vitro selection
In the late 1990’s, there were several questions and unmet needs in the management and treatment of prostate cancer (PCa). Many of these remain important today. What distinguishes higher risk patients from lower risk patients with Grey Zone Gleason Scores? Can novel prostate-targeted imaging agents find tumors undetectable by FDG PET? Can prostate-targeted agents selectively deliver therapeutics to tumors for enhanced therapeutic efficacy or reduced off-target toxicity? To address these questions we sought to apply a developing molecular screening technology, called in vitro selection. This technique had the promise to develop new tools capable of detecting and targeting specific receptors or cell types.
In vitro selection applies systematic evolution to a highly diverse library of molecules in order to identify unique molecules that can perform a specific function, such as bind cancer cells. This method begins with a polymer library, where each polymer is composed of consecutive subunits organized in a randomized fashion (Figure 1A). For example, a 10-mer random-sequence RNA aptamer library would be composed of ten consecutive ribonucleotides arranged in random order [1,2]. With four different nucleotides (A, G, C, and U), this library could encompass 410 or 1 × 106 different RNA aptamers. The random arrangement of nucleotides creates a unique folding potential and shape for each aptamer sequence. In the same way, a 10-mer random peptide library would consist of 20 different amino acids, encompassing 2010 or 1 × 1013 different peptides [3-5]. By chance, a very small percentage of these polymers will fold into unique shapes that bind a target of interest, such as a cell surface receptor, with high specificity and affinity. To begin the in vitro selection process, the polymer library and the intended target are incubated together under physiologic (or desired) conditions for a defined time. Through partitioning, the target-bound and non-target-bound polymers are separated (Figure 1B). Target immobilization, for example on magnetic beads, provides a partitioning platform to capture the highest affinity polymers and to wash away weak or non-bound ligands. Systematic evolution iteratively repeats these steps by amplifying the bound ligands and re-selecting for target binding, thereby increasing the selective pressures in each round of selection, and evolving the highest affinity binders from the library. RT-PCR or PCR can amplify target-bound RNA and DNA aptamers, while peptide libraries require an associated genomic template within a phage vector or RNA template bound to a ribosome [6]. Sequencing then identifies common polymers and consensus polymer families in the selected pool. Additional in vitro selection techniques, such as counter-selection, competitive elution, and target dilution offer a means to increase ligand specificity and affinity.
Figure 1.
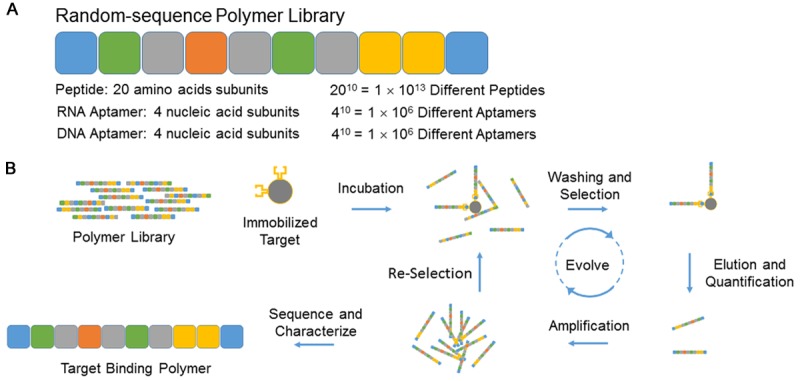
In vitro selection of random sequence polymer libraries. A. Schematic of a 10-mer random sequence polymer library with calculated diversities for peptide and aptamer libraries. B. Schematic of polymer library selection. Not drawn to scale.
Development of PSMA targeting RNA aptamers
In an effort to develop ligands capable of binding PCa cells for imaging agents and therapeutic delivery, we elected to generate RNA aptamers. RNA aptamers embody many attractive properties as targeting ligands: they are non-immunogenic, can be chemically synthesized under GMP guidelines, they typically bind with nanomolar or better affinities, they can be modified to include chemical linkers for conjugation to therapeutic or imaging agents, and their short circulating half-lives are suitable for radiologic imaging [7,8]. We chose PSMA (Prostate Specific Membrane Antigen) as the target for aptamer selection because previous studies had proven the feasibility of detecting metastatic PCa tumors in humans via anti-PSMA antibody imaging [9]. PSMA is a type II integral membrane protein and glutamate carboxypeptidase that is highly over-expressed on the surface of PCa cells [10]. The bulk of PSMA protein, 706 of 750 amino acids, is extracellular, providing a large surface for ligand binding. Estimates predict that PCa cells express between 100,000 to 1,000,000 copies of PSMA molecules per cell [11]. Moreover, PSMA naturally internalizes PSMA-bound ligands into cells [12].
To perform in vitro selection against PSMA, we first generated a fusion protein encoding the extracellular portion of PSMA using a baculovirus and insect cell system [13]. We intentionally applied a eukaryotic model, and a secreted fusion protein, because PSMA requires post-translational glycosylation for proper protein folding and enzymatic activity [14]. The purified PSMA fusion protein, xPSM, exhibited glutamate carboxypeptidase activity comparable to native human PSMA [13]. Native protein folding is often crucial for successful in vitro selections, because aptamers or peptides can bind within active sites or structural regions, rather than short amino-acids motifs. To prepare for aptamer selection, we removed the fusion protein tags and immobilized the xPSM protein onto commercial magnetic beads.
The dsDNA template for our aptamer library consisted of a 40-mer random sequence region, flanked by universal regions with sequences for PCR priming and T7 in vitro transcription. Our aptamer transcriptions applied 2’-Fluorpyrimidine, rather than 2’-hydroxypyrimidine, bases to produce nuclease stabilized RNA aptamer transcripts. 2’-modified nucleotides are crucial for the development of pharmacologic aptamers because they provide sufficient serum stability for intravenous injections [15]. Modified nucleotides also contribute to aptamer folding structures. Therefore, they are included throughout the in vitro selection process. Our random sequence region offered a potential diversity of 440 or 1 × 1024 different aptamers. It is not feasible to apply the full diversity of this library, because it would require over 50 milligrams of RNA. Instead we applied one nanomole of the library, representing a potential diversity of 6 × 1014 different RNA aptamers. To select PSMA-binding aptamers, we incubated the library with xPSM-coated magnetic beads for magnetic capture, washing, and purification [13]. We quantified the number of xPSM-bound aptamers for each round of in vitro selection by real time quantitative RT-PCR and determined library enrichment, relative to a non-selected library or uncoated magnetic beads. Six rounds of in vitro selection produced two independent PSMA-binding aptamers, xPSM-A9 and xPSM-A10 (Table 1). The xPSM-A10 aptamer binds native PSMA and competitively inhibits its glutamate carboxypeptidase enzymatic activity with a Ki of 11.9 nM, indicating active site binding. The xPSM-A9 aptamer binds human PSMA and non-competitively inhibits its glutamate carboxypeptidase activity with a Ki of 1.1 nM, suggesting an aptamer binding site away from the active site.
Table 1.
Characteristics of PSMA aptamers and truncated aptamers
| Aptamer | Sequence | Bases | Affinity | Ref |
|---|---|---|---|---|
| 40N7 Library | GGGAGGACGAUGCGGNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNCAGACGACUCGCCCGA | 71 | N.A. | [13] |
| xPSM-A9 | GGGAGGACGAUGCGGACCGAAAAAGACCUGACUUCUAUACUAAGUCUACGUUCCCAGACGACUCGCCCGA | 70 | 1.1 nM (Ki) | [13,16] |
| 110 nM (Kd SBA) | ||||
| xPSM-A10 | GGGAGGACGAUGCGGAUCAGCCAUGUUUACGUCACUCCUUGUCAAUCCUCAUCGGCAGACGACUCGCCCGA | 72 | 11.9 nM (Ki) | [13,17] |
| 1.5 nM (Kd CBA) | ||||
| A10-3 | GGGAGGACGAUGCGGAUCAGCCAUGUUUACGUCACUCCUUGUCAAUCCUCAUCGGC | 56 | 1.1 nM (Ki) | [13,17] |
| 0.5 nM (Kd CBA) | ||||
| A10-3.2 | GGGAGGACGAUGCGGAUCAGCCAUGUUUACGUCACUCCU | 39 | 2.9 nM (Kd CBA) | [17] |
| A9g | GGGACGGAAAAAGACCUGACUUCUAUACUAAGUCUACGUUCCC | 43 | 130 nM (Kd SBA) | [16] |
| 5 nM (Kd SPR) | ||||
| A9L | GGGCCGAAAAAGACCUGACUUCUAUACUAAGUCUACGUCCC | 41 | N.D. | [16] |
Ki, Determined by enzymatic Inhibition assay; SBA, Saturation Filter Binding Assay; CBA, Cell Binding Assay; SPR, Surface Plasmon Resonance.
PSMA aptamer truncation: identifying the smallest functional unit
RNA aptamers fold into unique three-dimensional shapes for target binding. Specific hairpin structures within the sequence may facilitate target binding, while other regions mays be dispensable. Consequently, it common practice to truncate aptamers, from either end, to identify the minimal functional unit. In our truncation studies, xPSM-A9 lost the ability to inhibit PSMA enzymatic activity after removal of ten or more nucleotides from the 3’ end [13]. In later studies, Rockey, Hernandez, Huang, Giangrande and colleagues used rational truncation, based on predicted aptamer folding and protein/RNA docking algorithms, to identify a smaller 41 nucleotide isoform of A9, termed A9g [16]. The A9g aptamer retains the ability to inhibit PSMA enzymatic activity. Saturation filter binding studies determined an affinity of 130 nM for A9g, while Surface Plasmon Resonance indicated an affinity of 5 nM (Table 1). We truncated the A10 aptamer, from the 3’-end, to identify a smaller 56-nucleotide isoform, A10-3, that competitively inhibits PSMA enzymatic activity with a Ki of 20.5 nM [13]. Removal of 20 or more nucleotides from the 3’-end abolished the ability of A10 to inhibit PSMA enzymatic activity. Dassie and colleagues later identified a smaller isoform of A10-3, termed A10-3.2, which replaced the 3’-end of A10-3 with a therapeutic siRNA duplex [17]. The 39-nucleotide A10-3.2 aptamer binds PSMA-positive cells with an affinity of 2.9 nM, while still lacking the ability to inhibit PSMA enzymatic activity [18]. These studies suggest that A10-3 binds PSMA near the glutamate carboxypeptidase active site, with the 3’end competitively inhibiting substrate binding. Upon further 3’-truncation, competitive enzymatic inhibition activity is lost without affecting PSMA binding activity.
Applications of PSMA aptamers to nanotechnology
The chemical synthesis of 2’-modified RNA aptamers has been limited to approximately 60 or fewer nucleotides. Therefore, original syntheses focused on the 56 nucleotide A10-3 aptamer, with a modified 5’-terminus to include a primary hexyl-amine for chemical crosslinking [13]. Farokhzad, Langer, and colleagues first demonstrated the use of aptamers in nanoparticle (NP) targeting by conjugating the 5’-end of A10-3 to poly (D,L-lactic-co-glycolic acid) (PLGA) controlled-release NPs (Figure 2) [19]. A10-3 conjugation significantly increased particle binding, uptake, and drug delivery to PSMA-positive PCa cells, in vitro, and PSMA positive tumors, in vivo [19-21]. This approach has been successful with other NP formulations including A10-3 targeted quantum dots [22], superparamagnetic iron oxide NPs [23], and ChemoRad NPs [24]. The A9 aptamer, which is too large for chemical synthesis, has also been successful in NP targeting through a clever annealing strategy. Javier and colleagues added a 3’-extension to in vitro transcribed A9 aptamers, providing a means to anneal the aptamers to a complementary oligonucleotide sequence attached to the surface of gold NPs (Figure 2) [25]. Kim and colleagues used a similar approach to generate A9-targeted gold NPs that enhanced the detection of PSMA-positive PCa cells by CT imaging and delivered cytotoxic doxorubicin payloads [26]. This strategy of extending and hybridizing the A9 aptamers to complementary oligonucleotides has also been successful with drug-loaded liposomes [27]. Collectively, these studies support the potential utility of PSMA aptamers to deliver NPs and NP payloads to PCa cells and tumors.
Figure 2.
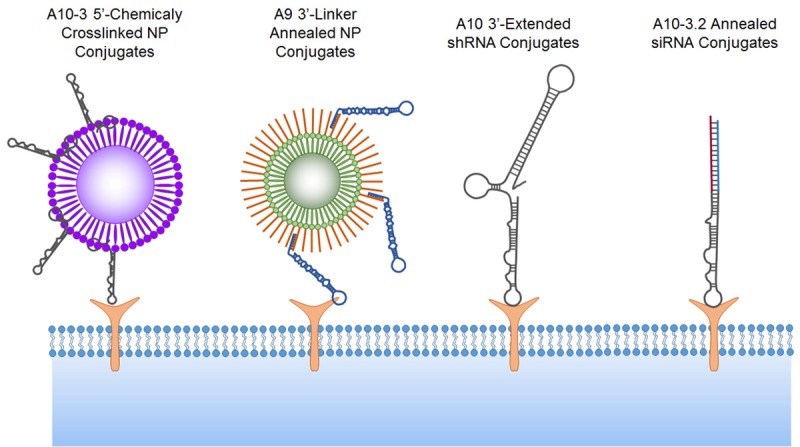
PSMA Aptamer conjugates. Schematic of aptamer-nanoparticle, aptamer-shRNA, and aptamer-siRNA conjugates. Not drawn to scale. Schematic emphasizes conjugation strategies and aptamers truncations and is not representative of PSMA binding sites. NP, nanoparticle.
Applications of PSMA aptamers to RNA interference therapy
RNA interference (RNAi) is a natural biologic process of gene regulation mediated by small RNAs that are complementary to endogenous protein-coding mRNAs. The RNA-induced silencing complex (RISC) recognizes these cytoplasmic double-stranded RNA complexes and suppresses gene expression by direct mRNA cleavage, mRNA destabilization, or inhibition of mRNA translation. RNAi therapeutics exploit this pathway by applying artificially engineered small interfering RNAs (siRNAs) or natural microRNAs (miRNAs) that target and inhibit protein coding genes that drive pathologic process, such as oncogenes in cancer or viral gene products in infectious disease [28,29]. RNAi therapy holds great promise in cancer because it has the ability to inhibit traditionally undrugable targets, such as transcription factors or targets within complex protein-protein interactions [30]. In spite of this, the clinical translation of RNAi therapy has been challenging, partially because it is difficult to systemically deliver a sufficient amount RNAi therapeutics deep into tissues, such as tumors [31].
It was soon recognized that RNA aptamers could serve as unique RNA-based tools for the delivery of siRNAs and miRNAs to specific cell types. The first applications of this concept applied the xPSM-A9 and xPSM-A10 aptamers and PSMA-positive prostate cancer models [32,33]. PSMA is an ideal target for siRNA and miRNA delivery because it encompasses tissue-selective expression and the ability to internalize bound ligands into cancer cells. Chu, Ellington, Levy, and colleagues applied biotin-streptavidin chemistry to conjugate siRNAs to xPSM-A9. The resulting aptamer-siRNA chimera (AsiC) selectively knocked-down target genes in PSMA-positive cells [32]. In a different strategy, McNamara, Sullenger, Giangrande, and colleagues extended the xPSM-A0 aptamer to include a 3’-terminal sequence complementary to Plk-1 or Bcl-2 siRNAs. After annealing the antisense siRNA, the resulting AsiCs demonstrated selective targeting, internalization, and target gene knockdown with efficient cell killing and tumor lysis of PSMA-expressing cells and tumors [33].
The second generation AsiCs successfully reduced the PSMA aptamer size and pursued new siRNA targets for cancer therapy. Wullner and colleagues extended the shorter A10-3 aptamer, from the 3’-end, to include short hairpin RNAs (shRNAs). This approach produced a single contiguous AsiC and eliminated the need for siRNA annealing (Figure 2) [34]. Monovalent and bivalent A10-3 AsiCs targeting EEF2 showed selective PSMA-positive cell binding, gene knock down, and cytotoxicity. Using a similar strategy, our laboratory developed a monovalent A10-3 AsiC for the targeted knockdown of DNA repair machinery in prostate tumors. These AsiCs knocked down the catalytic subunit of DNA-PK, PRKDC, and sensitized PSMA-positive PCa cells and established xenograft tumors to ionizing radiation therapy [35].
These initial AsiCs were too large for chemical synthesis, and therefore production required in vitro transcription and purification. Size-based purification is an important step in generating in vitro transcribed 2’-Fluoro- and 2’-Amino-modified RNA aptamers. The 2’-modified transcription reactions generate a significant amount of incomplete products, which may compete for target binding and thereby inhibit the delivery of 3’-linked siRNAs and shRNAs. Through run-off transcription and polyacrylamide gel purification, full-length RNA aptamer transcripts can be generated in the laboratory. This multistep process can be difficult and expensive to perform on a large scale, and it presents challenges for generating sufficiently pure material for clinical applications. In light of this, the field has moved towards chemically synthesized AsiC. Dassie, Giangrande, and colleagues engineered a series of AsiCs based on the truncated A10-3.2 aptamer, with additional modifications for efficient RNAi processing (Figure 2) [17]. The resulting AsiCs were compatible with large-scale chemical synthesis, providing sufficient quantities for in vivo testing. Intraperitoneal administration of the optimized A10-3.2-Plk AsiC induced tumor volume reduction in mice bearing established PSMA-positive 22Rv1 tumors, while PSMA negative tumors were unaffected. In a separate approach, we applied a three-part annealing strategy to assemble AsiCs from the chemically synthesized A10-3 aptamer, an antisense siRNA targeting DNA-PK, and a complementary sense siRNA that bridged the anti-sense siRNA and 3’-end of the aptamer [36]. Intravenous administration of these annealed AsiCs selectively sensitized established PSMA-positive tumors to ionizing radiation therapy. These applications of AsiC to RNAi therapy are not unique to PSMA aptamers and PCa models. Additional aptamers, targeting multiple different cell surface targets, have been effective delivery vehicles for siRNAs, shRNAs, miRNAs, and RNAi-nanocomplexes in multiple disease models [37].
What is next for PSMA aptamers?
In summary, PSMA-targeted RNA aptamers have shown promise as potential targeting agents for the treatment and management of PCa. Recent work, by multiple laboratories, have advanced these reagents through truncation, extension, and modification. There is still potential for improvement through further modification of 2’-purine subunits. Additional modifications or sequence substitutions may improve aptamer folding, stability, or conjugation potential. New aptamer selection techniques or libraries may also identify new PSMA targeting aptamers with superior size or affinity, considering that only 0.00000006% of the potential 1024 different aptamers were applied to the original xPSM in vitro selection. New approaches for RNA synthesis or nucleotide modification may reduce the cost of aptamer synthesis, or simplify their GMP production, and thereby accelerate their use or translation into the clinic.
There are still unanswered questions remaining in PSMA aptamer research. Detailed structural studies of aptamer folding and docking remain unsolved. Crystallography requires highly purified aptamers, and protein, with homogenous RNA folding and three-dimensional structures for crystallization. Unfortunately, these crystals have been challenging to obtain, possibly due to heterogeneous folding confirmations of the A10-3 aptamer. The detailed mechanisms of AsiC internalization, endosomal escape, and processing for efficient RNAi also remain unexplained. Finally, it is important to acknowledge that PSMA aptamers are only a small part of the larger effort to develop PSMA-targeted therapeutics and imaging reagents for PCa patients. Small-molecule-based PSMA-targeted reagents, which recognize the carboxypeptidase active site of PSMA, show great promise as PCa imaging and therapeutic agents in pre-clinical models and in human clinical trials [38,39]. PSMA-targeting antibodies, including the widely applied J591, have also shown great promise in pre-clinical and clinical studies of PCa imaging and targeted therapeutics [40,41]. Each of these targeting agents encompass unique advantages and disadvantages to PSMA-targeted imaging or therapeutic agents. It will be important to understand the advantages and disadvantages of each of these PSMA targeting agents, and their conjugates, in order to maximize their potential benefit for men suffering from PCa.
Don Coffey - the legacy
Don Coffey was a powerful inspirational, supportive, and driving force in my laboratory training, in my research as a faculty member, and in my life. While it will never be possible to emulate him, we all know that his stories, teachings, and research will continue on forever, inspiring the next generation of scientists and apple pie chefs.
Acknowledgements
This article is dedicated to Donald and Eula Coffey. I am thankful for their boundless love, support, and encouragement. I am also thankful for the Society of Basic Urological Research and the American Journal of Clinical and Experimental Urology for providing this opportunity to honor Don Coffey and to acknowledge his research and leadership. This work was supported by the National Institutes of Health (5P50CA058236, P30CA006973).
Disclosure of conflict of interest
None.
References
- 1.Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 2.Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 3.Scott JK, Smith GP. Searching for peptide ligands with an epitope library. Science. 1990;249:386–390. doi: 10.1126/science.1696028. [DOI] [PubMed] [Google Scholar]
- 4.Cwirla SE, Peters EA, Barrett RW, Dower WJ. Peptides on phage: a vast library of peptides for identifying ligands. Proc Natl Acad Sci U S A. 1990;87:6378–6382. doi: 10.1073/pnas.87.16.6378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Devlin JJ, Panganiban LC, Devlin PE. Random peptide libraries: a source of specific protein binding molecules. Science. 1990;249:404–406. doi: 10.1126/science.2143033. [DOI] [PubMed] [Google Scholar]
- 6.Mattheakis LC, Bhatt RR, Dower WJ. An in vitro polysome display system for identifying ligands from very large peptide libraries. Proc Natl Acad Sci U S A. 1994;91:9022–9026. doi: 10.1073/pnas.91.19.9022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keefe AD, Pai S, Ellington A. Aptamers as therapeutics. Nat Rev Drug Discov. 2010;9:537–550. doi: 10.1038/nrd3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou J, Rossi J. Aptamers as targeted therapeutics: current potential and challenges. Nat Rev Drug Discov. 2017;16:440. doi: 10.1038/nrd.2017.86. [DOI] [PubMed] [Google Scholar]
- 9.Sodee DB, Malguria N, Faulhaber P, Resnick MI, Albert J, Bakale G. Multicenter ProstaScint imaging findings in 2154 patients with prostate cancer. The ProstaScint imaging centers. Urology. 2000;56:988–993. doi: 10.1016/s0090-4295(00)00824-4. [DOI] [PubMed] [Google Scholar]
- 10.Israeli RS, Powell CT, Fair WR, Heston WD. Molecular cloning of a complementary DNA encoding a prostate-specific membrane antigen. Cancer Res. 1993;53:227–230. [PubMed] [Google Scholar]
- 11.Ma D, Hopf CE, Malewicz AD, Donovan GP, Senter PD, Goeckeler WF, Maddon PJ, Olson WC. Potent antitumor activity of an auristatin-conjugated, fully human monoclonal antibody to prostate-specific membrane antigen. Clin Cancer Res. 2006;12:2591–2596. doi: 10.1158/1078-0432.CCR-05-2107. [DOI] [PubMed] [Google Scholar]
- 12.Liu H, Rajasekaran AK, Moy P, Xia Y, Kim S, Navarro V, Rahmati R, Bander NH. Constitutive and antibody-induced internalization of prostate-specific membrane antigen. Cancer Res. 1998;58:4055–4060. [PubMed] [Google Scholar]
- 13.Lupold SE, Hicke BJ, Lin Y, Coffey DS. Identification and characterization of nuclease-stabilized RNA molecules that bind human prostate cancer cells via the prostate-specific membrane antigen. Cancer Res. 2002;62:4029–4033. [PubMed] [Google Scholar]
- 14.Ghosh A, Heston WD. Effect of carbohydrate moieties on the folate hydrolysis activity of the prostate specific membrane antigen. Prostate. 2003;57:140–151. doi: 10.1002/pros.10289. [DOI] [PubMed] [Google Scholar]
- 15.Pagratis NC, Bell C, Chang YF, Jennings S, Fitzwater T, Jellinek D, Dang C. Potent 2’-amino-, and 2’-fluoro-2’-deoxyribonucleotide RNA inhibitors of keratinocyte growth factor. Nat Biotechnol. 1997;15:68–73. doi: 10.1038/nbt0197-68. [DOI] [PubMed] [Google Scholar]
- 16.Rockey WM, Hernandez FJ, Huang SY, Cao S, Howell CA, Thomas GS, Liu XY, Lapteva N, Spencer DM, McNamara JO, Zou X, Chen SJ, Giangrande PH. Rational truncation of an RNA aptamer to prostate-specific membrane antigen using computational structural modeling. Nucleic Acid Ther. 2011;21:299–314. doi: 10.1089/nat.2011.0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dassie JP, Liu XY, Thomas GS, Whitaker RM, Thiel KW, Stockdale KR, Meyerholz DK, McCaffrey AP, McNamara JO 2nd, Giangrande PH. Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors. Nat Biotechnol. 2009;27:839–849. doi: 10.1038/nbt.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dassie JP, Hernandez LI, Thomas GS, Long ME, Rockey WM, Howell CA, Chen Y, Hernandez FJ, Liu XY, Wilson ME, Allen LA, Vaena DA, Meyerholz DK, Giangrande PH. Targeted inhibition of prostate cancer metastases with an RNA aptamer to prostate-specific membrane antigen. Mol Ther. 2014;22:1910–1922. doi: 10.1038/mt.2014.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Farokhzad OC, Jon S, Khademhosseini A, Tran TN, Lavan DA, Langer R. Nanoparticle-aptamer bioconjugates: a new approach for targeting prostate cancer cells. Cancer Res. 2004;64:7668–7672. doi: 10.1158/0008-5472.CAN-04-2550. [DOI] [PubMed] [Google Scholar]
- 20.Dhar S, Gu FX, Langer R, Farokhzad OC, Lippard SJ. Targeted delivery of cisplatin to prostate cancer cells by aptamer functionalized Pt(IV) prodrug-PLGA-PEG nanoparticles. Proc Natl Acad Sci U S A. 2008;105:17356–17361. doi: 10.1073/pnas.0809154105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng J, Teply BA, Sherifi I, Sung J, Luther G, Gu FX, Levy-Nissenbaum E, Radovic-Moreno AF, Langer R, Farokhzad OC. Formulation of functionalized PLGA-PEG nanoparticles for in vivo targeted drug delivery. Biomaterials. 2007;28:869–876. doi: 10.1016/j.biomaterials.2006.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bagalkot V, Zhang L, Levy-Nissenbaum E, Jon S, Kantoff PW, Langer R, Farokhzad OC. Quantum dot-aptamer conjugates for synchronous cancer imaging, therapy, and sensing of drug delivery based on bi-fluorescence resonance energy transfer. Nano Lett. 2007;7:3065–3070. doi: 10.1021/nl071546n. [DOI] [PubMed] [Google Scholar]
- 23.Wang AZ, Bagalkot V, Vasilliou CC, Gu F, Alexis F, Zhang L, Shaikh M, Yuet K, Cima MJ, Langer R, Kantoff PW, Bander NH, Jon S, Farokhzad OC. Superparamagnetic iron oxide nanoparticle-aptamer bioconjugates for combined prostate cancer imaging and therapy. ChemMedChem. 2008;3:1311–1315. doi: 10.1002/cmdc.200800091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang AZ, Yuet K, Zhang L, Gu FX, Huynh-Le M, Radovic-Moreno AF, Kantoff PW, Bander NH, Langer R, Farokhzad OC. ChemoRad nanoparticles: a novel multifunctional nanoparticle platform for targeted delivery of concurrent chemoradiation. Nanomedicine (Lond) 2010;5:361–368. doi: 10.2217/nnm.10.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Javier DJ, Nitin N, Levy M, Ellington A, Richards-Kortum R. Aptamer-targeted gold nanoparticles as molecular-specific contrast agents for reflectance imaging. Bioconjug Chem. 2008;19:1309–1312. doi: 10.1021/bc8001248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim D, Jeong YY, Jon S. A drug-loaded aptamer-gold nanoparticle bioconjugate for combined CT imaging and therapy of prostate cancer. ACS Nano. 2010;4:3689–3696. doi: 10.1021/nn901877h. [DOI] [PubMed] [Google Scholar]
- 27.Baek SE, Lee KH, Park YS, Oh DK, Oh S, Kim KS, Kim DE. RNA aptamer-conjugated liposome as an efficient anticancer drug delivery vehicle targeting cancer cells in vivo. J Control Release. 2014;196:234–242. doi: 10.1016/j.jconrel.2014.10.018. [DOI] [PubMed] [Google Scholar]
- 28.Tang G. siRNA and miRNA: an insight into RISCs. Trends Biochem Sci. 2005;30:106–114. doi: 10.1016/j.tibs.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 29.Sridharan K, Gogtay NJ. Therapeutic nucleic acids: current clinical status. Br J Clin Pharmacol. 2016;82:659–672. doi: 10.1111/bcp.12987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dang CV, Reddy EP, Shokat KM, Soucek L. Drugging the ‘undruggable’ cancer targets. Nat Rev Cancer. 2017;17:502–508. doi: 10.1038/nrc.2017.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aagaard L, Rossi JJ. RNAi therapeutics: principles, prospects and challenges. Adv Drug Deliv Rev. 2007;59:75–86. doi: 10.1016/j.addr.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chu TC, Twu KY, Ellington AD, Levy M. Aptamer mediated siRNA delivery. Nucleic Acids Res. 2006;34:e73. doi: 10.1093/nar/gkl388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McNamara JO 2nd, Andrechek ER, Wang Y, Viles KD, Rempel RE, Gilboa E, Sullenger BA, Giangrande PH. Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nat Biotechnol. 2006;24:1005–1015. doi: 10.1038/nbt1223. [DOI] [PubMed] [Google Scholar]
- 34.Wullner U, Neef I, Eller A, Kleines M, Tur MK, Barth S. Cell-specific induction of apoptosis by rationally designed bivalent aptamer-siRNA transcripts silencing eukaryotic elongation factor 2. Curr Cancer Drug Targets. 2008;8:554–565. doi: 10.2174/156800908786241078. [DOI] [PubMed] [Google Scholar]
- 35.Ni X, Zhang Y, Ribas J, Chowdhury WH, Castanares M, Zhang Z, Laiho M, DeWeese TL, Lupold SE. Prostate-targeted radiosensitization via aptamer-shRNA chimeras in human tumor xenografts. J Clin Invest. 2011;121:2383–2390. doi: 10.1172/JCI45109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ni X, Zhang Y, Zennami K, Castanares M, Mukherjee A, Raval RR, Zhou H, DeWeese TL, Lupold SE. Systemic administration and targeted radiosensitization via chemically synthetic aptamer-siRNA chimeras in human tumor xenografts. Mol Cancer Ther. 2015;14:2797–2804. doi: 10.1158/1535-7163.MCT-15-0291-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kruspe S, Giangrande PH. Aptamer-siRNA chimeras: discovery, progress, and future prospects. Biomedicines. 2017:5. doi: 10.3390/biomedicines5030045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lutje S, Heskamp S, Cornelissen AS, Poeppel TD, van den Broek SA, Rosenbaum-Krumme S, Bockisch A, Gotthardt M, Rijpkema M, Boerman OC. PSMA ligands for radionuclide imaging and therapy of prostate cancer: clinical status. Theranostics. 2015;5:1388–1401. doi: 10.7150/thno.13348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maurer T, Eiber M, Schwaiger M, Gschwend JE. Current use of PSMA-PET in prostate cancer management. Nat Rev Urol. 2016;13:226–235. doi: 10.1038/nrurol.2016.26. [DOI] [PubMed] [Google Scholar]
- 40.Vallabhajosula S, Nikolopoulou A, Jhanwar YS, Kaur G, Tagawa ST, Nanus DM, Bander NH, Goldsmith SJ. Radioimmunotherapy of metastatic prostate cancer with (1)(7)(7)Lu-DOTAhuJ591 anti prostate specific membrane antigen specific monoclonal antibody. Curr Radiopharm. 2016;9:44–53. doi: 10.2174/1874471008666150313114005. [DOI] [PubMed] [Google Scholar]
- 41.Nanus DM, Milowsky MI, Kostakoglu L, Smith-Jones PM, Vallabahajosula S, Goldsmith SJ, Bander NH. Clinical use of monoclonal antibody HuJ591 therapy: targeting prostate specific membrane antigen. J Urol. 2003;170:S84–88. doi: 10.1097/01.ju.0000095151.97404.7c. discussion S88-89. [DOI] [PubMed] [Google Scholar]